Back to Archived Journals » Advances and Applications in Bioinformatics and Chemistry » Volume 13
Current Challenges and Opportunities in Designing Protein–Protein Interaction Targeted Drugs
Authors Shin WH ![]() , Kumazawa K
, Kumazawa K ![]() , Imai K, Hirokawa T, Kihara D
, Imai K, Hirokawa T, Kihara D ![]()
Received 1 September 2020
Accepted for publication 22 October 2020
Published 12 November 2020 Volume 2020:13 Pages 11—25
DOI https://doi.org/10.2147/AABC.S235542
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Juan Fernandez-Recio
Woong-Hee Shin,1 Keiko Kumazawa,2 Kenichiro Imai,3 Takatsugu Hirokawa,3 Daisuke Kihara4– 7
1Department of Chemical Science Education, Sunchon National University, Suncheon 57922, Republic of Korea; 2Pharmaceutical Discovery Research Laboratories, Teijin Pharma Limited, Tokyo 191-8512, Japan; 3Cellular and Molecular Biotechnology Research Institute, National Institute of Advanced Industrial Science and Technology (AIST), Tokyo 135-0064, Japan; 4Department of Biological Sciences, Purdue University, West Lafayette, IN 47906, USA; 5Department of Computer Science, Purdue University, West Lafayette, IN 47906, USA; 6Center for Cancer Research, Purdue University, West Lafayette, IN 47906, USA; 7Department of Pediatrics, Cincinnati Children’s Hospital Medical Care, University of Cincinnati, Cincinnati, OH 45229, USA
Correspondence: Daisuke Kihara Email [email protected]
Abstract: It has been noticed that the efficiency of drug development has been decreasing in the past few decades. To overcome the situation, protein–protein interactions (PPIs) have been identified as new drug targets as early as 2000. PPIs are more abundant in human cells than single proteins and play numerous important roles in cellular processes including diseases. However, PPIs have very different physicochemical features from the conventional drug targets, which make targeting PPIs challenging. Therefore, as of now, only a small number of PPI inhibitors have been approved or progressed to a stage of clinical trial. In this article, we first overview previous works that analyzed differences between PPIs with PPI targeting ligands and conventional drugs with their binding pockets. Then, we constructed an up-to-date list of PPI targeting drugs that have been approved or are currently under clinical trial and have bound drug–target structures available. Using the dataset, we analyzed the PPIs and their ligands using several scores of druggability. Druggability scores showed that PPI sites and their drugs targeting PPIs are less druggable than conventional binding pockets and drugs, which also indicates that PPI drugs do not follow the conventional rules for drug design, such as Lipinski’s rule of five. Our analyses suggest that developing a new rule would be beneficial for guiding PPI-drug discovery.
Keywords: protein–protein interaction, PPI, PPI drugs, drug discovery
Introduction
A core concept in modern drug discovery has been “one drug, one target”, which indicates that a drug will be designed to bind its specific target. Following this concept, a usual drug discovery process is initiated by identifying a proper target protein that has a well-defined binding pocket, so that a compound can be developed that fits the binding pocket to modulate the function of the protein.
Although the conventional paradigm has led to many successful drug development projects,1 the efficiency of the process has been decreasing drastically in the last 60 years.2 One of the main reasons for this crisis is that the target space for the current paradigm is almost saturated. Therefore, from early 2000, researchers in drug discovery have been eagerly seeking new types of therapeutic targets, where protein–protein interactions (PPIs) have been identified as one of such new paradigms in the quest.3–7 PPIs play vital roles in various cellular processes, including many important diseases such as various cancers7 and Alzheimer’s disease.8
As a PPI is formed by two or more proteins, the total number of PPIs (interactome) is much larger than the individual human protein target space, which is estimated to be around 19,000.9 The Center for Cancer Systems Biology (CCSB) Interactome Database contains 13,993 human PPIs,10 and 3,787 viral protein-human protein interactions,11 which were determined by experiments. The Human Reference Interactome (HuRI),12 which contains 64,006 binary interactions. The STRING database13 contains 505,116 high-confidence experimentally determined or predicted human PPIs. Venkatesan et al14 estimated the size of human interactome as ~130,000 by comparing PPIs determined by them using yeast-two-hybrid method with known human PPIs. Stompf et al15 estimated the interactome size as 650,000 using a statistical approach that estimates the size of the entire network from sub-network data.
As experimental methods are known to have false positives, it is important to detect high-confidence interactions. Tyagi et al16 proposed to use homology against PPIs with experimentally solved structures to select high-confidence data. Karagoz et al17 developed a score for selecting high-confidence PPIs by integrating multiple different types of information, such as the number of experimental methods performed, the number of publications, the number of data sources, and domain compositions of proteins in PPIs.
From 2004 to 2014, there were more than 40 PPIs that have been targeted, and several of them have further proceeded to clinical trials.18 Moreover, more than 800 drug development projects related to PPIs were published in PubChem in 2012.19 The p53/Mdm2 complex is a typical example of a PPI target. P53 is a tumor suppressor protein that is strongly downregulated by MDM2 in cancerous cells. Therefore, re-activating p53 is considered as a promising anti-cancer drug design strategy.20 Since Mdm2 interacts with p53 to inhibit the protein, protein–protein interaction drugs (PPIDs) have been developed to modulate the p53/Mdm2 interaction.21 In ChEMBL,22 there are 1,854 molecules that inhibit p53/Mdm2 interaction. Among them, 304 molecules have affinities less than 1 nM (IC50); and interestingly, 303 of them violate the Lipinski’s rule of 5 (RO5),23 which implies that PPIDs tend to have different physicochemical features from typical drug targets reflecting the unique nature of PPIs. In recent years, drug repurposing (DR) (also known as drug repositioning) has become a popular strategy. DR will be useful for targeting PPIs, too. There are computational methods developed for PPI targeted drugs. These methods enable us to extract similar PPI surfaces and to understand physical/functional interactions of proteins.24–26
In this article, we will discuss the difference of physicochemical features of PPIDs and conventional drug molecules. First, we review existing articles that discuss different natures of PPIs and PPIDs from conventional small chemical compound drugs designed for binding pockets. Then, to understand the current status of the PPI drug developments, we discuss the current list of PPI-PPID pairs that have experimentally solved tertiary structures, which we collected from databases and literature. Next, we investigate the differences and the similarities of the PPIDs and typical small chemical compound drugs and their binding interfaces by using SiteScore,27 Quantitative Estimate of Druglikeness (QED),28 and FTMap.29 From the SiteScore and FTMap results, we observed that PPI sites have lower scores and less binding compound fragments than typical binding pockets. Our analysis also showed that PPIDs have less druggability (QED) score than non-PPI, regular drugs. The analyses clearly showed that the PPI sites and their drugs are less druggable from the viewpoint of the conventional drug discovery. Finally, we discuss the challenges and potential future directions for developing PPIDs.
Comparison Between PPI Interfaces and Binding Pockets, PPIDs and Conventional Drugs
In this section, we review existing works that compare PPI interfaces relative to binding pockets as well as PPIDs against conventional small chemical compound drugs.
Earlier works focused on the geometrical properties of PPIs with an interest in the structural biology of PPIs before the era of PPIDs. Janin and co-workers30,31 studied structures of protein–protein complexes, which were collected from the Protein Data Bank (PDB).32 The interface area of PPIs they analyzed ranged from 1,000 Å2 to 4,000 Å2 with an average of 1,600 Å2, which were larger than typical binding pockets (300 Å2 to 1,000 Å2). They also found that PPIs usually have a planar shape, whereas drug-binding pockets have a concave shape so that drug molecules can bind. From the chemistry point of view, typical PPIs have a hydrophobic region, which is often called the PPI core.33,34 Amino acids that make up the core regions are more conserved than the rest of the protein–protein complex surfaces, which govern hydrophobic interaction between the proteins.35 The core region is surrounded by a rim region, which has a similar amino acid composition to the rest of a protein surface, whereas the core region mostly consists of aromatic residues.36
From late 2000, works have started to appear that analyze PPIs and pockets in the context of PPIDs. Fuller et al used Q-SiteFinder to analyze the structure of PPI sites bound with PPIDs in comparison with conventional drug-binding pockets.37 Q-SiteFinder is a software for predicting ligand-binding pockets by examining interactions between the methyl probe and the query protein surface. The program predicts binding pockets on protein surface by ranking pockets according to the van der Waals energy between the methyl probe and the input protein structure, based on the assumption that a putative ligand could bind the site which have favorable van der Waals energy. For comparison, the authors used four datasets: 134 protein-ligand complexes from their previous work,38 50 bound protein-marketed-drug complexes from DrugBank,39 97 protein–protein complexes,40 and 24 crystal structures of PPI sites with bound PPIDs collected by Wells and McClendon.41 The most distinguished feature of PPIs was that the average volume of top-ranked pockets of PPIs (261 Å3) was only about half of typical binding pockets (524 Å3). The authors also found that PPIDs tend to occupy six small pockets of an average size of 55 Å3, while typical drugs bind to the top- or second-ranked pockets with a larger volume (~260 Å3).37
Since the PPI binding sites have different physicochemical properties relative to conventional drug pockets, PPIDs also have contrasting features from the typical small compound drugs. For the typical ligands, a well-known rule called Lipinski’s RO5, describes drug-likeness of a small compound.23 It states that a compound is suitable for a drug if it has the molecular weight <500, logP <5, the number of hydrogen bond donors <10, and the number of hydrogen bond acceptors <5. Morelli et al analyzed ligands in the 2P2I database, a dataset of structures of PPI sites with bound PPIDs.42 2P2I contains structures of protein–protein complexes and complexes of PPI sites and bound PPIDs that are stored in PDB.43 They found that properties of small-molecule PPIDs often substantially deviate from RO5. Instead of RO5, they proposed a new rule, the rule-of-four (RO4) for PPIDs: the molecular weight >400, logP >4, the number of rings >4, and the number of hydrogen bond acceptors >4. RO4 indicates that PPIDs are relatively heavier and more hydrophobic than traditional drug compounds. This is consistent with Sperandio et al, who analyzed the small compounds in the i-PPI database,44 which is a manually curated database of non-peptide PPI modulators. The database contains the target PPI information, the molecular structure of compounds, physicochemical features, and the pharmacological data of PPIDs, which were extracted from literature.45 The authors compared 66 experimentally validated PPIDs from the i-PPI database and 557 compounds from DrugBank. The average molecular weight, logP values, and topological polar surface area of PPIDs in the i-PPI database were, 421, 3.58, and 89 Å2, respectively, which were higher than compounds bound to typical pockets (341, 2.61, and 71 Å2, respectively) all with statistical significance (p-value < 0.001).
Morelli et al analyzed PPIDs using two ligand efficiency metrics, the binding efficiency index (BEI), which is computed as the pKi divided by the molecular weight, and the surface efficiency index (SEI), the binding free energy per unit of polar surface area.42 BEI and SEI are similar to the original concept of the ligand efficiency (LE), which is computed by dividing binding free energy of a compound with its number of heavy atoms.46 The affinity of a compound generally increases with the number of heavy atoms and thus, it correlates with the molecular weight. However, for marketed drugs, reducing the molecular weight is important for lowering attrition rates. Thus, LE was introduced for selecting a good lead compound in high-throughput screening for developing commercial drug compounds. BEI and SEI, instead, introduced the molecular weight and the polar surface area as denominators. To balance the molecular size and potency, both values should be optimized during the drug development process. The ideal values for BEI and SEI for idealized drug compounds are 27 and 18, respectively.47 Morelli et al compared 92 marketed drugs and 39 PPIDs in the 2P2I database and showed that conventional drugs have mean BEI and SEI values of 25.8 and 14.5, respectively, while PPIDs have smaller values, 11.7 for BEI and 7.2 for SEI. The smaller value means that a compound has a large molecular weight (BEI) and polar surface area (SEI), thus not good for the general drug discovery process. The authors map the compounds to two-dimensional space of BEI and SEI. Most of the PPIDs fell to the region called “sub-optimal series that could not get optimized”.47
Turnbull and his colleagues analyzed PPIs and PPIDs from the viewpoint of fragment-based drug discovery (FBDD).48 FBDD can explore a larger chemical space as a compound is assembled from fragments.49 FBDD would be very suitable for developing PPIDs because PPIs have more subpockets, to each of which a binding fragment can be designed.37 It is known that bound fragments generally follow a rule-of-three (RO3) for conventional binding pockets: the molecular weight <300, the number of hydrogen bond donors and acceptors ≤3, logP ≤3, the number of rotatable bonds ≤3, and polar surface area ≤60 Å2.50 Comparing 100 fragments that inhibit PPIs and 100 active fragments for non-PPI targets, the authors found that the properties of the two datasets generally follow RO3 but they have distinct value ranges. PPI fragments had a higher molecular weight on average, 278, than non-PPI targeting fragments (221), a higher hydrophobicity (2.48 versus 1.77 for typical fragments), and more flexible, ie rotatable bonds (4.01 in contrast to 2.50 for typical fragments). PPIDs are generally heavier and more hydrophobic than conventional drugs, which is consistent with the observation by Morelli et al.42
To summarize, PPIs have distinguished features from traditional binding pockets. PPIs have larger, flatter, and more hydrophobic surface respect to the traditional binding pockets, and PPIs consist of a larger number of subpockets. To interact with PPIs, PPIDs also have different physicochemical characteristics from conventional drugs. PPIDs and their fragments have larger molecular weight, higher logP (more hydrophobic), and a larger number of hydrogen bond donors/acceptors than the typical drugs. The findings in the earlier works discussed above are summarized in Table 1.
 |
Table 1 Summary of Earlier Works That Analyzed PPIs and PPI Drugs |
PPI Drugs That are Approved or in Clinical Trials
To understand the progress and the current status of PPI drug developments, we collected PPIDs that are approved by the US Food and Drug Administration (FDA) and those currently in clinical trials from two databases, 2P2I,43 TIMBAL,51 and two recent review articles52,53 (Table 2). In the table, we focused on modality (types of molecules used as drugs),54 clinical phase, the mode of action, and the target structure availability. Particularly, we confirmed that the drugs/drug candidates act on PPI interfaces but not at a distant site from the PPI interfaces. Table 2 is likely the most comprehensive list of PPIDs and drug candidates that have the tertiary structure information of their binding pose.
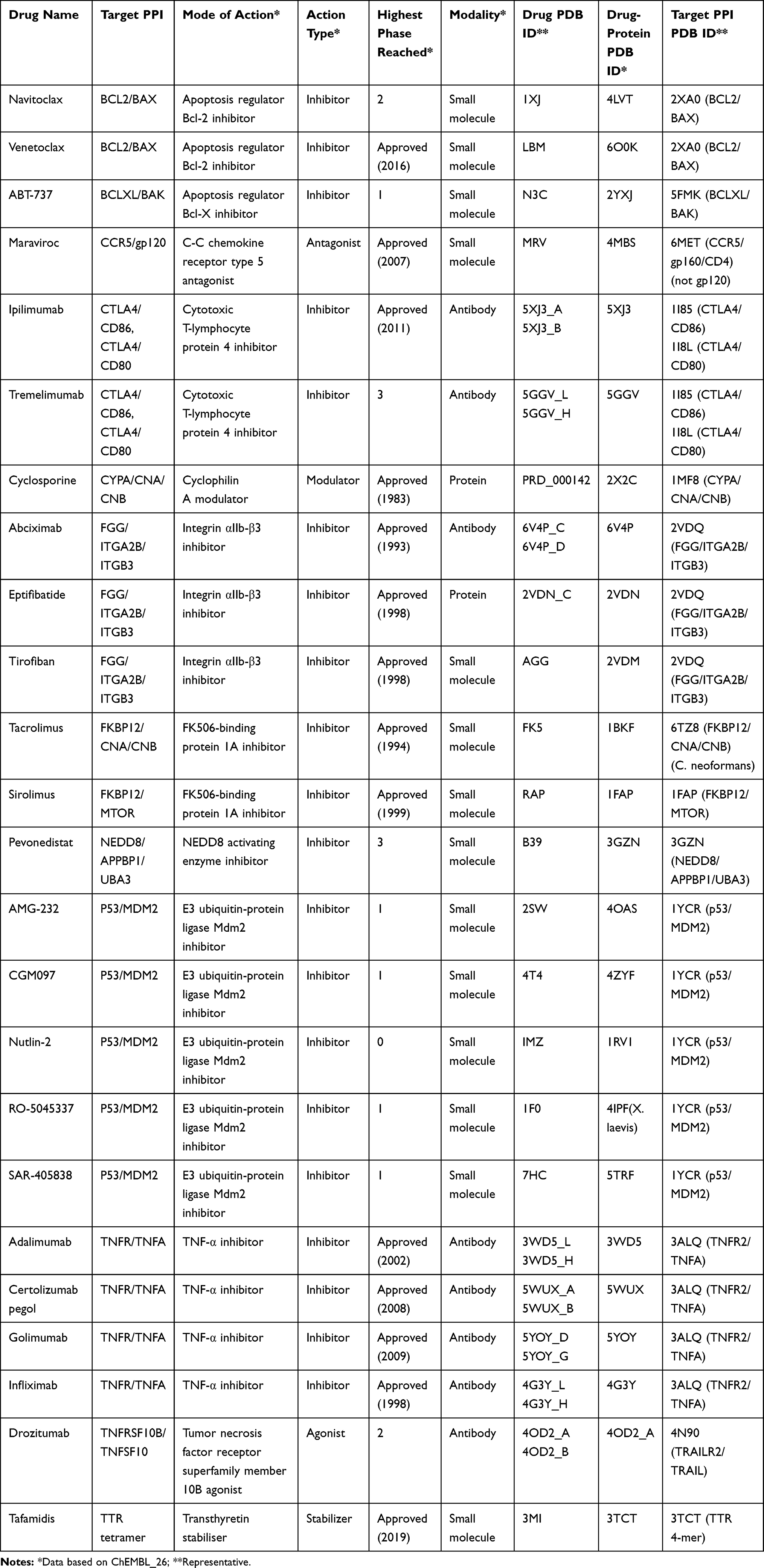 |
Table 2 PPI Drugs/Candidates with Structure Information in PDB |
In constructing Table 2, we selected known PPIDs and drug candidates as follows: (i) we first extracted UniProt ID or target names from 2P2I, TIMBAL, and the recent review articles.52,53 This process yielded 30 UniProt IDs from 2P2I, 105 UniProt IDs from TIMBAL, and 40 targets from the review articles including duplicates from different sources. (ii) Next, we searched ChEMBL2622 with the identified UniProt ID and target names, and then obtained additional information of drugs/drug candidates from ChEMBL26 such as modality, clinical phase, mode of action from “APPROVED DRUG/PESTICIDE DATA” and the structure availability of the target proteins and PPI complexes from “TARGET INFORMATION”. After removing duplicated drugs/drug candidates, we obtained 146 drugs/drug candidates for 57 PPI targets; 61 small-molecule drugs, 59 antibody drugs, 24 other protein drugs like peptide, one oligosaccharide drug, and one RNA aptamer. (iii) Finally, we checked whether the drugs/drug candidates act on PPI surfaces or not by visually checking available complex structures. From the list of drugs/drug candidates constructed in the previous step, we removed entries if they do not have available structures or if the complex structures show that they do not act on PPI surfaces. This remained the list in Table 2, which has 24 drugs/drug candidates for 14 PPI targets. The 24 compounds include 14 approved drugs for 10 PPI targets and ten drug candidates for seven PPI targets that are currently in clinical trials. Additionally, we also provide Supplementary Table S1, which lists all the 146 entries found in the step (ii).
We note that drug candidates under preclinical stages are not included the list even though those candidates often target typical PPIs in the field of drug discovery. For example, β-catenin/BCL9 complex is one of the such PPIs, which are not included in Table 2. It is currently considered as a key PPI in the Wnt/β-catenin signaling pathway, which is highly activated in several human cancers such as colorectal cancer, colon cancer, and breast cancer.55,56 For inhibiting the β-catenin/BCL9 PPI, a few stapled peptide inhibitors have been designed.57,58 Sang et al recently designed a series of novel α-helical sulfono-γ-AApeptide inhibitors mimicking binding mode of the α-helical HD2 domain of BCL9.59 Also, several small-molecule inhibitors have been designed based on structure-based approaches as well by mimicking the binding mode of side chains of hot spots of the α-helical HD2 domain.60–62
In Table 2, there are six small-molecule drugs (four inhibitors, one antagonist, and one stabilizer), six antibody drugs (six inhibitors), and two protein drugs (one inhibitor and one modulator), which were already approved by FDA. Drugs in clinical trials are eight small-molecule inhibitors and two antibody drugs (one inhibitor and one agonist).
The platelet integrin receptor, αIIb-β3 (ITGA2B), is an interesting target because it is modulated by three different modalities types of drugs and, moreover, the mechanisms of the actions are well studied with the tertiary structure information. The three drugs developed for αIIb-β3 are Abciximab (antibody), Eptifibatide (protein), and Tirofiban (small molecule). αIIb-β3 is activated and binds to its ligand fibrinogen. αIIb-β3 binds specifically to the C-terminal intrinsically disordered region of the γ subunit of fibrinogen (γC peptide), which contains the sequence Lys-Gln-Ala-Gly-Asp-Val. There are some other physiologic ligands that have a similar motif, Arg-Gly-Asp (RGD), and interact with αIIb-β3 at the binding site of fibrinogen. Taking advantage of this interaction mechanism, a small molecule, Tirofiban, and a cyclic peptide, Eptifibatide, were developed, which mimic the RGD sequence63,64 to prevent the ligand binding. In addition, recently, the structure of the αIIb-β3-Abciximab complex was determined at 2.8 Å resolution by cryo-electron microscopy (cryo-EM).65 The cryo-EM structure revealed a novel mode of action for inhibition, which shows that Abciximab binds primarily to the specificity-determining loop (SDL) of β3 but lacks the effect on the fibrinogen-binding pocket. The cryo-EM model, molecular-dynamics simulations, and mutagenesis suggested Abciximab compresses and reduces the flexibility of the SDL.65
While constructing Table 2, we noticed that the target PPIs are still very limited. Considering that the estimated number of PPIs in the human cell is very large, ranging from 130,000 to 650,000 PPIs,14,15 the vast majority of PPIs are still not explored. A primary reason for this is that structural information of most PPIs is still lacking. It is expected that such remarkable advances in cryo-EM will lead to a further increase of available structures of target PPI complexes. Indeed, submissions to the Electron Microscopy Data Bank (EMDB) have increased exponentially in recent years, and the resolution of the density maps is also improving with the highest resolution maps reaching sub 2 Å resolution.66
Characterization of PPI Drugs from the Perspective of Druggability
To understand the characteristic features of PPIDs, we examined the PPIDs in Table 2 in terms of the druggability. Typically, druggability indicates the possibility of obtaining a small-molecule drug for a target.67 Here, we used the QED score28 for evaluating the ligand druggability and SiteScore27,68 for considering the protein pocket druggability. QED considers the molecular weight, AlogP, and the number of hydrogen bond acceptors and donors. Those descriptors are individually scored and weighted by their relative significance of druglikeness based on the distribution of approved drugs. The score ranges from 0 to 1 with 1 being the most druggable. SiteScore gauges the propensity of a protein local site, which will contribute to a tight binding of ligands. It is computed based on properties such as the exposure of the site to solvent, the degree of the enclosure by the protein, and the degree of hydrophilicity/hydrophobicity. SiteScore is computed as a weighted sum of those properties. The SiteScore above 1 indicates that a protein pocket is highly druggable and 0.8 distinguishes between drug-binding and non-drug-binding sites. Note that these two scores were originally developed for typical binding pockets and drugs. Currently, there are no scoring methods that are designed for evaluating PPIs and PPIDs.
In Figure 1, we see that SiteScore of many small molecules’ receptors (yellow circles) are higher than antibodies (blue squares). The highest SiteScore observed among antibodies’ receptors was 0.91, indicating that the site is druggable, which was computed for Integrin αIIb-β3 inhibitor. Indeed, the receptor also has known protein and small-molecule ligands as we described in the previous section. If we look only at small molecules (yellow circles), a positive correlation between QED and SiteScore was observed (R2: 0.73). The correlation indicates that more drug-like ligands bind more druggable protein pockets, which is reasonable. However, there are exceptions, which need further explanation. There are three PPIDs with a high SiteScore and a low QED (labeled with a in Figure 1) and a drug with a low SiteScore and a high QED (labeled with b). The three PPI drugs labeled as a are those which target the Bcl family (Bcl-2/BAX or Bcl-xL/BAK). These are relatively large compounds (Figure 1A), which were designed to mimic the partner protein. The molecular weight of the three compounds is over 800, where the average of PPI small-molecule drugs/candidates in Table 2 is 663. The Bcl-2 antagonist Venetoclax (PDB ID: 6O0K) approved for cancer therapy, is one of the three drugs circled with the label a. Bcl-2 is a member of the pro-survival class of proteins that includes Bcl-xL, Bcl-W, A1/BFL-1, and Mcl-1. Their pro-survival function is caused by binding and restraining related members of a family of pro-apoptotic proteins such as the sensors of cellular stress, like BH3-only proteins, and the effectors of apoptosis, like BAX or BAK.69 As shown in Figure 2A–E, Venetoclax mimics a part of the alpha-helix of the partner protein. The bound form of Venetoclax with BCL2 is shown in Figure 2A, and the complex of BAX BH3 peptide with BCL2 is shown in Figure 2B. In Figure 2C and D, the structure of Venetoclax and the peptide structure is shown separately. In Figure 2E, the SiteMap result for the BCL2 binding site is shown. Pharmacophoric features were detected in the corresponding part to the side chain of amino acids in the helix. To form interactions at a distance about 3 turns of helix, the size of the compound needed to be large, and therefore, their QED turned low.
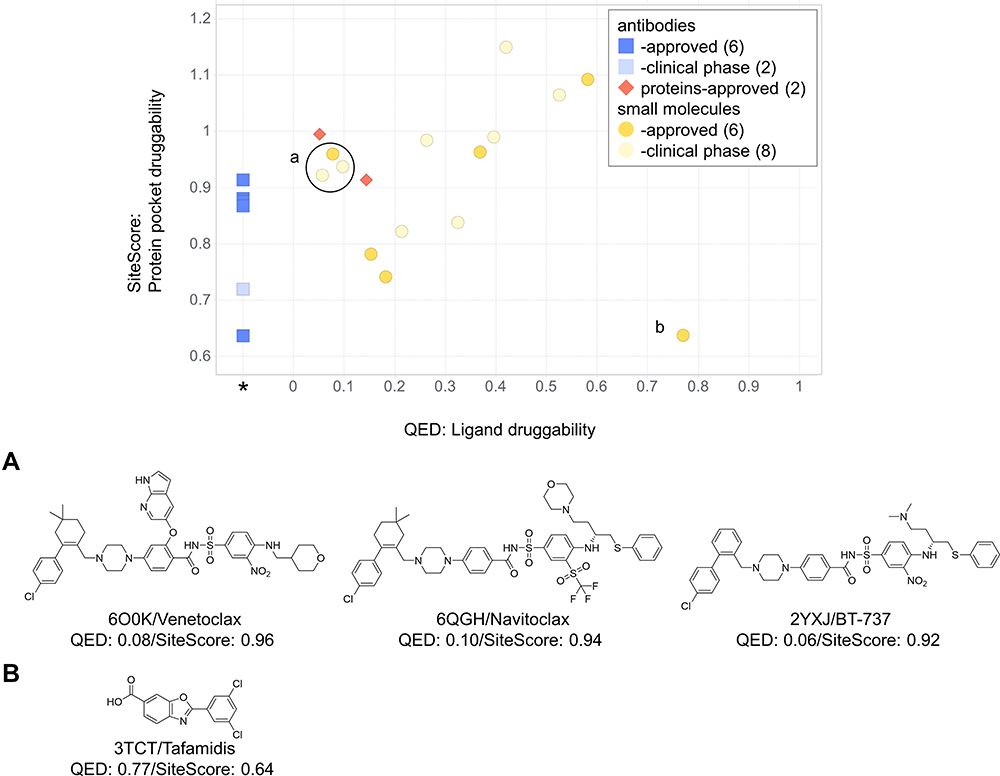 |
Figure 1 The ligand druggability relative to the protein pocket druggability for PPI drugs/candidates. QED and SiteScore were computed for PPI drugs and candidates in Table 2. Yellow circles, small-molecule drugs; blue squares, antibodies; red diamonds, proteins. Approved drugs are shown in a darker color. Compounds labeled as a and b are shown below in the panel. QED was not computed for antibodies and shown at an extra column marked with *SiteMap (ver. 5.0.011), the software we used for computing SiteScore, offers two modes. As default, we used the option of “Identify top-ranked potential receptor binding sites”, which automatically detects a pocket and computes SiteScore. Scores were properly computed with this option for small compound binding pockets but often failed for PPI surfaces because of their flat shape. In such cases, we used another option, “Evaluate a single binding site region”, which computes SiteScore for a user-specified region. For binding sites for antibodies, we combined it with an additional option of “detect shallow binding site” option. (A) The chemical structures of the three compounds specified by a circle in the graph. (B) The chemical structure of the compound specified at the right bottom in the graph. |
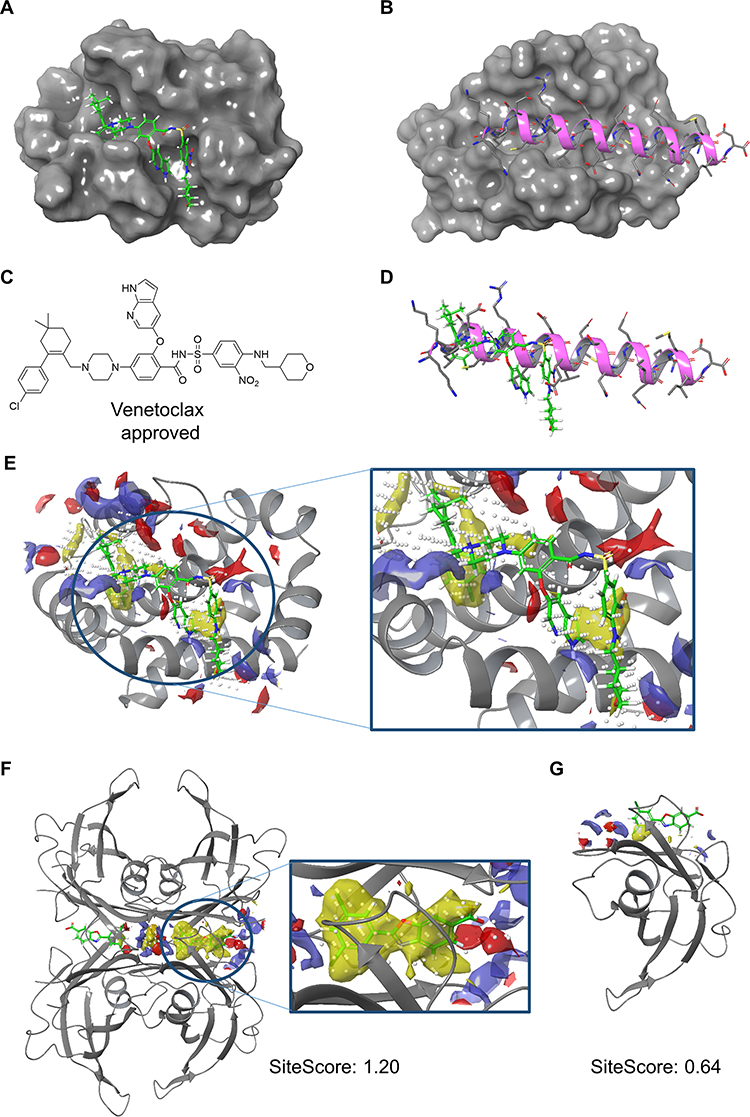 |
Figure 2 A few structures of PPI drugs in complex with target proteins. (A) The crystal structure of BCL2 with Venetoclax (PDB ID: 6O0K). (B) The crystal structure of BCL2 in complex with a BAX BH3 peptide (PDB ID: 2XA0). (C) The 2D structure of Venetoclax; (D) Superposition of Venetoclax and BAX BH3 peptide (magenta). (E) The SiteMap result calculated under the “Evaluate a single binding site region” condition with “detect shallow binding site” option for the BCL2/venetoclax complex (PDB ID: 6O0K). The white dots indicate the size of the pocket that is reflected in SiteScore. The color represents pharmacophoric features: yellow, hydrophobic; blue hydrogen bond donor; red, hydrogen bond acceptor. (F) The SiteMap result calculated with the “Identify top-ranked potential receptor binding sites” condition without the “detect shallow binding site” option for the biological assembly of TTR in complex with two Tafamidis units (green) (PDB ID: 3TCT). (G) The SiteMap result calculated with the “Evaluate a single binding site region” option with the “detect shallow binding site” option for the monomer of TTR and Tafamidis (green) (PDB ID: 3TCT). |
On the other hand, the drug labeled with b at the right bottom corner of Figure 1, Tafamidis (PDB ID: 3TCT) (Figure 1B), showed a low SiteScore with a high QED. It acts as a stabilizer and the target protein is a homo tetramer. Transthyretin, a natively tetrameric protein, is involved in the transport of thyroxine and the vitamin A–retinol-binding protein complex. Tetramer dissociation causes aggregation of transthyretin, which triggers transthyretin amyloidoses (ATTR). ATTR is a fatal disease characterized by progressive neuropathy and/or cardiomyopathy.70 Since the transthyretin dimer has a flat PPI surface, as shown in Figure 2G, no pocket was detected around the ligand when SiteScore was calculated with a transthyretin monomer. Interestingly, however, a druggable pocket was detected around the ligand at the interface when SiteScore was calculated with the transthyretin tetramer (Figure 2F). That is how the small molecule, with the molecular weight of 308, functioned as a drug against the target protein with a flat surface as a monomer. Because of the flat surface, the SiteScore was low. This example shows that understanding how the target protein works under in vivo condition is critical to develop PPIDs.
Next, we compared the PPI small-molecule drugs in Table 2 with a dataset of non-PPI ligands. We used the non-PPI ligand dataset constructed by Soga et al,71 which consists of 41 non-redundant protein-ligand complexes determined by X-ray crystallography at a high resolution. As shown in Figure 3A and B, it is very clear that PPI drugs/candidates have lower druggability scores, both QED and SiteScore, than non-PPI ligands. Since the two metrics were originally developed for assessing typical pockets and drugs, the results indicate that the PPIs and the PPIDs are different and not suitable as conventional drugs and binding sites. In Figure 3C, we compared the four descriptors used in the RO4, the molecular weight, AlogP, the number of rings, and the number of hydrogen bond acceptors for PPI drugs (yellow) and the non-PPI ligands (green). For all four descriptors, PPIDs tend to have larger values than non-PPI ligands, which is the main reason why the PPIDs showed lower QED scores in our analysis. Figure 3C also shows that most of the PPIDs follow the RO4, which is in principle the opposite from the RO5, the empirical rule of drug-likeness of compounds. Accumulating examples of active ligands that are outside the general parameters of the RO5 will be crucial to exploring new drug candidates, and expanding the concept of drugs as a whole.
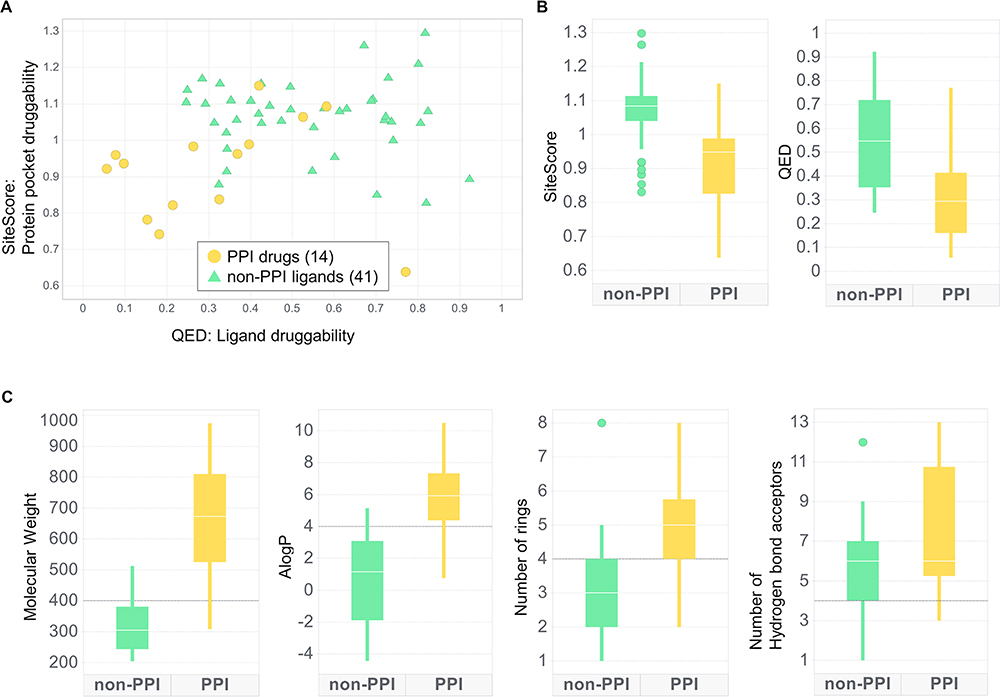 |
Figure 3 Distributions of druggability scores and small-molecule 2D descriptors of PPI drugs/candidates and non-PPI ligands. PPI drug/candidates are only limited to small molecules. Non-PPI ligands include natural ligands and are not limited to drug and drug candidates. (A) The ligand druggability relative to the protein pocket druggability. PPI drugs include drug candidates. (B) Box plots of druggability score distribution. A line through each box shows the median. (C) Distributions of small-molecule 2D descriptors used for QED and the RO4. Black lines on the plots show the criteria of the RO4. |
Extended Analysis with the 2P2I Database
We further analyzed the characteristics of PPIs in comparison with typical drug-binding pockets using the FTMap server.29 In this analysis, we extended the dataset of PPIs by adding entries from the 2P2I database.50 2P2I classifies its entries into three categories: Class 1 for peptide–protein interactions, Class 2 for protein–protein structures, and histone-bromodomain complexes (BRD). We removed the BRD class from our study since they have high sequence and structure similarities among them. We also removed Menin/MLL complex, because it does not have a protein-PPID structures. In Table 2, there are four entries that are not included in 2P2I, which were CCR5/gp120, NEDD8/NEDD8’s heterodimeric E1, FKBP1A/Calcineurin, and transthyretin-tetramer. Among them, we only added CCR5/gp120 and FKBP1A/Calcineurin to the dataset since they have both the PPI structure and the protein-PPID structure available in PDB. The total number of PPIs used was 21. For traditional drug target data, we used the DUD set.72 The DUD set contains active compounds and decoys for 40 target proteins. Decoys are compounds that do not bind to the target but have similar chemical characteristics to active compounds. The 40 proteins consisted of popular drug targets, including kinases, proteases, nuclear hormone receptors. Among them, 21 targets that do not have cofactors and missing residues were used for this study. The list of PDB IDs of the dataset is shown in Supplementary Table S2.
The analysis method we used, FTMap, predicts drug-binding sites on a protein surface by identifying energetically favorable binding places for small compound fragments on the protein surface via docking. FTMap uses sixteen organic fragments shown in Table 3. If bound fragments form a cluster on a PPI site, the site could function similarly to a traditional ligand-binding pocket. Thus, from an FTMap result, the pocket-likeness of PPIs can be evaluated. We defined binding site residues in the receptor proteins as those which have at least one heavy atom within 4.5 Å to any heavy atom from ligands.
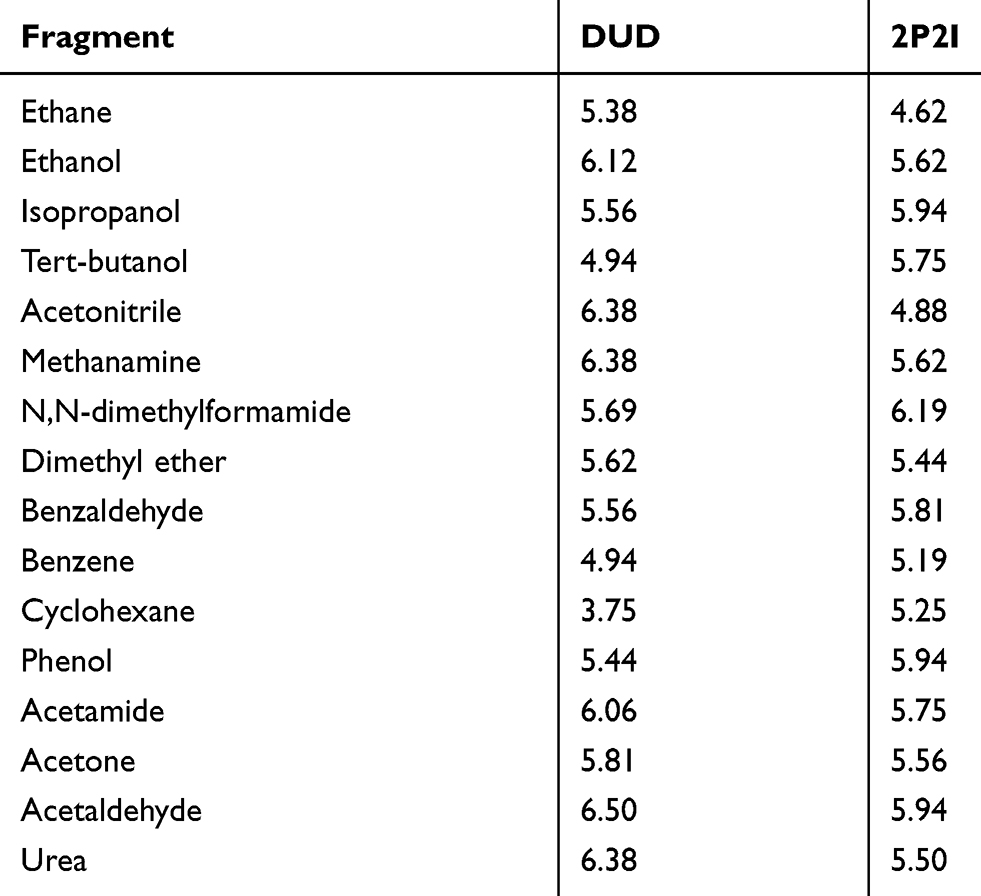 |
Table 3 The Average Number of Fragments Binding to the Interface |
In Figure 4A, we compared the number of interacting compound fragments for PPI sites and conventional pockets. The X-axis is the total number of fragments interacting with the receptor protein at the binding interface and the Y-axis shows the number of interacting fragments per binding site residue. The total number of interacting fragments (X-axis) of PPI sites (blue circles) ranged from 0 to 81, similar to the typical binding pockets (red circles), which was 6 to 87. Although there are a few PPI sites that have a large number of interacting fragments, overall the PPIs tend to have less interaction than the typical binding pockets. The average number of interacting fragments on the interface of the PPI set was 39.95 (standard deviation: 26.68), which was smaller than that of the binding pocket set (50.33, standard deviation: 19.74). The difference of the distribution of number of binding fragments for PPI sites were different with a statistically significance against the typical pockets at p-value 0.1 level (p-value of 0.07 by Student’s t-test), Turning our attention now to the Y-axis, on average PPI sites had a lower number of interacting fragments per residue (1.55, standard deviation: 1.09) than ligand-binding pockets (2.07, standard deviation: 0.77). This difference is statistically significant at p < 0.05 level (0.048). For the typical binding pockets, the highest value of the average number of fragments per residue is 2.89 and more than two-thirds (71.4%) of them have higher than 2.0. On the other hand, for the PPI set, even if the largest value was 4.05 and more than half (58.3%) cases are less than 2.0. These results show that PPI sites tend to have less concentration of bound fragments than typical binding pockets, indicating that PPIs do not form ideal pockets for drug binding. This observation is consistent with the conclusion from the previous section that PPIs have smaller SiteScore values than RO5 drug-binding pockets.
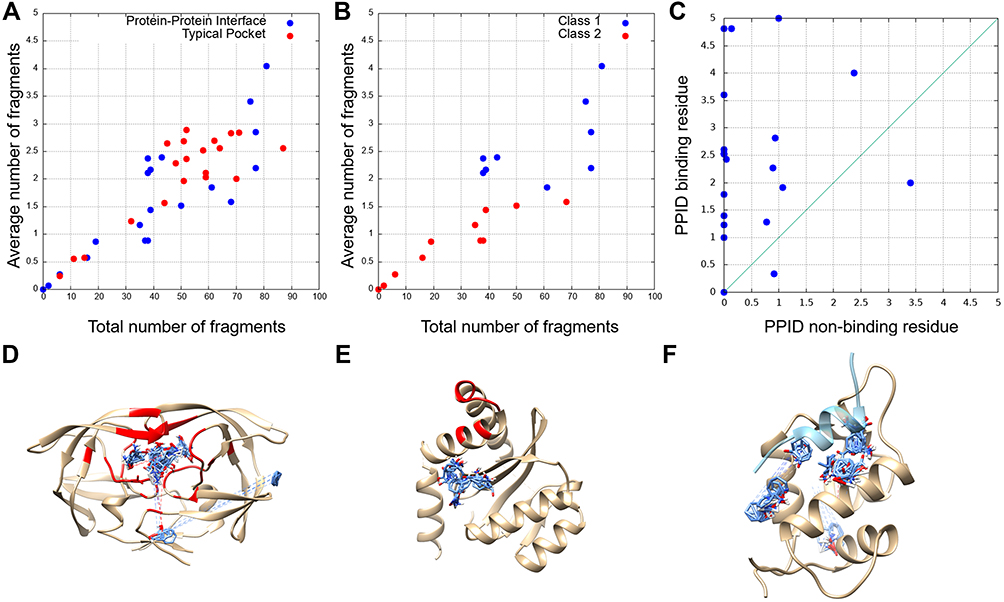 |
Figure 4 The number of bound fragments computed by FTMap at PPI-drug-binding sites and drug-binding pockets. (A) The total number of binding fragments and the average number of binding fragments per residue for PPI drug binding sites (blue) and drug-binding pockets (red). (B) PPI-drug-binding sites were further split into two classes: Blue, Class 1, protein-peptide interaction complexes; red, Class 2, protein–protein interaction complexes. (C) The average number of binding fragments per drug non-binding residues (x-axis) and drug-binding residues (y-axis). (D) The FTMap result for HIV protease (PDB ID: 1HPX), selected as a typical drug-binding pocket. The docking poses of fragments are colored in blue and binding pocket residues defined by considering the cognate binding ligand are colored red. (E) The FTMap results for integrase from the integrase/LEDGF complex (PDB ID: 2B4J) as an example of PPI interfaces. (F) The fragment docking result for MDM from the MDM/P53 complex (PDB ID: 1YCQ). A 17 amino-acids-long helical peptide from P53 is colored in light blue, and fragments bound to the receptor structure are shown in blue. |
Next, we analyzed two classes of PPI drug binding sites separately, peptide–protein interaction (Class 1, 9 entries) and protein–protein interaction (class 2, 12 entries including the CCR5/gp120 and FKBP1A/Calcineurin from Table 2) (Figure 4B). Interestingly, a clear separation was observed between the two classes. Class 1 (blue) tends to have a larger number for both the total number of fragments and the average number of fragments per binding site residue than class 2 (red). This implies that peptide binding PPIs tend to form pockets similar to typical ligand-binding pockets and are thus likely easier to develop drugs than protein–protein interaction sites. Naturally, compound fragments bind more to the drug-binding residues than non-drug binding residues at the PPI sites (Figure 4C). The result also implies that computational methods, protocols, and scores that are originally developed for typical binding pockets may be readily applicable to peptide binding PPIs.
Figure 4D and E show typical examples of FTMap analysis for conventional drug-binding pockets and PPI interfaces. Figure 4D is a result of HIV protease (PDB ID: 1HPX), one of the pockets from the DUD set. Even though FTMap tries to dock fragments for all the surface regions, most fragments (87 out of 92) were bound to the binding pocket of the receptor (residues colored in red). Figure 4E is a result for PPI interface, integrase (PDB ID: 2B4J). FTMap placed 95 fragments, most of the fragments (blue) were clustered together, but none of them were placed near the PPI interface (red). Figure 4F is an example of peptide binding sites, Mdm2 (PDB ID: 1YCQ). The protein gives the largest value of the average number of fragments per binding interface residue (4.05) among the PPI set. The ligand protein, p53, is a 17 residue-long helical peptide (light blue, Figure 4F). FTMap docked all the fragments at the binding site, which have a concave shape formed with 20 residues.
At last, in Table 3, we examined the breakdown of fragment types that bound on the PPI drug binding sites and conventional binding pockets used in Figure 4. The top five fragments that bind to the typical pockets were acetaldehyde, urea, methylamine, acetonitrile, and ethanol. All of the top five fragments have hydrogen bond donors or acceptors. On the other hand, the top five fragments for PPIs were N,N-dimethylformamide, isopropanol, phenol, acetaldehyde, and benzaldehyde. These five are relatively larger and more hydrophobic than the top five fragments for conventional pockets. In addition, phenol and benzaldehyde have benzene rings, which is consistent with the hydrophobic characteristic of PPI core regions.32
Conclusions
In this paper, we surveyed several previous works analyzing physicochemical properties of PPIs and PPIDs. Then, we gathered structural information of PPIDs which have been approved or under clinical trial. With the list, characteristics of PPIDs and their binding interface were computed using QED, SiteScore and FTMap. In comparison to typical protein-drug complexes, PPIs (and their PPIDs) have lower druggability scores (QED and SiteScore) and are less favored by molecular fragments (FTMap), as expected.
Since the explored drug target space is being saturated, PPIs are highlighted as new and potent targets. However, as listed in Table 2 and Supplementary Table S1, only a few number of PPIDs have been developed and are under clinical trial. More than 90% of human interactome still remains. Although PPIs are challenging targets for drug discovery, there is ample room for discovery and opportunities to treat orphan diseases.
Overall, existing rules describing druggability seem not to be optimized for PPIDs. Thus, new rules are necessary for PPI drug discovery to assess their druggability properly. To design a new rule, many structures of PPIDs binding to target PPIs would be needed, however, currently only limited structure information is available. Recent remarkable advances in emerging technologies such as cryo-EM and machine learning techniques would make up for the gap between the available and the required structure information, which will contribute to efficient advances of drug discovery research.
Acknowledgments
We thank Atsushi Hijikata and Tsuyoshi Shirai for their discussion on dataset construction and Jacob Verburgt for proofreading the manuscript. WHS acknowledges support from the National Research Foundation of Korea (NRF) grant funded by the Korea government (MSIT) (No. 2020R1F1A1075998). DK acknowledges supports from the National Institutes of Health (R01GM123055, R01GM133840), the National Science Foundation (DMS1614777, CMMI1825941, MCB1925643, DBI2003635) and the Purdue Institute of Drug Discovery. KI acknowledges support from JSPS KAKENHI Grant Numbers 18K11543. The following grant is also acknowledged; Platform Project for Supporting Drug Discovery and Life Science Research (Basis for Supporting Innovative Drug Discovery and Life Science Research (BINDS)) from AMED under grant number JP20am0101114 (to KI and TH).
Disclosure
The authors report no conflicts of interest in this work.
References
1. Santos R, Ursu O, Gaulton A, et al. A comprehensive map of molecular drug targets. Nat Rev Drug Discov. 2017;16:19–34. doi:10.1038/nrd.2016.230
2. Scannell JW, Balnckey A, Boldon H, Warrington B. Diagnosing the decline in pharmaceutical R&D efficiency. Nat Rev Drug Discov. 2012;11:191–200. doi:10.1038/nrd3681
3. Jin L, Wang W, Fang G. Targeting protein-protein interaction by small molecules. Annu Rev Pharmacol Toxicol. 2014;54:435–456. doi:10.1146/annurev-pharmtox-011613-140028
4. Arkin MR, Wells JA. Small-molecule inhibitors of protein-protein interactions: progress towards the dream. Nat Rev Drug Discov. 2004;3:301–317. doi:10.1038/nrd1343
5. Toogood PL. Inhibition of protein-protein association by small molecules: approaches and progress. J Med Chem. 2002;45:1543–1558. doi:10.1021/jm010468s
6. Dev KK. Making protein interaction druggable: targeting PDZ domains. Nat Rev Drug Discov. 2004;3:1047–1056.
7. Ivanov AA, Khuri FR, Fu H. Targeting protein-protein interactions as an anticancer strategy. Trends Pharmacol Sci. 2013;34:393–400. doi:10.1016/j.tips.2013.04.007
8. Mao Y, Fisher DW, Yang S, Keszycki RM, Dong H. Protein-protein interactions underlying the behavioral and psychological symptoms of dementia (BPSD) and Alzheimer’s disease. PLoS One. 2020;15:e0226021.
9. Ezkurdia I, Juan D, Rodriguez JM, et al. Multiple evidence strands suggest that there may be as few as 19000 human protein-coding genes. Hum Mol Genet. 2014;23:5866–5878. doi:10.1093/hmg/ddu309
10. Rolland T, Tasan M, Charloteaux B, et al. A proteome-scale map of the human interactome network. Cell. 2014;159:1212–1226. doi:10.1016/j.cell.2014.10.050
11. Rozenblatt-Rosen O, Deo RC, Padi M, et al. Interpreting cancer genomes using systematic host perturbations by tumour virus proteins. Nature. 2012;487:491–495. doi:10.1038/nature11288
12. Luck K, Kim DK, Lambourne L, et al. A reference map of the human binary interactome. Nature. 2020;508:402–408. doi:10.1038/s41586-020-2188-x
13. Szklarczyk D, Gable AL, Lyon D, et al. STRING v11: protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 2019;47:D607–D613. doi:10.1093/nar/gky1131
14. Venkatesan K, Rual JF, Vazquez A, et al. An empirical framework for binary interactome mapping. Nat Methods. 2009;6:83–90. doi:10.1038/nmeth.1280
15. Stumpf MPH, Thorne T, de Silva E, et al. Estimating the size of human interactome. Proc Natl Acad Sci U S A. 2008;105:6959–6964. doi:10.1073/pnas.0708078105
16. Tyagi M, Hashimoto K, Shoemaker B, Wuchty S, Panchenko AR. Large-scale mapping of human protein interactome using structural complexes. EMBO Rep. 2012;13:266–271. doi:10.1038/embor.2011.261
17. Karagoz K, Sevimoglu T, Arga KY. Integration of multiple biological features yields high confidence human protein interactome. J Theor Biol. 2016;403:85–96. doi:10.1016/j.jtbi.2016.05.020
18. Arkin MR, Tang Y, Wells JA. Small-molecule inhibitors of protein-protein interactions: progressing toward the reality. Chem Biol. 2014;21:1102–1114. doi:10.1016/j.chembiol.2014.09.001
19. Ni D, Lu S, Zhang J. Emerging roles of allosteric modulators in the regulation of protein-protein interactions (PPIs): a new paradigm for PPI drug discovery. Med Res Rev. 2019;39:2314–2342. doi:10.1002/med.21585
20. Sanz G, Singh M, Peuget S, Selivanova G. Inhibition of p53 inhibitors: progress, challenges and perspectives. J Mol Cell Biol. 2019;11:586–599. doi:10.1093/jmcb/mjz075
21. Vassilev LT, Vu BT, Graves B, et al. In vivo activation of the p53 pathway by small-molecule antagonists of MDM2. Science. 2004;303:844–848. doi:10.1126/science.1092472
22. Davies M, Nowotka M, Papadatos G, et al. ChEMBL web services: streamlining access to drug discovery data and utilities. Nucleic Acids Res. 2015;43:W612–620. doi:10.1093/nar/gkv352
23. Lipinski CA. Lead- and drug-like compounds: the rule-of-five revolution. Drug Discov Today Technol. 2004;1:337–341. doi:10.1016/j.ddtec.2004.11.007
24. E. Sila O, Farideh H, Ruth N, Attila G, Ozlem K. Methods for discovering and targeting druggable protein-protein interfaces and their application to repurposing. Methods Mol Biol. 2019;1903:1–21.
25. Vella D, Marini S, Vitali F, Di Silvestre D, Mauri G, Bellazzi R. MTGO: PPI network analysis via topological and functional module identification. Sci Rep. 2018;8:5499. doi:10.1038/s41598-018-23672-0
26. Shin WH, Christoffer CW, Kihara D. In silico structure-based approaches to discover protein-protein interaction-targeting drugs. Methods. 2017;131:22–32. doi:10.1016/j.ymeth.2017.08.006
27. Halgren TA. Identifying and characterizing binding sites and assessing druggability. J Chem Inf Model. 2009;49:377–389. doi:10.1021/ci800324m
28. Bickerton GR, Paolini GV, Besnard J, Muresan S, Hopkins AL. Quantifying the chemical beauty of drugs. Nat Chem. 2012;4(2):90–98. doi:10.1038/nchem.1243
29. Kozakov D, Grove LE, Hall DR, et al. The FTMap family of web servers for determining and characterizing ligand-binding hot spots of proteins. Nat Protoc. 2015;10:733–755. doi:10.1038/nprot.2015.043
30. Wodak SJ, Janin J. Structural basis of macromolecular recognition. Adv Protein Chem. 2002;61:9–73.
31. LoConte L, Clothia C, Janin J. The atomic structure of protein-protein recognition sites. J Mol Biol. 1999;285:2177–2198. doi:10.1006/jmbi.1998.2439
32. Berman HM, Westbrook J, Feng Z, et al. The protein data bank. Nucleic Acids Res. 2000;29:235–242. doi:10.1093/nar/28.1.235
33. Tunchag N, Kor G, Keshin O, Gursoy A, Nussinov R. A survey of available tools and web servers for analysis of protein-protein interactions and interfaces. Brief Bioinform. 2009;10:217–232. doi:10.1093/bib/bbp001
34. Chakrabarti P, Janin J. Dissecting protein-protein recognition sites. Proteins. 2002;47:334–343. doi:10.1002/prot.10085
35. David A, Sternberg MJE. The contribution of missense mutations in core and rim residues of protein-protein interfaces to human disease. J Mol Biol. 2015;427:2886–2898. doi:10.1016/j.jmb.2015.07.004
36. Sable R, Jois S. Surfing the protein-protein interaction surface using docking methods: application to the design of PPI inhibitors. Molecules. 2015;20:11569–11603. doi:10.3390/molecules200611569
37. Fuller JC, Burgoyne NJ, Jackson RM. Predicting druggable binding sites at the protein-protein interface. Drug Discov Today. 2009;14:155–161. doi:10.1016/j.drudis.2008.10.009
38. Laurie ATR, Jackson RM. Q-SiteFinder: an energy-based method for the prediction of protein-ligand binding sites. Bioinformatics. 2005;21:1908–1916. doi:10.1093/bioinformatics/bti315
39. Wishart DS, Feunang YD, Guo AC, et al. DrugBank 5.0: a major update to the DrugBank database for 2018. Nucleic Acids Res. 2018;46:D1074–D1082. doi:10.1093/nar/gkx1037
40. Burgoyne NJ, Jackson RM. Predicting protein interaction sites: binding hot-spots in protein-protein and protein-ligand interfaces. Bioinformatics. 2006;22:1335–1342. doi:10.1093/bioinformatics/btl079
41. Wells JA, McClendon CL. Reaching for high-hanging fruit in drug discovery at protein-protein interfaces. Nature. 2007;450:1001–1009. doi:10.1038/nature06526
42. Morelli X, Bourgeas R, Roche P. Chemical and structural lessons from recent successes in protein-protein interaction inhibition (2P2I). Curr Opin Chem Biol. 2011;15:475–481. doi:10.1016/j.cbpa.2011.05.024
43. Basse MJ, Betzi S, Morelli X, Roche P. 2P2Idb v2: update of a structural database dedicated to orthosteric modulation of protein-protein interactions. Database. 2016;2016:baw007. doi:10.1093/database/baw007
44. Sperandio O, Reynès CH, Camproux CA, Villoutreix BO. Rationalizing the chemical space of protein-protein interaction inhibitors. Drug Discov Today. 2010;15:220–229. doi:10.1016/j.drudis.2009.11.007
45. Labbe CM, Kuenemann MA, Zarzycka B, et al. iPPI-DB: an online database of modulators of protein-protein interactions. Nucleic Acids Res. 2016;44:D542–D547. doi:10.1093/nar/gkv982
46. Hopkins AL, Groom CR, Alex A. Ligand efficiency: a useful metric for lead selection. Drug Discov Today. 2004;3:660–672.
47. Abad-Zapatero C, Metz J. Ligand efficiency indices as guideposts for drug discovery. Drug Discov Today. 2005;10:464–469. doi:10.1016/S1359-6446(05)03386-6
48. Turbull A, Boyd S, Walse B. Fragment-based drug discovery and protein-protein interactions. Res Rep Biochem. 2014;4:13–26.
49. Morrison CN, Prosser KE, Stokes RW, Cordes A, Metzler-Nolte N, Cohen SM. Expanding medicinal chemistry into 3D space: metallofragments as 3D scaffolds for fragment-based drug discovery. Chem Sci. 2020;11:1216–1225. doi:10.1039/C9SC05586J
50. Congreve M, Carr R, Murray C. A ‘Rule of Three’ for fragment-based lead discovery? Drug Discov Today. 2003;8:876–877. doi:10.1016/S1359-6446(03)02831-9
51. Higueruelo AP, Schreyer A, Richard G, et al. Atomic interactions and profile of small molecules disrupting protein–protein interfaces: the TIMBAL database. Chem Biol Drug Des. 2009;74:457–467. doi:10.1111/j.1747-0285.2009.00889.x
52. Mabonga L, Kappo AP. Protein-protein interaction modulators: advances, successes and remaining challenges. Biophys Rev. 2019;11:559–581. doi:10.1007/s12551-019-00570-x
53. Macalino SJY, Basith S, Clavio NAB, Chang H, Kang S, Choi S. Evolution of in silico strategies for protein-protein interaction drug discovery. Molecules. 2018;23:1963. doi:10.3390/molecules23081963
54. Valeur E, Gueret SM, Adihou H, et al. New modalities for challenging targets in drug discovery. Angew Chem Int Ed. 2017;56:10294–10323.
55. Krishnamurthy N, Kurzrock R. Targeting the Wnt/beta-catenin pathway cancer: update on effectors and inhibitors. Cancer Treat Rev. 2018;62:50–60. doi:10.1016/j.ctrv.2017.11.002
56. Nusse R, Clevers H. Wnt/β-catenin signaling, disease, and emerging therapeutic modalities. Cell. 2017;169:985–999. doi:10.1016/j.cell.2017.05.016
57. Kawamoto SA, Coleska A, Ran X, Yi H, Yang CY, Wang S. Design of triazole-stapled BCL9 α-helical peptides to target the β-catenin/B-cell CLL/lymphoma 9 (BCL9) protein-protein interaction. J Med Chem. 2012;55:1137–1146. doi:10.1021/jm201125d
58. Takada K, Zhu D, Bird GH, et al. Targeted disruption of the BCL9/β-catenin complex inhibits oncogenic Wnt signaling. Sci Transl Med. 2012;4:148ra117. doi:10.1126/scitranslmed.3003808
59. Sang P, Zhang M, Shi Y, et al. Inhibition of β-catenin/B cell lymphoma 9 protein-protein interaction using α-helix-mimicking sulfono-γ-AApeptide inhibitors. Proc Natl Acad Sci U S A. 2019;116:10757–10762. doi:10.1073/pnas.1819663116
60. Hoggard LR, Zhang Y, Zhang M, Panic V, Wisniewski JA, Ji H. Rational design of selective small-molecule inhibitors for β-catenin/B-cell lymphoma 9 protein-protein interactions. J Am Chem Soc. 2015;137:12249–12260. doi:10.1021/jacs.5b04988
61. Zhang M, Wang Z, Zhang Y, Guo W, Ji H. Structure-based optimization of small-molecule inhibitors for the β-catenin/B-cell lymphoma 9 protein-protein interaction. J Med Chem. 2018;61:2989–3007. doi:10.1021/acs.jmedchem.8b00068
62. Wisniewski JA, Yin J, Teuscher KB, Zhang M, Ji H. Structure-based design of 1,4-dibenzoylpiperazines as β-catenin/B-cell lymphoma 9 protein-protein interaction inhibitors. ACS Med Chem Lett. 2016;7:508–513. doi:10.1021/acsmedchemlett.5b00284
63. Xiao T, Takagi J, Coller BS, Wang JH, Springer TA. Structural basis for allostery in integrins and binding to fibrinogen-mimetic therapeutics. Nature. 2004;432:59–67. doi:10.1038/nature02976
64. Springer TA, Zhu J, Xiao T. Structural basis for distinctive recognition of fibrinogen γC peptide by the platelet integrin αIIbβ3. J Cell Biol. 2008;182:791–800. doi:10.1083/jcb.200801146
65. Nešić D, Zhang Y, Spasic A, et al. Cryo-electron microscopy structure of the αIIbβ3-abciximab complex. Arterioscler Thromb Vasc Biol. 2020;40:624–637. doi:10.1161/ATVBAHA.119.313671
66. Callaway E. Revolutionary cryo-EM is taking over structural biology. Nature. 2020;578:201. doi:10.1038/d41586-020-00341-9
67. Owens J. Determining druggability. Nat Rev Drug Discov. 2007;6:187. doi:10.1038/nrd2275
68. Halgren T. New method for fast and accurate binding-site identification and analysis. Chem Biol Drug Des. 2007;69:146–148. doi:10.1111/j.1747-0285.2007.00483.x
69. Birkinshaw RW, Gong J, Luo CS, et al. Structures of BCL-2 in complex with venetoclax reveal the molecular basis of resistance mutations. Nat Commun. 2019;10:2385. doi:10.1038/s41467-019-10363-1
70. Bulawa CE, Connelly S, DeVit M, et al. Tafamidis, a potent and selective transthyretin kinetic stabilizer that inhibits the amyloid cascade. Proc Natl Acad Sci U S A. 2012;109:9629–9634. doi:10.1073/pnas.1121005109
71. Soga S, Shirai H, Kobori M, Hirayama N. Use of amino acid composition to predict ligand-binding sites. J Chem Inf Model. 2007;47:400–406. doi:10.1021/ci6002202
72. Huang N, Shoichet BK, Irwin JJ. Benchmarking sets for molecular docking. J Med Chem. 2006;49:6789–6801. doi:10.1021/jm0608356
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.