Back to Journals » International Journal of Women's Health » Volume 13
Current Knowledge on Endometriosis Etiology: A Systematic Review of Literature
Authors Mikhaleva LM, Radzinsky VE, Orazov MR, Khovanskaya TN, Sorokina AV ![]() , Mikhalev SA, Volkova SV, Shustova VB, Sinelnikov MY
, Mikhalev SA, Volkova SV, Shustova VB, Sinelnikov MY
Received 15 February 2021
Accepted for publication 4 May 2021
Published 1 June 2021 Volume 2021:13 Pages 525—537
DOI https://doi.org/10.2147/IJWH.S306135
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Elie Al-Chaer
Lyudmila M Mikhaleva,1 Victor E Radzinsky,2 Mekan R Orazov,2 Tatyana N Khovanskaya,1 Anastasia V Sorokina,1 Sergey A Mikhalev,3 Snezhana V Volkova,2 Victoria B Shustova,1 Mikhail Y Sinelnikov1
1Laboratory of Clinical Morphology, Research Institute of Human Morphology, Moscow, Russia; 2Peoples’ Friendship University (RUDN), Moscow, Russia; 3Family Planning and Reproduction Center, Moscow, Russia
Correspondence: Mikhail Y Sinelnikov Email [email protected]
Objective: To review the mechanisms of endometriosis development, including those related to epigenetic mutations, cellular dysregulation, inflammatory processes, and oxidative stress.
Methods: A systematic literature review regarding current aspects of endometriosis etiology, genesis and development was performed using the PubMed, Google Scholar, and eLibrary databases. Keywords included endometriosis, etiology, development, genesis, associations and mechanisms. A multilingual search was performed.
Results: Several mechanisms underline the pathophysiological pathways for endometriosis development. Epigenetic mutations, external and internal influences, and chronic conditions have a significant impact on endometriosis development, survival and regulation. Several historically valid theories on endometriosis development were discussed, as well as updated findings.
Conclusion: Despite recent advances, fundamental problems in understanding endometriosis remain unresolved. The identification of unknown circulating epithelial progenitors or stem cells that are responsible for epithelial growth in both the endometrium and endometriotic foci seems to be the next step in solving these questions.
Keywords: endometriosis etiology, endometriosis progression, endometriosis markers, genesis
Introduction
Endometriosis is an inflammatory, estrogen-dependent condition associated with a variety of clinical manifestation.1 Estimates regarding the incidence of endometriosis in the populous vary widely. This multiform and complex disease, affecting approximately 10% of women in reproductive age, has profound influence on the wellbeing of many patients.2 The ovaries, fallopian tubes, and pelvic peritoneum are most commonly affected in endometriosis. The nature of the disease remains unknown. Although the exact etiology of endometriosis remains unclear, it usually involves various genetic, ecologic, immune, angiogenic, and endocrine processes. In this review, we aim to explore historically significant and substantial new data regarding endometriosis etiology and development.
Materials and Methods
A systematic literature review was based on search for information regarding etiology, genesis, development aspects of endometriosis, using the PubMed, Google Scholar, eLibrary databases. We studied the references and conducted a citation search. Two co-authors independently selected, evaluated, and extracted data. Keywords of the strategy of the search were the combination of the following terms in English and Russian languages: endometriosis, etiology, development, genesis, associations, mechanisms. Foreign language material was included in this study. The criteria for inclusion of the literature source were limited to the presence of information regarding the etiology, pathogenesis and developmental aspects of endometriosis, including original studies, structured reviews. The criterion for the exclusion of the article was based on study design (case reports, unstructured literature reviews, surgical studies, pharmaceutical studies were excluded), availability of the study (subscription access, open access), date of publication (later than 2000 studies included, and historically important studies) and relevance.
Primary evaluation indicated that our search strategy had identified a large number of irrelevant studies. Criteria to eliminate studies that did not address our research question were developed throughout article processing in order to narrow the ongoing search. Primarily, we included original research articles and reviews that discussed findings on endometriosis epidemiology and etiology. We later included short communications and conference publications and excluded literature reviews written with case reports and non-conventional studies. These criteria identified a total of 341 articles for review. Next, we narrowed our inclusion criteria to articles published after 2000, with the exception of articles of significant historical value, which provided us with a total of 88 articles for review.
Results
Endometriosis Development Theory
Many different histological models have been suggested to explain the origin of endometriosis. The metaplastic theory suggests the possibility of endometriosis foci coming from multipotent peritoneum. Metaplasia in this theory is regarded as transformation of a differentiated cell type into another differentiated cell type. Similar to Müllerianosis,3 metaplastic foci of endometriotic lesions are thought to be derivatives of embryonic tissue. This theory bases its relevance on the fact that peritoneal and endometrial cells come from a common embryologic predecessor - the coelomic epithelium. Evidence suggests that endometrioid foci may be found not only in the mesothelial pleura, but also in the respiratory and urinary tract epithelium.4,5 The support for this theory comes from the presence of a transitional form occurring out of the mesothelium to the foci of the local ovarian endometriosis.6 Ovarian endometriosis is considered an outcome of mesothelial introduction into the ovarian cortex, coming from primordial follicle evolution.7 In this theory, the main role primarily centers within the epithelial component.
Studies regarding ectopic endometriosis showed that most cancer-associated mutations were found in epithelial cells.8 Mutations in the PIK3CA, KRAS, ARID1A, and other cancer-driving genes in benign pelvic endometriosis tissue, including the ovaries, are well-determined predisposing factors.9–15 Surprisingly, the epithelium of clinically and histologically normal endometrium samples appeared mutated as observed in endometrioid epithelium and in some types of cancer.16 Overtime, endometrioid disorders may analogously obtain genetic mutations, which are commonly associated with cancer, such as those caused by oxidative stress, replicative aging, or other, yet unidentified, factors.17 To date, endometriosis-related epithelial mutations have not been proven to increase the cancer risk of their carriers. However, it is clear that an endometrioid transformation exists in neoplastic lesions, which occur in 0.7–1.6% patients.18 The clear-cell and endometrioid carcinomas are the most common malignancies in such cases.19
Notably, some animals who do not have menstrual cycles do not suffer from endometriosis.20 Only menstruating primate females suffer from endometriosis, which evolves spontaneously, thereby suggesting the etiopathogenetic association of this disease with uterine specificity and mechanisms involved in menstruation.20 Compared to most placental mammals, the endometrial stroma differentiates into decidual cells with post-ovulatory increase in progesterone levels, which is an evolutionary unique mechanism for humans and mammals.21 This differentiation is not induced by blastocyst implantation.22 Decidualization in females is introduced with every ovulatory cycle, regardless of the onset of pregnancy.23 The following decidual generation and its support depends on progesterone levels. If decreased levels are not associated with pregnancy, apoptosis is provoked as well as the rejection of endometrium functional layer.24
The Sampson model attempts do include the clinical, historical, and evolutionary aspects of endometriosis.25 It proposed that endometriosis occurs due to retrograde menstruation with subsequent endometrioid tissue implantation and growth outside of the uterus.26 The existence of the menstrual reflux is the most common etiologic theory of endometriosis and explains the spread of endometriosis throughout the pelvis (peritoneum and ovaries). Artificially explanted endometrioid tissue supports this theory demonstrated by the ability of endometriosis to evolve in mice.23,27 The spontaneous frequency of endometriosis in women increases with prolonged life expectancy and increased number of ovulation cycles.28 However, this model has some contradictions. Only 10% of women develop endometriosis, whereas partial retrograde menstruations are normally present in the majority of women. This model does not include endometriosis cases which occur randomly out of the menstrual cycle or located outside of the pelvic region. The later has been linked to a possible dissemination of menstrual tissue from the endometrial cavity through veins or the lymphatic system; however, this assumption is not commonly accepted and remained experimentally unproven.25
Recently, the Sampson model was expanded to include molecular characteristics. It aims to explain why all retrograde menstruation does not lead to endometriosis. Several changes within the genetic regulatory pathways have been found in some endometrial stromal cells, which control hormonal triggers in menstruating primates and initiated the development of endometriosis.23,29 Epigenetic defects, which trigger the uterine physiological reaction to ovarian hormones, in turn affect endometriosis appearance and progression, shown by the detection of several methylated genes in endometrial cells.23,30 Etiologically, a major role is given to the stromal cell, where the pathogenetic mechanisms are turned on through epigenetic defects. This creates a very specific endometrial phenotype for applicable progesterone resistance, apoptosis and the dysregulation of other molecular processes involved in progesterone-dependent endometrial differentiation.31,32 Additionally, stromal component is prevalent in endometriosis foci, and particularly, stromal cells show the majority of key molecular endometriosis-related abnormalities, such as the production of estradiol, cytokines, and prostaglandins.27 Theoretically, this cellular component in the pelvis during retrograde menstruation likely leads to its implantation with tissue growth resembling endometrioid morphology.23 Changes in gene expression in the eutopic endometrium of mice after induction of endometriosis led researchers to speculate about connection via endometrioid signals, which altered ectopic endometrial gene expression by epigenetic programming.33
Epigenetic Defects
The role of epigenetic factors has been linked to endometriosis progression.23 Epigenetic modifications are reversible modifications of the DNA or histone proteins, which regulate gene expression with no sequencing changes to DNA, unlike genome or genetic mutations. Cytosine methylation and the methylation or acetylation of the specific histones, are two ways to obtain the most specific epigenetic (involving specific genes) or epigenomic (involving genetic aggregates) modifications. DNA methylation is an important regulator of genetic expression and differences in DNA methylation influence many pathological processes in endometriosis. Particularly, research on gene expression proliferation showed that endometriosis-related stromal cells had abnormal patterns of DNA methylation. They expressed changing DNA-methyltransferase (DNMT) −1, −3A, and −3B enzyme levels, which catalyze the methyl group addition to C5 cytosine within the target cytosine-phosphate-guanine (CpG) dinucleotides.34 DNMT-3B aberrantly expressed itself inside eutopic endometrial and endometriotic stromal cells during decidualization, linking to genetic areas of the steroidogenic factor 1 (NR5A) and estrogen alpha-receptor (ESR1).35,36 Also, abnormal DNA methylation in endometriosis activates the expression of several genes, including homeobox A10 (HOXA10), estrogen beta-receptor gene (ESR2), progesterone receptor (PGR), aromatase (CYP19A1, HOXC6, and ALDH1A2) (Figure 1).23,31,37,38
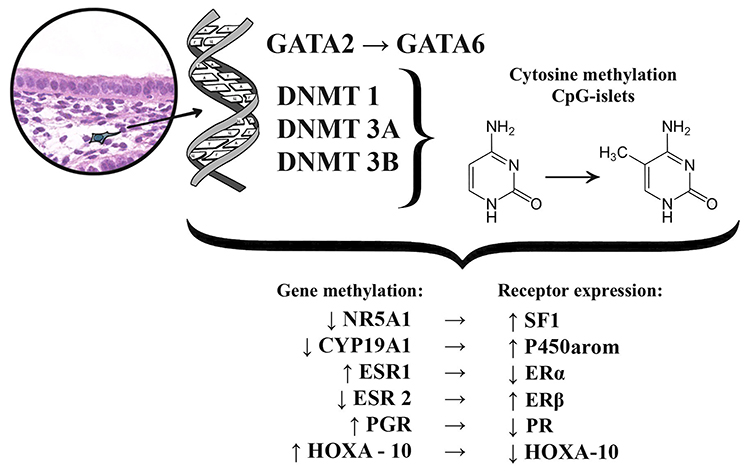 |
Figure 1 Schematic representation of epigenetic changes associated with endometriosis. |
Pathologic DNA methylation is a very important component of endometriosis transformation. The uterine physiological transcription factor GATA, which belongs to the HOXA gene, plays a crucial role leading to the expression shift of the GATA isoform resulting in specific stromal cell endometrioid phenomenon.39 GATA-related functional analysis showed that GATA2 regulates key genes necessary for hormonal differentiation of normal stromal cells; however, it is hypermethylated and repressed within the endometrioid cells.23,40 Simultaneously GATA6, which is hypomethylated and abundant in endometrioid cells, blocks the hormonal sensitivity as well as GATA2 (overexpression of GATA6 leads to a 2-fold decrease in the GATA2 mRNA expression), and induces endometriosis markers.23
A recent study showed an 8.7-fold reduction in GATA2 mRNA levels and a simultaneous 9.2-fold GATA2 mRNA increase within endometrial stromal cells.23 Immunofluorescence of GATA2 positive cells turned out very specific for the ectopic areas, which was characterized by a weak reaction and a unique punctate appearance. GATA6-induced overexpression within endometrial stroma shows that stromal cells demonstrate a pronounced change in gene expression, which was common in endometrioid cells: ESR1 and PGR mRNA decreased by 1.6-fold and 2.6-fold, respectively. In contrast, mRNA expression of ESR2, NR5A1, and CYP19A1 increased by 2.5-fold, 22.4-fold, and 8.5-fold, respectively. Finally, GATA6 overexpression reduced the endometrial stromal cellular ability for decidualization by blocking FOXO1, HAND2, PRL, and IGFBP1 genes. Additionally, neither endogenous GATA6 inhibition, nor GATA2 expression within the endometriosis-related cells improved the hormonal status and did not replace the cellular ability for decidualization. This way, epigenetic changes in the HOXA gene clusters, which are accompanied by GATA2 regulation and hypermethylation, as well as the GATA6 hypomethylation and activation, are the starting points in the development of progesterone resistance and changing estrogen reactions in endometriosis.23
Another HOXA gene cluster representative is HOXA-10, a well-known transcription regulator belonging to the homeobox gene subfamily, which is the transcription factor GATA. It plays a central role in adult uterine organogenesis, embryogenesis and endometrial functional differentiation.41 HOXA-10 expression is usually increased in the human endometrium at the beginning of the implantation period, and its levels increase drastically in the middle of the secretory phase of the menstrual cycle.42 HOXA-10 is the progesterone target, and it participates in downstream signal transmission in the endometrium. HOXA-10 deficient mice are infertile due to the loss in the ability of stromal cells to react to progesterone.31 HOXA-10 is one of the important proteins determining the receptivity of the endometrium, as it regulates the pinopodia formation, integrin αvβ3 production activity, and prostaglandin receptors.41,43
Gene expression proliferation studies showed that HOXA-10 is an important regulator of two critical events in implantation: stromal cell proliferation and local immunosuppression.41 Most studies report that HOXA-10 expression declines within the implantation period in infertile female patients for different reasons.41–47 The defective HOXA-10 expression in the endometrium during the middle-secretory phase may be related to endometriosis, adenomyosis, polycystic ovarian disease, submucosal uterine leiomyomas, and hydrosalpinx; these conditions are associated with endometrial implantation insolvency. However, at this point a consensus regarding its role in the implantation process in women with endometriosis is lacking. Moreover, the connection between the HOXA-10 gene aberrations and a lack of implantation in endometriosis remains unclear.42 HOXA-10 gene expression in the eutopic endometrium in endometriosis is significantly lower than in women with no endometriosis due to the hypermethylation of its promoter.39 Additionally, HOXA-10 levels correlate with endometriosis severity. In female patients with tougher endometriosis, the expression is much lower compared to women with moderate and mild levels of the disease.42 Furthermore, HOXA-10 gene expression in eutopic endometrium samples is quite different in patients with endometriosis but without fertility loss compared to cases with endometriosis-related infertility, showing a 1.8 and 3.5-fold decrease compared to healthy women (without infertility or endometriosis).42
Aberrant Estrogen Synthesis
As previously mentioned, the characteristic disorders involving DNA methylation in endometriosis include the hypomethylation of the CpG-area within the proximal promoter NR5A1. Following activation by the ascending stimulating factor 2 (upstream stimulatory factor 2: USF2), an aberrant expression of steroidogenic factor-1 (SF-1) is achieved.48,49 SF-1, also known as Ad4BP, belongs to the nuclear receptor family of intracellular transcription factors.48 In the beginning of the 1990s, it was identified as a steroidogenic enzyme regulator.49 SF-1 controls many aspects of the adrenal glands and reproductive system, and plays an important role in embryonic growth and sexual differentiation. Additionally, it participates in multilayer gene expression regulation of many hormone-producing enzymes.49–51 SF-1 induces the expression of the steroidogenic acute regulatory protein (StAR), which is another key enzyme playing a major role in estrogen synthesis. StAR mediates cholesterol translocation from the cytoplasm within the mitochondria and supports cells with substrates for estrogen synthesis.50,52 Moreover, SF-1 increases the expression of aromatase, inducing transcription factors of the aromatase II promoter, including the CCAAT/enhancer-binding protein, unidentified enhancers and co-activators 1 and 2.49,50
P450arom, which is coded by the CYP19A1 gene, is also hypomethylated in endometriosis and is a key estrogen biosynthesis enzyme. P450arom is responsible for the androstenedione transformation into estrone, and is further converted into the biologically active estradiol by the 17β-Hydroxysteroid dehydrogenase 1 (17β-HSD1) enzyme.52 P450arom activation may also result from prostaglandin E2 (PGE2) stimulation. PGE2 synthesis increases following the increase of cyclic adenosine monophosphate (cAMP) and the activation of some downstream signaling pathways within stromal cells.50 At the same time, PGE2 induces SF-1, an aromatase transcription regulator. Simultaneously, aromatase overexpression supports the sustainable production of PGE2, which mediates endometriosis-related inflammation (Figure 2).53–56
 |
Figure 2 Schematic representation of aberrant estrogen synthesis pathological functional system. |
SF-1 is expressed within endometriotic cells and is not detected in normal endometrium.39 SF-1 and StAR expression levels in ectopic endometrium correlate with endometriosis progression stage.50 An experimental model which conditionally expresses SF-1 within eutopic endometrium in vivo, showed weakening sensitivity to estrogen and progesterone. Auto-transplantation of uterine tissue into the pelvic membrane lead to ectopic disorder growth and the induction of endometriosis.21 Reports have suggested that the expression of SF-1 within an eutopic endometrium is associated with a high risk of endometriosis; however, some studies have indicated that SF-1 was not detected in eutopic endometrium of women with endometriosis.57–59 Nevertheless, endometrial stromal cells contain the full steroid genome for de novo synthesis of estradiol out of cholesterol, which aggravates and supports the disruption of endometrial steroid regulation in endometriosis.
Estrogen-Induced Inflammation
Endometrial estrogen receptors are represented by two forms: estrogen alpha receptors (ERα), which are coded by the ESR1 gene; and estrogen beta receptors (ERβ), which are coded by the ESR2 gene. Although they are transcribed by different genes, estrogen receptors possess very similar primary structures.31 Due to the DNA methylation pattern disorder within stromal cells, the balanced expression of estrogen receptors becomes disrupted. CpG hypomethylation within the ESR2 promoter area leads to a high level of ERβ expression within the endometrioid stromal cells, with the ESR1 gene becoming hypermethylated. Subsequently, the ERα mRNA level and the ERβ protein levels increase significantly within the endometrioid stromal cells.60 ERβ becomes a dominant isoform and a key estrogen mediator in ectopic foci and eutopic endometrium in endometriosis.60–63
ERβ mediates estradiol-stimulated cyclooxygenase-2 (COX-2) and leads to the hyperproduction of PGE2, resulting in increased angiogenesis and inflammatory reactions in endometriosis (Figure 2).56,64 PGE2 maintains the P450arom chronic overexpression as well as stable production of estradiol, supporting the fact that inflammation stimulates estrogen expression, and that estrogens promote inflammation.65 These conditions create a pathological functional system, which exacerbates the effects of each other.
Endometriosis-Related Immune Inflammation
According to most studies, immunologic dysfunction also leads to the development of endometriosis.64,66 The levels of immunocompetent cells such as monocytes, macrophages, natural killer cells, T- and B-lymphocytes, and regulatory T-cells as well as inflammation-derived cytokine and chemokine concentrations within the peritoneal fluid are elevated in patients with endometriosis.67 Among the main the inflammation-related factors, macrophages are usually present and are thought to play a critical role in endometriosis. Moreover, macrophages produce important inflammatory factors including monocyte chemoattractant proteins (MCPs), plasminogen activators, adhesion molecules, heme-oxygenase molecules, cytokine molecules, prostaglandins, and COX-2.68 These factors induce cell proliferation by fibroblasts in the endothelium, which in turn participate in further inflammation, tissue recovery, and neovascularization through the secretion of cytokines.1,69,70 Peritoneal macrophages can produce lytic enzymes, which increase and stimulate the implantation and proliferation of endometrioid cells within the abdomen and reinforce angiogenesis within the ectopic foci.71,72 The activation of macrophages during endometriosis has already been proven. It used to be aberrant to believe that macrophages are active within the ectopic foci;66 however, currently it is regarded that this activity is natural and not a redundant reaction to the endometrial tissue located outside of uterus. The macrophages perceive signals such as hypoxia, cellular injury, and iron overload within the injury foci and tend to reestablish local homeostasis.73 In other words, the macrophages perceive the endometriosis focus as a wound and activate adequate processes which lead to the ectopic cell survival and tissue vascularization. This way, they support the estrogen-provoked inflammation and become another member of the pathophysiologic process.
The role of macrophages in the growth and development of eutopic endometriosis remains debatable. On one hand, the macrophages are the leukocytes located within the tissue, and these leukocytes are key components of immune response and produce inflammatory and anti-inflammatory substances. In contrast, the exact number of macrophages within eutopic endometrium in endometriosis is variable. A study showed that the number of CD68 positive cells (a marker of mature and active macrophages) in the endometrium decreased in the early proliferative phase, and did not significantly differ in other phases.74 Another report showed that a significant increase in the number of macrophages in the proliferative phase of an eutopic endometrium in patients with endometriosis.75 Also, there is data suggesting that the number of macrophages within an eutopic endometrium increase during the menstrual cycle.76 However, it is unclear whether eutopic immune changes are primary or secondary.
It is not the quantity of macrophages, but rather the quality of research, in particular the ratio of macrophage subpopulations within eutopic endometrium that causes so much interest. Macrophages are polarized functionally into the M1 and M2 cell lines. M1 macrophages express inducible nitric oxide synthase (iNOS) and tumor necrosis factor (TNF). They produce a large amount of nitric oxide under oxidative stress. The main function of M1 macrophages is to protect against local infection with bacteria, viruses, and fungi. M2 macrophages play an important role in the response to parasitic infection and also in tissue remodeling, angiogenesis, and neoplastic progression. Endometrial macrophages are normally of the M2 lineage,77 although in endometriosis, the macrophage population ratio is shifted toward the M1-active form.76 In this manner, the M1-M2 macrophage polarization disorder in eutopic endometrium may play an essential role in disease progression.78
Steroid Hormone Receptor Expression Changes
The cyclic changes in endometrial tissues are controlled primarily by ovarian steroid hormones. The main effects of estrogen and progesterone indirectly self-realize through their connection with corresponding nuclear receptors of the glands and endometrial stroma. For this reason, many researchers assign a decisive role in the interaction of the functionally reliable endometrial receptors, rather than the impact of steroid hormones on the endometrium. The estrogen receptors (ER) and the progesterone receptors (PR) are located closely related and are co-regulated.79 In addition to the proliferation and stimulation of endometrial cells and the increased expression of an own receptor, upon estradiol activation, the ER is translocated to the nucleus and binds to a PGR promoter, thereby increasing the expression of PR.27,60,80 Therefore, the endometrium should be susceptible to estrogen, so that it eventually reacts to progesterone hormone action. Progesterone not only increases the synthesis of its own receptors, it also decreases ER and PR expression as part of its own biological activity. This way, the specific receptor expression levels depend on specific hormonal blood levels as well as the concentrations of other steroid hormones.
As mentioned above, the endometrial estrogen receptor is represented by two forms, ERα and ERβ, which are different from each other in their endometrial functions. An α-receptor expression is highest in the stroma and endometrial glands during the entire proliferative phase of the menstrual cycle. ERβ primarily plays an important functional role in ovulation, plus it participates within the decidualization and “maturation” of the cervix during pregnancy. There is usually a switch between ERα and ERβ expression during pregnancy, with the disappearance of ERβ accompanying the onset of labor.31 The highest β-receptor expression level is detected in endometrial epithelial cells within the preovulatory period, and the last secretory phase within the stromal and endothelial cells.31
Progesterone receptors are presented by two ligand-binding forms: PR-A and PR-B. They differ in the N-ends, and the PR-A protein is 164 amino acids shorter than the PR-B, because these isoforms are transcribed by two promoters of the same gene. It is suggested that the progesterone action in the endometrium, during the secretory phase of the menstrual cycle and early period of pregnancy, is carried out through the activation of A-subtype receptors, located in endometrial stromal cells.31 The progressive differentiation of endometrial stromal cells into decidual cells, which are unique in their trophoblast invasion regulation and local immune reactions, is the main result of the progesterone action during the second phase of the menstrual cycle.
The main function of the endometrium is the fixation of the blastocyst and following thromboblast invasion. The period of maximal endometrial susceptibility, when the probability of the blastocyst implantation is the highest, is called the implantation window. The implantation window opens up when the progesterone plasma concentration is greatest, within 7–10 days after the ovulation period, and it involves 20–24 days of a 28-day menstrual cycle, which corresponds to the middle stage of the secretion phase.43 The synchronous complementary expression of growth factors, extracellular matrix components, and membrane bound receptors during this time plays a decisive role in the fixation of the blastocyst implantation on top of the luminal epithelial apical surface, and the trophoblast invasion direction through such barrier.81–83
Since steroid hormones play a key role in the functioning of the endometrium, many researchers have evaluated the levels of ER and PR expression within the implantation window period. Changes in the expression of steroid hormones within the implantation window period are thoroughly reported in healthy reproductive women, and also in patients with different pathologic conditions, which can influence endometrial receptivity. The decrease in ERα expression within the endometrial glands and stroma provides proof of an implantation window discovery.83,84 ERα overexpression is related to an induction of its subordinate genes: mucin-1 (MUC-1), insulin-like growth factor-1 (IGF-1), and leukemia-inhibitory factor (LIF), which leads to endometrial receptivity disturbance.85,86 Progesterone-regulated ERα disappearance in the middle of the luteal phase supports the ability of progesterone to solely influence the stroma through the specific activation of its related receptors. In response to progesterone, paracrine stromal activity leads to gene expression in epithelial cells, which is necessary for implantation.84 Within an ectopic focus, ERβ mRNA and receptor levels are significantly higher than ERα and PR and PR-B, in particular, decreases significantly.60 An eutopic endometrium in patients with endometriosis also shows much higher expression of ERβ, which suggests that extremely high levels of endometrial ERβ may predispose women to endometriosis progression.60 Moreover, a decreased coordinated process for steroid hormone receptor expression becomes destroyed in the luteal phase cycle; therefore, ERα expression increases during the middle stage of the secretion phase.87 In endometriosis, this index may also surpass the PR expression level. Progesterone resistance, a hypersensitivity to estrogen and a local increase in estrogen production may become a reason for ER hyperexpression within an eutopic endometrium in the middle of luteal phase.84,87
Notably, progesterone blood levels do not affect the expression of progesterone receptors, and plasma progesterone levels in various females are not significantly different.81,88 Many researchers have noticed that PR expression in the nuclei of stromal cells in patients with endometriosis is lower compared to normal endometrium.81 This may be related to both hypermethylation of the PGR promoter area and to an abnormally high ERβ/ERα ratio in the endometrioid stromal cells, which may disturb the PGR gene by inducing estradiol.27 Through its own receptors, progesterone causes a resistance to endometrial growth, which is induced by estrogen. Insufficient progesterone action sharply increases the risk of endometrium hyperplasia. Simultaneously, progesterone promotes growth through an increase in proliferation, cellular hypertrophy, and the formation of an extracellular matrix in the uterine leiomyoma; in the mammary gland, particularly in breast cancer, progesterone has a proliferative effect and is a carcinogenic hormone.89 The key difference between these tissue types and the endometrium, which could explain the very different effects of progesterone, is based on the stroma paracrine interaction with the epithelium. Here, a normal endometrium is composed of stromal cells which express an abundant amount of the PR and results in a paracrine effect in epithelium proliferation and differentiation.90,91 Even though stroma decidualization is the main result of the actions of progesterone, the most pronounced difference between PR expression in infertile females and fertile controls occurs in epithelial cells.92
While most studies suggest a characteristic change in the receptor expression of steroid hormones in the eutopic endometrium (endometriosis), a study from the Sao-Paulo University (Brazil) showed no significant differences in PR expression compared to the control group.93 A global analysis of endometrial gene expression in patients with endometriosis revealed disparities in the regulation of some target progesterone genes during the implantation window time period.94,95 Here, the progesterone resistance may be explained by a possible PR deficit in poorly differentiated stromal cells, and by following the transfer of progesterone signals through a molecular cascade.96
Genetic/Epigenetic Theory of Endometriosis Pathogenesis
Due to the fact that existing theories of endometriosis pathogenesis cannot explain the specific mechanisms behind endometriotic transformation, exogenous survival and invasive patterns, a polygenetic/polyepigenetic theory of endometriosis pathogenesis was proposed.97 The theory of genetic/epigenetic pathogenesis serves as a unification theory including all previously discussed mechanisms of pathological transformation. This theory suggests that endometriosis development is a result of specific genetic and epigenetic incidence that are transmitted at birth, accounting for predisposition to the disease, endometriosis-associated endometrium changes, immunological peculiarities and placentation. The genetic/epigenetic theory provides a review of DNA sequence chromosomal alterations, occurring either due to genetic mutations or noxious agents, and their effect on multimodal transmission of endometriosis predisposition. More so, this theory explains the previously discussed effects of oxidative stress, hormonal imbalance and other influencing factors on endometriosis progression, which damage native DNA and promote pathological reorganization of genetic information, warranting endometriosis associated changes.
As such, the genetic/epigenetic theory states that endometriosis is brought about by a cumulative set of genetic and epigenetic incidents, which lead to defects in normal cellular physiology. These incidents are a result of specific pathological influence: both genetic and not genetic. A major influencing factor is inflammation,98 resulting from both bacterial and viral presence. Recently, it has been discussed that the genetic-epigenetic factors supporting endometriotic cellular transformation are potentiated by environmental pollution, oxidative stress and inflammation-inducing agents.99
Cancer and Endometriosis
Many of the above described pathogenetic mechanisms are commonly seen in tumor progression patterns. Two scenarios leading to endometriosis-associated cancer have been proposed.100 The first involves damage from recurrent hemorrhages from extracellular hemoglobin, iron, and heme, which cause cellular oxidative damage by with consequent DNA damage and malignant-transforming mutations. The second scenario assumes constant production of antioxidants, which help to maintain the tumor environment. Both options support the redox imbalance theory.
It have been shown that overexpression of HNF-1β is seen in endometriotic lesions regardless of signs of epithelial atypia. HNF-1β presence confirms a histogenetic relationship between ovarian endometriosis and adenocarcinoma cells.101 Such processes as oxidative stress, prevalence of certain cytokines, genetic mutations, and hyperestrogenism, seen in recurrent endometriosis are often seen in ovarian and cervical cancers, showing a connection between endometriosis and cancer.102 The balance of oxidants and endogenous antioxidants can play an important role in malignant transformation of endometriotic lesions. Atypical endometriosis is especially dangerous and is considered by some researchers as an intermediate precursor linking classical endometriosis and clear cell adenocarcinoma.103–105
The genetic aspects of endometriotic oncotransformation play an important role in understanding the associations between endometriosis and cancer. In a study of genetic expression profiles in both endometriotic and normal epithelium, numerous somatic mutations were found in genes seen in ovarian cancer associated with endometriosis.106 Such cancer-associated mutations were identified in genes KRAS, ARID1A, PIK3CA, KRAS, PPP2R1A, ARID1B, PIK3R1, PTEN, MLL3, FBXW7 and ARHGAP35.11,14 Interestingly, a consensus regarding endometriosis association with cancer has still not been made, as such mutations are even regarded as a normality in endometriotic tissue.107 Despite this, endometriotic tissue has been pinpointed as the source of various forms of ovarian carcinomas, which are collectively known as endometriosis associated cancer.108
As of today, certain types of ovarian malignancies are associated with endometriosis, arising from a pre-existing endometriotic lesion. These tumors include ovarian clear cell carcinoma, ovarian endometrioid carcinoma, and ovarian seromucinous tumors, which are characterized by common molecular genetic changes including ARID1A, PI3K and PP2A pathways, but also have unique molecular changes such as microsatellite instability, CTNNB1 mutations and overexpression of HNF-1β.109 Nonetheless, the role of the endometriotic progenitor cell in oncotransformation has not yet been shown, and it is unclear, whether the cancer arises from the endometriotic lesion, or due to the influence of pathogenetic factors associated with the endometriotic lesion.
Conclusion
Despite recent advances, fundamental problems in understanding and diagnosing endometriosis remain unresolved. A key step in further understanding the ethological aspects of pathogenetic mechanisms of endometriosis will be the identification of unknown circulating epithelial progenitors or stem cells that are responsible for epithelial regeneration in both the endometrium and endometriotic foci. It will be equally important to determine the origin of these precursors and to explore their use as biomarkers for predicting endometriosis risk and response to treatment.
Data Sharing Statement
All relevant raw data are freely available to any researchers who wish to use them for non-commercial purposes while preserving any necessary confidentiality and anonymity. The datasets are available on request to the corresponding author.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
This research received no external funding.
Disclosure
The authors reported no conflicts of interest for this work.
References
1. Burney RO, Giudice LC. Pathogenesis and pathophysiology of endometriosis. Fertil Steril. 2012;98(3):511–519. doi:10.1016/j.fertnstert.2012.06.029
2. Viganò P, Parazzini F, Somigliana E, Vercellini P. ‘Endometriosis: epidemiology and aetiological factors’. Best Pract Res Clin Obstet Gynaecol. 2004;18:177–200. doi:10.1016/j.bpobgyn.2004.01.007
3. Browne H, Taylor H. HOXA10 expression in ectopic endometrial tissue. Fertil Steril. 2006;85(5):1386–1390. doi:10.1016/j.fertnstert.2005.10.072
4. Matsuura K, Ohtake H, Katabuchi H, Okamura H. Coelomic metaplasia theory of endometriosis: evidence from in vivo studies and an in vitro experimental model. Gynecol Obstet Invest. 1999;47(Suppl.1):18–22. doi:10.1159/000052855
5. Fukunaga M, Ushigome S. Epithelial metaplastic changes in ovarian endometriosis. Modern Pathol. 1998;11(8):784–788.
6. Zheng W, Li N, Wang J, et al. Initial endometriosis showing direct morphologic evidence of metaplasia in the pathogenesis of ovarian endometriosis. Int J Gynecol Pathol. 2005;24(2):164–172. doi:10.1097/01.rct.0000157091.37057.b4
7. Mikhaleva LM, Solomatina AA, Boltovskaya MN, et al.Clinical and morphological aspects of ovarian endometriosis depending on the severity of the disease. Clin Exp Morphol. 2016;3(19):15–21.
8. Patsap OI, Tairova MB, Davydov AI, Mikhaleva LM. The role of cytological evaluation in sclerotherapy of ovarian endometrioma. Gynecol Obstet Perinatol. 2020;19(2):5–10. doi:10.20953/1726-1678-2020-2-5-10.
9. Mikhaleva LM, Davydov AI, Patsap OI, et al. Malignant transformation and associated biomarkers of ovarian endometriosis: a narrative review. Adv Ther. 2020;37:2580–2603. doi:10.1007/s12325-020-01363-5
10. Anglesio MS, Papadopoulos N, Ayhan A, et al. Cancer-associated mutations in endometriosis without cancer. N Engl J Med. 2017;376(19):1835–1848. doi:10.1056/NEJMoa1614814
11. Suda K, Nakaoka H, Yoshihara K, et al. Clonal expansion and diversification of cancer-associated mutations in endometriosis and normal endometrium. Cell Rep. 2018;24(7):1777–1789. doi:10.1016/j.celrep.2018.07.037
12. Guo SW. Cancer-associated mutations in endometriosis: shedding light on the pathogenesis and pathophysiology. Hum Reprod Update. 2020;26(3):423–449. doi:10.1093/humupd/dmz047
13. Zou Y, Zhou JY, Guo JB, et al. The presence of KRAS, PPP2R1A and ARID1A mutations in 101 Chinese samples with ovarian endometriosis. Mutat Res. 2018;809:1–5. doi:10.1016/j.mrfmmm.2018.03.001
14. Suda K, Nakaoka H, Yoshihara K, et al. Different mutation profiles between epithelium and stroma in endometriosis and normal endometrium. Hum Reprod. 2019;34(10):1899–1905. doi:10.1093/humrep/dez155
15. Li X, Zhang Y, Zhao L, et al. Whole-exome sequencing of endometriosis identifies frequent alterations in genes involved in cell adhesion and chromatin-remodeling complexes. Hum Mol Genet. 2014;23(22):6008–6021. doi:10.1093/hmg/ddu330
16. Kyo S, Sato S, Nakayama K. Cancer-associated mutations in normal human endometrium: surprise or expected? Cancer Sci. 2020;111:3458–3467. doi:10.1111/cas.14571
17. Davydov AI, Mikhaleva LM, Patsap OI. On markers of early detection of endometriosis-associated ovarian tumors. Gynecol Obstet Perinatol. 2019;4:133–137.
18. Haidarali E, Vahedi A, Mohajeri S, Mostafidi E, Azimpouran M, Naghavi Behzad M. Evaluation of the pathogenesis of tumor development from endometriosis by estrogen receptor, P53 and Bcl-2 Immunohistochemical Staining. Asian Pac J Cancer Prev. 2016;17(12):5247–5250. doi:10.22034/APJCP.2016.17.12.5247
19. Davydov AI, Mikhaleva LM, Tairova MB, Patsap OI. Ovarian endometriosis: a form of genital endometriosis or a specific nosological unit. Gynecol Obstet Perinatol. 2019;5:5–12.
20. Ramathal CY, Bagchi IC, Taylor RN, Bagchi MK. Endometrial decidualization: of mice and men. Semin Reprod Med. 2010;28(1):17–26. doi:10.1055/s-0029-1242989
21. Vasquez YM, Wu SP, Anderson ML, et al. Endometrial expression of steroidogenic factor 1 promotes cystic glandular morphogenesis. Mol Endocrinol. 2016;30(5):518–532. doi:10.1210/me.2015-1215
22. Viger RS, Guittot SM, Anttonen M, Wilson DB, Heikinheimo M. Role of the GATA family of transcription factors in endocrine development, function, and disease. Mol Endocrinol. 2008;22(4):781–798. doi:10.1210/me.2007-0513
23. Dyson MT, Roqueiro D, Monsivais D, et al. Genome-wide DNA methylation analysis predicts an epigenetic switch for GATA factor expression in endometriosis. PLoS Genet. 2014;10(3):e1004158. doi:10.1371/journal.pgen.1004158
24. Zaret KS, Carroll JS. Pioneer transcription factors: establishing competence for gene expression. Genes Dev. 2011;25(21):2227–2241. doi:10.1101/gad.176826.111
25. Sampson JA. Metastatic or embolic endometriosis, due to the menstrual dissemination of endometrial tissue into the venous circulation. Am J Pathol. 1927;3(2):93–110.43.
26. D’Hooghe TM, Debrock S. Endometriosis, retrograde menstruation and peritoneal inflammation in women and in baboons. Hum Reprod Update. 2002;8(1):84–88. doi:10.1093/humupd/8.1.84
27. Bulun SE, Yilmaz BD, Sison C, et al. Endometriosis. Endocr Rev. 2019;40(4):1048–1079. doi:10.1210/er.2018-00242
28. Missmer SA, Hankinson SE, Spiegelman D, et al. Reproductive history and endometriosis among premenopausal women. Obstet Gynecol. 2004;104(5 Pt 1):965–974. doi:10.1097/01.AOG.0000142714.54857.f8
29. Izawa M, Taniguchi F, Harada T. GATA6 expression promoted by an active enhancer may become a molecular marker in endometriosis lesions. Am J Reprod Immunol. 2019;81(2):e13078. doi:10.1111/aji.13078
30. Iwafuchi-Doi M, Zaret KS. Pioneer transcription factors in cell reprogramming. Genes Dev. 2014;28(24):2679–2692. doi:10.1101/gad.253443.114
31. Marquardt RM, Kim TH, Shin JH, Jeong JW. Progesterone and estrogen signaling in the endometrium: what goes wrong in endometriosis? Int J Mol Sci. 2019;20(15):3822. doi:10.3390/ijms20153822
32. Monsivais D, Dyson MT, Yin P, et al. ERβ- and prostaglandin E2-regulated pathways integrate cell proliferation via Ras-like and estrogen-regulated growth inhibitor in endometriosis. Mol Endocrinol. 2014;28(8):1304–1315. doi:10.1210/me.2013-1421
33. Lee B, Du H, Taylor HS. Experimental murine endometriosis induces DNA methylation and altered gene expression in eutopic endometrium. Biol Reprod. 2009;80(1):79–85. doi:10.1095/biolreprod.108.070391
34. Wu Y, Strawn E, Basir Z, Halverson G, Guo SW. Aberrant expression of deoxyribonucleic acid methyltransferases DNMT1, DNMT3A, and DNMT3B in women with endometriosis. Fertil Steril. 2007;87(1):24–32. doi:10.1016/j.fertnstert.2006.05.077
35. Dyson MT, Kakinuma T, Pavone ME, et al. Aberrant expression and localization of deoxyribonucleic acid methyltransferase 3B in endometriotic stromal cells. Fertil Steril. 2015;104(4):953–963.e2. doi:10.1016/j.fertnstert.2015.06.046
36. Bernardi LA, Dyson MT, Tokunaga H, et al. The essential role of GATA6 in the activation of estrogen synthesis in endometriosis. Reprod Sci. 2019;26(1):60–69. doi:10.1177/1933719118756751
37. Naqvi H, Ilagan Y, Krikun G, Taylor HS. Altered genome-wide methylation in endometriosis. Reprod Sci. 2014;21(10):1237–1243. doi:10.1177/1933719114532841
38. Signorile PG, Severino A, Santoro M, Spyrou M, Viceconte R, Baldi A. Methylation analysis of HOXA10 regulatory elements in patients with endometriosis. BMC Res Notes. 2018;11(1):722. doi:10.1186/s13104-018-3836-1
39. Orazov MR, Khamoshina MB, Mikhaleva LM, et al. Molecular genetic features of the state of endometry in endometriosis-associated infertility. Difficult Patient. 2020;1–2:23–32.
40. Rubel CA, Wu SP, Lin L, et al. A Gata2-dependent transcription network regulates uterine progesterone responsiveness and endometrial function. Cell Rep. 2016;17(5):1414–1425. doi:10.1016/j.celrep.2016.09.093
41. Yang Y, Chen X, Saravelos SH, et al. HOXA-10 and E-cadherin expression in the endometrium of women with recurrent implantation failure and recurrent miscarriage. Fertil Steril. 2017;107(1):136–143.e2. doi:10.1016/j.fertnstert.2016.09.016
42. Özcan C, Özdamar Ö, Gökbayrak ME, Doğer E, Çakıroğlu Y, Çine N. HOXA-10 gene expression in ectopic and eutopic endometrium tissues: does it differ between fertile and infertile women with endometriosis? Eur J Obstet Gynecol Reprod Biol. 2019;233:43–48. doi:10.1016/j.ejogrb.2018.11.027
43. Orazov MR, Lokshin VN, Silantyeva EC, et al. Morphological and molecular biological determinants of repeated implantation failure. Bulletin of National Academy of Sciences of the Republic Of Kazakhstan. 2020;3(385):6–15. doi:10.32014/2020.2518-1467.63
44. Wang J, Huang C, Jiang R, et al. Decreased Endometrial IL-10 impairs endometrial receptivity by downregulating HOXA10 expression in women with adenomyosis. Biomed Res Int. 2018;2018:2549789. doi:10.1155/2018/2549789
45. Riyanti A, Febri RR, Zakirah SC, et al. Suppressing HOXA-10 Gene Expression by MicroRNA 135b during the window of implantation in infertile women. J Reprod Infertil. 2020;21(3):217–221.
46. Robles TG, Fernández RA, García-Palencia P, et al. Hoxa-10 and Cyclin D3 overexpression in the decidual reaction in a superovulation protocol in Young Adult C57BL/6J Mice. Vet Pathol. 2017;54(2):328–335. doi:10.1177/0300985816660748
47. Celik O, Unlu C, Otlu B, Celik N, Caliskan E. Laparoscopic endometrioma resection increases peri-implantation endometrial HOXA-10 and HOXA-11 mRNA expression. Fertil Steril. 2015;104(2):356–365. doi:10.1016/j.fertnstert.2015.04.041
48. Wu PL, Zhou Y, Zeng C, et al. Transcription factor 21 regulates expression of ERβ and SF-1 via upstream stimulatory factor-2 in endometriotic tissues. Biochim Biophys Acta Gene Regul Mech. 2018;1861(8):706–717. doi:10.1016/j.bbagrm.2018.06.008
49. Ferraz-de-souza B, Lin L, Achermann JC. Steroidogenic factor-1 (SF-1, NR5A1) and human disease. Mol Cell Endocrinol. 2011;336(1–2):198–205. doi:10.1016/j.mce.2010.11.006
50. Tian Y, Kong B, Zhu W, Su S, Kan Y. Expression of steroidogenic factor 1 (SF-1) and steroidogenic acute regulatory protein (StAR) in endometriosis is associated with endometriosis severity. J Int Med Res. 2009;37(5):1389–1395. doi:10.1177/147323000903700513
51. Fabbri HC, Ribeiro de Andrade JG, Maciel-Guerra AT, Guerra-Júnior G, de Mello MP. NR5A1 loss-of-function mutations lead to 46, XY partial gonadal dysgenesis phenotype: report of three novel mutations. Sex Dev. 2016;10(4):191–199. doi:10.1159/000448013
52. Attar E, Tokunaga H, Imir G, et al. Prostaglandin E2 via steroidogenic factor-1 coordinately regulates transcription of steroidogenic genes necessary for estrogen synthesis in endometriosis. J Clin Endocrinol Metab. 2009;94(2):623–631. doi:10.1210/jc.2008-1180
53. Noble LS, Takayama K, Zeitoun KM, et al. Prostaglandin E2 stimulates aromatase expression in endometriosis-derived stromal cells. J Clin Endocrinol Metab. 1997;82(2):600–606. doi:10.1210/jcem.82.2.3783
54. Tsai SJ, Wu MH, Lin CC, Sun HS, Chen HM. Regulation of steroidogenic acute regulatory protein expression and progesterone production in endometriotic stromal cells. J Clin Endocrinol Metab. 2001;86(12):5765–5773. doi:10.1210/jcem.86.12.8082
55. Sun HS, Hsiao KY, Hsu CC, Wu MH, Tsai SJ. Transactivation of steroidogenic acute regulatory protein in human endometriotic stromalcells is mediated by the prostaglandin EP2 receptor. Endocrinology. 2003;144(9):3934–3942. doi:10.1210/en.2003-0289
56. Tamura M, Deb S, Sebastian S, Okamura K, Bulun SE. Estrogen up-regulates cyclooxygenase-2 via estrogen receptor in human uterine microvascular endothelial cells. Fertil Steril. 2004;81(5):1351–1356. doi:10.1016/j.fertnstert.2003.09.076
57. Xue Q, Bai L, Li T, Dong Y, Zhang Y, Zhou YF.[Expression of steroidogenic factor 1 in ovarian endometriosis and adenomyosis]. Zhonghua Fu Chan Ke Za Zhi. 2011;46(11):831–833.
58. Noël JC, Anaf V, Borghese B, Vaiman D, Fayt I, Chapron C. The steroidogenic factor-1 protein is not expressed in various forms of endometriosis but is strongly present in ovarian cortical or medullary mesenchymatous cells adjacent to endometriotic foci. Fertil Steril. 2011;95(8):2655–2657. doi:10.1016/j.fertnstert.2011.01.131
59. Noël JC, Borghese B, Vaiman D, Fayt I, Anaf V, Chapron C. Steroidogenic factor-1 expression in ovarian endometriosis. Appl Immunohistochem Mol Morphol. 2010;18(3):258–261. doi:10.1097/PAI.0b013e3181c06948
60. Xue Q, Lin Z, Cheng YH, et al. Promoter methylation regulates estrogen receptor 2 in human endometrium and endometriosis. Biol Reprod. 2007;77(4):681–687. doi:10.1095/biolreprod.107.061804
61. Juhasz-Böss I, Fischer C, Lattrich C, et al. Endometrial expression of estrogen receptor β and its splice variants in patients with and without endometriosis. Arch Gynecol Obstet. 2011;284(4):885–891. doi:10.1007/s00404-010-1768-7
62. Han SJ, Hawkins SM, Begum K, et al. A new isoform of steroid receptor coactivator-1 is crucial for pathogenic progression of endometriosis. Nat Med. 2012;18(7):1102–1111. doi:10.1038/nm.2826
63. Hu X, Zhou Y, Feng Q, et al. Association of endometriosis risk and genetic polymorphisms involving biosynthesis of sex steroids and their receptors: an updating meta-analysis. Eur J Obstet Gynecol Reprod Biol. 2012;164(1):1–9. doi:10.1016/j.ejogrb.2012.05.008
64. Bellelis P, Barbeiro DF, Rizzo LV, Baracat EC, Abrão MS, Podgaec S. Transcriptional changes in the expression of chemokines related to natural killer and T-regulatory cells in patients with deep infiltrative endometriosis. Fertil Steril. 2013;99(7):1987–1993. doi:10.1016/j.fertnstert.2013.02.038
65. Jr MH, Haddad C, Coelho G, Casoy J. Role of inflammation and aromatase expression in the eutopic endometrium and its relationship with the development of endometriosis. Womens Health (Lond). 2012;8(6):647–658. doi:10.2217/whe.12.52
66. Podgaec S, Barbeiro DF, Gueuvoghlanian-Silva BY, Bellelis P, Abrão MS, Baracat EC. Foxp3 expression in deep rectosigmoid endometriosis lesions and its association with chronic pelvic pain. J Reprod Immunol. 2014;104:96–99. doi:10.1016/j.jri.2014.05.002
67. Waiyaput W, Pumipichet S, Weerakiet S, Rattanasiri S, Sophonsritsuk A. Effect of simvastatin on monocyte chemoattractant protein-1 expression in endometriosis patients: a randomized controlled trial. BMC Womens Health. 2017;17(1):89. doi:10.1186/s12905-017-0446-3
68. Bellelis P, Barbeiro DF, Gueuvoghlanian-Silva BY, Kalil J, Abrão MS, Podgaec S. Interleukin-15 and interleukin-7 are the major cytokines to maintain endometriosis. Gynecol Obstet Invest. 2019;84(5):435–444. doi:10.1159/000496607
69. Gueuvoghlanian-Silva BY, Bellelis P, Barbeiro DF, Hernandes C, Podgaec S. Treg and NK cells related cytokines are associated with deep rectosigmoid endometriosis and clinical symptoms related to the disease. J Reprod Immunol. 2018;126:32–38. doi:10.1016/j.jri.2018.02.003
70. Gazvani R, Templeton A. New considerations for the pathogenesis of endometriosis. Int J Gynaecol Obstet. 2002;76(2):117–126. doi:10.1016/s0020-7292(01)00577-x
71. Khan KN, Masuzaki H, Fujishita A, et al. Peritoneal fluid and serum levels of hepatocyte growth factor may predict the activity of endometriosis. Acta Obstet Gynecol Scand. 2006;85(4):458–466. doi:10.1080/00016340500432556
72. Rached MR, Coelho V, Marin MLC, et al. HLA-G is upregulated in advanced endometriosis. Eur J Obstet Gynecol Reproductive Biol. 2019;235:36–41. doi:10.1016/j.ejogrb.2019.01.030
73. Capobianco A, Rovere-Querini P. Endometriosis, a disease of the macrophage. Front Immunol. 2013;4:9. doi:10.3389/fimmu.2013.00009
74. Braun DP, Ding J, Shen J, Rana N, Fernandez BB, Dmowski WP. Relationship between apoptosis and the number of macrophages in eutopic endometrium from women with and without endometriosis. Fertil Steril. 2002;78(4):830–835. doi:10.1016/s0015-0282(02)03334-4
75. Berbic M, Schulke L, Markham R, Tokushige N, Russell P, Fraser IS. Macrophage expression in endometrium of women with and without endometriosis. Hum Reprod. 2009;24(2):325–332. doi:10.1093/humrep/den393
76. Takebayashi A, Kimura F, Kishi Y, et al. Subpopulations of macrophages within eutopic endometrium of endometriosis patients. Am J Reprod Immunol. 2015;73(3):221–231. doi:10.1111/aji.12331
77. Orazov MR, Mikhaleva LM, Semenov PA. Khronicheskiy endometrit: patogenez, diagnostika, lecheniye i yego svyaz’ s besplodiyem. Klin Eksp Morfologiya. 2020;9(2):16–25. doi:10.31088/CEM2020.9.2.16-25
78. Poli-Neto OB, Meola J, Rosa-E-Silva JC, Tiezzi D. Transcriptome meta-analysis reveals differences of immune profile between eutopic endometrium from stage I-II and III-IV endometriosis independently of hormonal milieu. Sci Rep. 2020;10(1):313. doi:10.1038/s41598-019-57207-y
79. Attia GR, Zeitoun K, Edwards D, Johns A, Carr BR, Bulun SE. Progesterone receptor isoform A but not B is expressed in endometriosis. J Clin Endocrinol Metab. 2000;85(8):2897–2902. doi:10.1210/jcem.85.8.6739
80. Babayev SN, Park CW, Keller PW, Carr BR, Word RA, Bukulmez O. Androgens upregulate endometrial epithelial progesterone receptor expression: potential implications for endometriosis. Reprod Sci. 2017;24(10):
81. Vasquez YM, Wang X, Wetendorf M, et al. FOXO1 regulates uterine epithelial integrity and progesterone receptor expression critical for embryo implantation. PLoS Genet. 2018;14(11):e1007787. doi:10.1371/journal.pgen.1007787
82. Marin MLC, Coelho V, Visentainer JEL, et al. Inhibitory KIR2DL2 gene: risk for deep endometriosis in euro-descendants. Reprod Sci. 2021;28(1):291–304. doi:10.1007/s43032-020-00255-x
83. Orazov MR, Ye.Orekhov R, Kamillova DP, Ye. SS. Tayny patogeneza povtornykh neudach implantatsii. Trudnyy Patsiyent. 2020;4:Tom 18. doi:10.24411/2074-1995-2020-10030
84. Dorostghoal M, Ghaffari HO, Marmazi F, Keikhah N. Overexpression of endometrial estrogen receptor-alpha in the window of implantation in women with unexplained infertility. Int J Fertil Steril. 2018;12(1):37–42. doi:10.22074/ijfs.2018.5118
85. Milesi MM, Varayoud J, Ramos JG, Luque EH. Uterine ERα epigenetic modifications are induced by the endocrine disruptor endosulfan in female rats with impaired fertility. Mol Cell Endocrinol. 2017;454:1–11. doi:10.1016/j.mce.2017.05.028
86. Camargo-Díaz F, García V, Ocampo-Bárcenas A, González-Marquez H, López-Bayghen E. Colony stimulating factor-1 and leukemia inhibitor factor expression from current-cycle cannula isolated endometrial cells are associated with increased endometrial receptivity and pregnancy. BMC Womens Health. 2017;17(1):63. doi:10.1186/s12905-017-0418-7
87. Lessey BA, Palomino WA, Apparao KB, Young SL, Lininger RA. Estrogen receptor-alpha (ER-alpha) and defects in uterine receptivity in women. Reprod Biol Endocrinol. 2006;4 Suppl 1(Suppl1):S9. doi:10.1186/1477-7827-4-S1-S9
88. Petousis S, Prapas Y, Margioula-Siarkou C, et al. Expression of progesterone receptors is significantly impaired in the endometrium of infertile women during the implantation window: a prospective observational study. J Matern Fetal Neonatal Med. 2016;29(23):3912–3919. doi:10.3109/14767058.2016.1152244
89. Kim JJ, Kurita T, Bulun SE. Progesterone action in endometrial cancer, endometriosis, uterine fibroids, and breast cancer. Endocr Rev. 2013;34(1):130–162. doi:10.1210/er.2012-1043
90. Sletten ET, Arnes M, Lyså LM, Larsen M, Ørbo A. Significance of progesterone receptors (PR-A and PR-B) expression as predictors for relapse after successful therapy of endometrial hyperplasia: a retrospective cohort study. BJOG. 2019;126(7):936–943. doi:10.1111/1471-0528.15579
91. Jackson KS, Brudney A, Hastings JM, Mavrogianis PA, Kim JJ, Fazleabas AT. The altered distribution of the steroid hormone receptors and the chaperone immunophilin FKBP52 in a baboon model of endometriosis is associated with progesterone resistance during the window of uterine receptivity. Reprod Sci. 2007;14(2):137–150. doi:10.1177/1933719106298409
92. Petousis S, Prapas Y, Margioula-Siarkou C, et al. Unexplained infertility patients present the mostly impaired levels of progesterone receptors: prospective observational study. Am J Reprod Immunol. 2018;79(6):e12828. doi:10.1111/aji.12828
93. Broi MGD, Junior RCV, Meola J, et al. Expression of PGR, HBEGF, ITGAV, ITGB3 and SPP1 genes in eutopic endometrium of infertile women with endometriosis during the implantation window: a pilot study. JBRA Assist Reprod. 2017;21(3):196–202. doi:10.5935/1518-0557.20170038
94. Kao LC, Germeyer A, Tulac S, et al. Expression profiling of endometrium from women with endometriosis reveals candidate genes for disease-based implantation failure and infertility. Endocrinology. 2003;144(7):2870–2881. doi:10.1210/en.2003-0043
95. Burney RO, Talbi S, Hamilton AE, et al. Gene expression analysis of endometrium reveals progesterone resistance and candidate susceptibility genes in women with endometriosis. Endocrinology. 2007;148(8):3814–3826. doi:10.1210/en.2006-1692
96. Osteen KG, Bruner-Tran KL, Eisenberg E. Reduced progesterone action during endometrial maturation: a potential risk factor for the development of endometriosis. Fertil Steril. 2005;83(3):529–537. doi:10.1016/j.fertnstert.2004.11.026
97. Koninckx PR, Ussia A, Adamyan L, Wattiez A, Gomel V, Martin DC. Pathogenesis of endometriosis: the genetic/epigenetic theory. Fertil Steril. 2019;111(2):327–340.
98. Koninckx PR, Ussia A, Tahlak M, et al. Infection as a potential cofactor in the genetic-epigenetic pathophysiology of endometriosis: a systematic review. Facts, Views Vision ObGyn. 2019;11(3):209.
99. Koninckx PR, Ussia A, Adamyan L, et al. The genetic-epigenetic pathophysiology of endometriosis: a surgeon’s view. In: Endometriosis in Adolescents. Cham: Springer; 2020:173–193.
100. Bayramova NN, Protasova AE, Raskin GA, Sobivchak MS, Yarmolinskaya MI. ‘Endometrioz-assotsiirovannyye opukholi yaichnika: morfologicheskiye i immunogistokhimicheskiye osobennosti’. Zlokachestvennyye Opukholi. 2019;9.
101. Levakov SA, Gromova TA. Prognozirovaniye retsidiva endometrioza yaichnikov i yego neoplasticheskoy transformatsii. Rossiiskii Vestnik Akushera-Ginekologa. 2019;19.
102. Gromova TA, Levakov SA. ‘Prognozirovaniye razvitiya retsidiva endometrioza yaichnikov’. Akusherstvo I Ginekologiya Sankt-Peterburga. 2019;33–39.
103. Won S, Cho YJ, Lee N, et al. Atypical endometriosis is related to a higher recurrence rate. Eur J Obstet Gynecol Reproductive Biol. 2020;254:44–51. doi:10.1016/j.ejogrb.2020.08.054
104. Kajiyama H, Suzuki S, Yoshihara M, et al. Endometriosis and cancer. Free Radic Biol Med. 2019;133:186–192. doi:10.1016/j.freeradbiomed.2018.12.015
105. Kim N, Hong SR, Lim KT, Lee KH, Kim TJ, So KA. EP886 Atypical endometriosis associated with ovarian malignancy. International Journal of Gynecological Cancer. 2019.
106. Moore L, Leongamornlert D, Coorens TH, et al. The mutational landscape of normal human endometrial epithelium. Nature. 2020;580(7805):640–646. doi:10.1038/s41586-020-2214-z
107. Lac V, Nazeran TM, Tessier‐Cloutier B, et al. Oncogenic mutations in histologically normal endometrium: the new normal? J Pathol. 2019;249(2):173–181. doi:10.1002/path.5314
108. Kurman RJ, Shih IM. The dualistic model of ovarian carcinogenesis: revisited, revised, and expanded. Am J Pathol. 2016;186(4):733–747. doi:10.1016/j.ajpath.2015.11.011
109. Maeda D, Shih IM. Pathogenesis and the role of ARID1A mutation in endometriosis-related ovarian neoplasms. Adv Anat Pathol. 2013;20(1):45. doi:10.1097/PAP.0b013e31827bc24d
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.