Back to Journals » Clinical Epidemiology » Volume 7
Cushing's syndrome: epidemiology and developments in disease management
Authors Sharma S, Nieman L, Feelders R
Received 8 May 2014
Accepted for publication 10 November 2014
Published 17 April 2015 Volume 2015:7 Pages 281—293
DOI https://doi.org/10.2147/CLEP.S44336
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Henrik Toft Sørensen
Susmeeta T Sharma,1 Lynnette K Nieman,1 Richard A Feelders2
1Program in Reproductive and Adult Endocrinology, Eunice Kennedy Shriver National Institute of Child Health and Human Development, National Institutes of Health, Bethesda, MD, USA; 2Division of Endocrinology, Department of Internal Medicine, Erasmus Medical Center, Rotterdam, the Netherlands
Abstract: Cushing’s syndrome is a rare disorder resulting from prolonged exposure to excess glucocorticoids. Early diagnosis and treatment of Cushing’s syndrome is associated with a decrease in morbidity and mortality. Clinical presentation can be highly variable, and establishing the diagnosis can often be difficult. Surgery (resection of the pituitary or ectopic source of adrenocorticotropic hormone, or unilateral or bilateral adrenalectomy) remains the optimal treatment in all forms of Cushing’s syndrome, but may not always lead to remission. Medical therapy (steroidogenesis inhibitors, agents that decrease adrenocorticotropic hormone levels or glucocorticoid receptor antagonists) and pituitary radiotherapy may be needed as an adjunct. A multidisciplinary approach, long-term follow-up, and treatment modalities customized to each individual are essential for optimal control of hypercortisolemia and management of comorbidities.
Keywords: Cushing’s syndrome, hypercortisolemia, treatment, epidemiology
Introduction
Cushing’s syndrome is a rare disorder caused by prolonged exposure to excess glucocorticoids. The clinical presentation is highly variable. While the diagnosis can be straightforward in florid cases, establishing the diagnosis can be challenging in cases with mild hypercortisolism and subtle clinical features, especially given the overlap in symptoms in individuals with and without the syndrome.1 Here, we discuss the epidemiology, diagnosis, and advances in management of endogenous Cushing’s syndrome.
Epidemiology and prognosis
Administration of supraphysiologic doses of glucocorticoids is the most common cause of Cushing’s syndrome (exogenous or iatrogenic Cushing’s syndrome).2 As glucocorticoids are used to treat inflammatory, autoimmune, and neoplastic disorders, a detailed medication history is essential. Although the oral route is most commonly associated with iatrogenic Cushing’s syndrome, any mode of delivery, including inhaled, topical, or injectable glucocorticoids, should be sought.
Endogenous Cushing’s syndrome is rare, with an incidence of 0.7–2.4 per million population per year.1,3,4 A population-based study from Denmark reported a diagnosis of Cushing’s syndrome in 166 patients over an 11-year period (1985–1995), yielding an incidence of two cases per million inhabitants per year.3 Of the 139 patients with nonmalignant disease, 23 (16.5%) died during follow-up (median 8 years), yielding a standard mortality ratio of 3.7 (95% confidence interval 2.3–5.3), with the highest mortality during the first year after initial presentation. Eight deaths occurred before initiation of treatment. Causes of death included suicide (n=1), cardiac rupture (n=1), stroke (n=1), and severe infections (n=3; peritonitis, septicemia, and pneumonia). The cause of death could not be ascertained in two patients. A similar study from Spain reported 49 cases of Cushing’s syndrome over 18 years, yielding an incidence of 2.4 cases per million inhabitants per year, with a standard mortality ratio of 3.8.4 Although the mortality risk decreases with remission of hypercortisolism,5 it is not equivalent to that of the general population. Several studies have shown that increased morbidity, especially related to cardiovascular disease, persists for several years after biochemical remission.6–9 Furthermore, varying criteria for remission of hypercortisolemia across studies make it difficult to interpret these outcome measures accurately.
Although population-based studies demonstrate a low incidence of endogenous Cushing’s syndrome, more evaluations of patients with uncontrolled diabetes mellitus or hypertension suggest that this may be an underestimation.10–13 Leibowitz et al screened 90 obese subjects with uncontrolled diabetes mellitus (hemoglobin A1c >9%), and found three (3.3%) to have Cushing’s syndrome.10 Similarly, Catargi et al diagnosed Cushing’s syndrome in four (2%) of 200 obese subjects with uncontrolled diabetes mellitus.11 However, this reported prevalence of 2%–5% was not confirmed in other studies.14–17 Also, an evaluation of 369 overweight and obese subjects with at least two other features of Cushing’s syndrome did not identify any with Cushing’s syndrome, but 84 subjects had at least one abnormal screening test result.18 Widespread screening for Cushing’s syndrome in overweight individuals or patients with type 2 diabetes mellitus is therefore not recommended.18,19 Instead, a case-finding approach in patients with other features of Cushing’s syndrome or uncontrolled diabetes or hypertension despite appropriate treatment may be indicated.19
Currently, extensive use of computed tomography (CT) and magnetic resonance imaging (MRI) scans has led to an increasing number of incidentally found adrenal masses. The prevalence of these adrenal “incidentalomas” increases from 0.2% to 7% with increasing age.20–22 “Subclinical” or subtle Cushing’s syndrome has been reported in 5%–10% of these patients, who represent a population in which Cushing’s syndrome is more common.22,23
Classification of Cushing’s syndrome
Traditionally, endogenous Cushing’s syndrome is classified as adrenocorticotropic hormone (ACTH)-dependent or ACTH-independent. ACTH-dependent Cushing’s syndrome accounts for 80%–85% of cases. Of these, 75%–80% are due to ACTH production from a pituitary adenoma (Cushing’s disease [CD]), 15%–20% are due to ACTH production from nonpituitary tumors (ectopic ACTH syndrome [EAS]) and <1% are caused by corticotropin-releasing hormone (CRH)-producing tumors.1,23 Most pituitary tumors are sporadic, resulting from monoclonal expansion of a single mutated cell. Rarely, they may occur as part of a genetic syndrome, the most common being multiple endocrine neoplasia type 1 and familial isolated pituitary adenomas. Ectopic ACTH secretion most often derives from small-cell carcinoma of the lung or pulmonary carcinoid tumor. Other causes include pancreatic neuroendocrine tumors (NETs), thymic NETs, gastrinomas, medullary thyroid cancer, and pheochromocytoma.24
ACTH-independent Cushing’s syndrome accounts for 15%–20% of endogenous Cushing’s syndrome in adults; 90% are unilateral adrenal tumors. Of these, adenomas are the cause in ~80% of the cases, while the others are adrenocortical carcinoma.25 Rare adrenal causes of Cushing’s syndrome include macronodular adrenal hyperplasia, primary pigmented nodular adrenal disease (sporadic or as part of Carney’s complex) and McCune–Albright syndrome. In a recent study, Louiset et al described intra-adrenal production of ACTH and paracrine regulation of cortisol secretion in 30 cases of bilateral macronodular adrenal hyperplasia.26 Proopiomelanocortin messenger ribonucleic acid expression was detected in all hyperplastic tissue samples and ACTH was detected in steroidogenic cell clusters throughout adrenal tissue specimens. ACTH levels were also found to be higher in adrenal venous samples of two patients compared to the periphery.26 These findings bring into question the traditional classification of “ACTH-independent Cushing’s syndrome”.27
In childhood and adolescence, as in adults, exogenous glucocorticoids are the most common cause of Cushing’s syndrome. The causes of endogenous Cushing’s syndrome in children are similar to those in adults, with some differences. Cushing’s syndrome in infancy is commonly associated with McCune–Albright syndrome, adrenocortical tumors are usually the cause in children less than 5–7 years of age, CD is the commonest cause after 7 years of age, and EAS is extremely rare.28
Clinical features of Cushing’s syndrome
The clinical presentation of Cushing’s syndrome is variable (Table 1). It is influenced by age and sex and the severity and duration of the disease.1,24,28–31 No single sign or symptom is pathognomonic.
 | Table 1 Clinical features of Cushing’s syndrome |
While weight gain is the most common sign of Cushing’s syndrome, it is also extremely common in patients without the syndrome.18 Moreover, an underlying malignancy or paraneoplastic wasting syndrome can mask the weight gain associated with hypercortisolism. As may be concluded from Table 1, establishing a diagnosis based on clinical features alone can be difficult. Some of the features that are thought to distinguish Cushing’s syndrome more reliably from simple obesity include proximal muscle weakness, easy bruising, and violaceous striae greater than 1 cm wide. Decreased linear growth along with progressive weight gain is one of the hallmarks of pediatric Cushing’s syndrome.32 Detailed description of differences in presentation between pediatric and adult Cushing’s syndrome is beyond the scope of this review.28,33,34
The clinical presentation of Cushing’s syndrome can vary by sex. Men with CD are more likely to present at a younger age with more florid clinical features. Violaceous striae, muscle weakness and atrophy, osteoporosis, and kidney stones are much more common.29 Gonadal dysfunction is common in both men and women, presenting as decreased libido, infertility, menstrual irregularity or amenorrhea in women, and erectile dysfunction in men. While CD has a female preponderance (female:male ratio 3–4:1), EAS is equally common in both sexes among the adult population.24 Unlike adults, pediatric CD is characterized by significant male preponderance in the prepubertal years. With increasing age, the sex distribution of CD equalizes toward puberty, with a trend toward female preponderance in adulthood.28,33,34
Classically, patients with ectopic Cushing’s have severe hypercortisolemia associated with a rapid onset of symptoms and a florid presentation, hypokalemia, and severe or opportunistic infections.24,35,36 However, these features reflect the severity of hypercortisolism, and can be seen in patients with CD. By contrast, the clinical presentation of many ACTH-secreting carcinoid tumors may be indistinguishable from that of CD, reflecting less extreme hypercortisolism.24,37
Decreased bone mineral density, osteoporosis, and fractures are present in 50%–80% of patients with Cushing’s syndrome. After cure, bone mineral density improves, but additional specific treatment for fractures and related pain may be needed. Bone loss can be more severe in primary adrenal disease compared to CD. This may be related to a protective effect of the higher adrenal androgen levels in CD.38,39 However, these findings have not been reproduced in other studies, and the significance of disease etiology in bone loss and fractures remains controversial.40,41
Psychiatric and cognitive dysfunction is present in 70%–85% of patients with Cushing’s syndrome. Depression, emotional lability, and irritability are the most common manifestations; acute psychosis, mania, anxiety, panic attacks, suicidal ideation, and paranoia are rarer.42–45 Hypercortisolemia is also associated with a decrease in brain volume, particularly the hippocampus, and related impairment in learning and short-term memory.46,47 Although psychiatric and cognitive symptoms improve after remission, many symptoms may persist.45,46
Excess glucocorticoids have a catabolic effect on skeletal muscles, skin, and connective tissue. Increased protein wasting and type II muscle-fiber atrophy is associated with significant muscle weakness, with predominant involvement of the pelvic girdle musculature.48,49 Vertebral fractures, back pain, and depression further lead to decreased mobility and disuse muscle atrophy. A persistently impaired quality of life, primarily of the physical domain, and persistent muscle weakness have been documented even several years after remission.50–53
Dyslipidemia (increased low-density lipoprotein, decreased high-density lipoprotein) and glucose intolerance occur in 45%–70% of patients.54–56 This reported prevalence of glucose intolerance is likely an underestimate, as many patients with normal fasting glucose have underlying glucose intolerance, and not all patients with Cushing’s syndrome undergo glucose-tolerance testing. Hypertension (cortisol-mediated enhancement of vascular reactivity to vasoconstrictors and the mineralocorticoid effects of cortisol), though not invariably present, is frequently seen in patients with Cushing’s syndrome, with a prevalence of approximately 80%.35,55 Hepatic steatosis and increased visceral adipose tissue are common in patients with Cushing’s syndrome.57,58 These features of metabolic syndrome, along with a hypercoagulable state,59 lead to an increased cardiovascular risk that may not return to baseline after successful treatment.60–62
Diagnosis of Cushing’s syndrome
Clinical suspicion in patients with multiple and progressive signs and symptoms suggestive of Cushing’s syndrome should provoke diagnostic testing. Screening may be considered in patients with other features of Cushing’s syndrome, particularly with poorly controlled diabetes or hypertension, or unexplained osteoporosis. Patients with an incidentally discovered adrenal mass should be evaluated.
It is important to differentiate between the pathological hypercortisolemia of endogenous Cushing’s syndrome and that associated with pregnancy, glucocorticoid resistance, and pseudo-Cushing’s states like alcoholism, depression, severe obesity, anorexia nervosa, and bulimia. The hypercortisolism of pseudo-Cushing’s states is thought to be mediated via increased hypothalamic secretion of CRH. In contrast, hypothalamic CRH is suppressed in true Cushing’s syndrome.
Biochemical tests in Cushing’s syndrome are based on the cardinal features of increased endogenous secretion of cortisol, loss of normal feedback of the hypothalamic–pituitary–adrenal axis, and loss of the normal cortisol circadian rhythm. According to the 2008 Endocrine Society guidelines, the following tests should be used for the diagnosis of Cushing’s syndrome: 24-hour urinary free cortisol (UFC), late-night salivary cortisol, and/or a low-dose dexamethasone-suppression test (DST; 1 mg overnight or 2 mg/day over 48 hours).19 None of these tests has 100% diagnostic accuracy; each test has its own caveats, and multiple tests are usually needed to establish the diagnosis (Table 2).1,19,31,63
 | Table 2 Sensitivity, specificity, and caveats of screening tests for Cushing’s syndrome |
UFC is an index of circulating free (biologically active) cortisol. Unlike plasma cortisol, which measures total cortisol (bound and unbound), UFC results are not affected by factors that modulate the levels of corticosteroid-binding globulin (CBG). Inappropriate urine collection, increased fluid intake, and abnormal renal function can be associated with false-positive and -negative results.19,31,63 Review of urine volume and creatinine excretion can help assess the adequacy of urine collection. UFC values in Cushing’s syndrome are variable.64,65 Cushing’s syndrome is highly unlikely in the setting of three or more normal UFC levels, except in rare cases of cyclic Cushing’s syndrome. Values more than three- to fourfold the upper limit of normal are generally diagnostic of Cushing’s syndrome.1,19,31
DSTs rely on the loss of glucocorticoid negative-feedback inhibition of CRH and ACTH secretion. In the overnight test, 1 mg of dexamethasone is given at 11 pm, followed by measurement of fasting plasma cortisol between 8 and 9 am the next morning. In the 2-day test, 0.5 mg of dexamethasone is given every 6 hours (starting at 9 am) for 2 days, with cortisol measurement at the beginning and end of the test. A cortisol level of ≤1.8 μg/dL (50 nmol/L) at the end of either test excludes Cushing’s syndrome (Table 2).19 Variable absorption and metabolism of dexamethasone can affect the results of the DSTs. Estrogen treatment and pregnancy can increase CBG levels, leading to false-positive DST results. For accurate results, estrogen treatment should be stopped for 4–6 weeks to allow CBG levels to return to baseline.1,19 Some (3%–8%) CD patients show suppression of cortisol levels below 1.8 μg/dL (50 nmol/L) on DST.1,66 Multiple repeated tests may be needed to establish the diagnosis in these cases.
Salivary cortisol, like UFC, measures free cortisol, and is therefore not affected by CBG levels. Diagnostic ranges vary between studies, due to the different assays and comparison groups used to set cutoff points.63 Salivary cortisol can be measured by immunoassay or liquid chromatography–tandem mass spectrometry (LC/MS-MS). Given the lower sensitivity of LC/MS-MS (as it may not measure cortisol metabolites/precursors), immunoassay remains the preferred methodology for screening of Cushing’s syndrome.67–69 Educating the patient on optimal timing of specimen collection while taking into account their normal sleep–wake cycle, as well as avoiding exciting or stressful experiences on the evening of the test, are important for an accurate salivary cortisol result. False-positive results can also be seen, due to contamination of the saliva sample with widely available nonprescription topical hydrocortisone creams and ointments. However, overall, ease of collection, stability at room temperature, and a greater than 90% sensitivity and specificity make it a highly useful test, especially in outpatients, children, and in assessment of cyclic Cushing’s syndrome.67–69 Midnight plasma cortisol levels also can be used for screening. A single sleeping midnight plasma cortisol level of less than 1.8 μg/dL (50 nmol/L) excludes active Cushing’s syndrome.70 A midnight plasma cortisol level, drawn while the patient is resting but not fully asleep, of more than 7.5 μg/dL (207 nmol/L) is a more specific cutoff, but can miss up to 7% cases of mild Cushing’s syndrome.71,72
If the diagnosis remains unclear, dexamethasone-suppressed CRH stimulation (Dex-CRH) test73,74 or the desmopressin test75,76 can be performed to distinguish further between CD and pseudo-Cushing’s states. However, the exact diagnostic accuracy of these tests and the optimal cutoffs for diagnosis need further evaluation.77 Moreover, similarly to other dexamethasone tests, variable absorption and metabolism of dexamethasone, especially in the setting of concomitant use of other commonly prescribed medications, can affect the accuracy of the test.78 Recently, a prospective study of 73 patients with clinical features of hypercortisolism and an abnormal DST or UFC result reported a positive predictive value of 100% and a negative predictive value of 90% for the Dex-CRH test (sensitivity 94%, specificity 100%) using a 15-minute post-CRH cortisol cutoff of 3.2 μg/dL (87 nmol/L).79
Establishing the cause of Cushing’s syndrome
Once the diagnosis of Cushing’s syndrome has been established, the next step is to differentiate between the three causes. Measurement of plasma ACTH levels is the initial step in the differential diagnosis. A two-site immunoradiometric assay is preferred over radioimmunoassay, because it better discriminates low or suppressed ACTH levels.80 To avoid falsely low results, ACTH levels should be measured on multiple occasions, and samples should be collected in prechilled ethylenediaminetetraacetic acid tubes, transported in an ice bath, and processed immediately.1 Values <5 pg/mL (1.1 pmol/L) suggest ACTH-independent Cushing’s syndrome. Imaging studies of the adrenal gland can then identify unilateral or bilateral lesions. The glands may appear normal in primary pigmented nodular adrenal disease. Patients with proven ACTH-independent hypercortisolism but normal adrenal imaging studies should undergo screening for exogenous glucocorticoids, assessment for other features of Carney’s complex, and possibly genetic testing for PRKAR1A mutations.81 A paradoxical increase in glucocorticoid excretion in response to dexamethasone during the Liddle test (0.5 mg dexamethasone every 6 hours for 48 hours, followed by 2 mg every 6 hours for 48 hours) can further help identify patients with primary pigmented nodular adrenocortical disease.82 Recently, an inactivating germ-line mutation of ARMC5, a putative tumor-suppressor gene, has been implicated in the development of primary bilateral macronodular hyperplasia.83–85 Patients with this mutation show familial clustering and bilateral disease, and often present with a more severe clinical phenotype. Genetic testing for this mutation may help in earlier diagnosis and better management of these cases.85
An inappropriately normal or elevated ACTH level (>20 pg/ml, 4.4 pmol/L) is consistent with an ACTH-dependent form of Cushing’s syndrome. Patients with mild adrenal Cushing’s may not have suppressed ACTH levels. Moreover, recently, intra-adrenal production of ACTH has been reported in macronodular adrenal hyperplasia, although peripheral ACTH levels were suppressed.26 CRH stimulation and high-dose DSTs may help differentiate between adrenal and ACTH-dependent forms of Cushing’s syndrome when ACTH results are indeterminate.1
Although there is significant overlap in ACTH levels between CD and EAS, extremely high levels (>500 pg/mL, 110 pmol/L) usually reflect EAS.1,31 Sex may help: 90% of cases of ACTH-dependent Cushing’s syndrome in women are caused by CD.
A pituitary MRI should be obtained in ACTH-dependent cases. A large mass (>6 mm) strongly suggests CD. However, ACTH-secreting pituitary tumors are usually small and may not be detected, even with newer, more advanced MRI techniques (spoiled gradient-recalled acquisition or dynamic MRI sequences) in 20%–58% of patients with CD.86,87 Moreover, ~10% of “healthy” individuals can have incidental pituitary lesions up to 6 mm in size.88 Differential diagnosis of ACTH-dependent Cushing’s syndrome can therefore be very challenging.
The high-dose DST (2 mg given every 6 hours for 48 hours, or the overnight test with a single 8 mg dose) is based on the concept that corticotrope adenomas arise clonally from normal cells and retain some sensitivity to glucocorticoid negative feedback, while ectopic ACTH-secreting tumors do not. In the overnight test, a greater than 69% suppression of cortisol levels following 8 mg of dexamethasone suggests a pituitary source of ACTH.89 Logistic regression modeling has shown that the diagnostic accuracy of this test is less than the pretest likelihood based on clinical features alone.90 Due to the poor diagnostic accuracy (~80%) of a high-dose DST, many endocrinologists do not recommend performing the test unless inferior petrosal sinus sampling (IPSS) is not available.1,91
In the CRH-stimulation test, recombinant ovine or human sequence CRH (1 μg/kg, maximum 100 μg dose) is used to stimulate corticotrope tumors to secrete ACTH. Most patients with CD respond with an increase in ACTH (>34%) and/or cortisol (>20%) levels within 45 minutes of intravenous administration of ovine CRH (sensitivity 93%).92 Following administration of human CRH, most CD patients have at least a 14% increase in cortisol levels (sensitivity 85%, specificity 100%).93 While some reports have shown ovine CRH to be superior to recombinant human CRH,94 others have found similar responses.95 A systematic review of all published series on the CRH-stimulation test revealed that 7%–14% of patients with CD fail to respond to CRH.31 Some patients with ACTH-secreting pulmonary carcinoids (~10%) can respond to dexamethasone and/or CRH.24
If the CRH-stimulation test and high-dose DST are both consistent with a pituitary source and imaging studies identify a pituitary lesion consistent with an adenoma (>6 mm), no further testing is necessary. On the other hand, if biochemical testing is discordant and/or the pituitary MRI is normal or equivocal (lesion <6 mm), IPSS with ACTH measurements before and after CRH administration should be performed.1,96 A central-to-peripheral ACTH gradient of ≥2.0 before and/or ≥3.0 after CRH administration is consistent with CD. A systematic review of all published studies on IPSS showed an overall sensitivity of 96% and specificity of 100% using these criteria.31
Although IPSS is the gold-standard test to distinguish between a pituitary and ectopic source of ACTH, it is an invasive procedure requiring a high degree of skill, and thus is best performed in experienced centers. False-negative IPSS results of 1%–10% have been attributed to anomalous venous drainage, abnormal venous anatomy, lack of expertise, and technical problems.97–99 Review of the IPSS venogram and/or prolactin measurement during IPSS can improve diagnostic accuracy and decrease false-negative results.100–102 A baseline prolactin inferior petrosal sinus to peripheral (IPS/P) ratio (ipsilateral to the dominant post-CRH ACTH IPS/P ratio) of 1.8 or more suggests successful catheterization during IPSS. Prolactin-normalized ACTH IPS/P ratios can then be used to differentiate between a pituitary and ectopic source of ACTH. Values ≤0.7 suggest EAS, and those ≥1.3 suggest CD. Indeterminate values (0.7–1.3) need further study.102 False-positive IPSS results can occur in rare cases of ectopic CRH production103,104 and in patients with cyclic Cushing’s syndrome if the normal corticotropes are not completely suppressed.102 Documentation of sustained hypercortisolism prior to dynamic testing therefore remains crucial.
Localization of the source of ACTH
Cushing’s disease
As discussed earlier, advanced MRI techniques often do not identify pituitary adenomas. While IPSS localizes the tumor to the pituitary gland, its role in determining the precise tumor location remains controversial. Using an intersinus ratio of 1.4 or more before or after CRH to predict the location of pituitary adenomas,105 a review of 19 studies (313 cases) reported poor localizing accuracy (78% overall, 50%–100% in individual studies).31 More recently, Wind et al found that IPSS correctly predicted tumor location in 273 of 396 patients (positive predictive value 69%) with surgically proven CD and a lateral adenoma.87 Mulligan et al demonstrated that the use of prolactin-normalized ACTH intersinus ratios led to improvement in the lateralization accuracy of IPSS (75% versus 54%) in 28 patients with surgically proven CD.106 Although these data are promising, larger prospective studies are needed. Therefore, at present IPSS lateralization results can only be used as a guide for where to begin initial transsphenoidal exploration in MRI negative cases. A thorough exploration of the pituitary gland is essential before using lateralization results for hemihypophysectomy.
Ectopic ACTH syndrome
Structural (CT and MRI) and functional (somatostatin scintigraphy and positron emission tomography [PET] scans) imaging studies are used to identify the source of ACTH in EAS, but the optimal imaging strategy has not been well defined. Since no single imaging modality identifies all tumors, correlation of multiple studies is needed.24,107
Because the majority of these tumors are intrathoracic, initial imaging should focus on the chest, with additional studies obtained as needed. Thin-cut multislice CT scans and 3 T MRI improve tumor detection, but also can give false-positive results.107 As many NETs express somatostatin-subtype (sst) receptors, they may be detected via somatostatin scintigraphy (eg, octreotide scan). The ability of these scans to identify a tumor depends on the type (sst1–5) and degree of sst-receptor expression, size of the tumor, and the dose (6–18 mCi pentetreotide) of the radiopharmaceutical and the use of single photon emission CT.108 In vitro studies have shown that glucocorticoids downregulate sst-receptor expression in human neuroendocrine cells.109 This has been described in vivo, as well in two patients with ACTH-secreting pulmonary NETs, where mifepristone (a glucocorticoid receptor antagonist) treatment led to a change in the octreotide-scan status and correct localization of the tumor.110 This phenomenon of improved diagnostic ability of the octreotide scan after medical control of hypercortisolism needs to be further evaluated in larger studies. Ga68-DOTATATE and DOTATOC, PET radiopharmaceuticals with high affinity for sst2, have shown promise in identifying gastrointestinal–pancreatic and pulmonary NETs. However, patients with ACTH-secreting tumors were not included.111–113 The use of 68Ga-DOTATATE PET scans in identifying the source of ACTH in ectopic Cushing’s syndrome needs further investigation.
As most EAS tumors are slow growing and have low metabolic activity, fluorodeoxyglucose (18F) PET scans have limited utility in tumor localization; they may help define the extent of metastatic disease.114 Radiolabeled l-3,4- dihydroxyphenylalanine and (11C) 5-hydroxytryptamine PET scans can help with localization.107,115,116 Non-IPSS venous sampling usually does not provide any additional information, and is not considered necessary in the evaluation of ectopic Cushing’s syndrome.24,117
Management
Surgery
Surgical resection of the source of glucocorticoid excess (pituitary adenoma, nonpituitary tumor-secreting ACTH or adrenal tumor[s]) remains the first-line treatment of all forms of Cushing’s syndrome. The initial remission rate after transsphenoidal surgery is 60%–80% (<15% in macroadenomas),118 with a relapse rate of up to 20% within 10 years.1,119,120 However, the success rate depends on the skill and experience of the neurosurgeon, and can be as high as 90% at experienced centers. Patients with hypocortisolism in the immediate postoperative period need glucocorticoid replacement until the recovery of the hypothalamic–pituitary–adrenal axis (usually 6–18 months after surgery).1 Although long-term remission is more likely when postoperative cortisol levels are less than 2 μg/dL (<54 nmol/L), no cortisol value excludes the possibility of recurrence.120 These data emphasize the need for ongoing surveillance and alternative treatment modalities for CD.
As in CD, surgical removal of an ectopic ACTH-secreting tumor is the optimal treatment. However, occult or metastatic tumors require medical therapy or bilateral adrenalectomy.24
Laparoscopic unilateral or bilateral adrenalectomy is the treatment of choice in adrenal causes of Cushing’s syndrome, and has an excellent prognosis in benign cases.121 Bilateral adrenalectomy in ACTH-dependent Cushing’s syndrome may be used when surgery and medical therapy are unsuccessful, or based on patient preference. It leads to rapid resolution of hypercortisolemia and related morbidity.122 However, after bilateral adrenalectomy, patients need lifelong glucocorticoid and mineralocorticoid replacement. Another concern with bilateral adrenalectomy in patients with CD is the development of Nelson’s syndrome (local tumor growth with mass effects and increased ACTH levels causing hyperpigmentation). Modern imaging techniques allow early detection and management of corticotrope tumor progression after bilateral adrenalectomy in these patients.123 Some physicians advocate prophylactic pituitary radiotherapy to decrease the risk of development of Nelson’s syndrome.124
Pituitary radiotherapy
Persistent hypercortisolemia after transsphenoidal surgery due to residual tumor can be treated with radiotherapy. Adjunctive medical control of hypercortisolemia may be needed while awaiting the effects of radiotherapy. Conventional fractionated radiotherapy is very effective, but its effects may be delayed up to 10 years, and it can be associated with long-term hypopituitarism.125 Stereotactic radiosurgery is more rapidly effective, but has been associated with a relapse rate of 20%.126
Medical therapy
Medical control of hypercortisolemia may be needed in occult cases, while awaiting surgery, when surgery is contraindicated or unsuccessful, and while awaiting the effect of radiation treatment. Medical treatments for hypercortisolemia include agents that inhibit steroidogenesis (ketoconazole, metyrapone, mitotane, and etomidate), modulate ACTH release (somatostatin and dopamine agonists) or block glucocorticoid action at its receptor (mifepristone) (Table 3).127 A major concern with all medical therapies is the risk of overtreatment and adrenal insufficiency. Medical control of hypercortisolemia can be achieved in two ways: either by blocking cortisol production to achieve normal levels, or by blocking cortisol secretion completely along with glucocorticoid replacement (block and replace). Regardless of the strategy, all patients on medical therapy should be educated about the symptoms of adrenal insufficiency and the emergency use of glucocorticoids.
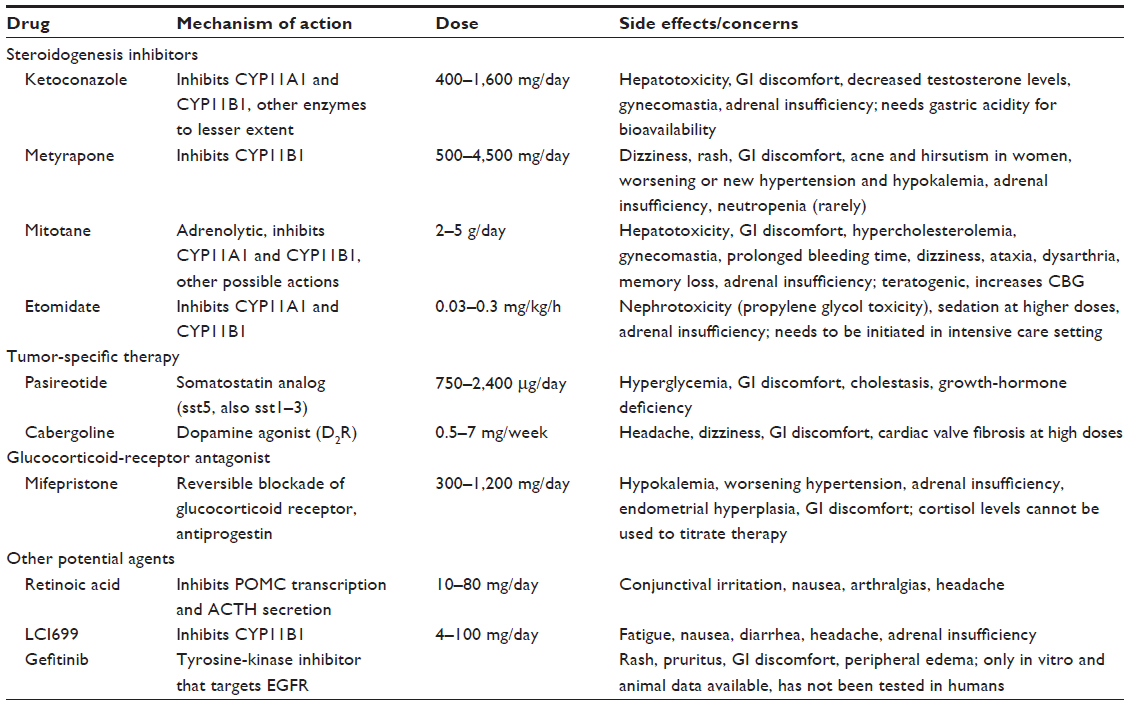 | Table 3 Drugs used to treat Cushing’s syndrome: mechanism of action, dosage, and important side effects and concerns |
Ketoconazole, a steroidogenesis inhibitor, has a rapid onset of action. It inhibits the first step in cortisol biosynthesis (side-chain cleavage) and to a lesser degree 11β-hydroxylase and 17,20-desmolase.128 It requires an acidic environment for maximal absorption, and thus has reduced efficacy if used in combination with proton-pump inhibitors. Although used off-label, ketoconazole is usually the first-line agent for medical control of hypercortisolism in the US. However, gastrointestinal side effects, hepatocellular dysfunction, gynecomastia, and decreased libido in men may limit its use.129,130 The European Medicines Agency (EMA) has recently withdrawn ketoconazole from the market because of hepatic dyscrasia in patients treated for fungal infections.131 A similar safety announcement from the US Food and Drug Administration (FDA) advised against its use for any fungal infection.132 When used for the control of hypercortisolemia, liver-function tests need to be monitored closely. Despite the risks associated, ketoconazole remains an important drug in the physician’s armamentarium for medical control of hypercortisolism. The EMA has therefore invited national authorities with expertise in this area to make ketoconazole available for patients with Cushing’s syndrome under controlled conditions.131
Metyrapone inhibits 11β-hydroxylase and has a rapid onset of action. In the US, it is primarily used as adjunctive therapy with other steroidogenesis inhibitors and with radiation therapy in CD.133,134 Increased deoxycorticosterone levels, due to inhibition of 11β-hydroxylase by metyrapone, can lead to salt retention and (rarely) hypertension, while increased adrenal androgens can lead to new or worsening hirsutism and acne in women. Both ketoconazole and metyrapone have been associated with an increase in ACTH secretion and loss of control of hypercortisolism over time in CD (escape phenomena).
Mitotane, another steroidogenesis inhibitor, is also adrenolytic. It has a slow-onset, long-lasting action. Mitotane increases CBG levels, leading to falsely high total cortisol levels, complicating titration of therapy. It is teratogenic, not well tolerated, and is primarily used in the treatment of adrenocortical carcinoma.135 During treatment, plasma mitotane concentrations must be monitored.135 Etomidate is a substituted imidazole anesthetic agent that inhibits 11β-hydroxylase. It has been used for rapid control of hypercortisolemia in hospitalized patients in an intensive care unit setting, and is the only available intravenous parenteral agent.136
Recently, somatostatin (sst5 and sst2) and dopamine (D2) receptors have been identified as therapeutic targets in CD.137 Pituitary-directed therapy with pasireotide (a somatostatin analog with high affinity for sst1, -2, -3, and -5) or cabergoline (a dopamine agonist) normalizes UFC in 20%–40% of patients with CD.138,139 This represents an off-label use for cabergoline. Pasireotide (Signifor), the first agent approved by the EMA and FDA for the treatment of patients who have failed surgery or are not surgical candidates, is most effective when UFC is less than twice the upper limit of normal. Seventy-three percent of patients treated with pasireotide develop glucose intolerance138 due to inhibition of incretin secretion, with a concomitant decrease in insulin secretion. Glucagon-like peptide-1 agonists or dipeptidyl peptidase-4 inhibitors can effectively control glucose levels in this setting.140
Mifepristone (RU-486, Korlym), a glucocorticoid and progesterone-receptor antagonist, has been approved in the US for control of hyperglycemia secondary to hypercortisolism in patients with Cushing’s syndrome who have failed surgery or are not surgical candidates. It has a rapid onset of action, and can be used to treat acute complications of Cushing’s syndrome, especially cortisol-induced psychosis.141,142 It is important to note that cortisol and ACTH levels remain unchanged or increase with the use of mifepristone, and cannot be used to gauge treatment success or to diagnose adrenal insufficiency. Moreover, increases in ACTH and cortisol levels can lead to (worsening of) mineralocorticoid effects (hypertension, edema, hypokalemia), and women can develop endometrial thickening during prolonged treatment.141,142
Other agents currently under (preclinical) study for the medical control of hypercortisolemia include LCI699 (an 11β-hydroxylase inhibitor), retinoic acid, and gefitinib (tyrosine-kinase inhibitor with EGFR [epidermal growth factor receptor] as target) (Table 3).143–145
Combination therapy with different agents may be needed to achieve normal plasma cortisol levels in patients with moderate-to-severe hypercortisolism. This can reduce drug-related adverse events if lower combined doses are effective. A small study of 17 patients showed an overall response rate of 88% after sequential addition of ketoconazole and/or cabergoline when initial pasireotide therapy was not successful.146 Overall, medical treatment in Cushing’s syndrome needs to be individualized according to patient characteristics, potential side effects, and other pharmacological properties of the drugs.127
Conclusion
Cushing’s syndrome, a rare disorder, is associated with significant morbidity and mortality. Clinical presentation can be broad, and establishing the diagnosis can be difficult. Early recognition and rapid control of hypercortisolemia is necessary to decrease morbidity and mortality in these patients. Surgery remains the optimal treatment in all forms of Cushing’s syndrome, but may not be curative. Individualized medical treatment and a multidisciplinary approach are needed for optimal control of hypercortisolemia and management of comorbidities. New alternative modalities of medical treatment are needed.
Disclosure
STS reports no conflicts of interest in this work. LKN has received funding from Laboratoire-HRA Pharma as part of a Cooperative Research and Development Agreement to conduct research on the antiglucocorticoid agent, mifepristone. RAF has received research grants from Novartis.
References
Newell-Price J, Bertagna X, Grossman AB, Nieman LK. Cushing’s syndrome. Lancet. 2006;367:1605–1617. | |
Hopkins RL, Leinung MC. Exogenous Cushing’s syndrome and glucocorticoid withdrawal. Endocrinol Metab Clin N Am. 2005;34:371–384. | |
Lindholm J, Juul S, Jørgenson JO, et al. Incidence and late prognosis of Cushing’s syndrome: a population-based study. J Clin Endocrinol Metab. 2001;86:117–123. | |
Etxabe J, Vazquez JA. Morbidity and mortality in Cushing’s disease: an epidemiological approach. Clin Endocrinol (Oxf). 1994;40:479–484. | |
Feelders RA, Pulgar SJ, Kempel A, Pereira AM. The burden of Cushing’s disease: clinical and health-related quality of life aspects. Eur J Endocrinol. 2012;167:311–326. | |
Ntali G, Asimakopoulou A, Siamatras T, et al. Mortality in Cushing’s syndrome: systematic analysis of a large series with prolonged follow-up. Eur J Endocrinol. 2013;169:715–723. | |
Colao A, Pivonello R, Spiezia S, et al. Persistence of increased cardiovascular risk in patients with Cushing’s disease after five years of successful cure. J Clin Endocrinol Metab. 1999;84:2664–2672. | |
Faggiano A, Pivonello R, Spiezia S, et al. Cardiovascular risk factors and common carotid artery caliber and stiffness in patients with Cushing’s disease during active disease and 1 year after disease remission. J Clin Endocrinol Metab. 2003;88:2527–2533. | |
Dekkers OM, Horváth-Puhó E, Jørgensen JO, et al. Multisystem morbidity and mortality in Cushing’s syndrome: a cohort study. J Clin Endocrinol Metab. 2013;98:2277–2284. | |
Leibowitz G, Tsur A, Chayen SD, et al. Pre-clinical Cushing’s syndrome: an unexpected frequent cause of poor glycaemic control in obese diabetic patients. Clin Endocrinol (Oxf). 1996;44:717–722. | |
Catargi B, Rigalleau V, Poussin A, et al. Occult Cushing’s syndrome in type-2 diabetes. J Clin Endocrinol Metab. 2003;88:5808–5813. | |
Omura M, Saito J, Yamaguchi K, Kakuta Y, Nishikawa T. Prospective study on the prevalence of secondary hypertension among hypertensive patients visiting a general outpatient clinic in Japan. Hypertens Res. 2004;27:193–202. | |
Terzolo M, Reimondo G, Chiodini I, et al. Screening of Cushing’s syndrome in outpatients with type 2 diabetes: results of a prospective multicentric study in Italy. J Clin Endocrinol Metab. 2012;97:3467–3475. | |
Mullan K, Black N, Thiraviaraj A, et al. Is there value in routine screening for Cushing’s syndrome in patients with diabetes? J Clin Endocrinol Metab. 2010;95:2262–2265. | |
Reimondo G, Pia A, Allasino B, et al. Screening of Cushing’s syndrome in adult patients with newly diagnosed diabetes mellitus. Clin Endocrinol (Oxf). 2007;67:225–229. | |
Newsome S, Chen K, Hoang J, Wilson JD, Potter JM, Hickman PE. Cushing’s syndrome in a clinic population with diabetes. Intern Med J. 2008;38:178–182. | |
Gagliardu L, Chapman IM, Loughlin PO, Torpy DJ. Screening for subclinical Cushing’s syndrome in type 2 diabetes mellitus: low false-positive rates with nocturnal salivary cortisol. Horm Metab Res. 2010;42:280–284. | |
Baid SK, Rubino D, Sinaii N, Ramsey S, Frank A, Nieman LK. Specificity of screening tests for Cushing’s syndrome in an overweight and obese population. J Clin Endocrinol Metab. 2009;94:3857–3864. | |
Nieman LK, Biller BM, Findling JW, et al. The diagnosis of Cushing’s syndrome: an Endocrine Society Clinical Practice Guideline. J Clin Endocrinol Metab. 2008;93:1526–1540. | |
Herrara MF, Grant CS, van Heerden JA, Sheedy PF, Ilstrup DM. Incidentally discovered adrenal tumors: an institutional perspective. Surgery. 1991;110:1014–1021. | |
Bovio S, Cataldi A, Reimondo G, et al. Prevalence of adrenal incidentaloma in a contemporary computerized tomography series. J Endocrinol Invest. 2006;29:298–302. | |
Nieman LK. Approach to the patient with an adrenal incidentaloma. J Clin Endocrinol Metab. 2010;95:4106–4113. | |
Mantero F, Terzolo M, Arnaldi G, et al. A survey on adrenal incidentaloma in Italy. Study Group on Adrenal Tumors of the Italian Society of Endocrinology. J Clin Endocrinol Metab. 2000;85:637–644. | |
Ilias I, Torpy DJ, Pacak K, et al. Cushing’s syndrome due to ectopic corticotrophin secretion: twenty years experience at National Institutes of Health. J Clin Endocrinol Metab. 2005;90:4955–4962. | |
Lacroix A, Bourdeau I. Bilateral adrenal Cushing’s syndrome: macronodular adrenal hyperplasia and primary pigmented nodular adrenocortical disease. Endocrinol Metab Clin N Am. 2005;34:441–458. | |
Louiset E, Duparc C, Young J, et al. Intraadrenal corticotrophin in bilateral macronodular adrenal hyperplasia. N Eng J Med. 2013;369:2115–2125. | |
Lacroix A. Heredity and cortisol regulation in bilateral macronodular adrenal hyperplasia. N Eng J Med. 2013;369:2147–2149. | |
Storr HL, Chan LF, Grossman AB, Savage MO. Paediatric Cushing’s syndrome: epidemiology, investigation and therapeutic advances. Trends Endocrinol Metab. 2007;18:167–174. | |
Pecori Giraldi F, Moro M, Cavagnini F. Gender-related differences in the presentation and course of Cushing’s disease. J Clin Endocrinol Metab. 2003;88:1554–1558. | |
Faggiano A, Pivonello R, Melis D, et al. Nephrolithiasis in Cushing’s disease: prevalence, etiopathogenesis, and modification after disease cure. J Clin Endocrinol Metab. 2003;88:2076–2080. | |
Newell-Price J, Trainer P, Besser M, Grossman A. The diagnosis and differential diagnosis of Cushing’s syndrome and pseudo-Cushing’s states. Endocr Rev. 1998;19:647–672. | |
Greening JE, Storr HL, McKenzie SA, et al. Linear growth and body mass index in pediatric patients with Cushing’s disease or simple obesity. J Endocrinol Invest. 2006;29:885–887. | |
Magiakou MA, Mastorakos G, Oldfield EH, et al. Cushing’s syndrome in children and adolescents. Presentation, diagnosis, and therapy. N Engl J Med. 1994;331:629–636. | |
Storr HL, Isidori AM, Monson JP, Besser GM, Grossman AB, Savage MO. Prepubertal Cushing’s disease is more common in males, but there is no increase in severity at diagnosis. J Clin Endocrinol Metab. 2004;89:3818–3820. | |
Torpy DJ, Mullen N, Ilias I, Nieman LK. Association of hypertension and hypokalemia with Cushing’s syndrome caused by ectopic ACTH secretion: a series of 58 cases. Ann N Y Acad Sci. 2002;970:134–144. | |
Sarlis NJ, Chanock SJ, Nieman LK. Cortisolemic indices predict severe infections in Cushing syndrome due to ectopic production of adrenocorticotropin. J Clin Endocrinol Metab. 2000;85:42–47. | |
Isidori AM, Kaltsas GA, Pozza C, et al. The ectopic adrenocorticotropin syndrome: clinical features, diagnosis, management, and long-term follow-up. J Clin Endocrinol Metab. 2006;91:371–377. | |
Minetto M, Reimondo G, Osella G, Ventura M, Angeli A, Terzolo M. Bone loss is more severe in primary adrenal than in pituitary-dependent Cushing’s syndrome. Osteoporosis Int. 2004;15:855–861. | |
Ohmori N, Nomura K, Ohmori K, Kato Y, Itoh T, Takano K. Osteoporosis is more prevalent in adrenal than in pituitary Cushing’s syndrome. Endocr J. 2003;50:1–7. | |
Trementino L, Appolloni G, Ceccoli L, et al. Bone complications in patients with Cushing’s syndrome: looking for clinical, biochemical, and genetic determinants. Osteoporos Int. 2014;25:913–921. | |
Valassi E, Santos A, Yaneva M, et al. The European Registry on Cushing’s syndrome: 2-year experience. Baseline demographic and clinical characteristics. Eur J Endocrinol. 2011;165:383–392. | |
Starkman MN, Schteingart DE. Neuropsychiatric manifestations of patients with Cushing’s syndrome. Relationship to cortisol and adrenocorticotropic hormone levels. Arch Intern Med. 1981;41:215–219. | |
Haskett RF. Diagnostic categorization of psychiatric disturbance in Cushing’s syndrome. Am J Psychiatry. 1985;142:911–916. | |
Dorn LD, Burgess ES, Dubbert B, et al. Psychopathology in patients with endogenous Cushing’s syndrome: ‘atypical’ or melancholic features. Clin Endocrinol (Oxf). 1995;43:433–442. | |
Pereira AM, Tiemensma J, Romijn JA. Neuropsychiatric disorders in Cushing’s syndrome. Neuroendocrinology. 2010;92 Suppl 1:65–70. | |
Bourdeau I, Bard C, Noel B, et al. Loss of brain volume in endogenous Cushing’s syndrome and its reversibility after correction of hypercortisolism. J Clin Endocrinol Metab. 2002;87:1949–1954. | |
Starkman MN, Gebarski SS, Berent S, Schteingart DE. Hippocampal formation volume, memory dysfunction, and cortisol levels in patients with Cushing’s syndrome. Biol Psychiatry. 1992;32:756–765. | |
Mandel S. Steroid myopathy. Insidious cause of muscle weakness. Postgrad Med. 1982;72:207–210, 213–215. | |
Rebuffé-Scrive M, Krotkiewski M, Elfverson J, Björntorp P. Muscle and adipose tissue morphology and metabolism in Cushing’s syndrome. J Clin Endocrinol Metab. 1988;67:1122–1128. | |
Lindsay JR, Nansel T, Baid S, Gumowski J, Nieman LK. Long-term quality of life in Cushing’s syndrome despite initial improvement after surgical improvement after surgical remission. J Clin Endocrinol Metab. 2006;91:447–453. | |
Pikkarainen L, Sane T, Reunsnen A. The survival and well-being of patients treated for Cushing’s syndrome. J Intern Med. 1999;245:463–468. | |
Mishra AK, Agarwal A, Gupta S, Agarwal G, Verma AK, Mishra SK. Outcome of adrenalectomy for Cushing’s syndrome: experience from a tertiary care center. World J Surg. 2007;31:1425–1432. | |
Feelders RA, Pulgar SJ, Kempel A, Pereira AM. The burden of Cushing’s disease: clinical and health-related quality of life aspects. Eur J Endocrinol. 2012;167:311–326. | |
Pivonello R, De Leo M, Vitale P, et al. Pathophysiology of diabetes mellitus in Cushing’s syndrome. Neuroendocrinology. 2010; 92 Suppl 1:77–81. | |
Sharma ST, Nieman LK. Cushing’s syndrome: all variants, detection, and treatment. Endocrinol Metab Clin North Am. 2011;40:379–391, viii–ix. | |
Arnaldi G, Scandali VM, Trementino L, Cardinaletti M, Appolloni G, Boscaro M. Pathophysiology of dyslipidemia in Cushing’s syndrome. Neuroendocrinology. 2010;92 Suppl 1:86–90. | |
Rockall AG, Sohaib SA, Evans D, et al. Hepatic steatosis in Cushing’s syndrome: a radiological assessment using computerized tomography. Eur J Endocrinol. 2003;149:543–548. | |
Rockall AG, Sohaib SA, Evans D, et al. Computed tomography assessment of fat distribution in male and female patients with Cushing’s syndrome. Eur J Endocrinol. 2003;149:561–567. | |
van der Pas R, Leebeek FW, Hofland LJ, de Herder WW, Feelders RA. Hypercoagulability in Cushing’s syndrome: prevalence, pathogenesis and treatment. Clin Endocrinol (Oxf). 2013;78:481–488. | |
Giordano R, Picu A, Marinazzo E, et al. Metabolic and cardiovascular outcomes in patients with Cushing’s syndrome of different aetiologies during active disease and 1 year after remission. Clin Endocrinol (Oxf). 2011;75:354–360. | |
Barahona MJ, Resmini E, Viladés D, et al. Coronary artery disease detected by multislice computed tomography in patients after long-term cure of Cushing’s syndrome. J Clin Endocrinol Metab. 2013;98:1093–1099. | |
Geer EB, Shen W, Strohmayer E, Post KD, Freda PU. Body composition and cardiovascular risk markers after remission of Cushing’s disease: a prospective study using whole-body MRI. J Clin Endocrinol Metab. 2012;97:1702–1711. | |
de Castro M, Moreira AC. Screening and diagnosis of Cushing’s syndrome. Arq Bras Endocrinol Metab. 2007;51:1191–1198. | |
Petersenn S, Newell-Price J, Findling JW, et al. High variability in baseline urinary free cortisol values in patients with Cushing’s disease. Clin Endocrinol (Oxf). 2014;80:261–269. | |
Alexandraki KI, Grossman AB. Is urinary free cortisol of value in the diagnosis of Cushing’s syndrome? Curr Opin Endocrinol Diabetes Obes. 2011;18:259–263. | |
Findling JW, Raff H, Aron DC. The low-dose dexamethasone suppression test: a reevaluation in patients with Cushing’s syndrome. J Clin Endocrinol Metab. 2004;89:1222–1226. | |
Raff H. Utility of salivary cortisol measurements in Cushing’s syndrome and adrenal insufficiency. J Clin Endocrinol Metab. 2009;94:3647–3655. | |
Gafni RI, Papanicolaou DA, Nieman LK. Nighttime salivary cortisol measurement as a simple, noninvasive, outpatient screening test for Cushing’s syndrome in children and adolescents. J Pediatr. 2000;137:30–35. | |
Raff H. Update on late-night salivary cortisol for the diagnosis of Cushing’s syndrome: methodological considerations. Endocrine. 2013;44:346–349. | |
Newell-Price J, Trainer P, Perry L, Wass J, Grossman A, Besser M. A single sleeping midnight cortisol has 100% sensitivity for the diagnosis of Cushing’s syndrome. Clin Endocrinol (Oxf). 1995;43:545–550. | |
Reimondo G, Allasino B, Bovio S, Paccotti P, Angeli A, Terzolo M. Evaluation of the effectiveness of midnight serum cortisol in the diagnostic procedures for Cushing’s syndrome. Eur J Endocrinol. 2005;153:803–809. | |
Papanicolaou DA, Yanovski JA, Cutler GB Jr, Chrousos GP, Nieman LK. A single midnight serum cortisol measurement distinguishes Cushing’s syndrome from pseudo-Cushing states. J Clin Endocrinol Metab. 1998;83:1163–1167. | |
Yanovski JA, Cutler GB Jr, Chrousos GP, Nieman LK. The dexamethasone-suppressed corticotropin-releasing hormone stimulation test differentiates mild Cushing’s disease from normal physiology. J Clin Endocrinol Metab. 1998;83:348–352. | |
Yanovski JA, Cutler GB Jr, Chrousos GP, Nieman LK. Corticotropin- releasing hormone stimulation following low-dose dexamethasone administration. A new test to distinguish Cushing’s syndrome from pseudo-Cushing’s states. JAMA. 1993;269:2232–2238. | |
Tirabassi G, Faloia E, Papa R, Furlani G, Boscaro M, Arnaldi G. Use of the desmopressin test in the differential diagnosis of pseudo-Cushing state from Cushing’s disease. J Clin Endocrinol Metab. 2010;95:1115–1122. | |
Moro M, Putignano P, Losa M, Invitti C, Maraschini C, Cavagnini F. The desmopressin test in the differential diagnosis between Cushing’s disease and pseudo-Cushing states. J Clin Endocrinol Metab. 2000;85:3569–3574. | |
Nieman L. Editorial: The dexamethasone-suppressed corticotropin-releasing hormone test for the diagnosis of Cushing’s syndrome: what we have learned in 14 years. J Clin Endocrinol Metab. 2007;92:2876–2878. | |
Valassi E, Swearingen B, Lee H, et al. Concomitant medication use can confound interpretation of the combined dexamethasone-corticotropin releasing hormone test in Cushing’s syndrome. J Clin Endocrinol Metab. 2009;94:4851–4859. | |
Alwani RA, Schmit Jongbloed LW, de Jong FH, Van der Lely AJ, de Herder W, Feelders RA. Differentiating between Cushing’s disease and pseudo-Cushing’s syndrome: comparison of four tests. Eur J Endocrinol. 2014;170:477–486. | |
Raff H, Findling JW. A new immunoradiometric assay for corticotropin evaluated in normal subjects and patients with Cushing’s syndrome. Clin Chem. 1989;35:596–600. | |
Horvath A, Stratakis C. Primary pigmented nodular adrenocortical disease and Cushing’s syndrome. Arq Bras Endocrinol Metabol. 2007;51:1238–1244. | |
Stratakis CA, Sarlis N, Kirschner LS, et al. Paradoxical response to dexamethasone in the diagnosis of primary pigmented nodular adrenocortical disease. Ann Intern Med. 1999;131:585–591. | |
Assié G, Libé R, Espiard S, et al. ARMC5 mutations in macronodular adrenal hyperplasia with Cushing’s syndrome. N Eng J Med. 2013;369:2105–2114. | |
Alencar GA, Lerario AM, Nishi MY, et al. ARMC5 mutations are a frequent cause of primary macronodular adrenal hyperplasia. J Clin Endocrinol Metab. 2014;99:E1501–E1509. | |
Faucz FR, Zilbermint M, Lodish MB, et al. Macronodular adrenal hyperplasia due to mutations in an armadillo repeat containing 5 (ARMC5) gene: a clinical and genetic investigation. J Clin Endocrinol Metab. 2014;99:E1113–E1119. | |
Patronas N, Bulakbasi N, Stratakis CA, et al. Spoiled gradient recalled acquisition in the steady state technique is superior to conventional postcontrast spin echo technique for magnetic resonance imaging detection of adrenocorticotropin-secreting pituitary tumors. J Clin Endocrinol Metab. 2003;88:1565–1569. | |
Wind JJ, Lonser RR, Nieman LK, Devroom HL, Chang R, Oldfield EH. The lateralization accuracy of inferior petrosal sinus sampling in 501 patients with Cushing’s disease. J Clin Endocrinol Metab. 2013;98:2285–2293. | |
Hall WA, Luciano MG, Doppman JL, Patronas NJ, Oldfield EH. Pituitary magnetic resonance imaging in normal human volunteers: occult adenomas in the general population. Ann Intern Med. 1994;120:817–820. | |
Dicheck AL, Nieman LK, Oldfield EH, Pass HI, Malley JD, Cutler GB Jr. A comparison of the standard high dose dexamethasone suppression test and the overnight 8-mg dexamethasone suppression test for the differential diagnosis of adrenocorticotropin-dependent Cushing’s syndrome. J Clin Endocrinol Metab. 1994;78:418–422. | |
Aron DC, Raff H, Findling JW. Effectiveness versus efficacy: the limited value in clinical practice of high dose dexamethasone suppression testing in the differential diagnosis of adrenocorticotropin-dependent Cushing’s syndrome. J Clin Endocrinol Metab. 1997;82:1780–1785. | |
Findling JW, Raff H. Diagnosis and differential diagnosis of Cushing’s syndrome. Endocrinol Metab Clin North Am. 2001;30:729–747. | |
Nieman LK, Oldfield EH, Wesley R, Chrousos GP, Loriaux DL, Cutler GB Jr. A simplified morning ovine corticotropin-releasing hormone stimulation test for the differential diagnosis of adrenocorticotropin-dependent Cushing’s syndrome. J Clin Endocrinol Metab. 1993;77:1308–1312. | |
Newell-Price J, Morris DG, Drake WM, et al. Optimal response criteria for the human CRH test in the differential diagnosis of ACTH-dependent Cushing’s syndrome. J Clin Endocrinol Metab. 2002;87:1640–1645. | |
Nieman LK, Cutler GB Jr, Oldfield EH, Loriaux DL, Chrousos GP. The ovine corticotropin-releasing hormone (CRH) stimulation test is superior to the human CRH stimulation test for the diagnosis of Cushing’s disease. J Clin Endocrinol Metab. 1989;69:165–169. | |
Trainer PJ, Faria M, Newell-Price J, et al. A comparison of the effects of human and ovine corticotropin-releasing hormone on the pituitary-adrenal axis. J Clin Endocrinol Metab. 1995;80:412–417. | |
Oldfield EH, Doppman JL, Nieman LK, et al. Petrosal sinus sampling with and without corticotropin-releasing hormone for the differential diagnosis of Cushing’s syndrome. N Eng J Med. 1991;325:897–905. | |
Swearingen B, Katznelson L, Miller K, et al. Diagnostic errors after inferior petrosal sinus sampling. J Clin Endocrinol Metab. 2004;89:3752–3763. | |
Doppman JL, Chang R, Oldfield EH, Chrousos G, Stratakis CA, Nieman LK. The hypoplastic inferior petrosal sinus: a potential source of false-negative results in petrosal sampling for Cushing’s disease. J Clin Endocrinol Metab. 1999;84:533–540. | |
López J, Barceló B, Lucas T, et al. Petrosal sinus sampling for diagnosis of Cushing’s disease: evidence of false negative results. Clin Endocrinol (Oxf). 1996;45:147–156. | |
Findling JW, Kehoe ME, Raff H. Identification of patients with Cushing’s disease with negative pituitary adrenocorticotropin gradients during inferior petrosal sinus sampling: prolactin as an index of pituitary venous effluent. J Clin Endocrinol Metab. 2004;89:6005–6009. | |
Mulligan GB, Eray E, Faiman C, et al. Reduction of false-negative results in inferior petrosal sinus sampling with simultaneous prolactin and corticotrophin measurement. Endocr Pract. 2011;17:33–40. | |
Sharma ST, Raff H, Nieman LK. Prolactin as a marker of successful catheterization during IPSS in patients with ACTH-dependent Cushing’s syndrome. J Clin Endocrinol Metab. 2011;96:3687–3694. | |
Shahani S, Nudelman RJ, Nalini R, Kim HS, Samson SL. Ectopic corticotropin-releasing hormone (CRH) syndrome from metastatic small cell carcinoma: a case report and review of the literature. Diagn Pathol. 2010;5:56. | |
Young J, Deneux C, Grino M, Oliver C, Chanson P, Schaison G. Pitfall of petrosal sinus sampling in a Cushing’s syndrome secondary to ectopic adrenocorticotropin-corticotropin releasing hormone (ACTH-CRH) secretion. J Clin Endocrinol Metab. 1998;83:305–308. | |
Oldfield EH, Chrousos GP, Schulte HM, et al. Preoperative localization of ACTH-secreting pituitary microadenomas by bilateral and simultaneous inferior petrosal venous sinus sampling. N Eng J Med. 1985;312:100–103. | |
Mulligan GB, Faiman C, Gupta M, et al. Prolactin measurement during inferior petrosal sinus sampling improves the localization of pituitary adenomas in Cushing’s disease. Clin Endocrinol (Oxf). 2012;77:268–274. | |
Zemskova MS, Gundabolu B, Sinaii N, et al. Utility of various functional and anatomic imaging modalities for detection of ectopic adrenocorticotropin-secreting tumors. J Clin Endocrinol Metab. 2010;95:1207–1219. | |
Kwekkeboom DJ, Krenning EP, Bakker WH, Oei HY, Kooij PP, Lamberts SW. Somatostatin analogue scintigraphy in carcinoid tumours. Eur J Nucl Med. 1993;20:283–292. | |
de Bruin C, Feelder RA, Waaijers AM, et al. Differential regulation of human dopamine D2 and somatostatin receptor subtype expression by glucocorticoids in vitro. J Mol Endocrinol. 2009;42:47–56. | |
de Bruin C, Hofland LJ, Nieman LK, et al. Mifepristone effects on tumor somatostatin receptor expression in two patients with Cushing’s syndrome due to ectopic adrenocorticotropin secretion. J Clin Endocrinol Metab. 2012;97:455–462. | |
Gabriel M, Decristoforo C, Kendler D, et al. 68Ga-DOTA-Tyr3-octreotide PET in neuroendocrine tumors: comparison with somatostatin receptor scintigraphy and CT. J Nucl Med. 2007;48:508–518. | |
Kayani I, Conry BG, Groves AM, et al. A comparison of 68Ga-DOTATATE and 18F-FDG PET/CT in pulmonary neuroendocrine tumors. J Nucl Med. 2009;50:1927–1932. | |
Jindal T, Kumar A, Venkitaraman B, Dutta R, Kumar R. Role of 68Ga-DOTATOC PET/CT in the evaluation of primary pulmonary carcinoids. Korean J Intern Med. 2010;25:386–391. | |
Pacak K, Ilias I, Chen CC, Carrasquillo JA, Whatley M, Nieman LK. The role of [(18)F]fluorodeoxyglucose positron emission tomography and [(111)In]-diethylenetriaminepentaacetate-D-Phe-pentetreotide scintigraphy in the localization of ectopic adrenocorticotropin-secreting tumors causing Cushing’s syndrome. J Clin Endocrinol Metab. 2004;89:2214–2221. | |
Kauhanen S, Seppänen M, Ovaska J, et al. The clinical value of [18F]fluoro-dihydroxyphenylalanine positron emission tomography in primary diagnosis, staging, and restaging of neuroendocrine tumors. Endocr Relat Cancer. 2009;16:255–265. | |
Orlefors H, Sundin A, Garske U, et al. Whole-body (11)C-5-hydroxytryptophan positron emission tomography as a universal imaging technique for neuroendocrine tumors: comparison with somatostatin receptor scintigraphy and computed tomography. J Clin Endocrinol Metab. 2005;90:3392–3400. | |
Doppman JL, Pass HI, Nieman LK, et al. Corticotropin-secreting carcinoid tumors of the thymus: diagnostic unreliability of thymic vein sampling. Radiology. 1992;184:71–74. | |
Woo YS, Isidori AM, Wat WZ, et al. Clinical and biochemical characteristics of adrenocorticotropin-secreting macroadenomas. J Clin Endocrinol Metab. 2005;90:4963–4969. | |
Rees DA, Hanna FW, Davies JS, Mills RG, Vafidis J, Scanlon MF. Long-term follow-up results of transsphenoidal surgery for Cushing’s disease in a single center using strict criteria for remission. Clin Endocrinol (Oxf). 2002;56:541–551. | |
Lindsay JR, Oldfield EH, Stratakis CA, Nieman LK. The postoperative basal cortisol and CRH tests for prediction of long-term remission from Cushing’s disease after transsphenoidal surgery. J Clin Endocrinol Metab. 2011;96:2057–2064. | |
Brunt LM, Moley JF, Doherty GM, Lairmore TC, DeBenedetti MK, Quasebarth MA. Outcomes analysis in patients undergoing laparoscopic adrenalectomy for hormonally active adrenal tumors. Surgery. 2001;130:629–635. | |
Ritzel K, Beuschlein F, Mickisch A, et al. Clinical review: outcome of bilateral adrenalectomy in Cushing’s syndrome: a systematic review. J Clin Endocrinol Metab. 2013;98(10):3939–3948. | |
Assié G, Bahurel H, Coste J, et al. Corticotroph tumor progression after adrenalectomy in Cushing’s disease: a reappraisal of Nelson’s syndrome. J Clin Endocrinol Metab. 2007;92:172–179. | |
Mehta GU, Sheehan JP, Vance ML. Effect of stereotactic radiosurgery before bilateral adrenalectomy for Cushing’s disease on the incidence of Nelson’s syndrome. J Neurosurg. 2013;119:1493–1497. | |
Estrada J, Boronat M, Mielgo M, et al. The long-term outcome of pituitary irradiation after unsuccessful transsphenoidal surgery in Cushing’s disease. N Eng J Med. 1997;336:172–177. | |
Sheehan JM, Vance ML, Sheehan JP, Ellegala DB, Laws ER Jr. Radiosurgery for Cushing’s disease after failed transsphenoidal surgery. J Neurosurg. 2000;93:738–742. | |
Feelders RA, Hofland LJ. Medical treatment of Cushing’s disease. J Clin Endocrinol Metab. 2013;98:425–438. | |
Engelhardt D, Weber MM. Therapy of Cushing’s syndrome with steroid biosynthesis inhibitors. J Steroid Biochem Mol Biol. 1994;49:261–267. | |
Lewis JH, Zimmerman HJ, Benson GD, Ishak KG. Hepatic injury associated with ketoconazole therapy. Analysis of 33 cases. Gastroenterology. 1984;86:503–513. | |
Castinetti F, Guignat L, Giraud P, et al. Ketoconazole in Cushing’s disease: is it worth a try? J Clin Endocrinol Metab. 2014;99:1623–1630. | |
European Medicines Agency. European Medicines Agency recommends suspension of marketing authorizations for oral ketoconazole throughout the European Union [press release]. London: EMA; July 26, 2013. Available from: http://www.ema.europa.eu/docs/en_GB/document_library/Press_release/2013/07/WC500146613.pdf. Accessed November 13, 2014. | |
Food and Drug Administration. FDA Drug Safety Communication: FDA limits usage of Nizoral (ketoconazole) oral tablets due to potentially fatal liver injury and risk of drug interactions and adrenal gland problems [press release]. Silver Spring (MD): FDA; July 26, 2013. Available from: http://www.fda.gov/Drugs/DrugSafety/ucm362415.htm. Accessed November 13, 2014. | |
Orth DN. Metyrapone is useful only as adjunctive therapy in Cushing’s disease. Ann Intern Med. 1978;89:128–130. | |
Valassi E, Crespo I, Gich I, Rodríguez J, Webb SM. A reappraisal of the medical therapy with steroidogenesis inhibitors in Cushing’s disease. Clin Endocrinol (Oxf). 2012;77:735–742. | |
Baudry C, Coste J, Bou KR, et al. Efficiency and tolerance of mitotane in Cushing’s disease in 76 patients from a single center. Eur J Endocrinol. 2012;167:473–481. | |
Preda VA, Sen J, Karavitaki N, Grossman AB. Etomidate in the management of hypercortisolaemia in Cushing’s syndrome: a review. Eur J Endocrinol. 2012;167:137–143. | |
de Bruin C, Pereira AM, Feelders RA, et al. Coexpression of dopamine and somatostatin receptor subtypes in corticotroph adenomas. J Clin Endocrinol Metab. 2009;94:1118–1124. | |
Colao A, Petersenn S, Newell-Price J, et al. A 12-month phase 3 study of pasireotide in Cushing’s disease. N Engl J Med. 2012;366:914–924. | |
Godbout A, Manavela M, Danilowicz K, Beauregard H, Bruno OD, Lacroix A. Cabergoline monotherapy in the long-term treatment of Cushing’s disease. Eur J Endocrinol. 2010;163:709–716. | |
Henry RR, Ciaraldi TP, Armstrong D, Burke P, Ligueros-Saylan M, Mudaliar S. Hyperglycemia associated with pasireotide: results from a mechanistic study in healthy volunteers. J Clin Endocrinol Metab. 2013;98:3446–3453. | |
Castinetti F, Fassnacht M, Johanssen S, et al. Merits and pitfalls of mifepristone in Cushing’s syndrome. Eur J Endocrinol. 2009;160:1003–1010. | |
Fleseriu M, Biller NM, Findling JW, Molitch ME, Schteingart DE. Mifepristone, a glucocorticoid receptor antagonist, produces clinical and metabolic benefits in patients with Cushing’s syndrome. J Clin Endocrinol Metab. 2012;97:2039–2049. | |
Bertagna X, Pivonello R, Fleseriu M, et al. LCI699, a potent 11β-hydroxylase inhibitor, normalizes urinary cortisol in patients with Cushing’s disease: results from a multicenter, proof-of-concept study. J Clin Endocrinol Metab. 2014;99:1375–1383. | |
Pecori Giraldi F, Ambrogio AG, Andrioli M, et al. Potential role for retinoic acid in patients with Cushing’s disease. J Clin Endocrinol Metab. 2012;97:3577–3583. | |
Fukuoka H, Cooper O, Ben-Shlomo A, et al. EGFR as a therapeutic target for human, canine, and mouse ACTH-secreting pituitary adenomas. J Clin Invest. 2011;121:4712–4721. | |
Feelders RA, de Bruin C, Pereira AM, et al. Pasireotide alone or with cabergoline and ketoconazole in Cushing’s disease. N Eng J Med. 2010;362:1846–1848. | |
Ross EJ, Linch DC. Cushing’s syndrome-killing disease: discriminatory value of signs and symptoms aiding early diagnosis. Lancet. 1982;2:646–649. | |
Yanovski JA, Cutler GB Jr. Glucocorticoid action and the clinical features of Cushing’s syndrome. Endocrinol Metab Clin North Am. 1994;23:487–509. | |
Weber A, Trainer PJ, Grossman AB, et al. Investigation, management and therapeutic outcome in 12 cases of childhood and adolescent Cushing’s syndrome. Clin Endocrinol (Oxf). 1995;43:19–28. | |
Soffer L, Iannaccone A, Gabrilove J. Cushing’s syndrome: a study of fifty patients. American Journal of Medicine. 1961; 30:129–146. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.