")
Back to Journals » OncoTargets and Therapy » Volume 12
CXCR4 antagonism in combination with IDO1 inhibition weakens immune suppression and inhibits tumor growth in mouse breast cancer bone metastases
Authors Zhang J, Pang Y, Xie T, Zhu L
Received 7 January 2019
Accepted for publication 13 May 2019
Published 28 June 2019 Volume 2019:12 Pages 4985—4992
DOI https://doi.org/10.2147/OTT.S200643
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Leo Jen-Liang Su
Jian Zhang,1 Yanxia Pang,2 Tao Xie,1 Liulong Zhu1
1Department of Orthopaedics, Hangzhou First People’s Hospital, Zhejiang University School of Medicine, Hangzhou 310009, People’s Republic of China; 2Taizhou University Medical School, Linhai 318000, People’s Republic of China
Purpose: To investigate whether inhibition of the CXCL12/CXCR4 axis or IDO1 could produce antitumor effects in a metastasized breast cancer immunocompetent animal model.
Methods: Breast cancer metastasis models were established in mice. CXCR4 inhibitor and IDO1 inhibitor were used to evaluate the anticancer effects.
Results: Combination treatment using the CXCR4 antagonist AMD3465 and the IDO1 inhibitor D1MT successfully delayed the progression of breast cancer bone metastases. AMD3465 reduced the number of intratumoral regulatory T-cells (Tregs) and myeloid-derived suppressor cells (MDSCs), while D1MT facilitated the antitumor effects of intratumoral CD8+ T-cells. IDO1 inhibition elevated the expression of perforin, granzyme-B, and IFN-γ in CD8+ T-cells, and AMD3465 treatment weakened the potential immune suppressive effects of Tregs and MDSCs. As a result, combination treatment significantly prolonged tumor-bearing mouse survival in two metastasis models, and these antitumor effects relied on overexpression of indoleamine 2, 3-dioxygenase 1 (IDO1), an enzyme that modulates the immune response and impairs immune attack in ovarian cancers CXCR3+ CD8+ cytotoxic T-cell function.
Conclusion: The current study provides preclinical evidence that AMD3465 treatment in combination with IDO1 inhibition may be a promising therapeutic regimen for refractory metastasized breast cancers.
Keywords: breast cancer, immune response, metastasis, IDO1, chemokine
Introduction
Breast cancer (BC) is threatening the health of women worldwide.1 Although the overall survival of BC patients has improved in recent decades, individuals with metastasized cancer, ie stage IV BC, still have an unfavorable 5-year-survival.1 The metastatic spread of BC cells is considered to be the primary reason for treatment failure. Bones are common metastatic sites for BC, and bone metastases can cause pathological fracture and severe pain, which profoundly impair quality of life and shorten survival.2 Therapeutic strategies that can shrink or control metastasized tumors could be significantly beneficial during subsequent surgery or radiotherapy. Therefore, investigating promising targets for nonsurgical treatment of BC bone metastases is important.
Chemokines and their receptors have been reported to be involved in mediating tumor cell proliferation and immune escape through multiple mechanisms. CXCL12 and its receptor CXCR4 are expressed by primary BC tissues,3,4 and can stimulate tumor cell proliferation and migration by activating downstream signaling transduction pathways, including the PI3K-AKT and MAPK pathways.5 Previous studies have also demonstrated that CXCR4 expression is prognostic for BCs.3,6,7 Furthermore, CXCL12 produced by tumor tissue and stroma can facilitate the recruitment of CXCR4+ endothelial progenitor cells.5 CXCL12 has been shown to recruit immunosuppressive cells including regulatory T-cells (Tregs) and myeloid-derived suppressor cells (MDSCs), and consequently establish an immunosuppressive microenvironment.8,9 CXCL12-CXCR4 axis blockade has shown promising anticancer effects in some cancers.10,11 CXCR4 antagonists or blocking peptides have shown potential in suppressing CXCL12-induced cancer cell proliferation and metastasis.12,13
Indoleamine 2, 3-dioxygenase 1 (IDO1) is an enzyme that regulates catabolic conversion of tryptophan (Trp) into kynurenine (Kyn). In the cancer microenvironment, IDO1 depletes local L-Trp and produces Kyn, which impairs cytotoxic T-cells and generates Treg cells, and consequently permits tumor cells to escape from immune attack.14,15 Multiple clinical trials are ongoing to investigate if IDO1 inhibitors can suppress Treg function and restore the immune response in cancers. IDO1 is also highly expressed in BCs,16,17 which can theoretically impair the local immune response in BC. Therefore, inhibition of IDO1 may provide a favorable environment for the anticancer processes of cytotoxic T-cells.
In the current study, we hypothesized that CXCL12-CXCR4 axis blockade in BC bone metastases, using the CXCR4 antagonist AMD3465 in combination with the IDO1 inhibitor D-1-methyl-tryptophan (D1MT), could reduce the number of intratumoral Treg cells and impair Treg function, and consequently suppress tumor progression in vivo. We established conventional metastasis mouse models and spontaneous metastasis models to investigate whether continuous treatment using AMD3465 in combination with D1MT could delay tumor progression and prolong survival by facilitating the immune response.
Materials and methods
Reagents and cell line
The highly metastatic 4T1 mouse BC cell line was purchased from the Cell Collection of the Chinese Academy of Science (Shanghai, China). Cells were cultured at 37 °C in DMEM supplemented with 1% L-glutamine, 1% penicillin-streptomycin, and 10% fetal bovine serum.
AMD3465 was obtained from Genzyme Corporation (Cambridge, MA) and diluted in PBS before use. The IDO1 inhibitor D1MT was purchased from Sigma Aldrich.
Animal models and treatment
Female BALB/c mice aged 4–5 w were obtained from Shanghai experimental animal center and maintained in a gnotobiotic animal facility in Zhejiang Chinese Medical University. Ethical and legal approval from the animal care and use committee of Hangzhou First People’s Hospital was obtained prior to the commencement of the study. All animal procedures and experiments followed protocols approved by the animal care and use committee of Hangzhou First People’s Hospital.
To establish spontaneous metastasis BC models, 5×106 4T1 cells were inoculated into the mammary gland fat pad area, and the primary tumors were resected after 3 w. For conventional metastasis models, 5×105 cells in 20 μL of PBS were administrated into the tibia. After tumor inoculation (day 7), the mice were randomized into four different treatment groups, each having at least 10 mice per group as follows: vehicle control (PBS), AMD3465 single-agent treatment (AMD3465), D1MT single-agent treatment (D1MT), and combination treatment (Combo). AMD3465 continuous treatment was realized using osmotic pumps starting at 3 d after tumor resection (spontaneous models) or 14 d after tumor inoculation (conventional models). D1MT was dissolved in sterile water, and the mice were fed with D1MT supplemented water at 100 mg/kg twice a day at 3 d after tumor resection (spontaneous models) or 14 d after tumor inoculation (conventional models), and the water was replaced every 3 d. The survival rate was monitored.
Tumor sample processing
For sample preparation for flow cytometry analysis, five mice from each group were anesthetized and euthanized using carbon dioxide. Tumor tissues in the tibia in conventional metastasis model mice were harvested, and then the tumors were disaggregated using a razor blade and incubated at 37 °C for 2 h in DMEM with collagenase type IV (2 mg/mL, Sigma), DNase (0.1 mg/mL, Sigma), and hyaluronidase (0.1 mg/mL, Sigma). Cell suspensions were passed through 70 μm cell strainers to remove aggregates. The cells were washed using cell staining buffer (Biolegend, San Diego, CA, USA) and were then ready for antibody incubation.
Flow cytometry
The following antibodies were purchased from Biolegend: anti-CD3 (clone 17A2), anti-CD45 (clone 30-F11), anti-CD4 (clone GK1.5), anti-CD8α (clone 53–6.7), anti-IFN-γ (clone XMG1.2), anti-TIM-3 (clone RMT3-23), anti-CD62L (clone MEL-14), anti-CD11b (clone M1/70), and anti-Gr-1 (clone RB6-8C5). Conjugated antibodies obtained from eBioscience were as follows: anti-granzyme B (clone NGZB), anti-perforin (clone eBioOMAK-D), and anti-PD-1 (clone J43). For cell surface markers, cells were stained using antibodies at room temperature for 30 min. For intracellular antigen detection, an intracellular staining kit containing fixation/permeabilization reagents from eBioscience was used. Flow cytometry analyses were performed using a BD LSRFortessa (BD Biosciences). Flowjo V10 was used to analyze the data. Compensation was performed using micro beads.
Statistical analysis
Survival curves were drawn using the Kaplan–Meier method. Statistical analysis was performed using one-way ANOVA with Dunnett’s multiple comparison. All the analysis was performed using GraphPad Prism 6. Data are presented as mean ± SEM. P<0.05 was considered to be statistically significant.
Results
AMD3465 in combination with D1MT prolonged survival in metastasized BC models
We used two metastasis models to determine if the CXCR4 antagonist AMD3465 or the IDO1 inhibitor D1MT could delay tumor progression (Figure 1A and B). In spontaneous metastasis models (Figure 1A), ie mice bearing intratibial inoculated 4T1 BC cells, AMD3465 treatment alone slightly prolonged mouse survival when compared with phosphate buffered saline (PBS) treated mice, but no statistically significant difference was found (medium survival, AMD3465 34 d vs control 32.5 d; P=0.0704). D1MT treatment alone did not show a substantial effect in suppressing tumor progression. However, when these two agents were used in combination, treated mice demonstrated significantly longer survival (medium survival, combination treatment 42 d vs control 32.5 d; P<0.0001). In conventional metastasis models (Figure 1B), AMD3465 treatment alone did not produce significant antitumor effects, but D1MT treatment prolonged mouse survival (medium survival, D1MT 29 d vs control 23 d; P=0.0372). Moreover, similar to the results in the conventional metastasis models, combination treatment significantly prolonged mouse survival (medium survival, combination treatment 38.5 d vs control 23 d; P=0.0002). Taken together, these results indicate that solo treatment using either AMD3465 or D1MT had limited ability to control tumor progression, while combination treatment displayed powerful effects in prolonging mouse survival.
 |
Figure 1 Combination treatment (ie, combo in the figures) with AMD3465 and D1MT significantly increased survival in mice bearing metastasized breast cancer. (A) The survival curves of mice bearing spontaneously metastasized breast cancer. (B) The survival curves of mice with intratibial injected breast cancer cells. n=10/group. *P<0.05, **P<0.01, ***P<0.001, and ****P<0.0001. |
Combination treatment increased the number of infiltrated lymphocytes in mouse models
We investigated the underlying mechanism through which combination treatment prolonged mouse survival. As AMD3465 and D1MT have been determined to be associated with the immune response, we analyzed the intratumoral immune status in tumors in the conventional metastasis models. We counted the number of intratumoral leukocytes and discovered that IDO inhibition had potential in increasing the number of intratumoral CD45+ leukocytes, while AMD3465 appeared to slightly decrease the number of leukocytes. However, in combination treated mice, the number of intratumoral leukocytes was significantly elevated (Figure 2A and B). We further analyzed intratumoral CD45+ leukocytes, and found that the proportion of CD3+ CD8+ T-cells was significantly higher in AMD3465 treated or combination treated groups (Figure 2C and D). IDO inhibition also had potential in elevating the proportion of CD3+ CD8+ T-cells, although a statistically significant difference was not observed.
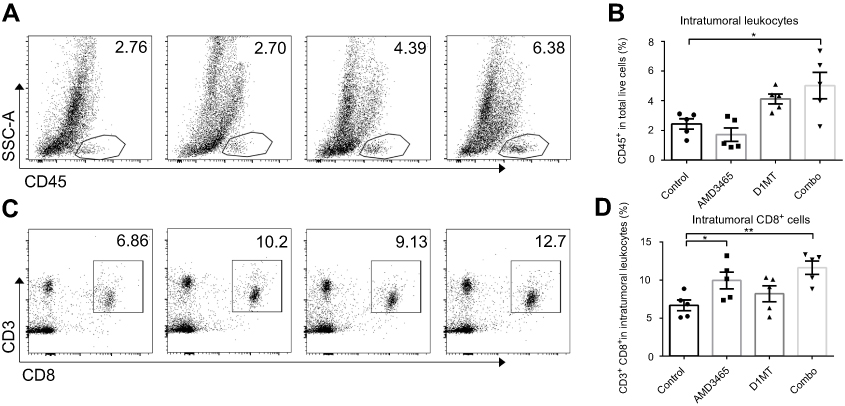 |
Figure 2 Combination treatment (combo) significantly elevated the number of intratumoral CD45+ leukocytes and CD3+ CD8+ T-cells. (A) Representative data of the proportions of intratumoral CD45+ leukocytes. (B) Pooled data of the proportions of CD45+ T-cells in the tibial breast metastases in different groups. n=5/group. (C) Representative data of the proportions of intratumoral CD3+ CD8+ T-cells (gated CD45+). (D) Pooled data of the proportions of intratumoral CD3+ CD8+ T-cells in different groups. n=5/group. *P<0.05, and **P<0.01. Data are presented as mean ± SEM. The different shapes in B and D represented samples in groups. |
IDO1 inhibition reinforced intratumoral CD8+ T-cell function
The activation status of intratumoral CD8+ cytotoxic cells is crucial in allowing them to attack cancer cells. We analyzed the expression of perforin, granzyme-B, and IFN-γ in intratumoral CD8+ cells. Both AMD3465 and D1MT showed potential in strengthening the cytokine secretion of intratumoral CD8+ cells, although there was no statistically significant increase in secretion. In the combination treatment group, the expression of perforin, granzyme-B, and IFN-γ demonstrated an over five-fold (Figure 3A and B), seven-fold (Figure 3C and D), and approximately two-fold (Figure 3E and F) increase, respectively. Many intratumoral cytotoxic cells become exhausted because of the tumor immunosuppressive microenvironment. Therefore, we evaluated the expression of TIM-3, a hallmark of CD8+ T-cell exhaustion, to determine if treatment could protect the cytotoxic cells from exhaustion. Intratumoral CD8+ T-cells from combination regimen treated mice had significantly lower expression of TIM-3 (Figure 3G and H), indicating that dual treatment maintained the activation status of intratumoral CD8+ T-cells.
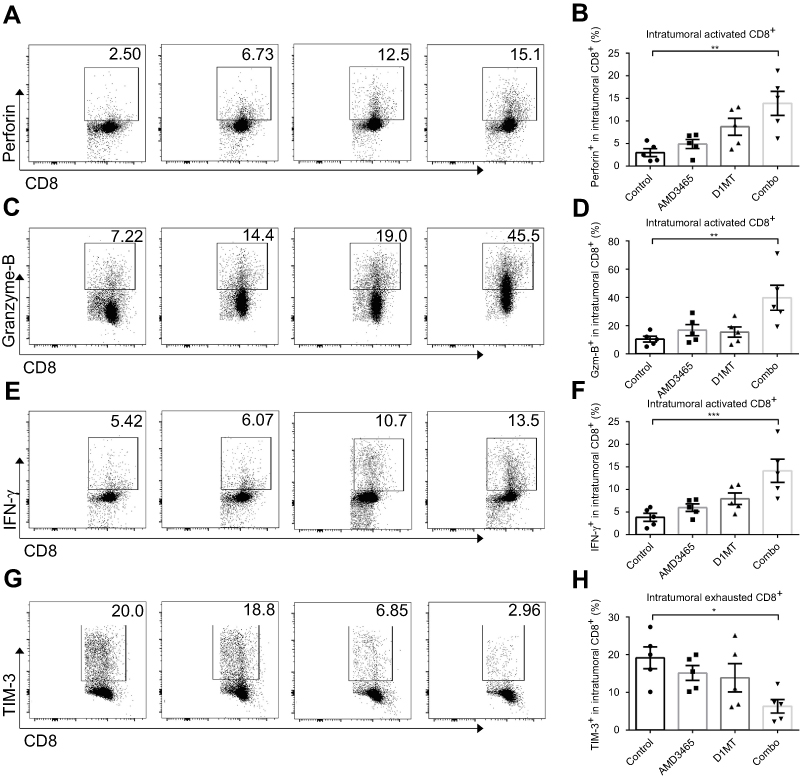 |
Figure 3 Combined treatment (combo) significantly strengthened the activity of intratumoral CD8+ T-cells. Representative and pooled data of the level of perforin (A and B), granzyme-B (C and D), and IFN-γ (E and F) in intratumoral CD8+ T-cells from different groups. The expression of TIM-3, an exhausted CD8+ T-cell marker, was also evaluated in different groups (G and H). n=5/group. *P<0.05, **P<0.01, and ***P<0.001. Data are presented as mean ± SEM. The different shapes in B, D, F, and H represented samples in groups. |
AMD3465 reduced the number of intratumoral Tregs and MDSCs
As a CXCR4 antagonist, AMD3465 was conjectured to impair the homing of CXCR4+ Treg cells and immunosuppressive MDSCs. We analyzed the proportion of CD25+ Foxp3+ cells in the intratumoral CD4+ T-cells. Unsurprisingly, AMD3465 treatment did decrease the number of Treg cells, and combination treatment also resulted in a lower proportion of Treg cells (Figure 4A and B). We also analyzed the number of CD11b+ Gr-1+ MDSCs in the CD45+ cells. As presented in Figure 4C and D, therapeutic regimens involving AMD3465 significantly reduced the number of MDSCs in tumors. Then we analyzed the ratio of CD8+ T-cells to Treg cells or MDSCs. AMD3465 and combination treatment resulted in a significantly higher ratio of CD8+ T-cells to Treg cells (Figure 4E) and CD8+ T-cells to MDSCs (Figure 4F). As shown above, IDO inhibition had potential in facilitating CD8+ T-cell function, and we therefore calculated the ratio of activated CD8+ T-cells (perforin positive) to functional Treg cells (CD62L- TIM-3+). We discovered that although IDO inhibition did not change the ratio of total CD8+ T-cells to Treg cells, it elevated the ratio of activated CD8+ T-cells to functional Treg cells (Figure 4G), indicating that IDO inhibition had strong effects in maintaining the tumor immune response.
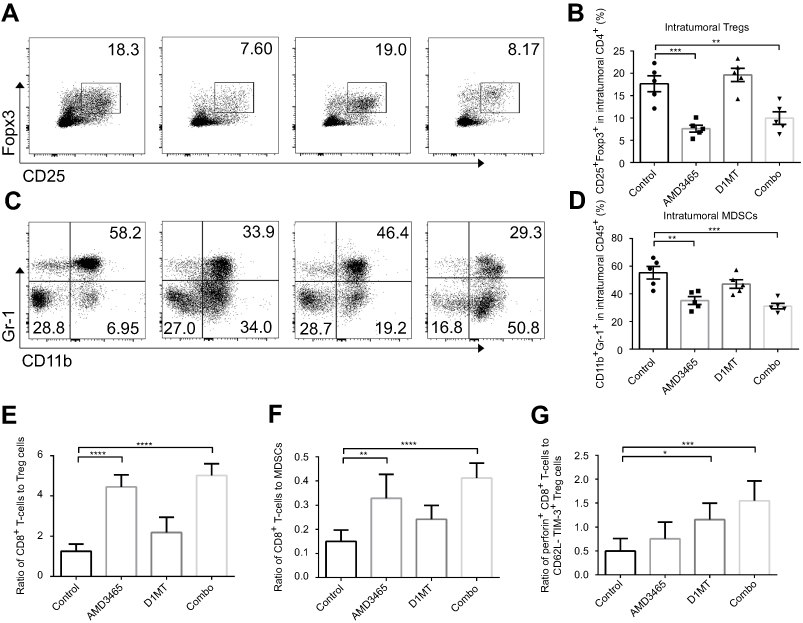 |
Figure 4 Combined treatment (combo) significantly decreased the number of intratumoral Treg cells and myeloid-derived suppressor cells (MDSCs). (A) Representative data of the proportion of CD25+ Foxp3+ Treg cells in intratumoral CD4+ cells. (B) Pooled data of the proportion of Treg cells in tumors in different groups. (C) Representative data of the proportion of CD11b+ Gr-1+ MDSCs in intratumoral CD45+ cells. (D) Pooled data of the proportion of MDSC cells in tumors in different groups. The ratio of CD8+ T-cells to Treg cells (E), or to MDSCs (F) was calculated. (G) The ratio of activated CD8+ T-cells to CD62L- TIM-3+ functional Treg cells was also calculated. n=5/group. * P<0.05, ** P<0.01, ***P<0.001, and ****P<0.0001. Data are presented as mean ± SEM. |
CXCR3+ CD8+ cells played key roles in suppressing tumor progression after combination treatment
As AMD3465 could theoretically impair the homing of CXCR4+ CD8+ T-cells, we investigated the subtype of CD8+ T-cells playing a major role in these antitumor procedures. As CXCR3+ CD8+ T-cells, which are responsive to TH1-type chemokine CXCL9 and CXCL10, are also one of the major subtypes of CD8+ T-cells, we used the CXCR3 antagonist AMG487 to inhibit the homing of CXCR3+ CD8+ T-cells to the tumor in both metastasis models. In addition, to determine whether the antitumor effects of combination treatment depend on the function of CD8+ T-cells or CD4+ T-cells, we used blocking antibodies to deplete these cells. As shown in Figure 5A, combination treatment could significantly prolong the survival of mice bearing spontaneous metastasis, and the addition of AMG487 significantly weakened these effects (medium survival, combination treatment 34.5 d vs combination treatment + AMG487 27 d; P<0.0001). Depletion of CD8+ or CD4+ T-cells also severely impaired combination treatment-mediated anticancer effects, but CD8+ T-cells appeared to play a more important role in this BC metastasis model. Similar results were also seen in the conventional metastasis models (Figure 5B). Combination treatment significantly prolonged mouse survival, while AMG487 almost abolished these effects (medium survival, combination treatment 41.5 d vs combination treatment + AMG487 31.5 d; P<0.0001). Depletion of CD4+ T-cells weakened the antitumor effects of combination therapy, but CD8+ T-cells appeared to be more dominant.
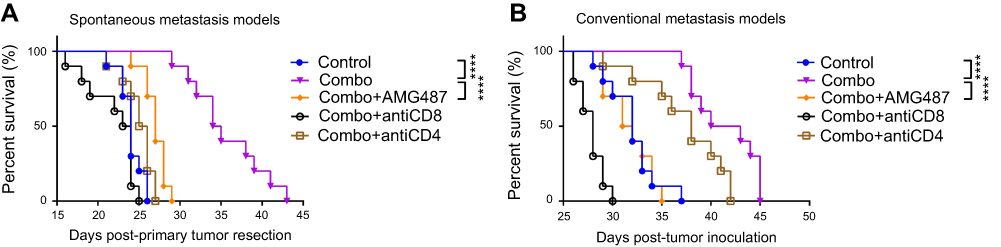 |
Figure 5 The anticancer effects of combination treatment (combo) relied on the function of CXCR3+ CD8+ T-cells. (A) Combination treatment (combo) significantly prolonged survival, while CXCR3 antagonist AMG487 could abrogate these effects. (B) In conventional metastasis models, AMG487 could also abolish the anticancer effects of combination treatment with AMD3465 and IDO1 inhibitor. ****P<0.0001. |
Discussion
BC metastasizing to remote organs is a devastating event which can lead to treatment failure. Although efforts have been made to improve treatment outcomes, current therapeutic strategies do not typically achieve favorable outcomes for individuals with metastasized BC. Orthopedic surgeons are also trying to improve the quality of life of BC bone metastasized patients by performing surgical resections, but micro metastases which cannot be discovered, or huge tumor burdens are still challenging for surgeons or medical oncologists. Immunotherapy is a promising non-surgical method for refractory cases, but more preclinical studies are still needed to discover more potential therapeutic regimens.
AMD3465, as a CXCR4 antagonist, can directly block the interaction of CXCL12 and CXCR4, and therefore inhibit tumor cell proliferation by impairing CXCL12-induced activation.18 The CXCL12-CXCR4 axis was found to be associated with the immune response in some solid malignancies, and the intervention of this axis might strengthen the immune attack.5 In the current study, we focused on the role AMD3465 plays in inhibiting the homing of CXCR4+ immunosuppressive cells, such as some Treg cells and MDSCs. We discovered that solo treatment using AMD3465 elevated the proportion of CD8+ T-cells in CD45+ cells, but reduced the total number of intratumoral CD45+ leukocytes, indicating that AMD3465 had effects that were more specific in other immune cells aside from CD8+ T-cells. Further assays demonstrated that AMD3465 significantly reduced the number of Treg cells and MDSCs, which provided a favorable environment for cytotoxic CD8+ T-cells to fight against cancer cells. Mouse survival assays showed that using AMD3465 alone produced limited effects in prolonging survival. Cancers can impair the immune response in various ways. Combination treatment is needed to reach a favorable outcome.
BC cells can also weaken the immune attack by overexpressing IDO1, which can establish a CD8+ T-cell-hostile environment through metabolic routes. In clinical trials, some IDO1 inhibitors such as D1MT or epacadostat have already been investigated for their presumed anticancer effects. However, according to published articles, using IDO1 inhibitors alone had limited effects in prolonging patient survival.19,20 Therefore, we investigated the effects of a combination treatment regimen in the current animal study. In addition to AMD3465, we employed an IDO1 inhibitor, D1MT, to suppress the function of IDO1. Single agent treatment using D1MT had modest effects in prolonging survival, but IDO1 inhibition showed promising effects in facilitating CD8+ T-cell function. Once used in combination with AMD3465, the intratumoral CD8+ T-cells were “refreshed,” and secreted significantly higher levels of cytokines which could kill cancer cells. Moreover, combination treatment protected the CD8+ T-cells from exhaustion. AMD3465 could theoretically prevent the homing of CXCR4+ T-cells, and therefore we investigated the role of CXCR3+ CD8+ T-cells in the immune response, and provided evidence that they play key roles in this process. Depletion assays demonstrated that the antitumor effects of the combination strategy largely relied on the function of CD8+ T-cells, and CD4+ T-cells also played a role in this process.
AMD3465 also appeared to elevate the proportion of CD11b+ Gr-1- cells in the CD45+ leukocytes. Quite a few subtypes of leukocytes have the CD11b+ Gr-1- phenotype, and we currently are not sure if AMD3465 facilitated the proliferation or survival of some CD11b+ cells, or if this occurred because of indirect effects after the inhibition of CXCR4+ MDSCs. Further studies are needed to clarify the role that AMD3465 and IDO1 inhibition play in cancer treatment.
Conclusion
Taken together, this is the first study providing preclinical in vivo evidence that AMD3465, in combination with the IDO1 inhibitor D1MT, can suppress metastasized BC progression and prolong mouse survival. Combination treatment triggered the immune response based on the activation of CD8+ T-cells and the inhibition of Treg cells and MDSCs. Our results suggest that this combination regimen may be a promising strategy for the treatment of metastasized BCs.
Disclosure
The authors declare no potential conflict of interest in this work.
References
1. Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D. Global cancer statistics. CA Cancer J Clin. 2011;61(2):69–90. doi:10.3322/caac.20107
2. Vacchelli E, Enot DP, Pietrocola F, Zitvogel L, Kroemer G. Impact of pattern recognition receptors on the prognosis of breast cancer patients undergoing adjuvant chemotherapy. Cancer Res. 2016;76(11):3122–3126. doi:10.1158/0008-5472.CAN-16-0294
3. Rhodes LV, Short SP, Neel NF, et al. Cytokine receptor CXCR4 mediates estrogen-independent tumorigenesis, metastasis, and resistance to endocrine therapy in human breast cancer. Cancer Res. 2011;71(2):603–613. doi:10.1158/0008-5472.CAN-10-3185
4. Muller A, Homey B, Soto H, et al. Involvement of chemokine receptors in breast cancer metastasis. Nature. 2001;410(6824):50–56. doi:10.1038/35065016
5. Scala S. Molecular pathways: targeting the CXCR4-CXCL12 axis–untapped potential in the tumor microenvironment. Clin Cancer Res. 2015;21(19):4278–4285. doi:10.1158/1078-0432.CCR-14-0914
6. Hiller DJ, Meschonat C, Kim R, Li BD, Chu QD. Chemokine receptor CXCR4 level in primary tumors independently predicts outcome for patients with locally advanced breast cancer. Surgery. 2011;150(3):459–465. doi:10.1016/j.surg.2011.07.005
7. Chu QD, Holm NT, Madumere P, Johnson LW, Abreo F, Li BD. Chemokine receptor CXCR4 overexpression predicts recurrence for hormone receptor-positive, node-negative breast cancer patients. Surgery. 2011;149(2):193–199. doi:10.1016/j.surg.2010.05.016
8. Noman MZ, Janji B, Hu S, et al. Tumor-promoting effects of myeloid-derived suppressor cells are potentiated by hypoxia-induced expression of miR-210. Cancer Res. 2015;75(18):3771–3787. doi:10.1158/0008-5472.CAN-15-0405
9. Li B, Zeng Y, Reeves PM, et al. AMD3100 augments the efficacy of mesothelin-targeted, immune-activating VIC-008 in mesothelioma by modulating intratumoral immunosuppression. Cancer Immunol Res. 2018;6(5):539–551. doi:10.1158/2326-6066.CIR-17-0530
10. Righi E, Kashiwagi S, Yuan J, et al. CXCL12/CXCR4 blockade induces multimodal antitumor effects that prolong survival in an immunocompetent mouse model of ovarian cancer. Cancer Res. 2011;71(16):5522–5534. doi:10.1158/0008-5472.CAN-10-3143
11. Li B, Wang Z, Wu H, et al. Epigenetic regulation of CXCL12 plays a critical role in mediating tumor progression and the immune response in osteosarcoma. Cancer Res. 2018;78(14):3938–3953.
12. Lu Z, O’Dell D, Srinivasan B, et al. Rapid diagnostic testing platform for iron and vitamin A deficiency. Proc Natl Acad Sci U S A. 2017;114(51):13513–13518. doi:10.1073/pnas.1711464114
13. Yoon Y, Liang Z, Zhang X, et al. CXC chemokine receptor-4 antagonist blocks both growth of primary tumor and metastasis of head and neck cancer in xenograft mouse models. Cancer Res. 2007;67(15):7518–7524. doi:10.1158/0008-5472.CAN-06-2263
14. Zhai L, Spranger S, Binder DC, et al. Molecular pathways: targeting IDO1 and other tryptophan dioxygenases for cancer immunotherapy. Clin Cancer Res. 2015;21(24):5427–5433. doi:10.1158/1078-0432.CCR-15-0420
15. Munn DH, Zhou M, Attwood JT, et al. Prevention of allogeneic fetal rejection by tryptophan catabolism. Science. 1998;281(5380):1191–1193.
16. Noonepalle SK, Gu F, Lee EJ, et al. Promoter methylation modulates indoleamine 2,3-dioxygenase 1 induction by activated T cells in human breast cancers. Cancer Immunol Res. 2017;5(4):330–344. doi:10.1158/2326-6066.CIR-16-0182
17. Soliman H, Rawal B, Fulp J, et al. Analysis of indoleamine 2-3 dioxygenase (IDO1) expression in breast cancer tissue by immunohistochemistry. Cancer Immunol Immunother. 2013;62(5):829–837. doi:10.1007/s00262-013-1393-y
18. Ling X, Spaeth E, Chen Y, et al. The CXCR4 antagonist AMD3465 regulates oncogenic signaling and invasiveness in vitro and prevents breast cancer growth and metastasis in vivo. PLoS One. 2013;8(3):e58426. doi:10.1371/journal.pone.0058426
19. Kristeleit R, Davidenko I, Shirinkin V, et al. A randomised, open-label, phase 2 study of the IDO1 inhibitor epacadostat (INCB024360) versus tamoxifen as therapy for biochemically recurrent (CA-125 relapse)-only epithelial ovarian cancer, primary peritoneal carcinoma, or fallopian tube cancer. Gynecol Oncol. 2017;146(3):484–490. doi:10.1016/j.ygyno.2017.07.005
20. Hong R, Zhou Y, Tian X, Wang L, Wu X. Selective inhibition of IDO1, D-1-methyl-tryptophan (D-1MT), effectively increased EpCAM/CD3-bispecific BiTE antibody MT110 efficacy against IDO1(hi)breast cancer via enhancing immune cells activity. Int Immunopharmacol. 2018;54:118–124. doi:10.1016/j.intimp.2017.10.008
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.