")
Back to Journals » Neuropsychiatric Disease and Treatment » Volume 15
Current Perspectives On The Role Of The Ketogenic Diet In Epilepsy Management
Authors Goswami JN, Sharma S
Received 27 August 2019
Accepted for publication 18 October 2019
Published 25 November 2019 Volume 2019:15 Pages 3273—3285
DOI https://doi.org/10.2147/NDT.S201862
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Roger Pinder
Jyotindra Narayan Goswami,1 Suvasini Sharma2
1Department of Pediatrics, Army Hospital (Research & Referral), New Delhi 110010, India; 2Neurology Division, Department of Pediatrics, Lady Hardinge Medical College and Associated Kalawati Saran Children’s Hospital, New Delhi 110001, India
Correspondence: Suvasini Sharma
Neurology Division, Department of Pediatrics, Lady Hardinge Medical College and Associated Kalawati Saran Children’s Hospital, Connaught Place 110001, New Delhi Tel +91-9910234344
Email [email protected]
Abstract: Drug-refractory epilepsy is a commonly prevalent pediatric neurological illness of global significance. Ketogenic diet (KD) is a time-tested therapeutic modality for refractory epilepsy, which has reemerged as a robust alternative to anti-epileptic pharmacotherapy. There is a growing body of evidence which supports the anti-seizure efficacy, safety profile and feasibility of KD use in childhood epilepsy. In addition, this modality has been recognized to reduce anti-epileptic exposure, improve cognition and behavioral profile of patients as well as improve the quality-of-life of care-givers. Current indications of KD include refractory epilepsy syndromes, selected metabolic disorders (such as pyruvate dehydrogenase deficiency) and a host of varied neurological entities. KD research has broadened the knowledge-base about its mechanisms of action. Four types of KD are in vogue currently with varying nutritional constitution, palatability, administration protocols and comparable efficacy. KD initiation and maintenance are the result of concerted effort of a team of pediatric neurologist/epileptologist, nutritionist and patient’s primary care-giver. Consensus is being formulated about various practical aspects of KD such as patient-selection, parental counseling, baseline work-up, dietary prescription, nutritional supplementation, concurrent anti-epileptic drug administration, follow-up and treatment-duration. Novel applications of KD include its use in neonatal epilepsy and super-refractory status epilepticus and tailor-made formulations such as cooking oil-based KD in predominantly rice-fed populations. Increasing body of clinical experience, improved nutritional designs and translational research are promoting KD as a major therapeutic modality. Currently, KD forms a core essence in the armamentarium against refractory epilepsy. In this review, we summarize the recent advances and current perspectives in the use of KD in refractory epilepsy.
Keywords: Modified Atkins Diet, low glycemic index diet, ketosis, intractable epilepsy, epileptic encephalopathies, ketogenic diet, epilepsy
Introduction
Epilepsy is a common neurological disorder encountered in pediatric practice, which accounts for nearly 1% of the global burden of disease across all ages.1 Nearly 30% of all children with epilepsy display drug-refractoriness.2 Management of children with drug-refractory epilepsy is a challenging proposition for health-care providers as well as primary caregivers. The adverse effects and often-unknown interactions between multiple drugs, frequent hospital visits, financial limitations and parental burnout are only few of the hurdles faced during the management of children with drug-refractory epilepsy. Epilepsy surgery is one of the the main treatment options for children and adults with refractory epilepsy. However, many children and adults with refractory epilepsy are not good surgical candidates. Also, epilepsy surgery is expensive, requires trained personnel and infrastructure, and hence is not easily available in many low resource settings.
Ketogenic diet (KD) is being increasingly accepted worldwide as a practical, effective and safe management alternative in children and adults with refractory epilepsy who are not good surgical candidates. KD is a high fat, low carbohydrate diet, which has been used for the last several decades for the treatment of refractory epilepsy in children.3 There has been a recent resurgence in its use. Now there are many less restrictive options available such as the Modified AtkinsDiet and the low glycemic index treatment. There is now emerging evidence of the use of KD and its variants in adults as well, and the use of KD in other non-epilepsy neurological conditions such as brain tumors, Alzheimer’s disease, Amyotrophic Lateral Sclerosis, Parkinson’s disease and autism. In this review, we discuss the current perspectives on the use of KD in children and adults with epilepsy.
Evidence Pertaining To The Use Of KD In Epilepsy
Preliminary evidence of efficacy of KD has been derived from uncontrolled studies of KD in drug-refractory epileptic children which revealed total seizure resolution in 16%, greater than 90% seizure-reduction in 32% and greater than 50% seizure reduction in 56%.4 Neal et al conducted an RCT wherein children with drug-resistant epilepsy were randomized to receive KD either at enrolment (study arm) or after three months (control-arm) in addition to the ongoing anti-epileptic drugs.5 on changes in the anti-epileptic drugs.5 Children who received early KD (n=54) had significantly decreased seizure burden after four months than the controls (n=49) with a 38% decrease in seizures compared to 37% increase in seizures (p< 0.0001).5
Current evidence pertaining to the benefits of KD in epilepsy has evolved from prospective and retrospective case-series, anecdotal reports and consensus opinions of experts. The 2018 Cochrane Review summarizes data from eleven Randomized Controlled Trials (RCTs) pertaining to KD. This encompasses a total of seven hundred and seventy eight patients including seven hundred and twelve children and sixty six adults.6 Maximum 'seizure freedom rate' and 'seizure reduction rate' seen with classical (4:1) KDwere 55% and 85% respectively noted three months after therapy initiation.6 Other less restrictive variants of KD, such as MAD, displayed similar seizure-control efficacy as classical KD, but the authors noted a need for more robust studies.6
Indications Of KD
KD initiation requires careful consideration of many factors, as it is a long-term therapy requiring concerted effort of health-care providers, parents and patient cooperation. Therefore, indications for starting KD need to be well defined.
KD is generally preferred as a therapeutic modality in childhood epilepsy after the failure of multiple drugs in controlling seizures. There is a growing opinion among epileptologists that KD should be considered after the failure of two adequately chosen anti-epileptic drugs used in appropriate dosage.7,8 It is preferred as the first line of therapy in deficiency of Glucose Transporter Type 1 and Pyruvate Dehydrogenase Deficiency.7 Other indications of KD include refractory epilepsy syndromes such as Ohtahara Syndrome, West Syndrome, Lennox-Gastaut Syndrome, Dravet Syndrome, Epilepsy with myoclonic-atonic seizures and Tuberous Sclerosis Complex.9 Multiple trials have demonstrated that KD is an effective treatment modality in children with Dravet Syndrome and hence it needs to be considered early in the course of therapy.8,10–12 Caraballo et al performed a retrospective study of children with Dravet Syndrome where it was observed that ten out of thirteen children on KD had significant seizure reduction at twelve months of therapy initiation.13 Dressler et al retrospectively studied the efficacy of KD in thirty-two children with Dravet Syndrome wherein they found that the efficacy of this modality was not significantly inferior to the gold standard anti-epileptic drug triple combination of stiripentol, clobazam and valproate and was significantly more effective than Levetiracetam alone.14 The Italian consensus guidelines advocate early initiation of KD in the treatment of Dravet Syndrome.8 KD has also been tried in variety of other conditions such as Febrile Infection Related Epilepsy Syndrome (FIRES), Angelman Syndrome, Rett Syndrome and tuberous sclerosis complex.7
Types Of KD In Current Practice
KD prescribed currently is of four main types (Table 1).
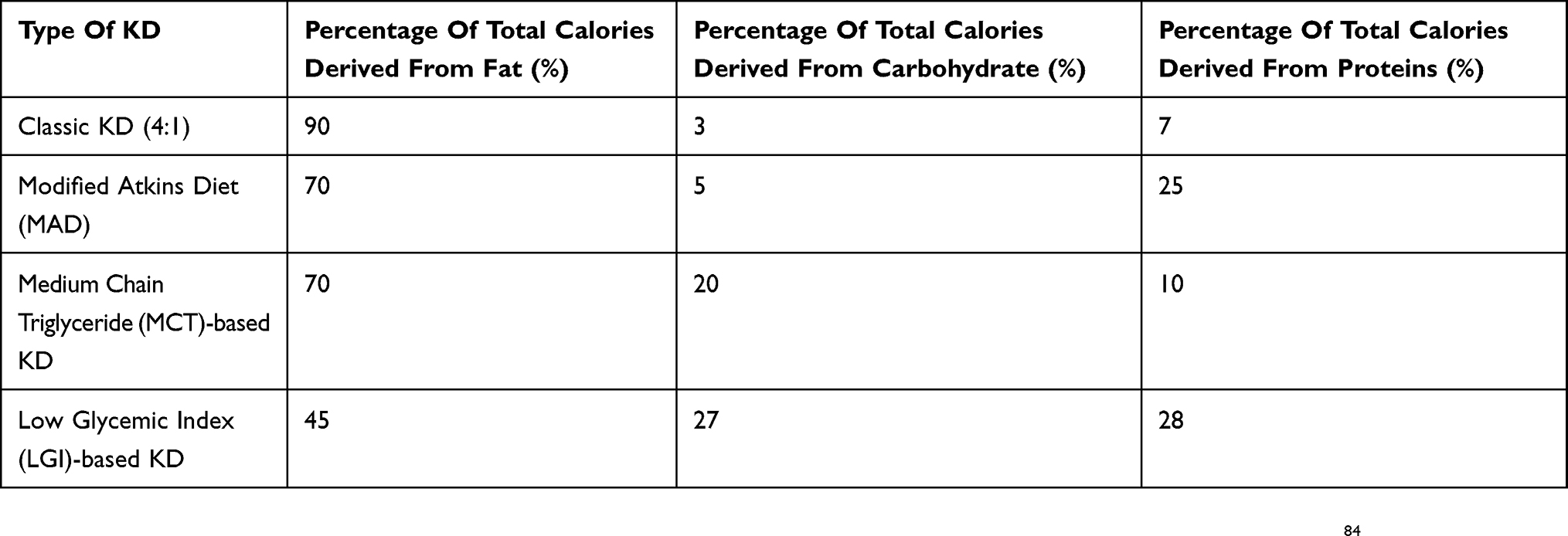 |
Table 1 Composition Of Various Types Of Ketogenic Diet |
Classic KD
The classic KD is the conventionally prescribed KD, which has a ketogenic ratio of 4:1. “Ketogenic ratio” refers to the ratio of weight of fat to the combined weight of carbohydrates and proteins in a diet.7,15 The primary challenges involved in prescribing classic KD account from its highly restrictive nature and the requirement of hospitalization at initiation that is practiced by most centers. Lower ratios such as 2.5:1 have been used and shown to have equivalent efficacy and better tolerability.7,16
Medium-Chain Triglyceride (MCT) KD
This variant of KD was developed by Huttenlocher et al and is based on the provision of calories predominantly through MCT.17 MCT-based KD, conceived in the 1950s, has more ketogenic potential than classic KD as MCT breaks down and gets absorbed rapidly, directly reaching the liver unlike long-chain triglycerides.15 MCT-based KD is less restrictive and more palatable than classic KD as it allows for consumption of more carbohydrates and calories.8,15,17,18 Neal EG et al compared classic KD with MCT-based KD in an RCT involving one hundred and forty-five children with intractable epilepsy, wherein both the dietary therapies were found to be comparable in their efficacy and tolerability.5 Common adverse effects of MCT-based KD are abdominal discomfort and bloating sensation, which is responsible for discontinuation of therapy in majority of children.9,19
Modified Atkins Diet (MAD)
MAD, initially described in 2003, is a less restrictive KD where fat provides about 65% of the calories.10,20 MAD initiation does not entail prior hospitalization, initial fasting or restriction of calories, fluids or proteins.8,15 In this form of dietary therapy, children are encouraged to consume large amount of fats in the form of butter, ghee or cream with no active protein restriction.15 In a randomized controlled trial conducted in India, the efficacy of MAD was evaluated in children with refractory epilepsy who were randomized to receive either MAD or no dietary therapy in addition to the ongoing medications for three months.21 MAD was found to be effective and well tolerated.21
MAD has also been found useful in children with Lennox-Gastaut Syndrome.22 Kim et al had found MAD to be of comparable efficacy as Classic KD in children with refractory epilepsy older than 2 years of age.23 However, classic KD was significantly more effective in children younger than two years of age.23
The liberal nature of MAD and its ease of administration makes it a promising choice, especially for resource-limited settings as has been demonstrated through an Indian study of a simplified MAD which could be used by parents with low literacy levels.24 In the West, the MAD has been used predominantly in adolescents and adults. However, it has been found useful even in infants and young children and is a more feasible option than KD in these populations in low resource settings.25,26
Low Glycemic Index (LGI) KD
LGI KD, pioneered by Pfeifer and Thiele in 2005, composed of 60% fats, 10%carbohydrates and 30% proteins.27 This is a less restrictive form of dietary therapy where children are allowed to take carbohydrates with LGI such as strawberries and whole grain, whereas foodstuffs like potatoes and rice are prohibited.27
LGI-KD produces minimal ketosis compared to classic KD with equivalent anti-seizure efficacy. There are no head-to-head trials of LGIT versus other KDs in children with refractory epilepsy.28 This variant of KD has been found to be particularly effective in controlling seizures in patients with Angelman Syndrome.29,30,31 Sondhi et al, in their RCT, randomized one hundred and seventy children (1–15 years) with drug-refractory epilepsy to receive KD, MAD or LGIT wherein they observed that MAD and LGIT were not non-inferior to KD though LGIT demonstrated >50% seizure reduction with a better safety profile.29 Kim et al found MAD may have similar seizure control as classic KD in children with refractory epilepsy older than 2 years of age.23
Current Perspectives About The Mechanisms Of Action Of KD
The exact mechanism of action of KD is open to controversy and hence, a subject of active research. Current perspectives pertaining to the mechanism of action of KD have been summarized in Table 2. Glucose is the chief source of energy in humans during basal conditions. During fasting or starvation, ketone bodies (acetone, acetoacetate and Gamma hydroxybutyrate) are formed in the liver, which converts to acetyl coenzyme A that enters the citric acid cycle and gets oxidized in mitochondria to provide energy as an alternative to glucose.32 The direct role of acetoacetate in seizure control has been demonstrated in animal models.32 Ketone bodies may act through activation of various synergistic pathways that are still under evaluation. Different mechanisms that have been hypothesized include potassium channel activation leading to membrane hyperpolarization, positive feedback to GABAergic channels, negative feedback to Glutamatergic channels.33 KD leads to elevated blood levels of polyunsaturated fatty acids (PUFAs) such as Docosahexaenoic acid, arachidonic acid and eicosapentaenoic acid. 33 As PUFAs are known to inhibit voltage-gated sodium channels and L-type calcium channels, it was presumed to be one of the possible mechanisms of action of KD in decreasing seizures.33 This hypothesis was refuted by studies that failed to reveal any additional improvement with KD fortified with PUFA.34,35 Notwithstanding the above, PUFAs are known to activate peroxisome proliferator-activated receptors (PPARs), which may have anti-seizure activity.36
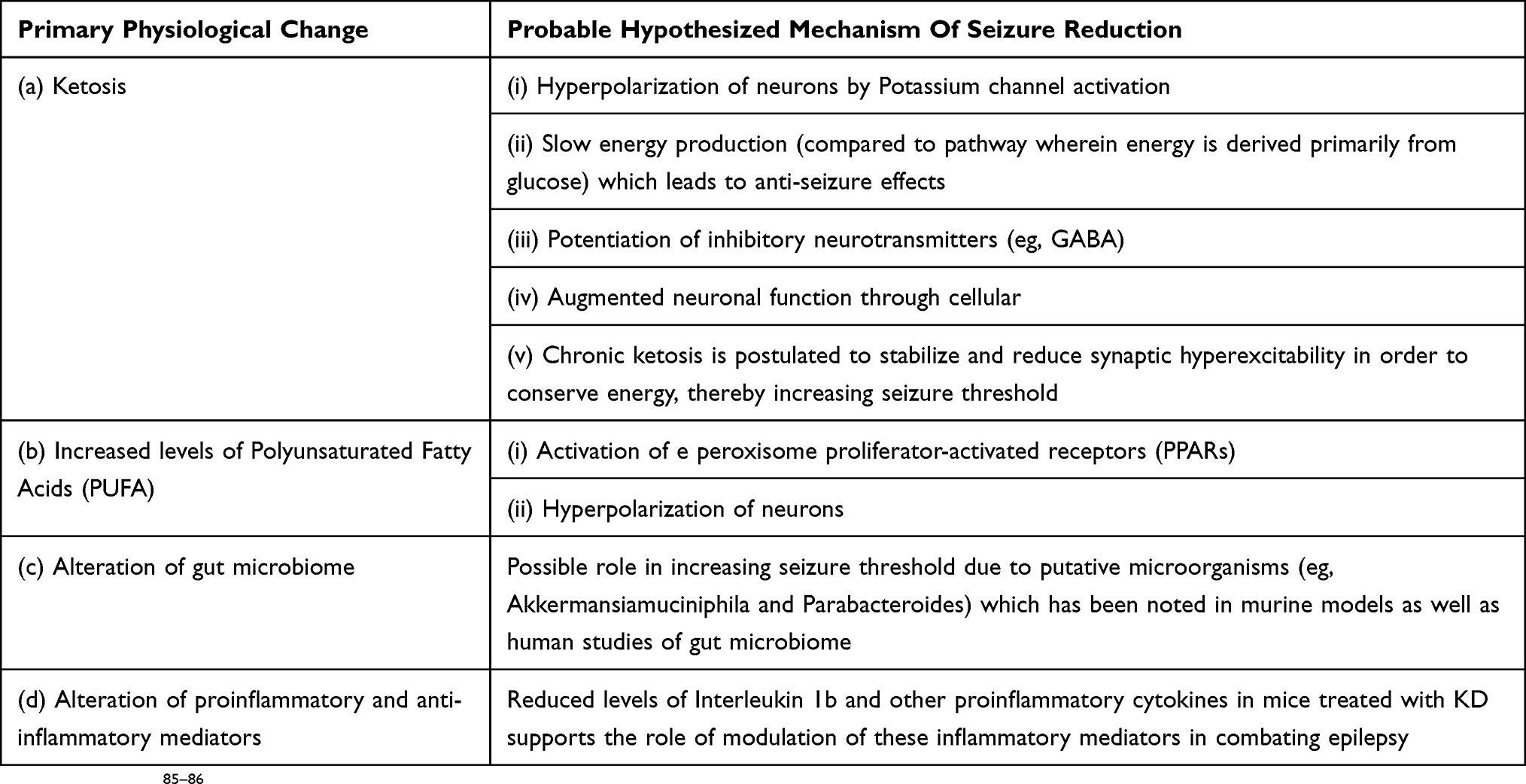 |
Table 2 Probable Anti-Seizure Mechanisms Of KD |
Gut microbiome has gained popularity as one of the major players that may possibly have anti-epileptic properties. Studies in mice and humans have shown alteration of gut microbiota with KD, namely decrease in Actinobacterial load, which might be responsible for anti-seizure effects.37–39 It has been demonstrated in mouse models that KD leads to increased gut population of Akkermansia muciniphila and Parabacteroides merdae which probably confer anti-seizure protection.41 When these bacteria are transplanted into germ-free mice, they may induce seizure protection.41 These bacteria have been found to decrease gamma-glutamylated ketogenic amino acids leading to neurotransmitter modulation, which has anti-seizure effects.37–39
Human studies have revealed that KD alters gut microbiome by varying degrees, notable being the selective increase in Bacteroides and decrease in Firmicutes and Actinobacteria in those individuals who have good seizure control.37,39 As these changes have been noted in persons having good seizure control with KD, it has been hypothesized that KD may exert anti-seizure effects through modulation of gut microbiota.
Dysregulation of the mammalian target of rapamycin (mTOR) signaling pathway has been found to be one of the key regulators responsible for intractable epilepsy, especially in conditions such as Tuberous Sclerosis Complex (TSC) and cortical malformations.40 It has been demonstrated through rat studies that KD inhibits mTOR pathway in healthy rats and prevents late mTOR activation after Kainic Acid induced status epilepticus.40 Hence, mTOR pathway inhibition may be an important additional mechanism of action of KD.
Contraindications For KD
KD can be fatal for children with metabolic disorders that impair energy generation from lipids.7 Hence, fatty acid oxidation disorders, primary carnitine deficiency, Carnitine Palmitoyl Transferase (CPT) deficiency Type I and Type II, Carnitine Translocase Deficiency, Pyruvate Carboxylase Deficiency and porphyria are absolute contraindications for initiation of KD.7,8 Parental and patient non-compliance, lesional epilepsy amenable to surgical resection and use of propofol are relative contraindications for KD.7 The contraindications for KD are summarized in Table 3.
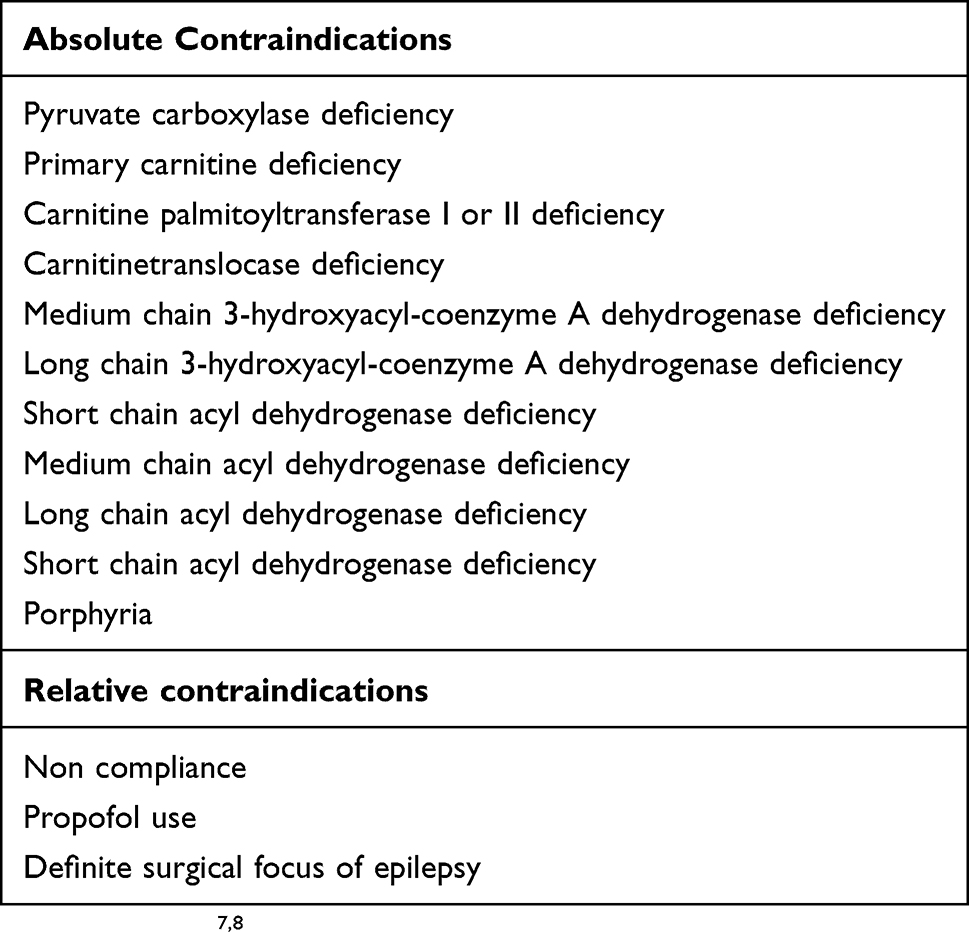 |
Table 3 Contraindications For Ketogenic Diet |
Pre-Requisites To Be Fulfilled Before KD Initiation
Prescription of KD is a concerted decision which requires interaction between the treating pediatric neurologist/epileptologist, nutritionist/dietary counselor and patient`s primary caregiver. The International Ketogenic Diet Study Group recommends structured counseling, meticulous nutritional assessment and certain mandatory laboratory investigations prior to initiation of KD.7 The pre-requisites that need to be fulfilled prior to initiation of KD are noted below:
Parental Counseling Prior To Initiation Of KD
Various factors that are taken into account before considering KD include patient characteristics such as age, epilepsy syndrome, medications used, underlying metabolic illness (if any), financial issues, social issues and parental motivation. Multiple counseling sessions are held in order to educate the parents and to assess their readiness and competence to handle KD therapy. Group counseling, handouts, training videos and KD support groups have an immense role in the counseling process. In resource-limited settings with high prevalence of illiteracy, counseling needs to be tailored accordingly.26 Counseling precedes the process of obtaining informed consent from parents prior to KD initiation.
Nutritional Assessment
Nutritionists constitute an integral part of any KD therapy center. The nutritionist assesses the dietary requirements of a prospective KD candidate and also looks into any food allergies, practicability and social issues before designing and advising a particular diet. Protocols may differ from center to center. Numerous diet calculators and mobile apps are available which aid in the process of obtaining individualized dietary calculation.
Baseline Investigations
Mandatory investigations that are obtained prior to initiation of KD include hemogram, liver and kidney function tests, serum electrolytes, serum vitamin D level complete lipid profile and serum acylcarnitine level.7,8,33,42 Additional investigations are advised as warranted on individualized basis.7
KD Initiation: Basic Protocols
Traditionally, most centers hospitalized children prior to initiation of classic KD. Admission is utilized to counsel caretakers, perform thorough nutritional estimation, do baseline biochemical monitoring and assess for the initial adverse effects of KD. In the earlier protocols, children were made to fast for 12 to 24 hrs prior to initiation of KD.7 However, current consensus recommendations allow for a liberal approach and do not advocate fluid and carbohydrate restriction at initiation of KD.7 Calories are progressively increased while maintaining the ratio of energy provided by fats and carbohydrates. The practice of calorie build-up in KD differs among centers. Conservative schools advocate gradual escalation of KD ratio from 1:1 to 4:1 by increasing relative quantity of fat in diet over few days. However, initiation of total fats on Day 1 may be equally efficacious and safe as per current evidence pertaining to KD in infants.42 The current trend shows a shift towards domiciliary KD therapy in view of the inherent shortfalls of hospitalization. European consensus is in favor of admission of infants prior to KD initiation.34,43 MAD and LGIT are usually initiated on outpatient basis. Primary caregivers are pivotal in the success of any dietary therapy and hence, these regimes are initiated after accessing the motivation, capability and interest of the primary-caregivers. They are initially called at close intervals till they gain confidence in sustaining the diet at home. MAD and LGIT are initiated without prior fasting or fluid restriction.
KD is usually tailor-made to a child depending on his age, food acceptance, socio-economic factors, parental convenience and experience of the center. There is a burgeoning growth of pre-fabricated, ready-to-eat KD formulations such as breakfast cereals and keto-snacks. These dietary options appear attractive but need to be validated by evidence and feasibility studies from an economic standpoint.
International KD Study Group recommendation advocates daily supplementation of multivitamin, calcium, vitamin D for children on KD.7 There are mixed opinions about the supplementation of oral citrate to counteract kidney stone formation during KD therapy.7,44
Follow-Up Plan During KD Therapy
KD is a long-term therapy, which warrants regular follow-up. Follow-up protocols vary from center to center. At the initiation phase, daily follow-up may be required. Serum ketone (especially, beta hydroxyl butyrate) monitoring, though more accurate, may not be feasible as it is more expensive. An exclusive KD clinic with epileptologist/pediatric neurologist and nutritionist is a pre-requisite for KD therapy. Additional aids like videos, app-based platforms and patient helpline numbers may make the maintenance of therapy more practical. The frequency of follow-up is commonly monthly at the onset, which may be subsequently spaced out to about three-monthly follow-ups.7 Infants may require prolonged close follow-up.7
At each clinic visit, special attention needs to be given to the frequency and pattern of seizures, difficulties faced in diet administration by parents, adverse effects and daily urinary ketone records. Parental concerns are addressed and diet is modified if required. Anti-seizure drugs are modified or continued as mandated. The child is examined in detail during every visit with special attention to anthropometry and signs of nutritional deficiency.
Laboratory tests that are obtained periodically include hemogram, liver and renal function tests, serum electrolytes, calcium, vitamin D level and lipid profile. These are advocated frequently at onset and less frequently subsequently. International KD study group recommends three-monthly investigations in the initial six months of KD therapy.7 These include complete hemogram, liver function test, renal function test, serum calcium, magnesium, serum vitamin D level and fasting lipid profile.7 Ultrasonographic screening for renal calculi, bone densitometry, electroencephalography (EEG) and serum carnitine level are performed at less frequent intervals.7
Adverse Effects Of KD
Various adverse effects of KD are as follows:
Gastrointestinal
Nearly 50% of the children started on KD have gastrointestinal adverse effects such as nausea, vomiting, diarrhea, pain abdomen, exacerbation of gastroesophageal reflux and constipation, especially during the initiation phase.7,45
Metabolic Abnormalities
Biochemical alterations that may be seen with KD include hypercholesterolemia, hypertriglyceridemia and depressed levels of low-density lipoprotein (LDL).46 Lin et al had noted hypoglycemia in about one-fourth of children during the first week of therapy of KD initiation in a retrospective study of one hundred and fifty-eight children.45
Renal
As high as 3–7% of the children on KD therapy may develop renal calculi, the risk of which is reportedly decreased by co-administration of oral citrates.47
Growth
The impact of KD on growth is a subject of intense research, which has yielded mixed findings.7,48 Decreased linear growth has been reported in children exposed to KD continuously for more than six months in six retrospective studies.48–53
On the contrary, Tagliabue et al studied eighteen children on KD prospectively for six months wherein they did not find any change in their weight or height.54
Cardiovascular
Speculations of detrimental cardiovascular outcomes in children on long-term KD have not been substantiated in studies on coronary intimal thickness.55
Prolonged QT interval and cardiomyopathy have been reported as an adverse effect of prolonged KD therapy with poorly understood causative mechanisms, relative selenium deficiency being one of them.59 Long-term adverse effects of KD have not been adequately studied.7
Skeletal
Osteoporosis and vitamin D deficiency may be observed in children on KD, especially when these children are on multiple anti-seizure medications.50,60
Rare Adverse Effects
Rare adverse effects of long-term KD exposure include pancreatitis and transaminitis especially with concurrent valproate use, which may be exacerbated by viral illnesses.7
The common adverse effects of KD have been depicted in Table 4. Patel et al observed that children on long-term KD had normalization of height, weight and serum cholesterol after a median period of six years of discontinuation of therapy.61 This may be indicative of the fact that there is resolution of adverse effects after discontinuation of KD.
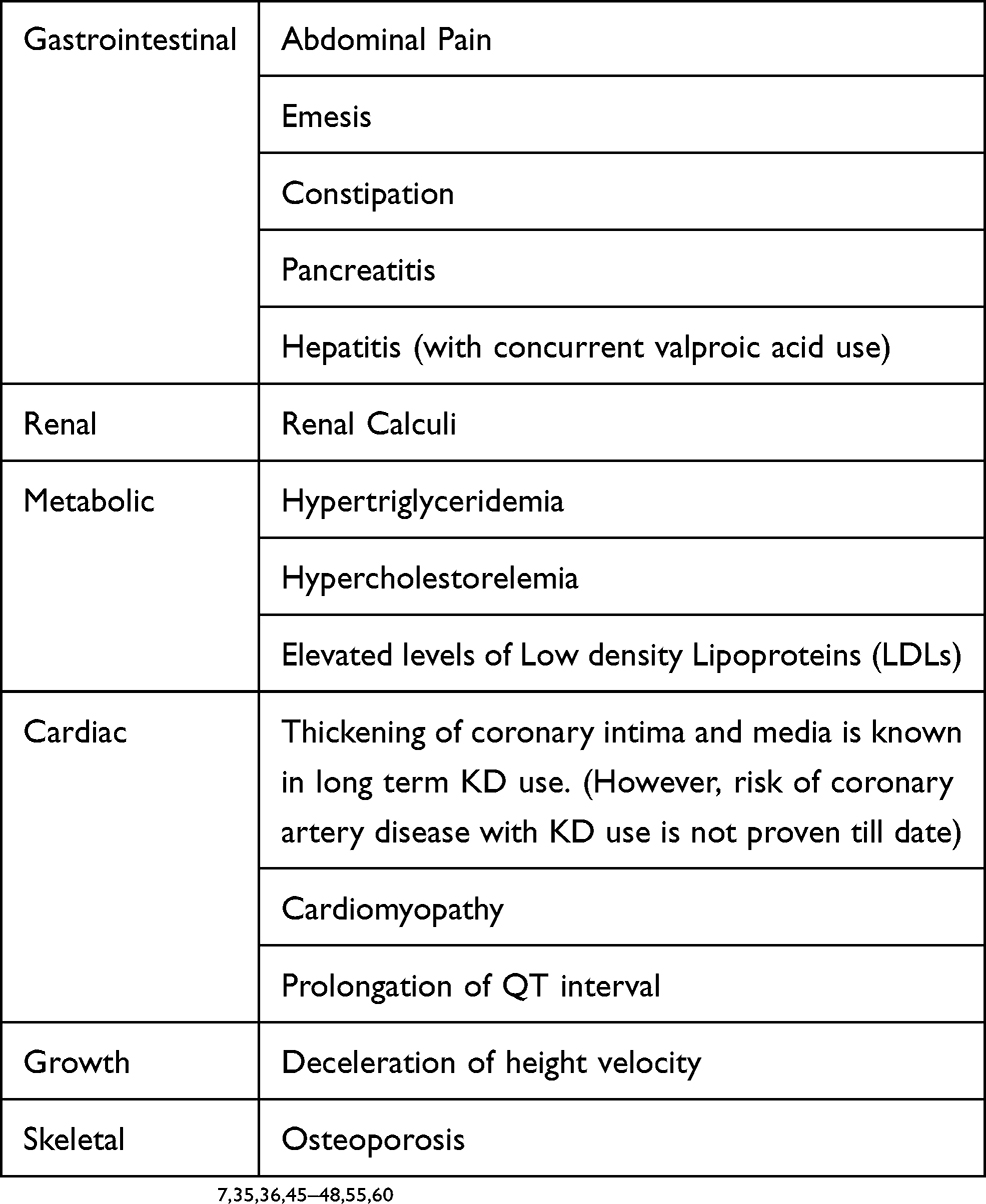 |
Table 4 Adverse Effects Reported With KD |
Antiepileptic Drugs And KD
It is recommended that anti-epileptic drug formulations should be converted from liquid to tablet form in children on KD, in order to restrict additional carbohydrate intake. Studies do not support dose modification of anti-epileptic drugs or serum drug level monitoring in children on KD.7 KD and Valproic acid both have the propensity to cause secondary carnitine deficiency and hence, when both agents are used concurrently, periodic serum carnitine assay is recommended.7 Administration of KD to a child on carbonic anhydrase inhibitors such as topiramate, zonisamide or acetazolamide may increase the chance of development of metabolic acidosis. Hence, periodic serum bicarbonate level monitoring of these children is warranted, especially at the initiation of therapy.62 Risk of urolithiasis multiplies with chronic administration of zonisamide and KD concurrently, whereas the same has not been demonstrated with concurrent use of topiramate and KD.63 This warrants periodic screening ultrasound of kidney and bladder. It is recommended to taper antiepileptic drugs after about one month of initiation of KD.7 Gradual tapering is advised, especially when benzodiazepines or phenobarbitone is used.7
Steroids And KD
Ville D et al conducted a pilot study on the role of combined therapy of KD and oral steroids in forty-two children with corticosteroid-resistant or dependent epileptic encephalopathy.64 All children attained desirable level of urinary ketosis and fourteen children had good seizure control for six months.64 KD helped in withdrawal of steroids in all the responders.64 The study proved the potentially beneficial role of combined therapy of KD and corticosteroids in epileptic encephalopathies.
Discontinuation Of KD
Discontinuation of KD is individualized. International KD Study Group recommends atrial of three months after initiation of KD in order to evaluate its efficacy.7 It is discontinued if there is initial worsening of seizures.68 Though there is no cut-off for the maximum period of KD use, two years are accepted as a fairly appropriate duration of KD use in children who have a reduction of seizures by more than 50%.68 It is also recommended that an informed decision of KD discontinuation should be taken based on risk–benefit ratio analysis.7 Prolonged KD therapy is indicated in Glucose Transporter Type I (GLUT 1) Deficiency till adulthood.65 On the other hand, short duration of KD for 6 months has been found to be effective in West Syndrome.66
Classic KD is conventionally tapered over four to six weeks prior to discontinuation. 4:1 KD is modified by gradual addition of regular foods containing carbohydrates. During this process, 4:1 KD is reinitiated anytime if there is a significant increase of seizure burden. Increased seizure frequency to the tune of 14% has been reported with KD stoppage.67
In situations where KD does not show marked benefit, rapid taper is tolerated. Studies have revealed that children with structural brain lesions, tuberous sclerosis and persistently abnormal electroencephalographs are at greater risk of seizure recurrence after stoppage of KD.69,70 Approximately 80% of the children with refractory epilepsy do not have recurrence after stoppage of KD.6
KD In Special Circumstances
KD In Infants
Neonatal onset epilepsy is a challenging entity wherein multiple drugs may not guarantee seizure freedom. KD use is controversial in neonatal age group in view of the unique physiology of infants and the recommendation of exclusive breast-feeding in this age group. The largest study of KD in infancy was performed by Le Pichon et al between 2005 and 2016 wherein nine infants with drug-resistant epilepsy between 1 and 13 months of age were treated with KD.71 All the infants were continued on breast-feed for at least one month after initiation of dietary therapy. All of them achieved significant ketosis with four having significant seizure freedom and none having side effects.70 The above study reinforced the role of KD as a safe, effective and practical management modality in breastfeeding infants with drug-refractory epilepsy. An international expert consensus pertaining to KD in infancy reinforces the various aspects of this therapy.43
KD In Resource-Limited Settings
MAD may be a more practical option for resource-limited settings compared to classic KD as it can be initiated as a domiciliary therapy with readily available home-based diets and less restrictive regimes. In an open-label randomized controlled study from India, the efficacy of a simplified Modified Atkins Diet (MAD) regime was evaluated in children with refractory epilepsy whose parents had poor literacy.24
In the study, forty-one children with refractory epilepsy (study group) were administered MAD for three months along with anti-epileptic drugs, while the control group comprising forty children received anti-epileptic drugs alone.24 The study group had significantly higher number of children with more than 50% seizure reduction and minimal adverse effects, thus underscoring the efficacy, feasibility and tolerability of MAD in resource-limited settings.24 The International League Against Epilepsy (ILAE) Task Force on Dietary Therapy have recommended certain practical steps for KD in resource-limited settings after due consideration of the unique challenges in such settings.71 Core recommendations of the Task Force included short-term training of manpower in KD therapy, mandatory multivitamin and trace element (including selenium) supplementation and baseline investigation guidelines.71
KD In FIRES And SRSE
Management of Super-Refractory Status Epilepticus (SRSE) is a therapeutic challenge. KD has been reported to be effective in controlling seizures in the PICU.72,73
Appavu B et al performed a retrospective study to evaluate the efficacy of KD in pediatric SRSE.73 Ten children (ages between two to sixteen years) with SRSE secondary to varying etiologies were identified among whom four had generalized and rest had focal status epilepticus.73 KD was initiated after a median duration of eighteen days. Nine children had cessation of SRSE within seven days of start of KD indicating the efficacy of KD for treating pediatric SRSE.51
Arya et al studied the efficacy of KD in pediatric convulsive Refractory Status Epilepticus (RSE) in children recruited through any of the eleven institutions participating in the Pediatric Status Epilepticus Research Group (pSERG) between January 2011 to December 2016.74 Fourteen children were administered KD, eleven of whom received it via enteral route.74 Electroencephalographic resolution of seizure was achieved within one week of starting KD in ten children while eleven children could be weaned off from intravenous anti-epileptic infusions within two weeks of initiation of KD.74 Adverse effects noted in three patients included gastroparesis and elevated triglyceride levels.74
KD In Adults
KD is gaining popularity as an effective therapeutic modality in adults with epilepsy.75 Many epileptic adolescents who have benefitted with KD have transitioned to adulthood who continue to prefer using KD.76 In addition, individuals with adult-onset epilepsy with drug-refractory seizures are increasingly turning towards KD in view of its better adverse effect profile. A meta-analysis of pooled data from twelve studies on the use of different types of KD in adults with drug-refractory epilepsy revealed that the efficacy of KD ranges from 13% to 70% in adults.76 Compliance is a major challenge in adults with studies showing an adherence rate of 45%.76 KD has proven to be effective in the management of SRSE in adults. In a recent multicentric phase I/II clinical trial, Classic KD was used in fifteen adults with SRSE, eleven of whom attained seizure resolution over a median period of five days.77
KD Administration In Children On Gastrostomy Tube Feeds
Children with refractory epilepsy and feeding issues mandating gastrostomy tube feeding may be administered KD through a gastrostomy tube effectively as portrayed through a case series of twelve children with static encephalopathy and intractable epilepsy on gastrostomy feeding.56,57
Socioeconomic Aspects Pertaining To KD Therapy
Any chronic medical management modality may impose various crucial repercussions on the socioeconomic aspects of the patients, their family members and society as a whole. Some of these salient aspects pertaining to KD have been highlighted through recent studies, as mentioned below.
Financial Implications Of KD
Financial viability is one of the key determinants of chronic therapy. Wijnen et al compared twenty-six children with intractable epilepsy receiving KD with twenty-two receiving other therapies in a study with sixteen-month follow-up wherein it was seen that KD leads to better seizure control with no significant difference in the Quality Adjusted Life Years (QALY) and cost-effectiveness per QALY of the two groups.57 Awaiting further studies on this aspect, KD may be accepted as a financially viable option for the management of refractory epilepsy in children.
Effect Of KD On Quality Of Life (QoL)
Implementation of KD is challenging. Right from the acceptance of this modality to procurement of the essential tools such as a weighing scale and glucometer, to the daily preparation and adhering to a dietary regime requires strict family support and motivation. These intangible stressors and gains reflected through seizure control and reduction of anti-epileptic drugs may ultimately decide the perceived quality of life of the immediate family members. Poelzer et al conducted a systematic review of literature from 2007 to 2014 pertaining to children on various forms of KD to evaluate the effect of KD on the QoL of their immediate family members.58 It emerged that no study directly addressed the issue of QoL.58 Surrogate markers like poor adherence to KD and high dropout rates may be indicative of poor QoL.58 Therefore, improvement of QoL may be ensured by pre-counseling and regular interaction between parents and health-care providers.58 It is also a case in point that studies need to fathom the effect of KD on the QoL of patients as well as their family members.
Effect Of KD On The Cognitive And Behavioral Profiles Of Children With Epilepsy
KD has been known for having positive cognitive and behavioral effects in children with epilepsy. This effect seems independent of seizure control and reduction in anti-epileptic drugs. Ijff et al compared the effects of KD on the cognition and behavior of twenty-eight children with refractory epilepsy treated with KD to that of twenty-two others who were treated with drugs during the first four months of therapy.78 Objective assessments proved that children in KD group had statistically significant improvement of cognition and behavior vis-à-vis the non-KD group, which was attributed to an independent effect of KD and not secondary to seizure-control itself.78
KD And Risk Of Sudden Unexpected Death In Epilepsy
Refractory epilepsy is a proven risk factor for Sudden Unexpected Death in Epilepsy (SUDEP).79 Kcna1-null mutant mouse model which lacks the potassium channel Kv1.1 alpha is studied frequently as it is at a high risk for SUDEP due to its physiological variables.80 Simeone KA et al carried out a proof of concept trial wherein they demonstrated the efficacy of KD in prolonging the life of Kcna1-null mice by 47%, thereby demonstrating that KD is likely to diminish rates of SUDEP and prolong life expectancy.80
New Frontiers In Dietary Therapy
Triheptanoin is a synthetic edible oil which is composed of a seven-carbon compound called heptanoic acid.81 It is used as an anaplerotic agent that replenishes the Intermediary metabolites of Tricarboxylic acid pathway.81 There are reports of triheptanoin being successfully used as for refractory seizure control in numerous mouse models of epilepsy and as a neuroprotectant in Huntington’s chorea and Glucose Transporter Type I deficiency.81 Its efficacy as an add-on trial for seizures has been highlighted in a pilot trial of twelve children with refractory epilepsy.82
Triheptanoin therapy is well tolerated except for gastrointestinal adverse effects.82 Poor palatability of KD owing to its extremely high fat content is a major reason for its discontinuation, which has prompted studies of alternative dietary therapies in epilepsy. The anti-seizure efficacy of a non-ketogenic diet comprising carbohydrates with low glycemic index, medium-chain fatty acids, PUFA with a high ratio of branched chain amino acids to aromatic amino acids was tried by Dallérac et al on the chronic kainate mouse model of epilepsy.83 They demonstrated good seizure and the study provided proof of concept that a non-ketogenic regimen may have effective seizure control.83
Conclusion
KD is rapidly gaining its pride of place as an effective management modality in management of drug-refractory epilepsy and certain metabolic disorders. There is a growing body of research pertaining to previously little known aspects about the mechanism of action of KD. RCTs have provided irrefutable evidence supporting the efficacy of KD in seizure control. Various other beneficial effects of KD such as improvement of cognitive and behavioral aspects of children and quality of life of parents are additional bonuses of KD. Early administration of KD is advocated in certain conditions such as Dravet Syndrome and GLUT-1 transporter deficiency. Besides classic KD, MAD, MCT-based KD and LGIT KD have been increasingly used in clinical practice. It is being increasingly used in adolescents and adults. Clinical and translational research may further the body of evidence pertaining to the use of KD in refractory epilepsy, thereby opening the doors to better management of this common neurological disorder. In view of the therapeutic efficacy, safety and economic feasibility, KD is likely to emerge as a popular therapy in resource-constrained as well as developed societies worldwide.
Author Contributions
JNG: Literature review and initial draft manuscript preparation, final approval of the version to be published.
SS: Literature review, critical review of manuscript for important intellectual content and final approval of the version to be published.
Both authors contributed to data analysis, drafting or revising the article, gave final approval of the version to be published, and agree to be accountable for all aspects of the work.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Kwan P, Arzimanoglou A, Berg AT, et al. Definition of drug resistant epilepsy: consensus proposal by the ad hoc task force of the ILAE commission on therapeutic strategies. Epilepsia. 2010;51(6):1069–1077. doi:10.1111/j.1528-1167.2009.02397.x
2. Winesett SP, Bessone SK, Kossoff EH. The ketogenic diet in pharmacoresistant childhood epilepsy. Expert Rev Neurother. 2015;15(6):621–628. doi:10.1586/14737175.2015.1044982
3. Sharma S, Jain P. The ketogenic diet and other dietary treatments for refractory epilepsy in children. Ann Indian AcadNeurol. 2014;17(3):253–258. doi:10.4103/0972-2327.138471
4. Lefevre F, Aronson N. Ketogenic diet for the treatment of refractory epilepsy in children: a systematic review of efficacy. Pediatrics. 2000;105(4):E46. doi:10.1542/peds.105.4.e46
5. Neal EG, Chaffe H, Schwartz RH, et al. The ketogenic diet for the treatment of childhood epilepsy: a randomised controlled trial. Lancet Neurol. 2008;7:500–506. doi:10.1016/S1474-4422(08)70092-9
6. Martin-McGill KJ, Jackson CF, Bresnahan R, Levy RG, Cooper PN. Ketogenic diets for drug-resistant epilepsy. Cochrane Database Syst Rev. 2018;11:CD001903. doi:10.1002/14651858.CD001903.pub4
7. Kossoff EH, ZupecKania BA, Auvin S, et al. Optimal clinical management of children receiving dietary therapies for epilepsy: updated recommendations of theInternational Ketogenic Diet Study Group. Epilepsia Open. 2018;3:175. doi:10.1002/epi4.12225
8. Veggiotti P, Burlina A, Coppola G, et al. The ketogenic diet for Dravet syndrome and other epileptic encephalopathies: an Italian consensus. Epilepsia. 2011;52(S 2):83–89. doi:10.1111/j.1528-1167.2011.03010.x
9. Sharma S, Tripathi M. Ketogenic diet in epileptic encephalopathies. Epilepsy Res Treat. 2013;2013:652052.
10. Yan N, Xin-Hua W, Lin-Mei Z, Yi-Ming C, Wen-Hui L. Prospective study of the efficacy of a ketogenic diet in 20 patients with Dravet syndrome. Seizure. 2018;60:144–148. doi:10.1016/j.seizure.2018.06.023
11. Wu Q, Wang H, Fan YY, Zhang JM, Liu XY, Fang XY. Ketogenic diet effects on 52 children with pharmacoresistant epileptic encephalopathy: a clinical prospective study. Brain Behav. 2018;8(5):e00973. doi:10.1002/brb3.2018.8.issue-5
12. Thammongkol S, Vears DF, Bicknell-Royle J, Nation J, Draffin K, Stewart KG. Efficacy of the ketogenic diet: which epilepsies respond? Epilepsia. 2012;53(3):e55–e59. doi:10.1111/epi.2012.53.issue-3
13. Caraballo RH, Cersósimo RO, Sakr D, Cresta A, Escobal N, Fejerman N. Ketogenic diet in patients with Dravet syndrome. Epilepsia. 2005;46(9):1539–1544. doi:10.1111/epi.2005.46.issue-9
14. Dressler A, Trimmel-Schwahofer P, Reithofer E, et al. Efficacy and tolerability of the ketogenic diet in Dravet syndrome - Comparison with various standard antiepileptic drug regimen. Epilepsy Res. 2015;109:81–89. doi:10.1016/j.eplepsyres.2014.10.014
15. Barzegar M, Afghan M, Tarmahi V, Behtari M, Rahimi KS, Raeisi S. Ketogenic diet: overview,types, and possible anti-seizure mechanisms. Nutr Neurosci. 2019;26:1–10. doi:10.1080/1028415X.2019.1627769
16. Raju KN, Gulati S, Kabra M, et al. Efficacy of 4: 1(classic) versus 2.5:1 ketogenic ratio diets in refractory epilepsy in young children: a randomized open labeled study. Epilepsy Res. 2011;96(1–2):96–100. doi:10.1016/j.eplepsyres.2011.05.005
17. Huttenlocher PR, Wilbourn AJ, Signore JM. Medium‐chain triglycerides as a therapy for intractable child epilepsy. Neurology. 1971;21:1097–1103. doi:10.1212/WNL.21.11.1097
18. Liu YM. Medium-chain triglyceride (MCT) ketogenic therapy. Epilepsia. 2008;49(Suppl 89):33–36. doi:10.1111/j.1528-1167.2008.01830.x
19. Zupec-Kania B, Neal E, Schultz R, Roan ME, Turner Z, Welborn M. An update on diets in clinical practice. J Child Neurol. 2013;28(8):1015–1026. doi:10.1177/0883073813487597
20. Sharma S, Jain P. The modified Atkins diet in refractory epilepsy. Epilepsy Res Treat. 2014;2014:404202.
21. Sharma S, Sankhyan N, Gulati S, Agarwala A. Use of the modified Atkins diet for treatment of refractory childhood epilepsy: a randomized controlled trial. Epilepsia. 2013;54(3):481–486. doi:10.1111/epi.2013.54.issue-3
22. Sharma S, Jain P, Gulati S, Sankhyan N, Agarwala A. Use of the modified Atkins diet in Lennox Gastaut syndrome. J Child Neurol. 2015;30(5):576–579. doi:10.1177/0883073814527162
23. Kim JA, Yoon JR, Lee EJ, et al. of the classic ketogenic and the modified Atkins diets in refractory childhood epilepsy. Epilepsia. 2016;57(1):51–58. doi:10.1111/epi.13256
24. Sharma S, Goel S, Jain P, Agarwala A, Aneja S. Evaluation of a simplified modified Atkins diet for use by parents with low levels of literacy in children with refractory epilepsy: a randomized controlled trial. Epilepsy Res. 2016;127:152–159. doi:10.1016/j.eplepsyres.2016.09.002
25. Mehta R. Efficacy and tolerability of the modified Atkins diet in young children with refractory epilepsy: Indian experience. Ann Indian Acad Neurol. 2016;19(4):523–527. doi:10.4103/0972-2327.194463
26. Sharma S. Use of the modified Atkins diet in infantile spasms refractory to first-line treatment. Seizure. 2012;21(1):45–48. doi:10.1016/j.seizure.2011.08.009
27. Pfeifer HH, Lyczkowski DA, Thiele EA. Low glycemic index treatment: implementation and new insights into efficacy. Epilepsia. 2008;49:42–45. doi:10.1111/epi.2008.49.issue-s8
28. Kim SH, Kang HC, Lee EJ, et al. Low glycemic index treatment inpatients with drug-resistant epilepsy. Brain Dev. 2017;39:687–692. doi:10.1016/j.braindev.2017.03.027
29. Sondhi V, Agarwala A, Chakrabarty B, et al. Dietary Therapy In EpilepsyTreatment (DIET-Trial): a randomised non-inferiority trial comparing KD, MAD & LGIT for drug resistant epilepsy. Neurology. 2018;90(15 S):
30. Grocott OR, Herrington KS, Pfeifer HH, et al. Low glycemic index treatment for seizure control in Angelman syndrome: a case series from the Center for Dietary Therapy of Epilepsy at the Massachusetts General Hospital. Epilepsy Behav. 2017;68:45–50. doi:10.1016/j.yebeh.2016.12.018
31. Freeman JM, Kossoff EH. Ketosis and the ketogenic diet, 2010: advances in treating epilepsy and other disorders. Adv Pediatr. 2010;57:315–329. doi:10.1016/j.yapd.2010.08.003
32. Likhodii SS, Serbanescu I, Cortez MA, Murphy P, Snead OC
33. Masino SA, Rho JM. Mechanisms of ketogenic diet action. In: Noebels JL, Avoli M, Rogawski MA,et al editors. Jasper’s Basic Mechanisms of the Epilepsies.
34. Bromfield E, Dworetzky B, Hurwitz S, et al. A randomized trial of polyunsaturated fatty acids for refractory epilepsy. Epilepsy Behav. 2008;12:187–190. doi:10.1016/j.yebeh.2007.09.011
35. Dahlin M, Hjelte L, Nilsson S, Åmark P. Plasma phospholipid fatty acids are influenced by a ketogenic diet enriched with n-3 fatty acids in children with epilepsy. EpilepsyRes. 2007;73:199–207.
36. Abdallah D. Anticonvulsant potential of the peroxisome proliferator activated receptor gamma agonist pioglitazone in pentylenetetrazole-induced acute seizures and kindling in mice. Brain Res. 2010;1351:246–253. doi:10.1016/j.brainres.2010.06.034
37. Zhang Y, Zhou S, Zhou Y, et al. Altered gut microbiome composition in children with refractory epilepsy after ketogenic diet. Epilepsy Res. 2018;145:163–168. doi:10.1016/j.eplepsyres.2018.06.015
38. Lindefeldt M, Eng A, Darban H, et al. The ketogenic diet influences taxonomic and functional composition of the gut microbiota in children with severe epilepsy. NPJ Biofilms Microbiomes. 2019;5:5. doi:10.1038/s41522-018-0073-2
39. Tagliabue A, Ferraris C, Uggeri F, et al. Short-term impact of a classical ketogenic diet on gut microbiota in GLUT1 deficiency syndrome: a 3-month prospective observational study. Clin Nutr ESPEN. 2017;17:33–37. doi:10.1016/j.clnesp.2016.11.003
40. McDaniel SS, Rensing NR, Thio LL, Yamada KA, Wong M. The ketogenic diet inhibits the mammalian target of rapamycin (mTOR) pathway. Epilepsia. 2011;52(3):e7–e11. doi:10.1111/j.1528-1167.2011.02981.x
41. Meira ID, Romão TT, Do Prado HJ, Krüger LT, Pires ME, da Conceição PO. Ketogenic diet and epilepsy: what we know so far. Front Neurosci. 2019;13:1–8. doi:10.3389/fnins.2019.00001
42. Bansal S, Cramp L, Blalock D, et al. The ketogenic diet: initiation at goal calories versus gradual caloric advancement. Pediatr Neurol. 2014;50:26–30. doi:10.1016/j.pediatrneurol.2013.08.006
43. van der Louw E, van den Hurk D, Neal E, et al. Ketogenic diet guidelines for infants with refractory epilepsy. Eur J Paediatr Neurol. 2016;20:798–809. doi:10.1016/j.ejpn.2016.07.009
44. McNally MA, Pyzik PL, Rubenstein JE, Hamdy RF, Kossoff EH. Empiric use of oral potassium citrate reduces symptomatic kidney stone incidence with the ketogenic diet. Pediatrics. 2009;124:e300–e304. doi:10.1542/peds.2009-0217
45. Lin A, Turner Z, Doerrer SC, Stanfield A, Kossoff EH. Complications during ketogenic diet initiation: prevalence, treatment, and influence on seizure outcomes. Pediatr Neurol. 2017;68:35–39. doi:10.1016/j.pediatrneurol.2017.01.007
46. Sharma S, Gulati S, Kalra V, Agarwala A, Kabra M. Seizure control and biochemical profile on the ketogenic diet in young children with refractory epilepsy-Indian experience. Seizure. 2009;18(6):446–449. doi:10.1016/j.seizure.2009.04.001
47. Sampath A, Kossoff EH, Furth SL, Pyzik PL, Vining EP. Kidney stones and the ketogenic diet: risk factors and prevention. J Child Neurol. 2007;22:375–378. doi:10.1177/0883073807301926
48. Peterson SJ, Tangney CC, Pimentel-Zablah EM, et al. Changes in growth and seizure reduction in children on the ketogenic diet as a treatment for intractable epilepsy. J Am Diet Assoc. 2005;105:718–725. doi:10.1016/j.jada.2005.02.009
49. Neal EG, Chaffe HM, Edwards N, Lawson MS, Schwartz RH, Cross JH. Growth of children on classical and medium chain triglyceride diets. Pediatrics. 2008;122:e334–e340. doi:10.1542/peds.2007-2410
50. Bergqvist AGC, Schall JI, Stallings VA, Stallings VA, Zemel BS. Progressive bone mineral content loss in children with intractable epilepsy treated with the ketogenic diet. Am J Clin Nutr. 2008;88:1678–1684. doi:10.3945/ajcn.2008.26099
51. Spulber G, Spulber S, Hagenas L, et al. Growth dependence on insulin- like growth factor-1 during the ketogenic diet. Epilepsia. 2009;50:297–303. doi:10.1111/j.1528-1167.2008.01769.x
52. Vining EP, Pyzik P, McGrogan J, et al. Growth of children on the ketogenic diet. DMCN. 2002;44:796–802.
53. Williams S, Basualdo-Hammond C, Curtis R, et al. Growth retardation in children with epilepsy on the ketogenic diet: a retrospective chart review. J Am Diet Assoc. 2002;102:
54. Tagliabue A, Bertoli S, Trentani C, Borrelli P, Veggiotti P. Effects of the ketogenic diet on nutritional status, resting energy expenditure, and substrate oxidation in patients with medically refractory epilepsy: a 6-month prospective observational study. Clin Nutr. 2012;31(2):246–249. doi:10.1016/j.clnu.2011.09.012
55. Coppola G, Natale F, Torino A, et al. The impact of the ketogenic diet on arterial morphology and endothelial function in children and young adults with epilepsy: a case-control study.n. Seizure. 2014;23:260–265. doi:10.1016/j.seizure.2013.12.002
56. Hosain SA, La VegaTalbott M, Solomon GE. Ketogenic diet in pediatric epilepsy patients with gastrostomy feeding. Pediatr Neurol. 2005;32:81. doi:10.1016/j.pediatrneurol.2004.09.006
57. Wijnen BFM, de Kinderen RJA, Lambrechts DAJE, et al. Long-term clinical outcomes and economic evaluation of the ketogenic diet versus care as usual in children and adolescents with intractable epilepsy. Epilepsy Res. 2017;132:91–99. doi:10.1016/j.eplepsyres.2017.03.002
58. Poelzer K, Mannion C, Ortiz MM, Bang R, Woods P. A systematic review of the quality of life for families supporting a child consuming the ketogenic diet for seizure reduction. Curr Dev Nutr. 2019;3(5):
59. Best TH, Franz DN, Gilbert DL, et al. Cardiac complications in pediatric patients on the ketogenic diet. Neurology. 2000;54:2328–2330. doi:10.1212/WNL.54.12.2328
60. Kang HC, Chung DE, Kim DW, Kim HD. Early and late onset complications of the ketogenic diet for intractable epilepsy. Epilepsia. 2004;45(9):1116–1123. doi:10.1111/epi.2004.45.issue-9
61. Patel A, Pyzik PL, Turner Z, Rubenstein JE, Kossoff EH. Long term outcomes of children treated with the ketogenic diet in the past. Epilepsia. 2010;51(7):1277–1282. doi:10.1111/(ISSN)1528-1167
62. Takeoka M, Riviello JJ, Pfeifer H, Thiele EA. Concomitant treatment with topiramate and ketogenic diet in pediatric epilepsy. Epilepsia. 2002;43:1072–1075. doi:10.1046/j.1528-1157.2002.00602.x
63. Paul E, Conant KD, Dunne IE, et al. Urolithiasis on the ketogenic diet with concurrent topiramate or zonisamide therapy. Epilepsy Res. 2010;90:151–156. doi:10.1016/j.eplepsyres.2010.04.005
64. Ville D, Chiron C, Laschet J, Dulac O. The ketogenic diet can be used successfully in combination with corticosteroids for epileptic encephalopathies. Epilepsy Behav. 2015;48:61–65. doi:10.1016/j.yebeh.2015.03.003
65. Leen WG, Taher M, Verbeek MM, Kamsteeg EJ, van de Warrenburg BP, Willemsen MA. GLUT1 deficiency syndrome into adulthood: a follow-up study. J Neurol. 2014. 261(3):589–599.
66. Hong AM, Turner Z, Hamdy RF, Kossoff EH. Infantile spasms treated with the ketogenic diet: prospective single-center experience in 104 consecutive infants. Epilepsia. 2010;51(8):1403–1407. doi:10.1111/j.1528-1167.2010.02586.x
67. Worden LT, Turner Z, Pyzik PL, Rubenstein JE, Kossoff EH. Is there an ideal way to discontinue the ketogenic diet? Epilepsy Res. 2011;95(3):232–236. doi:10.1016/j.eplepsyres.2011.04.003
68. Martinez CC, Pyzik PL, Kossoff EH. Discontinuing the ketogenic diet in seizure-free children: recurrence and risk factors. Epilepsia. 2007;48:187–190. doi:10.1111/epi.2007.48.issue-1
69. Caraballo R, Vaccarezza M, Cersósimo R, et al. Long-term follow up of the ketogenic diet for refractory epilepsy: multicenter Argentineanexperience in 216 pediatric patients. Seizure. 2011;20(8):640–645. doi:10.1016/j.seizure.2011.06.009
70. Le Pichon JB, Thompson L, Gustafson M, Abdelmoity A. Initiating the ketogenic diet in infants with treatment refractory epilepsy while maintaining a breast milk diet. Seizure. 2019;69:41–43. doi:10.1016/j.seizure.2019.03.017
71. Kossoff EH, Al-Macki N, Cervenka MC, et al. What are the minimum requirements for ketogenic diet services in resource-limited regions? Recommendations from the International League Against Epilepsy Task Force for Dietary Therapy. Epilepsia. 2015;56(9):1337–1342. doi:10.1111/epi.13039
72. Caraballo RH, Flesler S, Armeno M, et al. Ketogenic diet in pediatric patients with refractory focal status epilepticus. Epilepsy Res. 2014;108(10):1912–1916. doi:10.1016/j.eplepsyres.2014.09.033
73. Appavu B, Vanatta L, Condie J, Kerrigan JF, Jarrar R. Ketogenic diet treatment for pediatric super-refractory status epilepticus. Seizure. 2016;41:62–65. doi:10.1016/j.seizure.2016.07.006
74. Arya R, Peariso K, Gaínza-Lein M, et al. Efficacy and safety of ketogenic diet for treatment of pediatric convulsive refractory status epilepticus. Epilepsy Res. 2018;144:1–6. doi:10.1016/j.eplepsyres.2018.04.012
75. McDonald TJW, Cervenka MC. Ketogenic diets for adults with highly refractory epilepsy. Epilepsy Curr. 2017;17(6):346–350. doi:10.5698/1535-7597.17.6.346
76. Ye F, Li XJ, Jiang WL, Bin SH, Liu J. Efficacy of and patient compliance with a ketogenic diet in adults with intractable epilepsy: a meta-analysis. J Clin Neurol. 2015;11:26–31. doi:10.3988/jcn.2015.11.1.26
77. Cervenka MC, Hocker SE, Koenig M, et al. Phase I/II multicenter ketogenic diet study for adult super-refractory status epilepticus. Neurology. 2017;88(10):938–943. doi:10.1212/WNL.0000000000003690
78. Ijff DM, Postulart D, Lambrechts DAJE, et al. Cognitive and behavioral impact of the ketogenic diet in children and adolescents with refractory epilepsy: a randomized controlled trial. Epilepsy Behav. 2016;60:153–157. doi:10.1016/j.yebeh.2016.04.033
79. Abdel-Mannan O, Taylor H, Donner EJ, Sutcliffe AG. A systematic review of sudden unexpected death in epilepsy (SUDEP) in childhood. Epilepsy Behav. 2019;90:99–106. doi:10.1016/j.yebeh.2018.11.006
80. Simeone KA, Matthews SA, Rho JM, Simeone TA. Ketogenic diet treatment increases longevity in Kcna1-null mice, a model of sudden unexpected death in epilepsy. Epilepsia. 2016;57(8):178–182. doi:10.1111/epi.2016.57.issue-8
81. Kim TH, Borges K, Petrou S, Reid CA. Triheptanoin reduces seizure susceptibility in a syndrome-specific mouse model of generalized epilepsy. Epilepsy Res. 2013;103(1):101–105. doi:10.1016/j.eplepsyres.2012.09.016
82. Calvert S, Barwick K, Par M, Tan KN, Borges K. A pilot study of add-on oral triheptanoin treatment for children with medically refractory epilepsy. Eur J Paediatr Neurol. 2018;22(6):1074–1080. doi:10.1016/j.ejpn.2018.07.014
83. Dallérac G, Moulard J, Benoist JF, et al. Non-ketogenic combination of nutritional strategies provides robust protection against seizures. Sci Rep. 2017;7(1):5496. doi:10.1038/s41598-017-05542-3
84. Kossoff EH, Shields WD. Nonpharmacologic care for patients with Lennox-Gastaut syndrome: ketogenic diets and vagus nerve stimulation. Epilepsia. 2014;55(Suppl 4):29–33. doi:10.1111/epi.12546
85. Bough KJ, Rho JM. Anticonvulsant mechanisms of the ketogenic diet. Epilepsia. 2007;48(1):43–58. doi:10.1111/epi.2007.48.issue-1
86. Olson CA, Vuong HE, Yano JM, et al. The gut microbiota mediates the antiseizure effects of the ketogenic diet. Cell. 2018;173(7):1728–1741. doi:10.1016/j.cell.2018.04.027
87. Danial NN, Hartman AL, Stafstrom CE, Thio LL. How does the ketogenic diet work? Four potential mechanisms. J Child Neurol. 2013;28(8):1027–1033. doi:10.1177/0883073813487598
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.