")
Back to Journals » OncoTargets and Therapy » Volume 16
Current Evidence and Future Perspectives about the Role of PARP Inhibitors in the Treatment of Thoracic Cancers
Authors Parisi A , Rossi F, De Filippis C, Paoloni F , Felicetti C, Mammarella A, Pecci F, Lupi A , Berardi R
Received 18 January 2023
Accepted for publication 9 July 2023
Published 18 July 2023 Volume 2023:16 Pages 585—613
DOI https://doi.org/10.2147/OTT.S272563
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Geoffrey Pietersz
Alessandro Parisi, Francesca Rossi, Chiara De Filippis, Francesco Paoloni, Cristiano Felicetti, Alex Mammarella, Federica Pecci, Alessio Lupi, Rossana Berardi
Department of Clinical Oncology, Università Politecnica delle Marche, Azienda Ospedaliero Universitaria delle Marche, Ancona, 60126, Italy
Correspondence: Rossana Berardi, Department of Medical Oncology, Università Politecnica delle Marche, Azienda Ospedaliero Universitaria delle Marche, Ancona, 60126, Italy, Tel +39 715964169, Email [email protected]
Abstract: In recent years, poly (ADP-ribose) polymerase (PARP) inhibition has become a promising therapeutic option for several tumors, especially for those harboring a BRCA 1– 2 mutation or a deficit in the homologous recombination repair (HRR) pathway. Nevertheless, to date, PARP inhibitors are still not largely used for thoracic malignancies neither as a single agent nor in combination with other treatments. Recently, a deeper understanding of HRR mechanisms, alongside the development of new targeted and immunotherapy agents, particularly against HRR-deficient tumors, traced the path to new treatment strategies for many tumor types including lung cancer and malignant pleural mesothelioma. The aim of this review is to sum up the current knowledge about cancer-DNA damage response pathways inhibition and to update the status of recent clinical trials investigating the use of PARP inhibitors, either as monotherapy or in combination with other agents for the treatment of thoracic malignancies. We will also briefly discuss available evidence on Poly(ADP-Ribose) Glycohydrolase (PARG) inhibitors, a novel promising therapeutic option in oncology.
Keywords: lung cancer, PARP inhibitors, PARG inhibitors, homologous recombination repair, malignant pleural mesothelioma, targeted treatment
Introduction
Thoracic malignancies have emerged as models for development of novel targeted therapies due to a deeper understanding of key molecular alterations driving tumor development and progression, in particular considering oncogene-addicted non-small-cell lung cancer (NSCLC):1 it is estimated that around 70% of advanced NSCLC patients have druggable mutations in numerous genes, including epithelial growth factor receptor (EGFR), anaplastic lymphoma kinase (ALK), c-ros oncogene 1 (ROS1), Kirsten rat sarcoma virus (KRAS), V-raf murine sarcoma oncogene homolog B1 (BRAF), MET, human epidermal growth factor receptor (HER2), and others. On the other hand, the identification of novel targetable alterations is still an unanswered need considering small-cell lung cancer (SCLC) and malignant pleural mesothelioma (MPM).
In recent years interest in developing drugs targeting mutations in different mechanisms involved in DNA repair is growing: poly (ADP-ribose) polymerases (PARP) and genes related to DNA homologous recombination repair (HRR) are crucially involved in the process of protecting cells from DNA damaging agents such as chemotherapy or ionizing radiation.
In particular, the DNA repair mechanisms involved in maintaining genomic integrity can be divided into base-excision repair (BER), nucleotide-excision repair (NER), mismatch repair (MMR), homologous recombination (HR) and non-homologous end-joining (NHEJ) recombination repair. Poly (ADP-ribose) polymerase-1 and -2 (PARP-1 and PARP-2) are nuclear enzymes that synthesize ADP-ribose polymers using Nicotinamide Adenine Dinucleotide (NAD)+ as a substrate; PARP-1 and -2 are essential components of BER, responsible for the protection of DNA damage induced by radiation and monofunctional alkylating agents.2 In addition, some damage to DNA can be repaired directly; for example, methylation of guanine bases is directly reversed by the protein O6-methylguanine DNA methyltransferase (MGMT).
Poly(ADP-ribose) glycohydrolase (PARG) antagonizes the action of PARP enzymes, hydrolyzing the ribose–ribose bonds present in poly(ADP-ribose). Similarly to PARPs, PARG is involved in DNA replication and repair and PARG depleted/inhibited cells seem to be much more sensitive to DNA damaging agents exposure, thus providing an increased storage of perturbed replication intermediates which can lead to synthetic lethality. Moreover, PARG shares a main role in parthanatos, a caspase-independent cell death modality.3 Although agents able to specifically target PARP enzyme, universally known as PARP inhibitors (PARPi), have been initially developed for the treatment of BRCA1 (BReast CAncer gene 1) and BRCA2 (BReast CAncer gene 2) mutated tumors, they are now being explored in a wide variety of cancers presenting somatic deficiencies within the HRR pathways (Figure 1).4
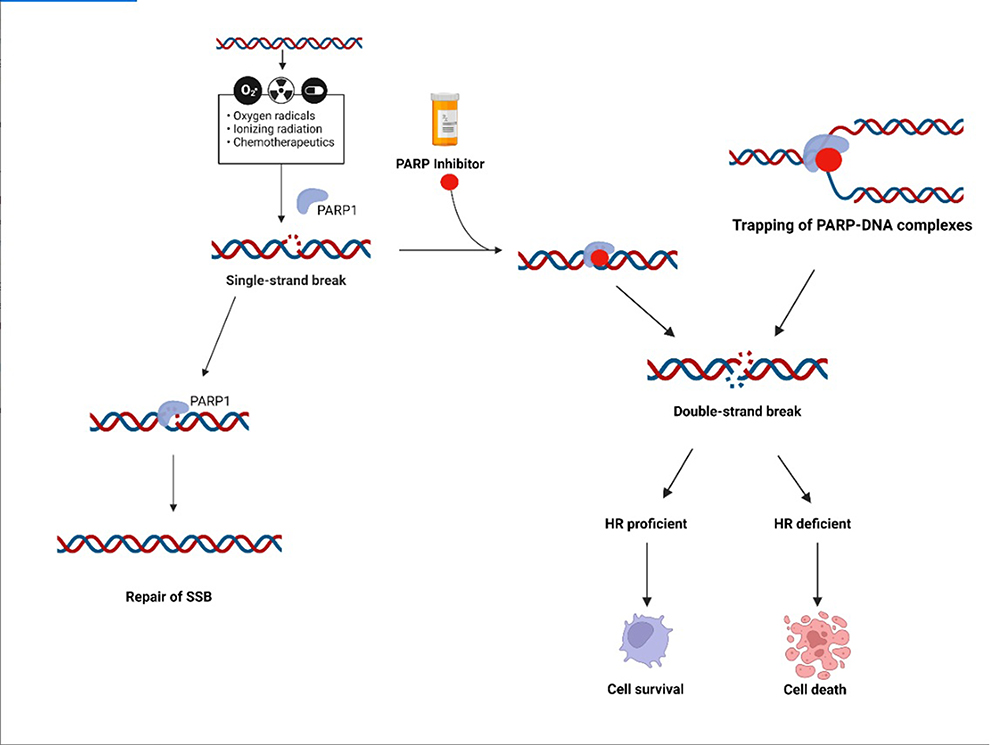 |
Figure 1 PARP inhibitor, mechanism of action. Chemotherapy, ionizing radiation can promote single-strand breaks (SSB) in the DNA. PARP, through the pathway of base excision repair (BER), is involved in SSB repair. In the presence of a PARP inhibitor the SSB cannot be repaired by BER pathway and can be turned into a double-strand break (DSB). The homologous recombination (HR) pathway is involved in DSB repair. A HR-deficient cell cannot repair DSB, the DSB accumulation is toxic and causes apoptosis and cell death. Created with BioRender.com. |
Therefore, BRCA mutation and HRR status are exploited as biomarkers of PARPi response and the evaluation for the HRR-signature is currently of great interest in scientific research given its promising therapeutic implications.
The synthetic lethal interaction between PARP inhibitors and BRCA1/2 mutations has been documented since the first human clinical trial of PARP inhibitor olaparib in 2009. In the next few years olaparib was approved for ovarian, breast, and pancreatic cancer in 2014, rucaparib for ovarian cancer was approved in 2016, and niraparib for ovarian cancer was approved in 2017. In 2018, talazoparib was approved for breast cancer treatment.5
Although the introduction of PARPi as monotherapy or in combination with chemotherapy has proven to be ineffective in thoracic tumors, a thorough exploration of these new drugs in the biological and immunological landscape underlying HRR-deficient cancers is auspicable considering the advent of new targeted therapy and immunotherapeutic agents.
In this regard, although nowadays immunotherapy represents a solid therapeutic option in most thoracic malignancies, there are still a lot of patients getting no benefit from it due to a “cold” tumor microenvironment of unknown reasons. Novel combinations with unexploited therapeutic agents such as PARPi may help to significantly increase their number.
In this review, we briefly provide an overview of current clinical trials investigating several therapeutic strategies of different PARPi in combination with chemotherapy, radiotherapy, immunotherapy, and other targeted agents, in patients with small cell lung cancer (SCLC), non-small cell lung cancer (NSCLC), and malignant pleural mesothelioma (MPM).
In contrast to PARPi, the therapeutic potential of PARG inhibitors (PARGi) has been largely ignored. Nevertheless, interest in these molecules is rising as they may represent a novel valuable option in cancer treatment, both as single agents and in combination with cytotoxic drugs or radiotherapy.
Non-Small Cell Lung Cancer (NSCLC)
PARPi with or without Chemotherapy
Biological Background and Preclinical Data
Although early clinical trials suggested that only tumors with germline and somatic mutations in BRCA 1/2 genes would benefit from PARP inhibition, new evidence has recently shown that these targeted agents can also be effective in other somatic deficiencies of the HRR pathway that altogether define the “BRCAness” phenotype.4
In particular, both BRCA mutations and BRCAness alterations have been detected in patients with NSCLC6,7 and studies on NSCLC models have already shown that mutations of genes involved in DNA repair, for example ERCC1 (excision repair cross-complementation group 1) deficiencies, led to PARPi sensitivity.8,9
However, a higher prevalence of loss of heterozygosis (LOH) of the mutant allele was identified in lung cancer patients with germline BRCA mutations.7 The process of LOH, in which a wild-type allele is lost and only an inactivated allele is left in the cancer genome, is frequently involved in the function loss of tumor suppressor genes and a large number of potential tumor suppressors were identified by analyzing sites of prevalent LOH in human cancers.10 Therefore, molecular features other than BRCA mutation also need to be considered.
The SAFIR02-Lung is an open-label, randomized, Phase II trial investigating the efficacy of targeted therapies and immune checkpoint inhibitors (ICIs) compared with standard-of-care in patients with advanced epithelial growth factor receptor (EGFR) and anaplastic lymphoma kinase (ALK) wild-type NSCLC without progression after first-line chemotherapy, based on high-throughput genome analysis.11
Considering the 379 patients with a druggable alteration identified, BRCA mutations were found in 20 patients (5.3%), though only 2.1% harbored a pathogenic BRCA1/2 mutation, with 75% of somatic mutations and 75% targeting BRCA 2 gene. Interestingly, while many genomic alterations in NSCLC are more common in females as they are usually related with non-smoking habits,12 all patients with pathogenic BRCA mutations were men and mainly heavy smokers. The overall response rate to chemotherapy was 13%. In 12 patients (3.2%), BRCA mutations of uncertain relevance were found, leading to an 8.3% overall response rate to treatment. Biallelic inactivation with a high HRD score were seen in one-third of tumors carrying pathogenic BRCA mutations or variations of uncertain relevance and overall survival of this cohort was 12.8 months.
In conclusion, pathogenic BRCA1/2 mutations occur in 2.1% of patients with advanced NSCLC and the predictive role of BRCA mutation for making treatment decisions in NSCLC seems limited based on clinical response (low platinum sensitivity) and molecular features (discrepancy between biallelic inactivation and high HRD score).
These results, although from a small cohort, surely contribute to determining BRCA mutation prevalence in a real-world population of NSCLC patients and are also coherent with literature: Heeke et al,13 who considered a DNA sequencing database of around 52,000 patients that underwent NGS or Sanger sequencing panel testing between July 2013 and September 2017, found a BRCA mutation in around 1–2% of patients analyzed.
Focusing on other HRR genes, changes in coding sequence of ATM (ataxia-telangiectasia mutated) were discovered in 12% of lung adenocarcinomas during large-scale genomics research.14
ATR (Ataxia-telangectasia and Rad3 related) kinase inhibitors sensitize lung cancer cell lines to cisplatin in vitro and in cell line and patient-derived xenografts in vivo15–18: cisplatin and ATR kinase inhibitors have been demonstrated to cooperate in vitro to kill ATM-deficient lung cancer cells and in vivo to resolve ATM-deficient xenografts.15,16,18 A multicenter Phase 1/1b clinical trial with AZD6738 (NCT02264678) is currently ongoing to test this hypothesis.19
Jette et al20 tested the effects of PARP inhibitor olaparib and ATR inhibitor VE-821 in ATM-knockout A549 cells lung adenocarcinoma cells: considering drug sensitivity data from the Genomics of Drug Sensitivity in Cancer (GDSC) project in lung cancer cell lines, IC50 (half-maximal inhibitory concentration) values for both olaparib and talazoparib were positively linked with ATM mRNA levels and gene amplification status. Conversely, ATM loss was linked to a considerable drop in the IC50 for olaparib. In cells with ATM deletion, olaparib caused phosphorylation of DNA damage markers and reversible G2 arrest, whereas olaparib with VE-821 caused cell death, evidencing the potential efficacy of PARP inhibitors in ATM mutated tumors.
PTEN (Phosphatase and tensin homolog) loss caused by a variety of inherited germline mutations, somatic mutations, epigenetic and transcriptional silencing, post-translational modifications, and protein–protein interactions occurs in 4–8% of NSCLC and seems to predict a response to PARPi in lung cancer cell lines.21
Sargazi et al22,23 found that the combination of valproic acid, an histone deacetylase inhibitor capable of downregulating the DNA repair genes pathway, and AZD2461, a novel PARP1, PARP2, and PARP3 inhibitor, effectively induces apoptosis in prostate cancer cell cultures (PC-3) harboring mutations in PTEN. Although the same anti-proliferative effect has already been evidenced in breast cancer cells (MCF-7), there is still no evidence of similar activity in NSCLC models.24
Considering the combination of PARPi with other therapeutic agents, chemotherapy with platinum compounds or other alkylating drugs and ionizing radiation are considered quite promising as they act on tumor cells by damaging DNA; on the other hand, the existence of DNA repairing abnormalities is a possible mechanism involved in the tumor sensitivity to therapies25,26 and activation of DNA repair pathways induced by these treatment modalities contributes to tumor resistance,27 thus offering a strong biological rationale for combining DNA-damaging agents with PARPi.
In preclinical studies, veliparib has demonstrated increased cytotoxicity when administered with platinum compounds, topoisomerase inhibitors, and alkylating agents in general.28,29 The combination of veliparib plus temozolomide (TMZ) was investigated in multiple xenograft models: tumor burden, expression of poly (ADP-ribose) polymer, and O6-methylguanine methyltransferase were used to assess the combination effectiveness in xenografts from divergent histologic tumor types, including B-cell lymphoma, SCLC, NSCLC, ovarian, breast, pancreatic, and prostate models implanted in subcutaneous, orthotopic, and metastatic sites. Veliparib (25 mg/kg/day, orally, twice daily for 5–6 days) seems to enhance TMZ efficacy (50 mg/kg/day, orally, once daily for 5 days), although different levels of effectiveness were reported (from 55 to 100% of tumor growth inhibition), including several regressions. Most interestingly, the addition of PARPi to TMZ seems to overcome both TMZ primary and secondary resistance in cancer cells.
Clinical Evidence and Predictive Factors of Response and Efficacy
Starting from this preclinical rationale, several clinical trials are ongoing in investigating PARPi either as a single agent or in combination with other therapies in the treatment of thoracic malignancies.
A randomized phase II trial investigated the addition of iniparib to a standard treatment with gemcitabine and cisplatin. One hundred and nineteen Stage IV NSCLC patients were randomly assigned to receive gemcitabine (1,250 mg/m2, days 1–8) and cisplatin (75 mg/m2, day 1) with or without iniparib (5.6 mg/kg, days 1, 4, 8, and 11) every 3 weeks for six cycles. Even if the experimental arm improved activity (overall response rate (ORR)=25.6%; 95% Confidence Interval (CI)=13.0–42.1% vs 20%; 95% CI=11.9–30.4%), no statistically significant and clinically relevant benefit was found in terms of PFS (median PFS=4.3; 95% CI=2.8–5.6 vs 5.7; 95% CI=4.6–6.6 months, Hazard Ratio (HR)=0.89; 95% CI=0.56–1.40 and median OS=8.5; 95% CI=5.5 to not reached vs 12; 95% CI=8.9–17.1 months, HR=0.78; 95% CI=0.48–1.27). Toxicity was similar between the two cohorts, with no higher incidence of neutropenia/febrile neutropenia or serious systemic infections in the experimental arm.
The failure of adding iniparib to a chemotherapy backbone in this context may be related with its peculiar mechanism of action that differs from other PARPi. In fact, this is since iniparib acts by eliciting cell response by non-selective cysteine adduct alteration of multiple proteins.30 Moreover, this proof-of-concept trial was not powerful enough to properly assess efficacy of iniparib + chemotherapy combination, even if it is highly likely a single-arm study design would have produced similar results.
The combination of the PARPi veliparib (days 1–7 q21 schedule) with carboplatin and paclitaxel (AUC6 and 200 mg/m2, respectively, day 3 q21 schedule) in naïve stage IV NSCLC patients has been investigated in a phase II trial enrolling 158 patients. No differences in PFS, OS, and ORR between the veliparib and placebo group have been founded, even if an interesting exploratory analysis31 highlighted a trend toward a PFS benefit in patients with squamous histology (above 48% of the ITT population). In addition, the study also stratified patients by smoking history and revealed that recent smokers who were given veliparib had significantly better PFS and OS than former smokers (HR=2.09; p=0.02 and HR=1.62; p=0.23) and never-smokers (HR=1.02; p=0.97 and HR=1.30; p=0.63).
These results led to the design of a Phase III trial investigating the efficacy and safety of veliparib in combination with conventional chemotherapy for untreated advanced squamous NSCLC.32 Patients were randomized to receive carboplatin and paclitaxel with either veliparib 120 mg twice daily or placebo twice daily for up to six cycles. Primary outcome was OS in the veliparib arm among current smokers, biomarker analysis was performed on archival tumor samples using a 52-gene expression histology classifier (LP52) whose positive predictive value had been proved in two different trials evaluating veliparib in NSCLC patients. Unfortunateluy, no significant OS benefit with veliparib in current smokers was found (median OS=11.9 vs 11.1 months, HR=0.90; 95% CI=0.74–1.10; p=0.26) even if a trend toward better OS was found in the veliparib cohort in the overall population (median OS=12.2 vs 11.2 months, HR=0.85; 95% CI=0.75–0.97). Focusing on patients with biomarker-evaluable tumor samples, an OS benefit due to veliparib was confirmed in the LP52-positive population (median 14.0 vs 9.6 months; HR=0.66; 95% CI=0.49–0.89). To sum up, veliparib in combination with first-line chemotherapy provided no therapeutic advantage in current smokers with advanced squamous NSCLC, but the LP52 classifier might help to identify a subset of patients who could benefit from PARP inhibition.
Another phase II study (NCT02154490) evaluating the clinical efficacy of the PARP inhibitor talazoparib in advanced stage and platinum sensitive squamous NSCLC harboring HRR deficiency failed to demonstrate efficacy in the primary analysis population, so it was closed early for futility.
Finally, first results from the aforementioned SAFIR02-Lung trial have recently been published: among 116 patients receiving NGS-driven target therapies (including PARPi olaparib), no difference in PFS was found in any molecular subgroup. Median PFS was 2.7 months (95% CI=1.6–2.9) vs 2.7 months (95% CI=1.6–4.1) both in the experimental and in the SOC arm (HR=0.97; 95% CI=0.7–1.36; p=0.87). Though no subgroup analysis exclusively considering olaparib is currently available, the results of the target therapy arm are definitely quite disappointing.11
Ongoing clinical trials evaluating the role of PARPi with or without chemotherapy in NSCLC patients are listed in Table 1. Published clinical trials evaluating the role of PARPi with or without chemotherapy in NSCLC patients are listed in Table 2.
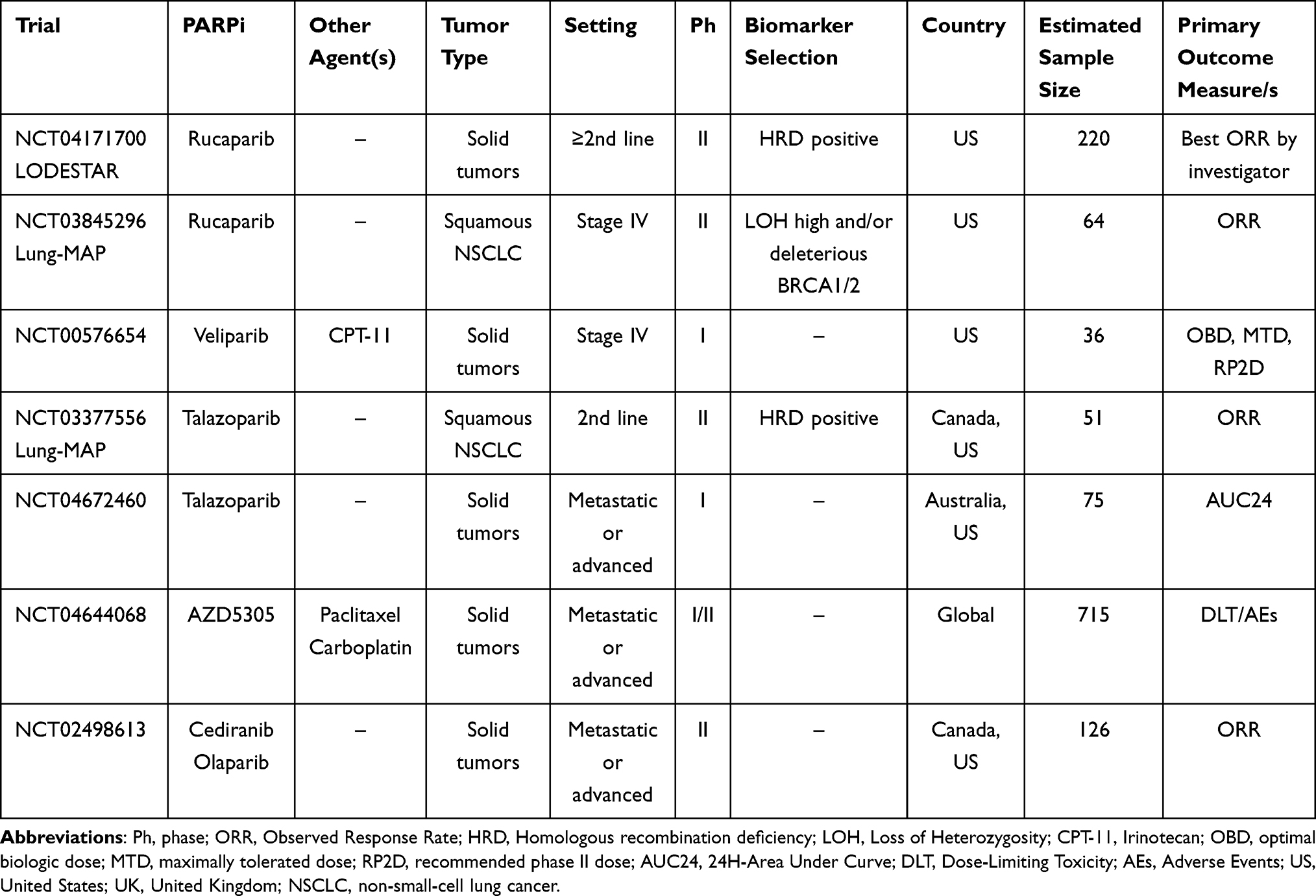 |
Table 1 Main Ongoing Clinical Trials Evaluating the Role of PARPi +/- Chemotherapy in NSCLC Treatment |
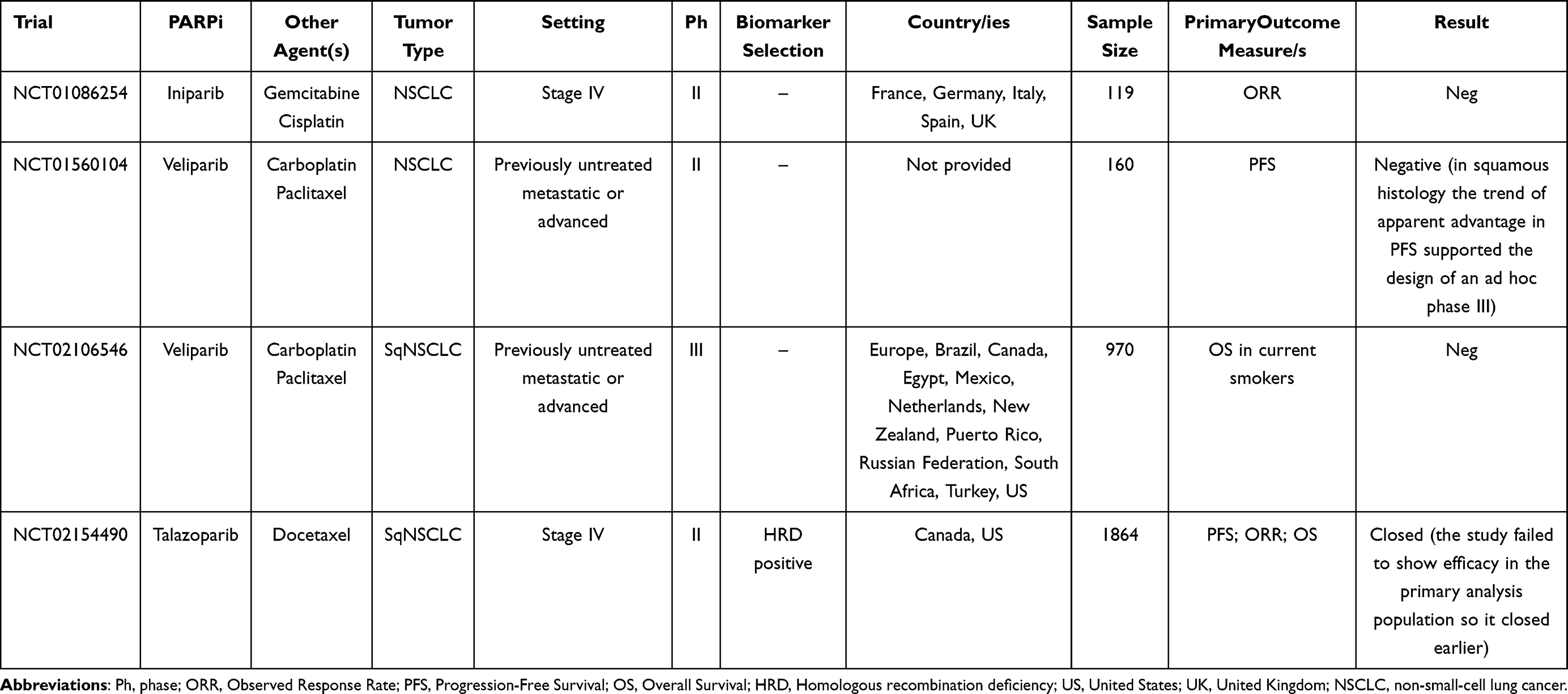 |
Table 2 Main Published Clinical Trials Evaluating the Role of PARPi +/- Chemotherapy in NSCLC Treatment |
PARPi + Radiotherapy
Biological Background and Preclinical Data
In the field of radiation therapy, ionizing radiation combined with radiotherapy-enhancing chemicals has the potential to improve the efficacy of radiotherapy as a therapeutic method while reducing harmful side-effects and potential damage to healthy surrounding tissues.
In particular, PARPi combination with radiotherapy is an interesting matter because of radiation's DNA disrupting effect and the paramount role of the PARP enzyme in DNA repair. It follows that PARPi may act as potential radiosensitizers, especially for BRCA mutated tumors exploiting PARP enzyme as their main tool to fix DNA alterations: there is evidence in literature that the absence of PARP-1 and -2 enzymes, that are both triggered by DNA damage and assist DNA repair, enhances tumor sensitivity to ionizing radiation by extending strand breaks and triggering a cell-death signaling cascade. This effect could be amplified in tumors with HRR abnormalities and through a synthetic lethality mechanism. However, the impact of PARPi is not limited to modifying DNA damage repair as PARPi show numerous properties that are essential for their radio-sensitizing action such as chromatin remodeling inhibition, G2/M arrest, and a vasodilatory action.33
A third of individuals with NSCLC are diagnosed at a locally advanced stage, a setting where chemo-radiotherapy (CRT) eventually followed by immunotherapy rather than surgery is the current standard of care. Despite technological progress in radiation therapy, tumor intrinsic radio-resistance still affects patients’ outcome and offers an interesting application for PARPi in this setting.
Albert et al34 found that veliparib and concomitant radiation reduced endothelial tubule development in vitro and reduced vessels formation in vivo, suggesting that this method may also target tumor angiogenesis. Veliparib hindered DNA repair in H460 lung cancer cells, as evidenced by increased production of the DNA strand break marker histone -H2AX (H2A histone family member X), seems to favor tumor cells death due to both apoptosis and autophagy. Veliparib also delayed tumor growth in murine models at well-tolerated doses: cancer cells proliferation was delayed by 1 day for veliparib alone, 7 days for radiation alone, and 13.5 days for combination treatment for a 5-fold increase in tumor volume. After the combined treatment, immunohistochemical staining of tumor sections suggested an increase in terminal deoxyribonucleotide transferase-mediated nick-end labeling apoptotic staining and a decrease in Ki-67 proliferative staining.
Another trial analyzed the effect of PARPi olaparib on H1299 lung cancer cell line with depletion or mutation of p53 gene using the γH2AX focus formation assay to examine the influence of olaparib on induction and repair of double-stranded DNA breaks after exposure to radiation. Even at 0.01 μM, and after a brief exposure interval (2 h) olaparib demonstrated a promising radio-sensitizing effect and, although p53-knockout H1299 cells were more radio-resistant than p53 wild-type, a benefit from olaparib administration was demonstrated in both subgroups. As tumor cells may be exposed to low amounts of olaparib and/or have varying levels of p53 mutation, these properties could be useful to guide therapeutic radiotherapy pre-planification.
Clinical Evidence and Predictive Factors of Response and Efficacy
A Phase I–II study (SWOG S1206) evaluated the addition of veliparib to CRT for patients affected by unresectable NSCLC35: in the Phase I dose-finding part of the trial, patients received weekly carboplatin (AUC 2) and paclitaxel (45 mg/m2) during concurrent thoracic radiotherapy (2 Gy fractions/day, total dose 60 Gy) and veliparib administered at three different dose levels (40, 80, and 120 mg, respectively) throughout radiotherapy duration. No dose-limiting toxicity (DLT) was seen at veliparib dose of 120 mg twice daily, which was selected for the next phase II part of the study. Thirty-one patients were subsequently randomized to receive veliparib or placebo during thoracic radiotherapy with concurrent weekly carboplatin and paclitaxel, followed by two cycles of consolidation treatment with carboplatin and paclitaxel plus either veliparib or placebo. No difference in PFS was detected between the two arms but an interesting benefit in 1-year OS (89% vs 54%) was found in the veliparib arm, though ORR slightly favored placebo (56% vs 69%).
In addition, another recent study with a similar design,36 the M14-360/AFT-07 phase I trial, confirmed the good tolerability with promising antitumor activity with a mPFS of 19.6 months for the combination of veliparib + CRT with carboplatin and paclitaxel followed by veliparib plus chemotherapy in 48 stage III NSCLC patients.
A phase 1 study evaluated the safety of olaparib in association with loco-regional radiotherapy, with and without concurrent cisplatin for locally advanced NSCLC.37 The dose of olaparib was increased in two groups respectively treated with radiation (66 Gy/24 fractions in 2.75 Gy/fraction) with and without daily cisplatin (6 mg/m2) using the time-to-event continuous reassessment technique with a 1-year DLT period. The maximum tolerable dose (MTD) was determined as the highest dose level having a DLT probability of less than 15%. The study enrolled 28 patients with loco-regional or oligometastatic disease, of whom 11 were treated with olaparib 25 mg twice daily and 17 with olaparib 25 mg once daily. Due to hematologic and late esophageal DLT, the lowest dose level with cisplatin was over the MTD, while 25 mg once daily was the MTD for olaparib without cisplatin. Severe pulmonary adverse events were detected in five patients across all dose levels with a latency of 1–2.8 years, probably due to radiation exposure to the lungs as observed in preliminary studies. Olaparib lowered PARP levels (determined in peripheral blood cells) by more than 95% and stopped radiation-induced PARylation at the MTD. After an average of 4.1 years, 2-year loco-regional control was 84% and median overall survival was 28 months. However, considering high rates of esophageal and hematologic toxicity, a combination of moderately hypofractionated radiation with low-dose daily cisplatin and olaparib was not acceptable and, even without cisplatin, severe pulmonary damage occurred, thus suggesting that more conformal radiation schedules saving the lungs and esophagus should be investigated.
Ongoing clinical trials evaluating the role of PARPi with or without radiotherapy in NSCLC patients are listed in Table 3. Published clinical trials evaluating the role of PARPi with or without radiotherapy in NSCLC patients are listed in Table 4.
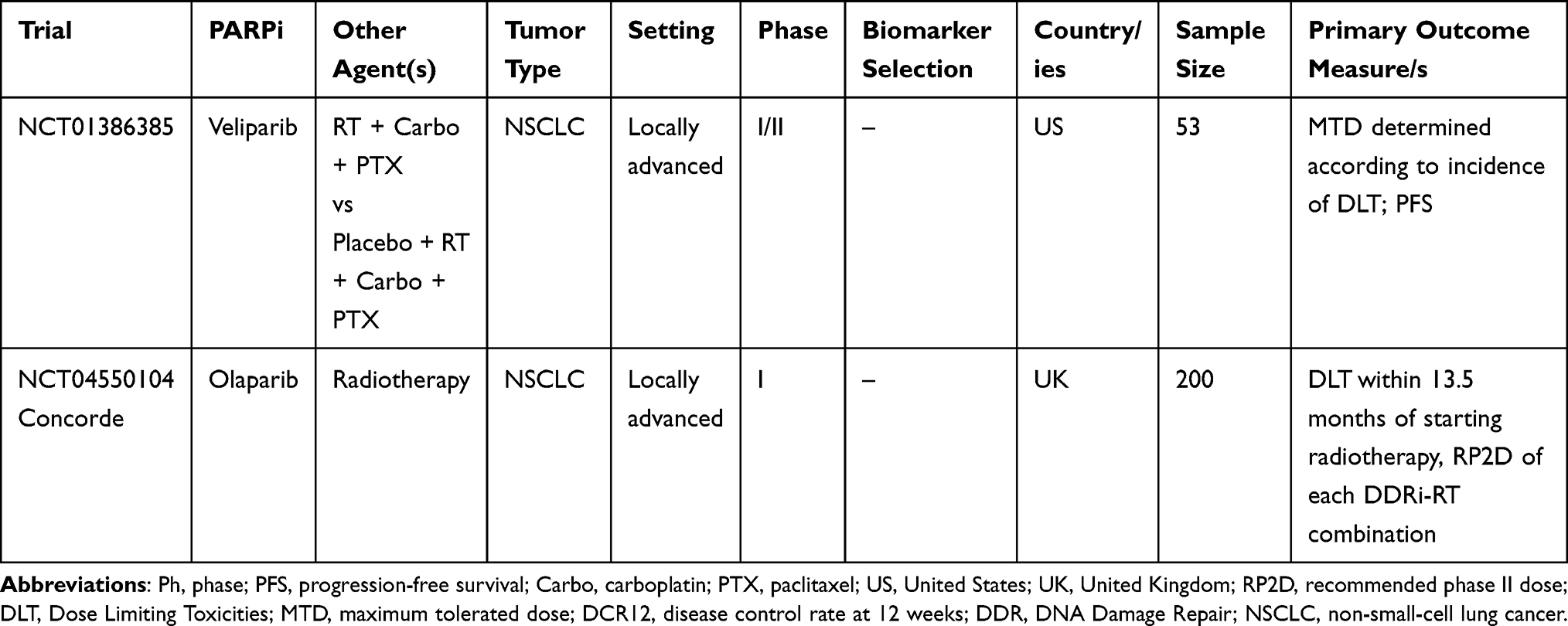 |
Table 3 Main Ongoing Clinical Trials Evaluating the Role of PARPi + Radiotherapy in NSCLC Treatment |
 |
Table 4 Main Published Clinical Trials Evaluating the Role of PARPi + Radiotherapy in NSCLC Treatment |
PARPi + Immunotherapy
Biological Rationale and Preclinical Data
In the last years the introduction of ICIs in the treatment algorithm of almost every tumor specimen has deeply changed clinical practice. NSCLC was among the first tumor type to receive the benefits from this therapeutic revolution.38–44
There is robust evidence about the deep correlation between inhibition of DNA repair pathways and ICIs activity:45,46 DDR alterations lead to an increase in the tumor mutational burden (TMB) and intrinsic immunogenicity due to tumor-specific neoantigen formation,46 while the stimulator of IFN genes (STING) pathway that activates innate antitumor immunity acts as an additional DNA damage recognition mechanism via neo-antigen independent pathways (Figure 2).47
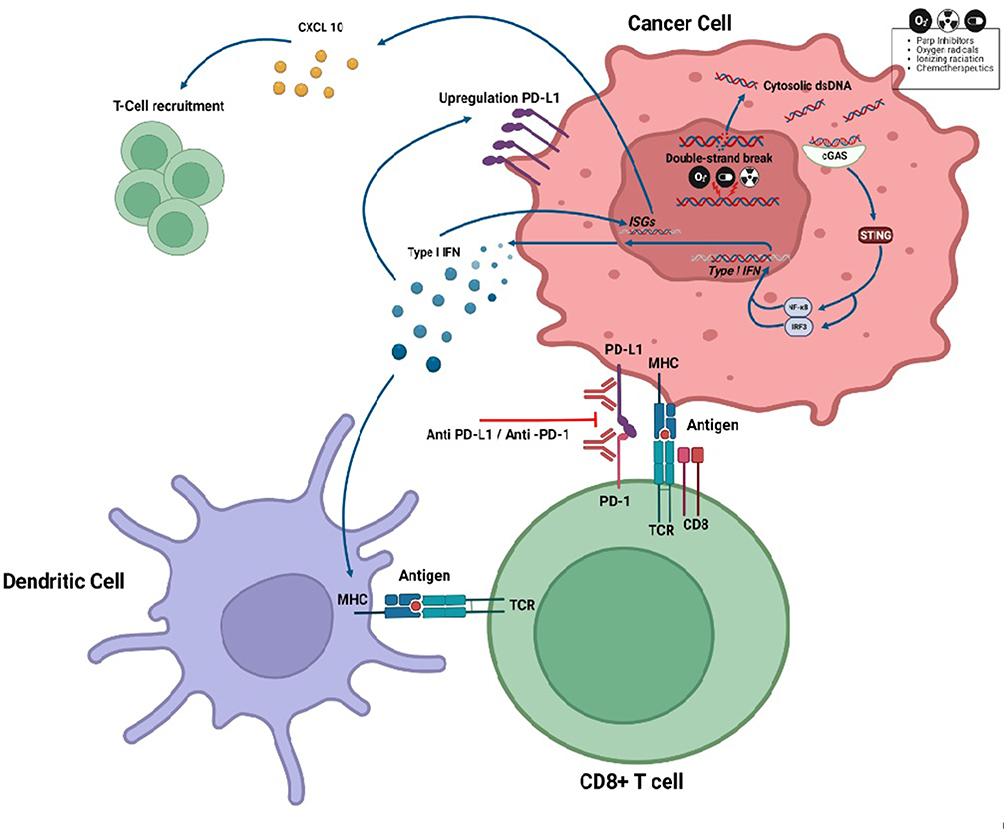 |
Figure 2 Interlink between DNA damage and immune checkpoint inhibitors (ICIs) via cGAS-STING activation. In homologous recombination (HR) proficient and HR deficient tumor cells the action of chemotherapeutics, ionizing radiation and PARPi generates cytosolic double-stranded DNA (dsDNA) fragments. These fragments of cytosolic dsDNA bind to cyclic GMP-AMP synthase (cGAS) and activate the stimulator of interferon genes (STING) that, through NF-kB and IRF-3, induce type I IFN and other transcriptional targets. The binding between the type I IFN to the IFN receptor stimulates the transcription of IFN-stimulated genes (ISGs) and production of cytokines like CXCL10 that promote the T-Cell recruitment. The IFN I also determines a PD-L1 upregulation on the cancer cell surface and promotes the tumor neoantigen presentation on the major histocompatibility complex (MHC) of dendritic cell to the T-cell receptor (TCR). Created with BioRender.com. |
The role of PARPi, such as niraparib, combined with anti-programmed cell death-1 (PD-1), has been evaluated in BRCA-proficient and BRCA-deficient preclinical models of sarcoma, lung squamous cell carcinoma, colon adenocarcinoma, breast cancer, and urothelial carcinoma. Following evidence supporting niraparib's role in increasing CD4+ and CD8+ immune cell infiltration and type I and type II interferon pathways activity,48 many clinical trials evaluating the combination of PARPi and ICIs have been designed, in particular including NSCLC patients where immunotherapy is a main option in the clinicians’ therapeutic arsenal.
Clinical Evidence and Predictive Factors of Response and Efficacy
The tumor mutational landscape seems to play an important role in predicting clinical response to ICIs. In NSCLC patients treated with anti-PD1 pembrolizumab, TMB has been associated with clinical efficacy in terms of PFS, ORR, OS, and durable clinical benefit,49,50 as from a biological standpoint higher TMB levels enhance CD8-positive T-cell intratumoral infiltration and inflammatory T-cell-mediated response.51 Finally, DDR mutations in NSCLC patients treated with ICIs have been associated with longer PFS (5.4 vs 2.2 months) and OS (18.8 vs 9.9 months) in a study by Ricciuti et al.52
Based on this rationale, several trials were designed to evaluate the association of PARPi and ICIs in NSCLC (Table 5). Among these, JASPER study is a multicenter, open‐label, 2‐stage Phase 2 trial evaluating the association of the PARPi niraparib in combination with the PD-1 inhibitor pembrolizumab as first-line treatment in patients affected by metastatic and/or locally advanced NSCLC. Patients were enrolled in two cohorts, according to PD-L1 expression evaluated by tumor proportion score (TPS): ≥50% (cohort 1) and 1–49% (cohort 2): ORR (primary endpoint) was 56.3% with two complete responses and 20.0% in cohort 1 and 2 while focusing on secondary endpoints, in the two groups median duration of response (DoR) was 19.7 and 9.4 months and PFS was 8.4 and 4.2 months, respectively. OS was not reached for patients with TPS ≥50 vs 7.7 months for the 1–49% group.53
 |
Table 5 Main Ongoing Clinical Trials Evaluating the Role of PARPi + Immunotherapy in NSCLC Treatment |
The safety profile detected on JASPER study was concordant with known safety profiles of single agents niraparib and pembrolizumab.53 With the strong bias of an indirect comparison with limited numbers, it is remarkable that, considering pembrolizumab monotherapy provided and ORR of 44.8% on patients with TPS ≥50% in the KEYNOTE-024 trial that led to its registration in the stage IV NSCLC setting,39 the addition of PARPi seems to improve the ORR to 56.3% in the same patients’ subgroup. The study was not powerful enough to compare efficacy between cohorts 1 and 2.
The clinical role of PARPi in NSCLC was also studied in a phase 1 open-label dose-escalation trial (NCT02944396).54 Twenty-five patients were treated with veliparib and nivolumab, a PD-1 inhibitor, plus chemotherapy with carboplatin and paclitaxel or carboplatin and pemetrexed, a four-drug combination that had never been studied before, and were subsequently divided into five dosing cohorts: the first one was treated with veliparib 120 mg twice daily in combination with nivolumab 360 mg (Q3W), carboplatin AUC 6 mg/mL∙min and paclitaxel 200 mg/m2 (C/PAC regimen), whereas the other four groups received veliparib, 80/120/200/240 mg, respectively, twice daily in combination with nivolumab 360 mg (Q3W), carboplatin AUC 6 mg/mL∙min, and pemetrexed 500 mg/m2 (C/PEM regimen).
Chemotherapy and veliparib were administered for six cycles as nivolumab and pemetrexed were then continued as maintenance therapy until disease progression or unacceptable toxicity occurred. The safety profile was concordant with known data available for the combination of nivolumab plus double chemotherapy55 as the addition of veliparib did not increase the rate of hematologic toxicities and the drug combination was well tolerated.54 ORR (primary endpoint) in the overall cohort was 40%, quite similar to the 38% showed in a CheckMate227 trial that considered NSCLC patients treated with nivolumab plus chemotherapy.56
Given the good safety profile shown in several trials with the combination of PARPi and immunotherapy, future studies with large sample size could better investigate the efficacy of PARPi in addition to immunotherapy in NSCLC.
Published clinical trials evaluating the role of PARPi with or without immunotherapy in NSCLC patients are listed in Table 6.
 |
Table 6 Main Published Clinical Trials Evaluating the Role of PARPi +/- Immunotherapy in NSCLC Treatment |
PARPi + Targeted Agents
Biological Rationale and Preclinical Data
Scientific research on PARPi acquired/innate resistance is based on studies with combinations of different DNA Damage Response Inhibitors (DDRi). In pre-clinical models with ATM deficiency, the combination of ATR inhibitor and PARPi improved the efficacy of PARPi.20
A member of the antiapoptotic BCL-2 proteins, MCL-1, is an important prosurvival agent due to its DNA repair function.57 Mattoo et al58 recently demonstrated both in vitro and in xenografts that mTOR inhibitors everolimus or AZD2014 deplete MCL-1 expression and lead to DNA repair suppression, thus increasing sensitivity to PARPi olaparib. In addition, inhibition of another important protein of the same axis as m-TOR, PI3K, caused DNA homologous recombination repair impairment and sensitization to PARP inhibitor olaparib in triple negative breast cancer cells in vivo59 and also in ex vivo cultured PIK3CA wild type ovarian cancer tissues independently of BRCA genes status.60
The role of vascular endothelial growth factor (VEGF) inhibition-driven hypoxia in providing DNA damage and genetic instability has already been highlighted.61 In particular, an interesting preclinical experience from Bizzarro et al62 demonstrated a wide anti-tumor activity due to the combination of olaparib and the VEGFR inhibitor cediranib on patient-derived ovarian cancer xenografts, regardless of the HRR status.
The role of PARPi therapy has also been studied in lung cell lines with EGFR mutation: the EGFR-mutant cells showed sensitivity to PARPi in vitro and in vivo due to an EGFR kinase-independent regulation of DNA repair.63
Clinical Evidence and Predictive Factors of Response and Efficacy
OLAPCO trial is a phase 2 basket trial recruiting patients in a range of tumor types with the potential to identify novel tumor indications for combination therapy with olaparib based on molecular markers from genetic profiling performed on their tumors prior to study entry. Patients with tumors harboring PTEN, PIK3CA, AKT, or ARID1A mutations or other molecular alterations leading to dysregulation of the PI3K/AKT pathway will be treated with target inhibitor AZD5363 plus olaparib. Another arm of the same trial, whose first results have already been published,64 has demonstrated an 8.3% overall response rate and 62.5% clinical benefit rate among a 25 patients cohort with different solid tumors, though none with NSCLC, treated with ATR selective inhibitor ceralasertib. The same drug in combination with carboplatin, durvalumab, or olaparib is also under evaluation in an ongoing phase I dose escalation study in patients affected by advanced solid tumors.65
Focusing on the PIK3CA-AKT-mTOR pathway, a phase Ib NCT04586335 clinical trial is currently evaluating safety, tolerability, and preliminary efficacy of novel PIK3CA inhibitor CYH33 in combination with olaparib in patients with DDR gene mutations and/or PIK3CA mutations. The study aims at recruiting at least 350 patients and comprehends a dose escalation part followed by a dose expansion phase.
Two different trials, NCT02588105 and NCT04267939, respectively, are evaluating PARP inhibitors olaparib and niraparib in combination with ATR inhibitor AZD0156 and ATM inhibitor BAY1895344 (Elimusertib). Both studies are still ongoing and no results have been published yet. On the other hand, first data from the NCT02723864VX trial by Mitra et al66 showed a promising activity producing two partial responses (6%) and 22 stable disease (65%, median four cycles, range=2–11) in 37 heavily pretreated patients with solid tumors though high toxicity (in particular anemia, 41% G3) prevented adequate veliparib delivery and the drug combination has not been studied further.
Following strong preclinical rationale and satisfactory results obtained in ovarian cancer, the phase II NCT02498613 trial is studying the mutual administration of PARPi olaparib with anti-VEGF target agent cediranib in patients with solid tumors. With the limitations of no report available focusing on NSCLC cohort, preliminary data considering other tumor specimens are encouraging (14% ORR in biomarker-unselected 37 patients with heavily pre-treated, metastatic triple-negative breast cancer)67 with a manageable toxicity, though 24% of patients reported hypertension often requiring prompt antihypertensives initiation.
Despite the combination of PARPi olaparib, EGFR inhibitor gefitinib in EGFR mutant NSCLC did not show a significant benefit in terms of PFS, compared to EGFR inhibitor alone in the phase 1–2 GOAL trial,68 further analysis evidenced that olaparib may instead be effective in a subset of patients with higher expression of BRCA1 mRNA (PFS 12.9 vs 9.2 months, p=0.0449).69
The tolerability and efficacy of the combination of niraparib and the 3rd generation EGFR inhibitor osimertinib, is under investigation in a phase I trial recruiting patients with EGFR-mutated advanced NSCLC (NCT03891615).
However, further studies are needed to better stratify those patients who might benefit from these therapeutic combinations.
Main ongoing clinical trials evaluating the role of PARPi with DDRi and other targeted agents in NSCLC patients are listed in Table 7.
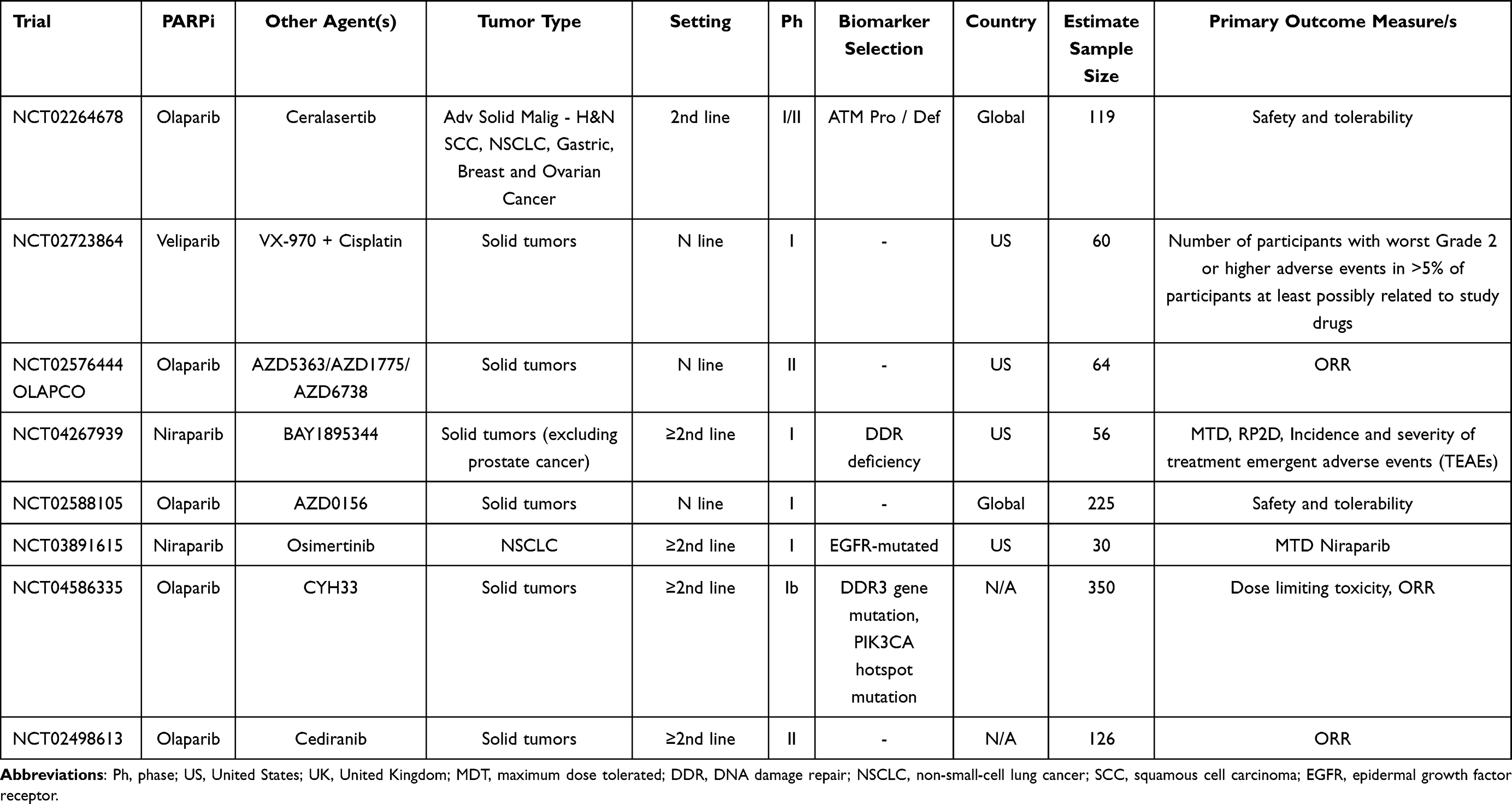 |
Table 7 Main Ongoing Clinical Trials Evaluating the Role of PARPi +/- Target Therapy in NSCLC Treatment |
Small Cell Lung Cancer (SCLC)
PARPi Alone or in Association with Chemotherapy
Biological Rationale and Preclinical Data
Despite the introduction in clinical practice of chemo-immunotherapy combining standard platinum etoposide with atezolizumab70 and, more recently, durvalumab,71 SCLC prognosis still remains abysmal, with a median overall survival of 13 months according to CASPIAN trial last update. It follows that there is much interest in novel drugs or alternative therapeutic strategies in order to improve patients’ outcomes.
There is preclinical evidence in the literature suggesting that SCLC is often characterized by alterations of the DNA damage repair pathways, in particular RB1 and TP53 lack of function72 or SOX2 and MYC genes amplification.73 Moreover, PARP enzyme and other DNA repair proteins,74 significant overexpression,75 and alterations among several HRR system genes identified in SCLC cell lines slightly contribute to tumorigenesis and growth,74 strongly supporting the idea of investigating PARPi in this disease setting treatment, alone or in combination with chemotherapy.76
Clinical Evidence and Predictive Factors of Response and Efficacy
PARPi alone as maintenance treatment after front-line chemotherapy has shown no significant benefit in terms of PFS and OS in the 220 patients recruited in the STOMP trial77 and randomized to receive PARPi olaparib or placebo after partial or complete response to first-line chemotherapy or chemoradiotherapy for SCLC proving no differences in PFS between olaparib and placebo cohorts for either twice-a-day schedule (HR=0.87; 90% CI=0.64–1.18; stratified logrank p=0.29) or the three-times-a-day schedule arm (HR=0.89; 90% CI=0.67–1.20; p=0.43). There was also no significant difference in OS between olaparib and placebo for both treatment schedules (HR=0.97; 90% CI=0.69–1.37; p=0.7 and HR=1.05; 90% CI=0.76–1.46; p=0.73, respectively).
Also, another phase III trial (NCT03516084) evaluating niraparib as maintenance therapy following first-line chemotherapy was stopped early due to the slow accrual.
It follows that interest in the potential role of PARPi as main actors in a maintenance strategy has declined and the most promising role for these therapeutic agents in SCLC seems to be in combination rather than following chemotherapy in the extended disease (ED) setting.
The phase II ECOG-ACRIN 2511 trial (NCT01642251)78 studied veliparib in association with standard chemotherapy with cisplatin and etoposide versus chemotherapy alone in 128 patients with treatment-naive ED-SCLC: median PFS was 6.1 months (95% CI=5.9–6.7 months) vs 5.5 months (95% CI=5.0–6.1 months) with an HR of 0.63 and a statistically significant p=0.001. However, no significant improvement in terms of both median OS (10.3 versus 8.9 months, HR=0.83; 80% CI=0.64–1.07), and ORR (71.9% vs 65.6%, two-sided p=0.57) were found.
A phase II trial investigated the efficacy of veliparib combined and following as a maintenance therapy with carboplatin and etoposide in first-line ED-SCLC treatment.79 One hundred and eighty-one patients were randomized 1:1:1 to receive veliparib plus chemotherapy followed by veliparib maintenance, veliparib plus chemotherapy followed by placebo, or placebo plus chemotherapy followed by placebo until unacceptable toxicity or progression, with PFS as the primary endpoint. An improvement in PFS (HR=0.67; 80% CI=0.50–0.88; p=0.059) with no OS benefit and an acceptable safety profile was shown in the veliparib arms.
Pietanza et al80–82
Several other clinical trials investigating the efficacy of PARPi combined with cytotoxic agents in ED-SCLC after first-line platinum-based chemotherapy are currently ongoing.
Clinical trials evaluating the role of PARPi with or without chemotherapy in SCLC patients are listed in Table 8.
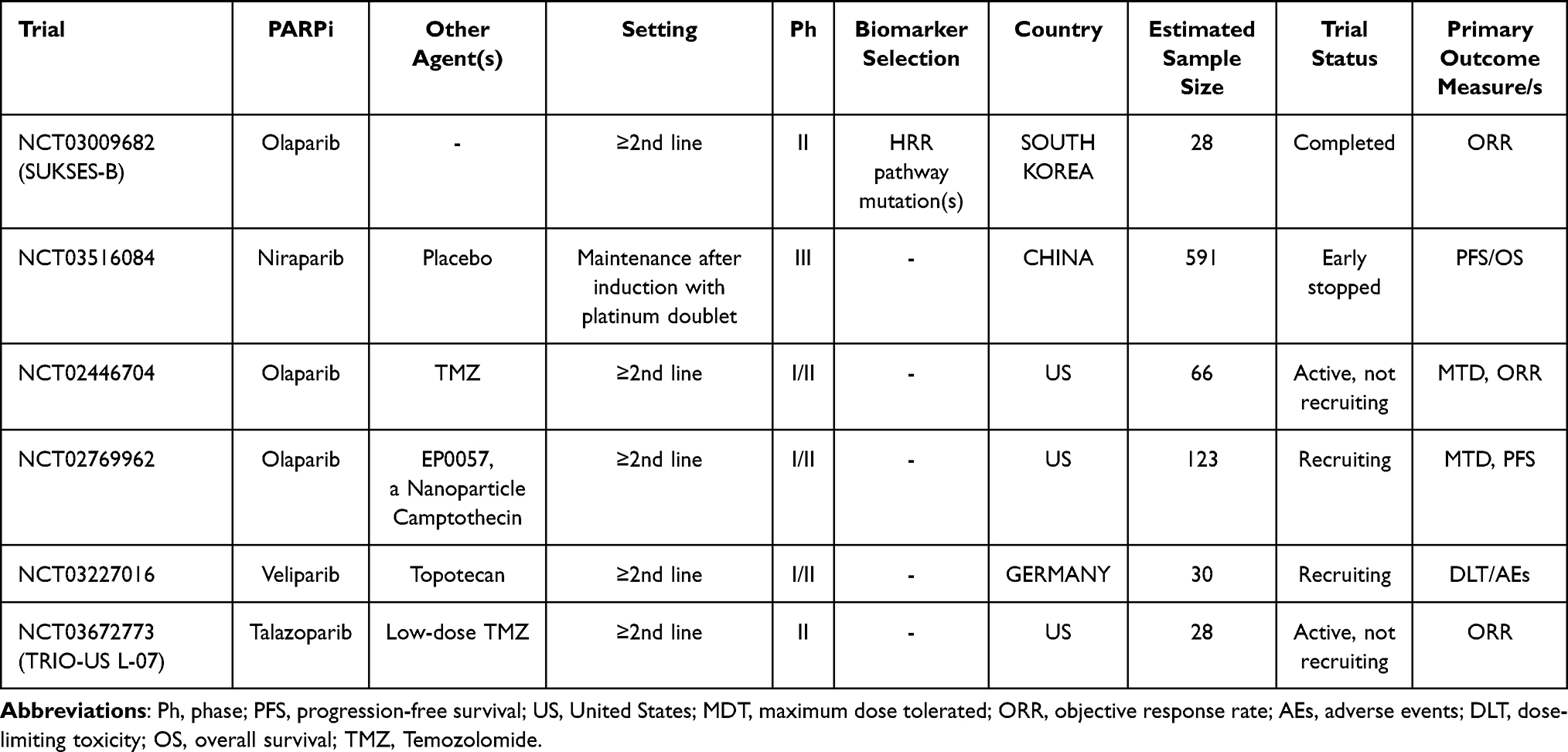 |
Table 8 Main Ongoing Clinical Trials Evaluating the Role of PARPi Alone or + Chemotherapy in SCLC Treatment |
PARPi + Radiotherapy
Biological Rationale and Preclinical Data
Radiotherapy plays an important role in the management of SCLC as thoracic CRT, typically with an etoposide and platinum-based regimen, is still is standard of care in limited stage (LS) SCLC.83 Results from two meta-analyses revealed that the addition of radiotherapy to chemotherapy provides a small but significant improvement of 3 years overall survival (5.4%),84 in particular in younger patients under 55 years of age. Moreover, radiation treatment also improved locoregional progression by 25% (95% CI=16.5–34.1%) compared with chemotherapy alone.85
Conversely, radiotherapy is not routinely adopted for the treatment of ED-SCLC, and the mainstay of treatment is a combination strategy of chemotherapy and immunotherapy. Traditionally, radiotherapy in this setting is primarily used with palliative intent of chest symptoms86 or as consolidation after response/stability to first line therapy. However, a systematic review found that consolidation radiotherapy provides a PFS benefit (HR=0.72, p<0.0001; 95% CI=0.61–0.83, I2=0%) but no improvement in OS (HR=0.88, p=0.36; 95% CI=0.66–1.18, I2=52%) compared to an exclusive chemo-immunotherapy regimen.87
With these varied results, new strategies need to be developed in order to better define the role of radiotherapy in the contest of ED-SCLC.
It is hypothesized that PARP inhibition in combination with other DNA damaging agents such as radiotherapy may result in increased sensitivity of tumor cells due to deficiency in DNA repair.88
SCLC has a very high level of PARP enzyme expression in comparison to other cancer types, thus suggesting a biologically relevant role for this protein in SCLC development and progression. In particular, it was demonstrated in SCLC lines that the sensitivity to PARPi is associated with elevated expression of PARP-189 and preclinical investigation also highlighted that PARP inhibition with veliparib enforces the cytotoxic effects of radiation in SCLC both in vitro and in vivo.90
Laird et al91 also showed that PARP trapping is effective in the radiosensitization of SCLC cell lines and patient-derived xenograft (PDX) models and that talazoparib, at the same concentration determined to cause equivalent enzymatic inhibition, may be an even better radiosensitizer than veliparib.
Clinical Evidence and Predictive Factors of Response and Efficacy
Several early phase trials are testing the combination of PARP inhibition and radiation in ED-SCLC.
An ongoing phase I trial (NCT03532880) is evaluating the safety of olaparib together with consolidative radiotherapy as well as reporting on the clinical outcomes for patients treated with this approach. This study plans to enroll 24 patients with SCLC receiving varying doses of olaparib orally twice daily for 3 weeks (administered at 50 mg to 300 mg until progression or unacceptable toxicity) and thoracic radiation once daily 5 days per week beginning 1 week after the initiation of olaparib for a total of 30 Gy in 10 fractions. The coprimary end-points are MTD of olaparib in association with radiotherapy and the safety of the combination based on the adverse events profile.
An additional analogous phase I trial (NCT04170946) will explore the same strategy with talazoparib in combination with the exactly alike consolidative radiation therapy regimen in ED-SCLC patients and another phase I/II trial (NCT04728230) of sequential treatment with olaparib, thoracic radiotherapy, and durvalumab or olaparib plus durvalumab as consolidation and maintenance therapy in ED-SCLC patients post-chemo-immunotherapy is also ongoing.
Finally, a phase I trial combining PARPi and CTLA-4 inhibitors and thoracic radiotherapy is currently underway. The trial specifically includes three arms to assess the activity and safety of durvalumab plus thoracic radiotherapy, durvalumab combined with tremelimumab plus thoracic radiotherapy, and durvalumab combined with olaparib plus thoracic radiotherapy in ED-SCLC after first-line chemotherapy (NCT03923270).
Ongoing clinical trials evaluating the role of PARPi with radiotherapy in SCLC patients are listed in Table 9.
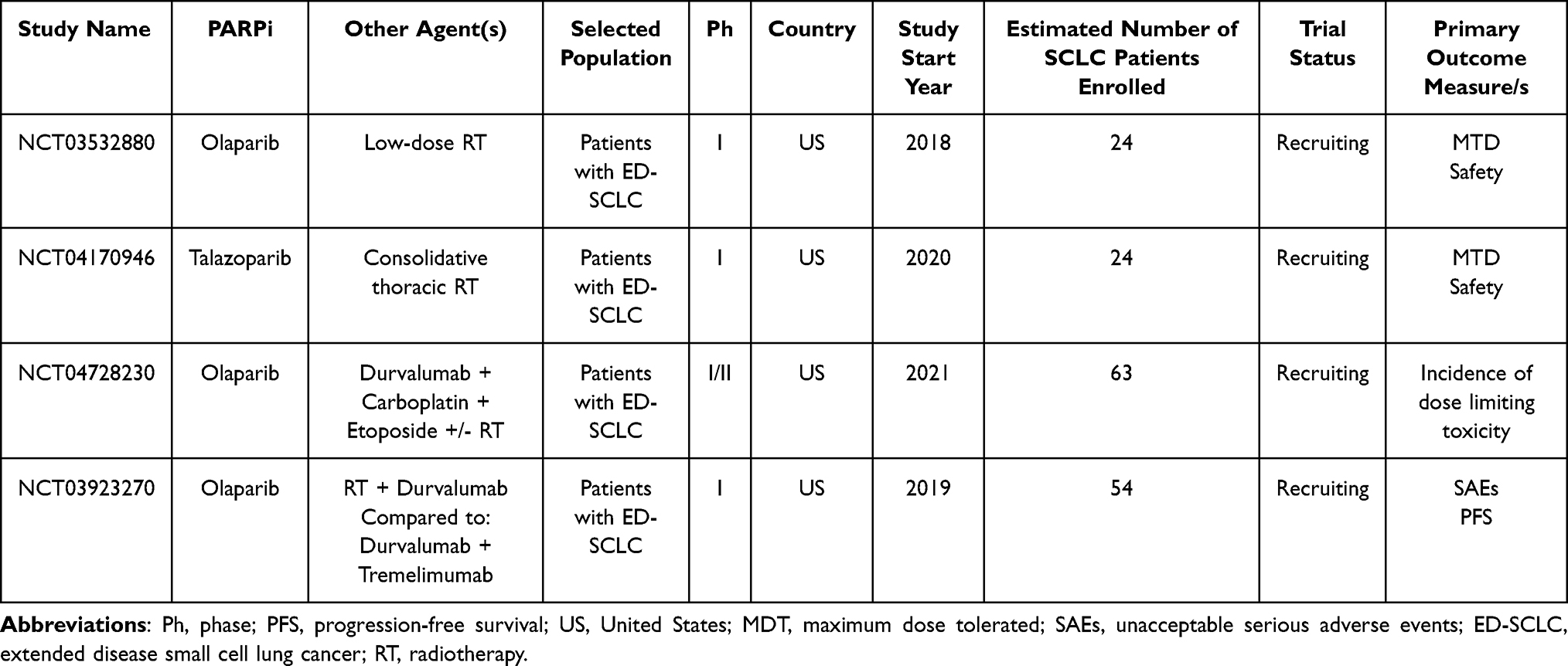 |
Table 9 Main Ongoing Clinical Trials Evaluating the Role of PARPi + Radiotherapy in SCLC Treatment |
PARPi + Immunotherapy
Biological Rationale and Preclinical Data
The recent integration of immunotherapy in the first-line treatment of SCLC has improved patients’ OS and provided a new therapeutic option in a disease with such an abysmal prognosis as SCLC.92
Due to the molecular signaling pathways between intracellular DNA and PD-L1 expression, interest in the potential synergy between PARP inhibitors and immune checkpoint inhibitors is rising.
The cGAS (cyclic GMP–AMP synthase)-STING pathway is the main cellular cytosolic double-stranded DNA (dsDNA) sensor, enabling innate immunity to respond to virus and bacteria attacks, inflammation, and eventually cancer.93,94 dsDNA interacts with cGAS, promoting a conformational change catalyzing the formation of 2′,3′-cyclic GMP-AMP (Cyclic guanosine monophosphate–adenosine monophosphate) and producing the activation of the STING pathway (Figure 2),95,96 thus leading to the recruitment and phosphorylation of TANK binding kinase 1 (TBK1) and interferon regulatory factor 3 (IRF-3)97 and inducing interferon stimulated genes expression including PD-L1.98,99
PARPi increase DNA damage and upregulate the PD-L1 expression by cGAS-STING pathway and there are preclinical studies suggesting that PARPi synergize with PD-1/PD-L1 blockade regardless of the BRCA status.48,100 Overall, these results provide a mechanistic rationale to exploit the PARPi immunomodulatory effect to reinforce immune-checkpoint blockade.
Sen et al101 have shown that olaparib activates the cGAS-STING pathway in SCLC, promoting the phosphorylation of TBK1 and IRF3 and the secretion of chemokines CCL5 and CXCL10 and increased surface expression levels of PD-L1 in SCLC models.
This study also highlighted that, as PARP inhibition with olaparib alone does not impact CD8+ cytototoxic T-cell infiltration, the simultaneous inhibition of PARP and PD-L1 leads to a clear rise in CD8+ cytotoxic T-cell infiltration and reduces regulatory T-cells levels in a triple-knockout RB-/-/p53-/-/p130-/- genetically engineered mouse model.
Clinical Evidence and Predictive Factors of Response and Efficacy
Despite these auspicious preclinical results, available evidence on PARPi plus ICIs combinations in SCLC patients has been questionable until today.
The results of a phase II trial by Thomas et al102 (NCT02484404) demonstrated that only two (10.5%) out of 19 evaluable relapsed ED-SCLC patients treated with durvalumab 1,500 mg every 4 weeks with olaparib 300 mg twice a day responded with a median PFS and OS of 1.8 months (95% CI=0.9–2.4) and 4.1 months (95% CI=2.4–9.2), respectively, and the most common treatment-related adverse event was cytopenia (45% G3–4). Of note, it has been observed that pre-existing tumor CD8+ T-cell infiltration predicts tumor response, suggesting that an inflamed-phenotype at baseline may help to identify patients who are most likely to respond to an ICI-based strategy in SCLC.
Similar results were reported in the phase II MEDIOLA study103 (NCT02734004) that analyzed 38 patients with relapsed SCLC receiving the same doses of olaparib and durvalumab but with a 4-week olaparib run-in period: only two patients had confirmed partial or complete responses and DCR at 12 weeks was 29%, below the futility boundary (<40%).
Another multicenter, open-label, phase 1a/b trial104 (NCT02660034) enrolled 49 patients with advanced solid tumors including patients with SCLC in order to assess the safety and activity of pamiparib, a novel oral PARP 1–2 inhibitor, combined with tislelizumab, an anti-PD-1. After a median follow-up of 8.3 months, ORR was 20% and treatment combination was well tolerated with anemia as the most common G3–4 adverse event.
An additional phase II trial (NCT03958045) including patients with pathological (biopsy) or cytologically confirmed stage IV SCLC achieving either partial or complete response after frontline chemotherapy with platinum doublet, is currently assessing survival and response rate of the combination of rucaparib and nivolumab as a maintenance therapy.
Other studies aiming to evaluate novel immunotherapy-based combinations as maintenance therapy following first-line treatment are currently underway (Table 10).
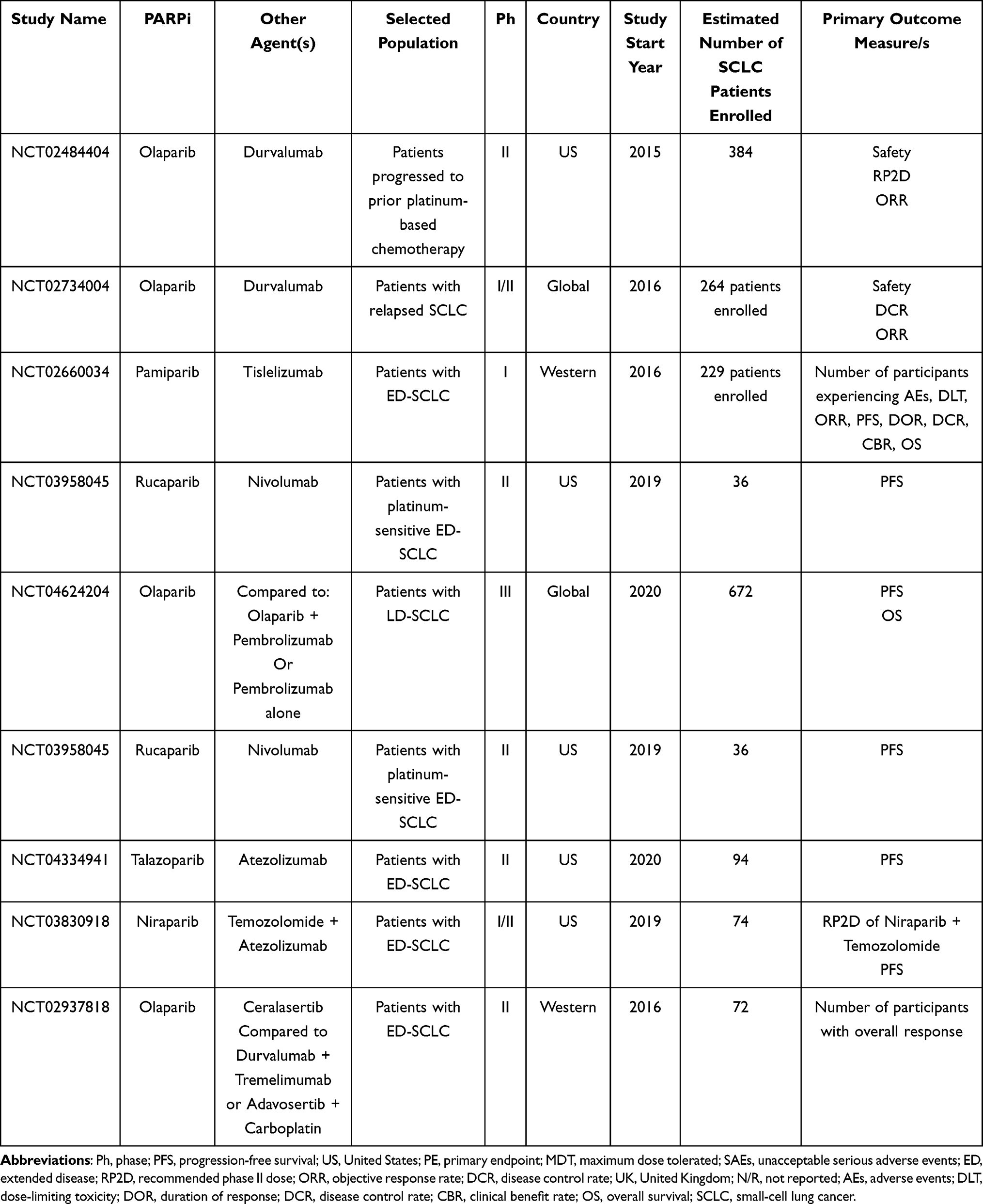 |
Table 10 Main Ongoing Clinical Trials Evaluating the Role of PARPi + Immunotherapy in SCLC Treatment |
PARPi + DDR Inhibitors
Biological Rationale and Preclinical Data
Both in vitro and in vivo SCLC models support the rationale of combining PARPi with other drugs targeting the HRR pathways, especially the ATR/CHK1 (Checkpoint kinase 1) axis.105 Preclinical models showed a higher activity of ATR inhibitors in tumors with TP53/ATM lack of function15 and CHK1 inhibition effectively overcome PARPi resistance in vitro, supporting the rationale for testing combination treatment.106
Clinical Evidence and Predictive Factors of Response and Efficacy
Though there are concerns regarding patients’ selection, safety profile and possible mechanisms of resistance, the rationale for combining different HRR inhibitors is strong and several ongoing clinical trials will provide clinical evidence about this novel strategy.
Adavosertib, a WEE1 kinase inhibitor, in association with olaparib has been tested in a phase Ib study (NCT02511795),107 showing a promising ORR of 30.8%. A phase II trial (NCT03428607, SUKSES-N2) investigating combination treatment with ceralasertib, a CHK1 inhibitor, and olaparib as second-line treatment of ED-SCLC has completed its enrollment, and primary analysis results are awaited soon.
Main ongoing clinical trials evaluating the role of PARPi with DDRi in SCLC patients are listed in Table 11.
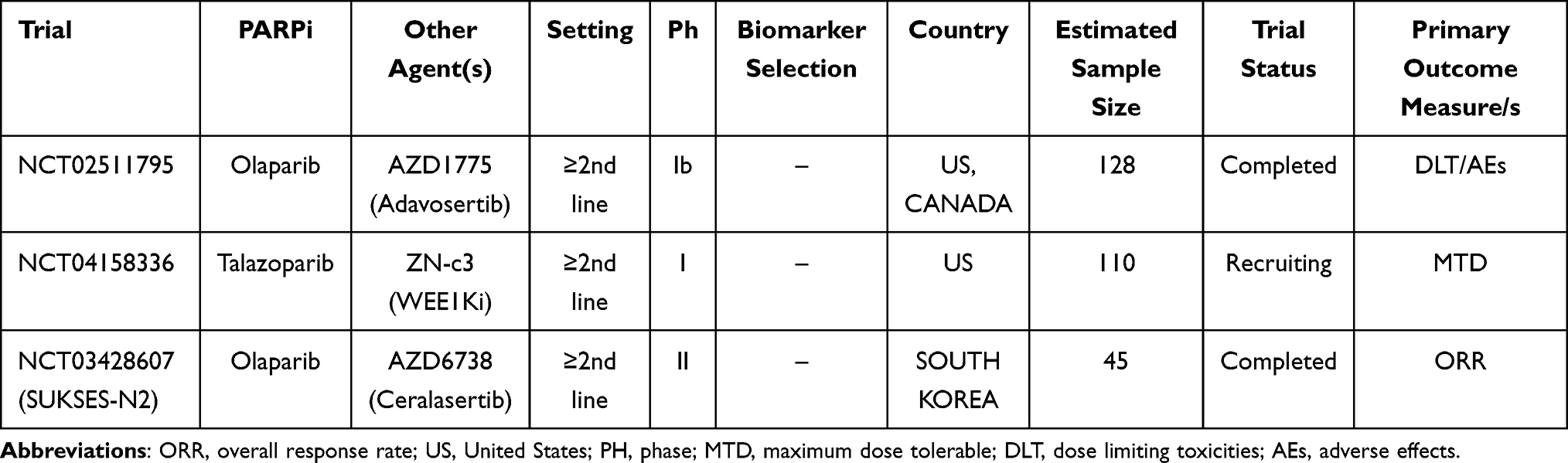 |
Table 11 Main Ongoing Clinical Trials Evaluating the Role of PARPi + DDR Inhibitors in SCLC Treatment |
Malignant Pleural Mesothelioma (MPM)
PARPi Alone or in Association with Chemotherapy
Biological Rationale and Preclinical Data
Until a few years ago, the only therapeutic option with proven effectiveness in the treatment of MPM was platinum and pemetrexed chemotherapy,108 but promising new options have recently emerged from the use of immunotherapy, in particular the combination of Nivolumab and Ipilimumab (Checkmate 743).109 In MPM, activating mutations are rare and genomic losses/alterations are more widespread, hence alternative treatments to current standards are still an open issue.
In addition, better understanding of cancer biology has opened the targeted therapies era also for neglected tumors such as MPM.
In particular, an important percentage (5–10%) of patients with MPM with germline mutations in the DDR genes pathway have shown a relevant sensitivity to asbestos110 and BRCA1/2 or mutations of other genes involved in the HRR pathway have been observed in many MPM patients.111 Moreover, more than 20% of MPM showed somatic inactivating mutations in the BRCA-1 associated gene (BAP1),112 which seem to play an important role in tumor pathogenesis.113 Hence, the idea that cells presenting mutated BAP1 may require PARP-1 enzyme to survive has led to the initiation of several preclinical studies exploring the potential efficacy of PARPi in this setting.114
Interestingly and partially negating the potential interaction between BAP1 gene and PARP enzyme specific inhibition, two different preclinical studies have shown that both niraparib and olaparib had potential efficacy against MPM cells, regardless of the presence of BAP1 mutations.115,116 Borchert et al,117,118 performing analysis with luminescence assays, also highlighted the promising combination of PARPi with cisplatin-based chemotherapy.
Clinical Evidence and Predictive Factors of Response and Efficacy
Further exploring evidence from preclinical studies, many trials have been evaluating the efficacy and tolerability of PARPi in MPM.112,119 Ghafoor et al120 studied olaparib in 23 patients with advanced MPM with somatic or germline mutations of DNA repair genes in a phase 2 single-center trial. Patients were given olaparib 300 mg twice daily in 21-day cycles until disease progression or intolerable toxicity. Definitive results were quite disappointing in particular in the germline mutation cohort: in germline and somatic BAP1 mutants, median PFS was 2.3 months (95% CI=1.3–3.6 months) versus 4.1 months (95% CI=2.7–5.5 months) with a median OS of 4.6 months (95% CI=3.1–4.9 months) vs 9.6 months (95% CI=5.5 months–not estimable) (p=0.0040). Among ongoing clinical trials, the UNITO-001, a prospective phase 2 single arm study, is evaluating the combination of niraparib and dostarlimab in metastatic mesotheliomas with both HRR deficiency and PD-L1 positivity (TPS>1%). The primary endpoint is PFS, secondary endpoints are overall survival, objective response, duration of response, and safety.121
The TALAMESO trial (NCT04462809) is an open-label phase II trial with three independent cohorts including patients with advanced malignant pleural (cohort A) or peritoneal (cohort B1 and B2) mesotheliomas without any sign of disease progression after four to six cycles of platinum-based chemotherapy (including a minimum one cycle of pemetrexed). The primary endpoint is the non-progression proportion (defined as the proportion of patients free of progression 6 months after talazoparib start); secondary endpoints are PFS, toxicity, and safety assessment.
The MiST trial is a Phase II, single arm study that is testing the efficacy of rucaparib in MPM patients with BAP1 or BRCA mutation.122 The primary endpoint is DCR, secondary endpoints are ORR, toxicity, and safety assessment. Moreover, new studies with PARPi started in mesothelioma with homologous recombination deficiency associated with BRCA1 mutation.123
The main ongoing clinical trials evaluating the role of PARPi in MPM patients are listed in Table 12.
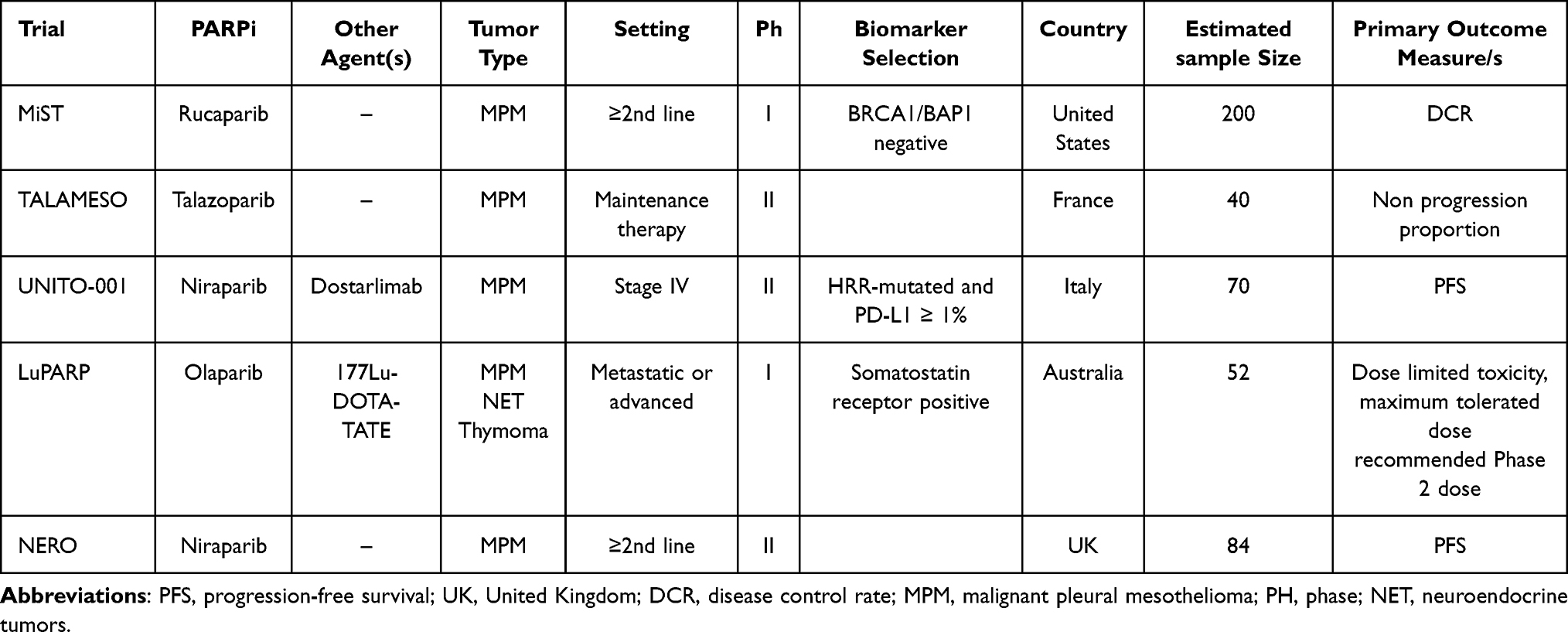 |
Table 12 Main Ongoing Clinical Trials Evaluating the Role of PARPi in MPM Treatment |
PARG Inhibitors in Thoracic Malignancies
Biological Rationale and Preclinical Data
Poly (ADP-ribose) glycohydrolase is the primary hydrolase involved in the degradation of PAR. PARG localizes at replication forks by binding PCNA and promotes recovery from prolonged replication stress.
PARG isoforms have a cytoplasmic and perinuclear distribution and seem to migrate to the nucleus when activated due to genotoxic insult. However, how the different isoforms fully contribute to PAR metabolism and its significance is yet to be elucidated.
The consensus opinion is that PARP and PARG share an important function in downstream cellular processes enhancement. Just like PARP, PARG inhibition weakens cancer cells’ resistance to DNA damaging agents due to PARG enzymes’ role in DNA repair mechanisms.
In contrast to PARP inhibitors, a clear correlation between HR deficiency and PARGi-related synthetic lethality still needs to be fully demonstrated.3–124
Clinical Evidence and Predictive Factors of Response and Efficacy
Depletion of HR involved proteins such as BRCA1, BRCA22, PALB2, ABRAXAS, and BARD1 in MCF7 breast cancer lines was shown to elicit synthetic lethal interactions with PARG inhibition.125
Pillay et al,126 evaluating PARGi PDD00017273 on ovarian cancer cell lines, found that cancer cells were differentially sensitive to PARG and PARP inhibition. In particular, a “replication catastrophe” event (identified as pan-nuclear γH2AX staining) due to underlying DNA replication vulnerabilities occurred upon PARGi administration; on the other hand, the same chain reaction was not seen with the PARPi olaparib. Despite the analysis being performed only on in vitro models, this study suggests that low expression of key replication factors promoting DNA fork stabilization may be a biomarker predictive of PARGi effectiveness also alternatively to PARPi. Moreover, BRCA1 mutated cells with acquired resistance to PARPi due to loss of 53BP-1 were still more sensitive to the PARGi COH34 than BRCA1 wildtype cells.127 PARGi have also been reported to be synthetic lethal in XRCC1 depleted and deficient cells.128 It follows that PARGi may have efficacy in XRCC1 tumors, probably because of the function of XRCC1 in stabilizing stalled forks. Furthermore, PARG depletion via siRNA was reported to be synthetic lethal with dual specificity phosphatase 22 (DUSP22) via suppression of the mTOR/PI3k/AKT in lung cancer.129
Nevertheless, PARG depletion did not show synthetic lethality with BRCA1 mutations in different cancer cell lines130 and PARG overexpression was even related with tumor-promoting genes downregulation and suppression in prostate cancer PC-3 cell lines.131
Focusing on the potential association of PARGi with other drugs, evidence supporting a chemosensitizing effect of PARG inhibition has already been reported in literature, in particular considering DNA-damaging agents. Chen and Yu127 described that a a novel small molecule identified from the NCI database, COH34, specifically inhibiting PARG can sensitize different tumor cell lines with DNA repair defects to topoisomerase I inhibitors and DNA-alkylating agents, which are widely used in cancer chemotherapy, such as cisplatin, temozolomide, and doxorubicine. Pillay et al126 demonstrated a similar effect of gemcitabine in combination with PARGi PDD00017273 in ovarian cancer cell lines.
Finally, Jain et al132 evaluated PARG as a target in ductal pancreatic adenocarcinoma models using both genetic silencing of PARG and established small-molecule PARGi PDDX-01/04. Homologous repair-deficient cells compared with homologous repair-proficient cells were more sensitive to PARGi in vitro, while in vivo silencing of PARG significantly decreased tumor growth. PARGi also seemed to synergize with DNA-damaging agents, in particular oxaliplatin and 5-fluorouracil, that are commonly used in pancreatic cancer treatment.
A potential synergistic effect between PARG inhibition and radiation therapy has been reported in different in vitro models due to the occurrence and accumulation of mitotic defects, finally culminating in cancer cell apoptosis.133
Most interestingly, Gravells et al134 provided a comparison of the potential radiosensitizing effect of PARP inhibitor olaparib and novel PARG inhibitor PDD00017273: both olaparib and PDD00017273 altered the repair of radiation-induced DNA damage, resulting in delayed resolution of RAD51 foci compared with control cells but only PARG inhibition was able to effectively induce a perturbed mitotic progression leading to cell death, thus suggesting that PARG plays different functions in the cell compared with inhibition of PARP1/2/3, likely via reversal of tankyrase enzyme activity. However, solid evidence of the potential application of PARGi radiosensitizing effect in clinical practice is still lacking.
Despite preclinical evidence in selected scenarios, PARG inhibition still represents a niche option in cancer treatment and we still lack a clinical trial demonstrating some sort of benefit. Nowadays, there is great expectation in the phase I–II NCT05787587 trial that is going to evaluate PARG Inhibitor IDE161 efficacy in patients with advanced solid tumors harboring BRCA1/2 loss of function alterations and/or other defects in the homologous recombination (HR) pathway.
Conclusions
Despite FDA approval in HR-deficient castration resistant prostate cancer and BRCA1/2 mutated ovarian, breast, and pancreatic tumors, the efficacy of PARPi is restricted by inevitable drug resistance, whereas dose-limiting toxicities are frequently linked to its use in conjunction with chemotherapy and targeted medicines. ICIs have shown persistent responses in a variety of solid tumors, however single agent action is only seen in a small proportion of patients and drug resistance is still a problem.135 Based on the success of both drug classes and considering the growing evidence in literature on the potential synergy between PARP inhibition and immunotherapy, a combination strategy may represent a turning point.135 Preclinical data revealed that the activation of the cGAS-sting pathway by PARP inhibition results in an important PD-L1 expression on cancer cells creating an appropriate tumor immune microenvironment to take advantage of ICB. The combination of ICB and PARPi has been encouraged by clinical trials showing improved survival outcomes in prostate, breast, ovarian, and SCLC, regardless of mutational status or even in the case of platinum sensitivity.
Considering NSCLC, trials investigating PARPi as monotherapy did not show improved outcomes, but associations with other therapies, such as immunotherapy, might result in better outcomes.136,137
PD-L1 expression has been identified as putative predictive biomarkers of response for ICB therapy selection in NSCLC; however, these have not been consistently shown to have predictive potential in the context of PARPi and ICB combination strategies.138 Similarly, in SCLC and prostate cancer, no clear correlation between PD-L1 expression and treatment response has been demonstrated.102,139
In this regard, new technologies such as Next Generation Sequencing (NGS) might be crucial in opening new paths as the identification of reliable molecular predictive factors other than MMR deficiency constitutes a critical issue that must be thoroughly addressed in future investigations. As most clinical trials designed so far lacked an adequate patient selection, new studies are underway and will help determine the potential role for PARPi in thoracic malignancies treatment.
Another open question is also the low oral bioavailability of PARPi, as these drugs effective and safe delivery during clinical cancer therapy still represents a challenge. In this regard, the development of drug delivery has advanced because of nanotechnology: this term commonly refers to structures that are up to several hundred nanometers in size.140 In recent decades, a number of nanoformulations, as drug delivery systems (DDSs), have been reported for the treatment of cancers, such as liposomes, polymeric micelles, hydrogels, nanoemulsions, nanosuspensions, and nanoparticles.141 Drug delivery systems could provide protection for drugs in blood circulation, increasing the amount of time of their circulation and they could gather in the leaking vasculature of the tumor and cancer cells releasing the chemotherapeutic moiety into the tumor microenvironment. At the same time nanoparticles are unable to pass through the body’s organs and tissues, so nanosystems may not only enhance drug efficiency but also reduce adverse side-effects.142
In conclusion, PARPi represent a solid reality in numerous solid tumors and several trials are exploring their potential employment in diseases such as thoracic malignancies where they might represent a novel therapeutic option. However, these drugs still face several serious downsides, including resistance mechanisms and unintended adverse effects and their efficacy as single agents still need to be fully demonstrated in the context of thoracic malignancies. In this regard, considering available evidence in the literature we already discussed in our review, interest is growing in exploring the potential synergistic effect and consequently combined administration of PARPi and PARGi with other agents such as chemotherapy, immunotherapy, or radiotherapy. Novel strategies such as tumor profiling with NGS might be helpful in determining reliable predictive factors guiding patients selection in clinical practice.
In addition, with the recent introduction of nanomedicine, off-site toxicity or drug resistance might be reduced in the next few years, thus opening new interesting scenarios for PARPi and PARGi in clinical practice.
Abbreviations
SCLC, Small Cell Lung Cancer; NSCLC, Non-Small Cell Lung Cancer; MPM, malignant pleural mesothelioma; EGFR, epithelial growth factor receptor; ALK, anaplastic lymphoma kinase; ROS1, c-ros oncogene 1; KRAS, Kirsten rat sarcoma virus (KRAS); BRAF, V-raf murine sarcoma oncogene homolog B1; HER2, human epidermal growth factor receptor; PARP, poly (ADP-ribose) polymerase; HRR, Homologous Recombination Repair; PARPi, poly (ADP-ribose) polymerase inhibitors; PARG, poly (ADP-ribose) glycohydrolase; PARGi, poly (ADP-ribose) glycohydrolas inhibitors; NAD, Nicotinamide Adenine Dinucleotide; BER, Base-Excision Repair; NER, Nucleotide-Excision Repair; MMR, Mismatch Repair; HR, Homologous Recombination; NHEJ, Non-Homologous End-Joining; PARP-1, poly (ADP-ribose) polymerase-1; PARP-2, poly (ADP-ribose) polymerase-2; MGMT, MethylGuanineDNA Methyltransferase; BRCA1, BReast CAncer gene 1; BRCA2, BReast CAncer gene 2; ERCC1, Excision repair cross-complementation group 1; LOH, Loss Of Heterozygosis; EGFR, Epithelial Growth Factor Receptor; ALK, Anaplastic Lymphoma Kinase; PFS, Progression Free Survival; ATM, Ataxia-Telangiectasia Mutated; ATR, Ataxia-Telangectasia and Rad3 related; GDSC, Genomics of Drug Sensitivity in Cancer; CRISPR, Clustered Regularly Interspaced Short Palindromic Repeats; CAS9, CRISPR Associated Protein 9; IC50, Half Maximal Inhibitory Concentration; PTEM, Phosphatase and Tensin Homolog; TMZ, Temozolomide; GCI, Gemcitabine/Cisplatin/Iniparib; GC, Gemcitabine/Cisplatin; ORR, Overall Response Rate; OS, Overall Survival; HR, Hazard Ratio; CI, Confidence Interval; WES, Whole Exome Sequencing; H2AFX, H2A Histone Family Member X; CRT, Chemoradiotherapy; DLT, Dose Limiting Toxicity; MTD, Maximum Tolerable Dose; ICIs, Immune Checkpoint Inhibitors; TMB, Tumor Mutational Burden; STING, Stimulator of IFN Genes; PD-1, Programmed Death-protein 1; PD-L1, Programmed Cell Death Ligand-1; TPS, Tumor Proportion Score; DOR, Duration of Response; DDRi, DNA Damage Response Inhibitors; ED, Extended Disease; LS, limited stage; PDX, Patient-Derived Xenograft; cRT, Consolidative Radiation Therapy; c-GAS, Cyclic GMP–AMP synthase; cGAMP, Cyclic Guanosine monophosphate–Adenosine Monophosphate; dsDNA, Double Stranded DNA; TBK1, TANK Binding Kinase 1; IRF-3, Interferon Regulatory Factor 3; CHK1, Checkpoint Kinase; BAP1, BRCA1-Associated Protein-1; DCR, Disease Control Rate; TPS, Tumor Proportion Score; VEGF, Vascular Endothelial Growth Factor; SSB, Single-strand breaks; ISGs, IFN-stimulated genes; MHC, major histocompatibility complex; TCR, T cell receptor; NGS, Next Generation Sequencing.
Disclosure
Professor Rossana Berardi reports grants from Lilly, grants from MSD, grants from Otsuka, grants from BI, grants from GSK, grants from Italfarmaco, grants from Roche, grants from AZ, outside the submitted work. The authors report no other conflicts of interest in this work.
References
1. Patel NT, Steuer CE. PARP-1 inhibitors and their emerging role in the treatment of lung cancer; 2015.
2. Farmer H, McCabe H, Lord CJ, et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature. 2005;434(7035):917–921. doi:10.1038/nature03445
3. Harrision D, Gravells P, Thompson R, Bryant HE. Poly(ADP-Ribose) glycohydrolase (PARG) vs. Poly(ADP-Ribose) Polymerase (PARP) - function in genome maintenance and relevance of inhibitors for anti-cancer therapy. Front Mol Biosci. 2020;7:191.
4. Chan CY, Tan KV, Cornelissen B. PARP inhibitors in cancer diagnosis and therapy. Clin Cancer Res. 2021;27(6):1585–1594. doi:10.1158/1078-0432.CCR-20-2766
5. Wang X, Zeng X, Li D, et al. PARP inhibitors in small cell lung cancer: the underlying mechanisms and clinical implications. Biomed Pharmacother. 2022;153:113458. doi:10.1016/j.biopha.2022.113458
6. Jordan EJ, Kim HR, Arcila ME, et al. Prospective comprehensive molecular characterization of lung adenocarcinomas for efficient patient matching to approved and emerging therapies. Cancer Discov. 2017;7(6):596–609. doi:10.1158/2159-8290.CD-16-1337
7. Jonsson P, Bandlamudi C, Cheng ML, et al. Tumour lineage shapes BRCA-mediated phenotypes. Nature. 2019;571(7766):576–579. doi:10.1038/s41586-019-1382-1
8. Paul I, Savage KI, Blayney JK, et al. PARP inhibition induces BAX/BAK-independent synthetic lethality of BRCA1-deficient non-small cell lung cancer. J Pathol. 2011;224(4):564–574. doi:10.1002/path.2925
9. Postel-Vinay S, Bajrami I, Friboulet L, et al. A high-throughput screen identifies PARP1/2 inhibitors as a potential therapy for ERCC1-deficient non-small cell lung cancer. Oncogene. 2013;32(47):5377–5387. doi:10.1038/onc.2013.311
10. Jiang G, Zhang S, Yazdanparast A, et al. Comprehensive comparison of molecular portraits between cell lines and tumors in breast cancer. BMC Genomics. 2016;17(Suppl 7):13–15. doi:10.1186/s12864-016-2911-z
11. Remon J, Besse B, Leary A, et al. Somatic and Germline BRCA 1 and 2 mutations in advanced NSCLC from the SAFIR02-lung trial. JTO Clin Res Rep. 2020;1(3):100068.
12. Barlesi F, Mazieres J, Merlio JP, et al. Routine molecular profiling of patients with advanced non-small-cell lung cancer: results of a 1-year nationwide programme of the French Cooperative Thoracic Intergroup (IFCT). Lancet. 2016;387(10026):1415–1426. doi:10.1016/S0140-6736(16)00004-0
13. Heeke AL, Pishvaian MJ, Lynce F, et al. Prevalence of homologous recombination–related gene mutations across multiple cancer types. JCO Precis Oncol. 2018;2(2):1–13.
14. Ding L, Getz G, Wheeler DA, et al. Somatic mutations affect key pathways in lung adenocarcinoma. Nature. 2008;455(7216):1069. doi:10.1038/nature07423
15. Reaper PM, Griffiths MR, Long JM, et al. Selective killing of ATM- or p53-deficient cancer cells through inhibition of ATR. Nat Chem Biol. 2011;7(7):428–430. doi:10.1038/nchembio.573
16. Vendetti FP, Lau A, Schamus S, Conrads TP, O’Connor MJ, Bakkenist CJ. The orally active and bioavailable ATR kinase inhibitor AZD6738 potentiates the anti-tumor effects of cisplatin to resolve ATM-deficient non-small cell lung cancer in vivo. Oncotarget. 2015;6(42):44289. doi:10.18632/oncotarget.6247
17. Hall AB, Newsome D, Wang Y, et al. Potentiation of tumor responses to DNA damaging therapy by the selective ATR inhibitor VX-970. Oncotarget. 2014;5(14):5674–5685. doi:10.18632/oncotarget.2158
18. Toledo LI, Murga M, Zur R, et al. A cell-based screen identifies ATR inhibitors with synthetic lethal properties for cancer-associated mutations. Nat Struct Mol Biol. 2011;18(6):721–727. doi:10.1038/nsmb.2076
19. O’Connor MJ. Targeting the DNA damage response in cancer. Mol Cell. 2015;60(4):547–560. doi:10.1016/j.molcel.2015.10.040
20. Jette NR, Radhamani S, Arthur G, et al. Combined poly-ADP ribose polymerase and ataxia-telangiectasia mutated/Rad3-related inhibition targets ataxia-telangiectasia mutated-deficient lung cancer cells. Br J Cancer. 2019;121(7):600–610. doi:10.1038/s41416-019-0565-8
21. Dillon L, Miller T. Therapeutic targeting of cancers with loss of PTEN function. Curr Drug Targets. 2014;15(1):65–79. doi:10.2174/1389450114666140106100909
22. Sargazi S, Saravani R, Zavar Reza J, et al. Novel Poly(Adenosine Diphosphate-Ribose) Polymerase (PARP) Inhibitor, AZD2461, down-regulates VEGF and induces apoptosis in prostate cancer cells. Iran Biomed J. 2019;23(5):312–323. doi:10.29252/ibj.23.5.2
23. Sargazi S, Saravani R, Zavar Reza J, et al. Induction of apoptosis and modulation of homologous recombination DNA repair pathway in prostate cancer cells by the combination of AZD2461 and valproic acid. EXCLI J. 2019;18:485–498. doi:10.17179/excli2019-1098
24. Sargazi S, Kooshkaki O, Zavar Reza J, et al. Mild antagonistic effect of Valproic acid in combination with AZD2461 in MCF-7 breast cancer cells. Med J Islam Repub Iran. 2019;33:29. doi:10.34171/mjiri.33.29
25. Mullane SA, Werner L, Guancial EA, et al. Expression levels of DNA damage repair proteins are associated with overall survival in platinum treated advanced urothelial carcinoma. Clin Genitourin Cancer. 2016;14(4):352. doi:10.1016/j.clgc.2015.12.029
26. Lord CJ, Ashworth A. PARP inhibitors: synthetic lethality in the clinic. Science. 2017;355(6330):1152–1158. doi:10.1126/science.aam7344
27. Kummar S, Chen A, Parchment RE, et al. Advances in using PARP inhibitors to treat cancer. BMC Med. 2012;10:10. doi:10.1186/1741-7015-10-10
28. Donawho CK, Luo Y, Luo Y, et al. ABT-888, an orally active Poly(ADP-Ribose) polymerase inhibitor that potentiates DNA-damaging agents in preclinical tumor models. Clin Cancer Res. 2007;13(9):2728–2737.
29. Palma JP, Wang YC, Rodriguez LE, et al. ABT-888 confers broad in vivo activity in combination with temozolomide in diverse tumors. Clin Cancer Res. 2009;15(23):7277–7290. doi:10.1158/1078-0432.CCR-09-1245
30. Liu X, Shi Y, Maag DX, et al. Iniparib nonselectively modifies cysteine-containing proteins in tumor cells and is not a Bona Fide PARP inhibitor. Clin Cancer Res. 2012;18(2):510–523. doi:10.1158/1078-0432.CCR-11-1973
31. Reck M, Blais N, Juhasz E, et al. Smoking history predicts sensitivity to PARP inhibitor veliparib in patients with advanced non–small cell lung cancer. J Thoracic Oncol. 2017;12(7):1098–1108. doi:10.1016/j.jtho.2017.04.010
32. Ramalingam SS, Novello S, Guclu SZ, et al. Veliparib in combination with platinum-based chemotherapy for first-line treatment of advanced squamous cell lung cancer: a randomized, multicenter Phase III study. J Clin Oncol. 2021;39(32):3633. doi:10.1200/JCO.20.03318
33. Lesueur P, Chevalier F, Austry JB, et al. Poly-(ADP-ribose)-polymerase inhibitors as radiosensitizers: a systematic review of pre-clinical and clinical human studies. Oncotarget. 2017;8(40):69105–69124. doi:10.18632/oncotarget.19079
34. Albert JM, Cao C, Kwang WK, et al. Inhibition of Poly(ADP-Ribose) polymerase enhances cell death and improves tumor growth delay in irradiated lung cancer models. Clin Cancer Res. 2007;13(10):3033–3042. doi:10.1158/1078-0432.CCR-06-2872
35. Cristea MC, Miao J, Argiris A, et al. SWOG S1206: a dose-finding study of veliparib (ABT-888) added to chemoradiotherapy (CRT) with carboplatin (C) and paclitaxel (P) for unresectable stage III non-small cell lung cancer (NSCLC). J Clin Oncol. 2016;34(15_suppl):8537.
36. Kozono DE, Stinchcombe TE, Salama JK, et al. Veliparib in combination with carboplatin/paclitaxel-based chemoradiotherapy in patients with stage III non-small cell lung cancer. Lung Cancer. 2021;159:56–65. doi:10.1016/j.lungcan.2021.06.028
37. de Haan R, van den Heuvel MM, van Diessen J, et al. Phase I and pharmacologic study of olaparib in combination with high-dose radiotherapy with and without concurrent cisplatin for non-small cell lung cancer. Clin Cancer Res. 2021;27(5):1256–1266. doi:10.1158/1078-0432.CCR-20-2551
38. Gandhi L, Rodríguez-Abreu D, Gadgeel S, et al. Pembrolizumab plus chemotherapy in metastatic non–small-cell lung cancer. N Engl J Med. 2018;378(22):2078–2092. doi:10.1056/NEJMoa1801005
39. Reck M, Rodríguez-Abreu D, Robinson AG, et al. Pembrolizumab versus chemotherapy for PD-L1-positive non-small-cell lung cancer. N Engl J Med. 2016;375(19):1823–1833. doi:10.1056/NEJMoa1606774
40. Antonia SJ, Villegas A, Daniel D, et al. Durvalumab after chemoradiotherapy in stage III non–small-cell lung cancer. N Engl J Med. 2017;377(20):1919–1929. doi:10.1056/NEJMoa1709937
41. Brahmer J, Reckamp KL, Baas P, et al. Nivolumab versus docetaxel in advanced squamous-cell non–small-cell lung cancer. N Engl J Med. 2015;373(2):123–135. doi:10.1056/NEJMoa1504627
42. Rittmeyer A, Barlesi F, Waterkamp D, et al. Atezolizumab versus docetaxel in patients with previously treated non-small-cell lung cancer (OAK): a Phase 3, open-label, multicentre randomised controlled trial. Lancet. 2017;389(10066):255–265. doi:10.1016/S0140-6736(16)32517-X
43. Borghaei H, Paz-Ares L, Horn L, et al. Nivolumab versus Docetaxel in Advanced Nonsquamous Non–Small-Cell Lung Cancer. N Engl J Med. 2015;373(17):1627–1639. doi:10.1056/NEJMoa1507643
44. Paz-Ares L, Luft A, Vicente D, et al. Pembrolizumab plus chemotherapy for squamous non–small-cell lung cancer. N Engl J Med. 2018;379(21):2040–2051. doi:10.1056/NEJMoa1810865
45. Peyraud F, Italiano A. Combined PARP inhibition and immune checkpoint therapy in solid tumors. Cancers. 2020;12(6):1502. doi:10.3390/cancers12061502
46. Mouw KW, Goldberg MS, Konstantinopoulos PA, D’Andrea AD. DNA damage and repair biomarkers of immunotherapy response. Cancer Discov. 2017;7(7):675–693. doi:10.1158/2159-8290.CD-17-0226
47. Barber GN. STING: infection, inflammation and cancer. Nat Rev Immunol. 2015;15(12):760. doi:10.1038/nri3921
48. Wang Z, Sun K, Xiao Y, et al. Niraparib activates interferon signaling and potentiates anti-PD-1 antibody efficacy in tumor models. Sci Rep. 2019;9(1):1853.
49. Rizvi NA, Hellmann MD, Snyder A, et al. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science. 2015;348(6230):124–128. doi:10.1126/science.aaa1348
50. Chae YK, Davis AA, Raparia K, et al. Association of Tumor Mutational Burden With DNA Repair Mutations and Response to Anti–PD-1/PD-L1 Therapy in Non–Small-Cell Lung Cancer. Clin Lung Cancer. 2019;20(2):88–96.e6. doi:10.1016/j.cllc.2018.09.008
51. Ricciuti B, Wang X, Alessi JV, et al. Association of high tumor mutation burden in non–small cell lung cancers with increased immune infiltration and improved clinical outcomes of PD-L1 blockade across PD-L1 expression levels. JAMA Oncol. 2022;8(8):1160. doi:10.1001/jamaoncol.2022.1981
52. Ricciuti B, Recondo G, Spurr LF, et al. Impact of DNA Damage Response and Repair (DDR) gene mutations on efficacy of PD-(L)1 immune checkpoint inhibition in non–small cell lung cancer. Clin Cancer Res. 2020;26(15):4135–4142. doi:10.1158/1078-0432.CCR-19-3529
53. Ramalingam SS, Thara E, Awad MM, et al. JASPER: Phase 2 trial of first‐line niraparib plus pembrolizumab in patients with advanced non–small cell lung cancer. Cancer. 2022;128(1):65. doi:10.1002/cncr.33885
54. Clarke JM, Patel JD, Robert F, et al. Veliparib and nivolumab in combination with platinum doublet chemotherapy in patients with metastatic or advanced non-small cell lung cancer: a phase 1 dose escalation study. Lung Cancer. 2021;161:180–188. doi:10.1016/j.lungcan.2021.09.004
55. Rizvi NA, Hellmann MD, Brahmer JR, et al. Nivolumab in combination with platinum‐based doublet chemotherapy for first-line treatment of advanced non–small-cell lung cancer. J Clin Oncol. 2016;34(25):2969. doi:10.1200/JCO.2016.66.9861
56. Hellmann MD, Paz-Ares L, Bernabe Caro R, et al. Nivolumab plus ipilimumab in advanced non–small-cell lung cancer. N Engl J Med. 2019;381(21):2020–2031. doi:10.1056/NEJMoa1910231
57. Jamil S, Stoica C, Hackett TL, Duronio V. MCL-1 localizes to sites of DNA damage and regulates DNA damage response. Cell Cycle. 2010;9(14):2843. doi:10.4161/cc.9.14.12354
58. Mattoo AR, Joun A, Milburn Jessup J. Repurposing of mTOR complex inhibitors attenuates Mcl-1 and sensitizes to PARP inhibition. Mol Cancer Res. 2019;17(1):42–53. doi:10.1158/1541-7786.MCR-18-0650
59. Ibrahim YH, García-García C, Serra V, et al. PI3K inhibition impairs BRCA1/2 expression and sensitizes BRCA-proficient triple-negative breast cancer to PARP inhibition. Cancer Discov. 2012;2(11):1036–1047. doi:10.1158/2159-8290.CD-11-0348
60. Wang D, Li C, Zhang Y, et al. Combined inhibition of PI3K and PARP is effective in the treatment of ovarian cancer cells with wild-type PIK3CA genes. Gynecol Oncol. 2016;142(3):548–556. doi:10.1016/j.ygyno.2016.07.092
61. Luoto KR, Kumareswaran R, Bristow RG. Tumor hypoxia as a driving force in genetic instability. Genome Integr. 2013;4(1):5. doi:10.1186/2041-9414-4-5
62. Bizzaro F, Fuso Nerini I, Taylor MA, et al. VEGF pathway inhibition potentiates PARP inhibitor efficacy in ovarian cancer independent of BRCA status. J Hematol Oncol. 2021;14(1):1–7. doi:10.1186/s13045-021-01196-x
63. Pfaäffle HN, Wang M, Gheorghiu L, et al. EGFR-activating mutations correlate with a fanconi anemia-like cellular phenotype that includes PARP inhibitor sensitivity. Cancer Res. 2013;73(20):6254–6263. doi:10.1158/0008-5472.CAN-13-0044
64. Mahdi H, Hafez N, Doroshow D, et al. Ceralasertib-mediated ATR inhibition combined with olaparib in advanced cancers harboring DNA damage response and repair alterations (Olaparib Combinations). JCO Precis Oncol. 2021;5(5):1432–1442. doi:10.1200/PO.20.00439
65. Krebs MG, Lopez J, El-Khoueiry A, et al. Abstract CT026: phase I study of AZD6738, an inhibitor of ataxia telangiectasia Rad3-related (ATR), in combination with olaparib or durvalumab in patients (pts) with advanced solid cancers. Cancer Res. 2018;78(13_Supplement):CT026–CT026. doi:10.1158/1538-7445.AM2018-CT026
66. Mittra A, Coyne GHO, Do KT, et al. Safety and tolerability of veliparib, an oral PARP inhibitor, and M6620 (VX-970), an ATR inhibitor, in combination with cisplatin in patients with refractory solid tumors. J Clin Oncol. 2019;37(15_suppl):3067.
67. Hafez N, Soliman HH, Fu S, et al. Preliminary efficacy data of triple-negative breast cancer cohort of NCI 9881 study: a phase II study of cediranib in combination with olaparib in advanced solid tumors. J Clin Oncol. 2020;38(15_suppl):1077.
68. Garcia-Campelo R, Arrieta O, Massuti B, et al. Combination of gefitinib and olaparib versus gefitinib alone in EGFR mutant non-small-cell lung cancer (NSCLC): a multicenter, randomized phase II study (GOAL). Lung Cancer. 2020;150:62–69. doi:10.1016/j.lungcan.2020.09.018
69. Karachaliou N, Arrieta O, Giménez-Capitán A, et al. BRCA1 Expression and Outcome in Patients With EGFR-Mutant NSCLC Treated With Gefitinib Alone or in Combination With Olaparib. JTO Clin Res Rep. 2021;2(3):100113. doi:10.1016/j.jtocrr.2020.100113
70. Horn L, Mansfield AS, Szczęsna A, et al. First-line atezolizumab plus chemotherapy in extensive-stage small-cell lung cancer. N Engl J Med. 2018;379(23):2220–2229. doi:10.1056/NEJMoa1809064
71. Paz-Ares L, Dvorkin M, Chen Y, et al. Durvalumab plus platinum–etoposide versus platinum–etoposide in first-line treatment of extensive-stage small-cell lung cancer (CASPIAN): a randomised, controlled, open-label, phase 3 trial. Lancet. 2019;394(10212):1929–1939. doi:10.1016/S0140-6736(19)32222-6
72. George J, Lim JS, Jang SJ, et al. Comprehensive genomic profiles of small cell lung cancer. Nature. 2015;524(7563):47–53. doi:10.1038/nature14664
73. Rudin CM, Durinck S, Stawiski EW, et al. Comprehensive genomic analysis identifies SOX2 as a frequently amplified gene in small-cell lung cancer. Nat Genet. 2012;44(10):1111–1116. doi:10.1038/ng.2405
74. Tlemsani C, Takahashi N, Pongor L, et al. Whole-exome sequencing reveals germline-mutated small cell lung cancer subtype with favorable response to DNA repair-targeted therapies. Sci Transl Med. 2021;13(578). doi:10.1126/scitranslmed.abc7488
75. Sen T, Tong P, Wang J, Byers LA. Abstract LB-132: proteomic profiling identifies ATM expression level as a predictive biomarker to ATR and PARP inhibition in small cell lung cancer (SCLC). Cancer Res. 2016;76(14_Supplement):LB–132. doi:10.1158/1538-7445.AM2016-LB-132
76. Byers LA, Wang J, Nilsson MB, et al. Proteomic profiling identifies dysregulated pathways in small cell lung cancer and novel therapeutic targets including PARP1. Cancer Discov. 2012;2(9):798–811. doi:10.1158/2159-8290.CD-12-0112
77. Woll P, Gaunt P, Steele N, et al. P1.07-015 STOMP: a UK national cancer research network randomised, double blind, multicentre Phase II trial of olaparib as maintenance therapy in SCLC. J Thoracic Oncol. 2017;12(1):S704–S705. doi:10.1016/j.jtho.2016.11.926
78. Owonikoko TK, Dahlberg SE, Sica GL, et al. Randomized phase II trial of cisplatin and etoposide in combination with veliparib or placebo for extensive-stage small-cell lung cancer: ECOG-ACRIN 2511 study. J Clin Oncol. 2019;37(3):222–229. doi:10.1200/JCO.18.00264
79. Byers LA, Bentsion D, Gans S, et al. Veliparib in combination with carboplatin and etoposide in patients with treatment-Naïve extensive-stage small cell lung cancer: a phase 2 randomized study. Clin Cancer Res. 2021;27(14):3884–3895. doi:10.1158/1078-0432.CCR-20-4259
80. Pietanza MC, Kadota K, Huberman K, et al. Phase II trial of temozolomide in patients with relapsed sensitive or refractory small cell lung cancer, with assessment of methylguanine-DNA methyltransferase as a potential biomarker. Clin Cancer Res. 2012;18(4):1138–1145. doi:10.1158/1078-0432.CCR-11-2059
81. Pietanza MC, Waqar SN, Krug LM, et al. Randomized, double-blind, phase II study of temozolomide in combination with either veliparib or placebo in patients with relapsed-sensitive or refractory small-cell lung cancer. J Clin Oncol. 2018;36(23):2386–2394. doi:10.1200/JCO.2018.77.7672
82. Lazzari C, Gregorc V, Bulotta A, Dottore A, Altavilla G, Santarpia M. Temozolomide in combination with either veliparib or placebo in patients with relapsed-sensitive or refractory small-cell lung cancer. Transl Lung Cancer Res. 2018;7(S4):S329–S333. doi:10.21037/tlcr.2018.12.02
83. Ndrew A, Urrisi TT, Onald R, et al. Twice-daily compared with once-daily thoracic radiotherapy in limited small-cell lung cancer treated concurrently with cisplatin and etoposide. J Med. 1999;340(4):265–271.
84. Pignon JP, Arriagada R, Ihde DC, et al. A meta-analysis of thoracic radiotherapy for small-cell lung cancer. N Engl J Med. 2010;327(23):1618–1624.
85. Warde P, Payne D, Ahn SH. Does thoracic irradiation improve survival and local control in limited-stage small-cell carcinoma of the lung? A meta-analysis. Gut and Liver. 2016;10(6):890–895. doi:10.5009/gnl15573
86. Tariq S, Kim SY, Monteiro de Oliveira Novaes J, Cheng H. Update 2021: management of Small Cell Lung Cancer. Lung. 2021;199(6):579–587. doi:10.1007/s00408-021-00486-y
87. Rathod S, Jeremic B, Dubey A, et al. Role of thoracic consolidation radiation in extensive stage small cell lung cancer: a systematic review and meta-analysis of randomised controlled trials. Eur J Cancer. 2019;110:110–119. doi:10.1016/j.ejca.2019.01.003
88. Powell C, Mikropoulos C, Kaye SB, et al. Pre-clinical and clinical evaluation of PARP inhibitors as tumour-specific radiosensitisers. Cancer Treat Rev. 2010;36(7):566–575. doi:10.1016/j.ctrv.2010.03.003
89. Cardnell RJ, Feng Y, Diao L, et al. Proteomic markers of DNA repair and PI3K pathway activation predict response to the PARP inhibitor BMN 673 in small cell lung cancer. Clin Cancer Res. 2013;19(22):6322–6328. doi:10.1158/1078-0432.CCR-13-1975
90. Owonikoko TK, Zhang G, Deng X, et al. Poly (ADP) ribose polymerase enzyme inhibitor, veliparib, potentiates chemotherapy and radiation in vitro and in vivo in small cell lung cancer. Cancer Med. 2014;3(6):1579–1594. doi:10.1002/cam4.317
91. Laird JH, Lok BH, Ma J, et al. Talazoparib is a potent radiosensitizer in small cell lung cancer cell lines and xenografts. Clin Cancer Res. 2018;24(20):5143–5152. doi:10.1158/1078-0432.CCR-18-0401
92. Esposito G, Palumbo G, Carillio G, et al. Immunotherapy in Small Cell Lung Cancer. Cancers. 2020;12(9):2522. doi:10.3390/cancers12092522
93. MacKenzie KJ, Carroll P, Martin CA, et al. cGAS surveillance of micronuclei links genome instability to innate immunity. Nature. 2017;548(7668):461–465. doi:10.1038/nature23449
94. Harding SM, Benci JL, Irianto J, Discher DE, Minn AJ, Greenberg RA. Mitotic progression following DNA damage enables pattern recognition within micronuclei. Nature. 2017;548(7668):466–470. doi:10.1038/nature23470
95. Wang Y, Luo J, Alu A, Han X, Wei Y, Wei X. CGAS-STING pathway in cancer biotherapy. Mol Cancer. 2020;19(1):1–16. doi:10.1186/s12943-020-01247-w
96. Ablasser A, Goldeck M, Cavlar T, et al. cGAS produces a 2′-5′-linked cyclic dinucleotide second messenger that activates STING. Nature. 2013;498(7454):380–384. doi:10.1038/nature12306
97. Tanaka Y, Chen ZJ. STING specifies IRF3 phosphorylation by TBK1 in the cytosolic DNA signaling pathway. Sci Signal. 2012;5(214). doi:10.1126/scisignal.2002521
98. Shen J, Zhao W, Ju Z, et al. PARPI triggers the STING-dependent immune response and enhances the therapeutic efficacy of immune checkpoint blockade independent of BRCANEss. Cancer Res. 2019;79(2):311–319. doi:10.1158/0008-5472.CAN-18-1003
99. Jiao S, Xia W, Yamaguchi H, et al. PARP inhibitor upregulates PD-L1 expression and enhances cancer-associated immunosuppression. Clin Cancer Res. 2017;23(14):3711–3720. doi:10.1158/1078-0432.CCR-16-3215
100. Robillard L, Nguyen M, Loehr A, et al. Abstract 3650: preclinical evaluation of the PARP inhibitor rucaparib in combination with PD-1 and PD-L1 inhibition in a syngeneic BRCA1 mutant ovarian cancer model. Cancer Res. 2017;77(13_Supplement):3650. doi:10.1158/1538-7445.AM2017-3650
101. Sen T, Rodriguez BL, Chen L, et al. Targeting DNA damage response promotes antitumor immunity through STING-mediated T-cell activation in small cell lung cancer. Cancer Discov. 2019;9(5):646–661. doi:10.1158/2159-8290.CD-18-1020
102. Thomas A, Vilimas R, Trindade C, et al. Durvalumab in combination with olaparib in patients with relapsed SCLC: results from a Phase II study. J Thoracic Oncol. 2019;14(8):1447–1457. doi:10.1016/j.jtho.2019.04.026
103. Krebs M, Ross K, Kim S, et al. P1.15-004 an open-label, Multitumor Phase II Basket Study of Olaparib and Durvalumab (MEDIOLA): results in Patients with Relapsed SCLC. J Thoracic Oncol. 2017;12(11):S2044–S2045. doi:10.1016/j.jtho.2017.09.1040
104. Friedlander M, Meniawy T, Markman B, et al. Pamiparib in combination with tislelizumab in patients with advanced solid tumours: results from the dose-escalation stage of a multicentre, open-label, phase 1a/b trial. Lancet Oncol. 2019;20(9):1306–1315. doi:10.1016/S1470-2045(19)30396-1
105. Doerr F, George J, Schmitt A, et al. Targeting a non-oncogene addiction to the ATR/CHK1 axis for the treatment of small cell lung cancer. Sci Rep. 2017;7(1):1–16. doi:10.1038/s41598-017-15840-5
106. Sen T, Tong P, Stewart CA, et al. CHK1 inhibition in small-cell lung cancer produces single-agent activity in biomarker-defined disease subsets and combination activity with cisplatin or olaparib. Cancer Res. 2017;77(14):3870–3884. doi:10.1158/0008-5472.CAN-16-3409
107. Hamilton E, Falchook GS, Wang JS, et al. Abstract CT025: phase Ib study of adavosertib in combination with olaparib in patients with refractory solid tumors: dose escalation. Cancer Res. 2019;79(13_Supplement):CT025–CT025. doi:10.1158/1538-7445.AM2019-CT025
108. Popat S, Baas P, Faivre-Finn C, et al. Malignant pleural mesothelioma: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up☆. Ann Oncol. 2022;33(2):129–142. doi:10.1016/j.annonc.2021.11.005
109. Baas P, Scherpereel A, Nowak AK, et al. First-line nivolumab plus ipilimumab in unresectable malignant pleural mesothelioma (CheckMate 743): a multicentre, randomised, open-label, phase 3 trial. Lancet. 2021;397(10272):375–386. doi:10.1016/S0140-6736(20)32714-8
110. Betti M, Casalone E, Ferrante D, et al. Germline mutations in DNA repair genes predispose asbestos-exposed patients to malignant pleural mesothelioma. Cancer Lett. 2017;405:38–45. doi:10.1016/j.canlet.2017.06.028
111. Panou V, Gadiraju M, Wolin A, et al. Frequency of germline mutations in cancer susceptibility genes in malignant mesothelioma. J Clin Oncol. 2018;36(28):2863–2871. doi:10.1200/JCO.2018.78.5204
112. Bueno R, Stawiski EW, Goldstein LD, et al. Comprehensive genomic analysis of malignant pleural mesothelioma identifies recurrent mutations, gene fusions and splicing alterations. Nat Genet. 2016;48(4):407–416. doi:10.1038/ng.3520
113. Guo G, Chmielecki J, Goparaju C, et al. Whole-exome sequencing reveals frequent genetic alterations in BAP1, NF2, CDKN2A, and CUL1 in malignant pleural mesothelioma. Cancer Res. 2015;75(2):264–269. doi:10.1158/0008-5472.CAN-14-1008
114. Cantini L, Pecci F, Murrone A, et al. Questioning the prognostic role of BAP-1 immunohistochemistry in malignant pleural mesothelioma: a single center experience with systematic review and meta-analysis. Lung Cancer. 2020;146:318–326. doi:10.1016/j.lungcan.2020.06.024
115. Bott M, Brevet M, Taylor BS, et al. The nuclear deubiquitinase BAP1 is commonly inactivated by somatic mutations and 3p21.1 losses in malignant pleural mesothelioma. Nat Genet. 2011;43(7):668–672. doi:10.1038/ng.855
116. Srinivasan G, Sidhu GS, Williamson EA, et al. Synthetic lethality in malignant pleural mesothelioma with PARP1 inhibition. Cancer Chemother Pharmacol. 2017;80(4):861–867.
117. Borchert S, Wessolly M, Schmeller J, et al. Gene expression profiling of homologous recombination repair pathway indicates susceptibility for olaparib treatment in malignant pleural mesothelioma in vitro. BMC Cancer. 2019;19(1):1–12. doi:10.1186/s12885-019-5314-0
118. Gabano E, Pinton G, Balzano C, et al. Unsymmetric cisplatin-based Pt(IV) conjugates containing a PARP-1 inhibitor pharmacophore tested on malignant pleural mesothelioma cell lines. Molecules. 2021;26(16):4740.
119. Hmeljak J, Sanchez-Vega F, Hoadley KA, et al. Integrative molecular characterization of malignant pleural mesothelioma. Cancer Discov. 2018;8(12):1549–1565. doi:10.1158/2159-8290.CD-18-0804
120. Ghafoor A, Mian I, Wagner C, et al. Phase 2 study of olaparib in malignant mesothelioma and correlation of efficacy with germline or somatic mutations in BAP1 gene. JTO Clin Res Rep. 2021;2(10):100231. doi:10.1016/j.jtocrr.2021.100231
121. Passiglia F, Bironzo P, Righi L, et al. A prospective Phase II single-arm study of niraparib plus dostarlimab in patients with advanced non–small-cell lung cancer and/or malignant pleural mesothelioma, positive for PD-L1 expression and germline or somatic mutations in the DNA repair genes: rationale and study design. Clin Lung Cancer. 2021;22(1):e63–e66. doi:10.1016/j.cllc.2020.07.014
122. Fennell DA, King A, Mohammed S, et al. Rucaparib in patients with BAP1-deficient or BRCA1-deficient mesothelioma (MiST1): an open-label, single-arm, phase 2a clinical trial. Lancet Respir Med. 2021;9(6):593–600. doi:10.1016/S2213-2600(20)30390-8
123. Pastorino S, Yoshikawa Y, Pass HI, et al. A subset of mesotheliomas with improved survival occurring in carriers of BAP1 and other germline mutations. J Clin Oncol. 2018;36(35):3485–3494.
124. Slade D. PARP and PARG inhibitors in cancer treatment. Genes Dev. 2020;34(5–6):360–394. doi:10.1101/gad.334516.119
125. Gravells P, Grant E, Smith KM, James DI, Bryant HE. Specific killing of DNA damage-response deficient cells with inhibitors of poly(ADP-ribose) glycohydrolase. DNA Repair. 2017;52:81–91. doi:10.1016/j.dnarep.2017.02.010
126. Pillay N, Tighe A, Nelson L, et al. DNA replication vulnerabilities render ovarian cancer cells sensitive to Poly(ADP-Ribose) glycohydrolase inhibitors. Cancer Cell. 2019;35(3):519–533.e8. doi:10.1016/j.ccell.2019.02.004
127. Chen SH, Yu X. Targeting dePARylation selectively suppresses DNA repair-defective and PARP inhibitor-resistant malignancies. Sci Adv. 2019;5(4):eaav4340. doi:10.1126/sciadv.aav4340
128. Leenus M, Tzuling C, Dominic IJ, et al. Abstract 1943: PARG inhibitors exhibit synthetic lethality with XRCC1 deficiency and a cellular mechanism of action that is distinct from PARP inhibition. Cancer Res. 2018;78(13_Supplement):1943. doi:10.1158/1538-7445.AM2018-1943
129. Sasaki Y, Fujimori H, Hozumi M, et al. Dysfunction of Poly (ADP-Ribose) glycohydrolase induces a synthetic lethal effect in dual specificity phosphatase 22-deficient lung cancer cells. Cancer Res. 2019;79(15):3851–3861. doi:10.1158/0008-5472.CAN-18-1037
130. Noll A, Illuzzi G, Amé JC, Dantzer F, Schreiber V. PARG deficiency is neither synthetic lethal with BRCA1 nor PTEN deficiency. Cancer Cell Int. 2016;16(1):53. doi:10.1186/s12935-016-0333-2
131. Karpova Y, Johnson SJ, Bordet G, et al. Upregulation of PARG in prostate cancer cells suppresses their malignant behavior and downregulates tumor-promoting genes. Biomed Pharmacother. 2022;153:113504. doi:10.1016/j.biopha.2022.113504
132. Jain A, Agostini LC, McCarthy GA, et al. Poly (ADP) Ribose Glycohydrolase Can Be Effectively Targeted in Pancreatic Cancer. Cancer Res. 2019;79(17):4491–4502. doi:10.1158/0008-5472.CAN-18-3645
133. Nakadate Y, Kodera Y, Kitamura Y, Tachibana T, Tamura T, Koizumi F. Silencing of poly(ADP-ribose) glycohydrolase sensitizes lung cancer cells to radiation through the abrogation of DNA damage checkpoint. Biochem Biophys Res Commun. 2013;441(4):793–798. doi:10.1016/j.bbrc.2013.10.134
134. Gravells P, Neale J, Grant E, et al. Radiosensitization with an inhibitor of poly(ADP-ribose) glycohydrolase: a comparison with the PARP1/2/3 inhibitor olaparib. DNA Repair. 2018;61:25–36. doi:10.1016/j.dnarep.2017.11.004
135. Pham MM, Ngoi NYL, Peng G, Tan DSP, Yap TA. Development of poly(ADP-ribose) polymerase inhibitor and immunotherapy combinations: progress, pitfalls, and promises. Trends Cancer. 2021;7(10):958–970. doi:10.1016/j.trecan.2021.05.004
136. Fennell DA, Porter C, Lester J, et al. Olaparib maintenance versus placebo monotherapy in patients with advanced non-small cell lung cancer (PIN): a multicentre, randomised, controlled, phase 2 trial. EClinicalMedicine. 2022;52:101595. doi:10.1016/j.eclinm.2022.101595
137. Riess JW, Redman MW, Wheatley-Price P, et al. A phase II study of rucaparib in patients with high genomic LOH and/or BRCA 1/2 mutated stage IV non-small cell lung cancer (Lung-MAP Sub-Study, S1900A). J Clin Oncol. 2021;39(15_suppl):9024. doi:10.1200/JCO.2021.39.15_suppl.9024
138. Farkkila A, Gulhan D, Casado J, et al. Immunogenomic profiling determines responses to combined PARP and PD-1 inhibition in ovarian cancer. Nat Commun. 2020;11(1):1459. doi:10.1038/s41467-020-15315-8
139. Karzai F, VanderWeele D, Madan R, et al. Activity of durvalumab plus olaparib in metastatic castration-resistant prostate cancer in men with and without DNA damage repair mutations. J Immunother Cancer. 2018;6(1):141. doi:10.1186/s40425-018-0463-2
140. Farokhzad OC, Langer R. Impact of nanotechnology on drug delivery. ACS Nano. 2009;3(1):16–20. doi:10.1021/nn900002m
141. Alotaibi BS, Buabeid M, Ibrahim NA, et al. Potential of nanocarrier-based drug delivery systems for brain targeting: a current review of literature. Int J Nanomed. 2021;16:7517–7533. doi:10.2147/IJN.S333657
142. Sargazi S, Mukhtar M, Rahdar A, Barani M, Pandey S, Díez-Pascual AM. Active targeted nanoparticles for delivery of Poly(ADP-ribose) polymerase (PARP) inhibitors: a preliminary review. Int J Mol Sci. 2021;22(19):10319. doi:10.3390/ijms221910319
 © 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.