Back to Archived Journals » Vaccine: Development and Therapy » Volume 5
Current and future prophylactic vaccines for hepatitis C virus
Authors Dunlop J, Owsianka A, Cowton V, Patel A ![]()
Received 27 February 2015
Accepted for publication 12 May 2015
Published 11 August 2015 Volume 2015:5 Pages 31—44
DOI https://doi.org/10.2147/VDT.S48437
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 6
Editor who approved publication: Professor Don Diamond
James I Dunlop, Anna M Owsianka, Vanessa M Cowton, Arvind H Patel
MRC-University of Glasgow Centre for Virus Research, Glasgow, Scotland, UK
Abstract: The development of a vaccine is necessary to combat the global hepatitis C virus (HCV) epidemic. The key to the development of a prophylactic vaccine is understanding the immune response in those who spontaneously resolve HCV infections versus those who develop chronic disease. Several promising vaccine candidates based on the use of viral vectors are currently in Phase I and Phase II clinical trials. The recently solved structures of the E2 glycoprotein have greatly aided epitope- and antibody-based vaccine design.
Keywords: antibody, E1E2 glycoproteins, epitope, HCV, neutralizing antibody, viral vector
Introduction
Approximately 185 million people or 2.5% of the world’s population are currently infected with hepatitis C virus (HCV), and there are 3–4 million new infections each year. A total of 60%–80% of infections will develop into chronic disease resulting in 350,000–500,000 deaths annually due to HCV-related conditions.1,2 However, many more suffer with liver cirrhosis and approximately 3% will develop hepatocellular carcinoma.3
HCV is a blood-borne virus transmitted by exchange of bodily fluids. Therefore, it is most prevalent in certain populations: intravenous drug users, those receiving unscreened blood products, newborns, and those engaging in higher risk sexual activity.4–6 However, in approximately 10% of patients, the route of transmission is unclear.7
A complicating factor in the treatment of the disease is the genetic diversity of the virus, which has seven distinct genotypes and at least 30% genetic sequence variability. Genotype 1 is the most common in North America, Europe, and Asia, followed by genotype 3 and then genotype 2. Genotype 4 predominates in Africa and the Middle East but has recently spread to several European countries. Genotype 6 is less prevalent and is found mainly in South and East Asia. Genotypes 5 and 7 are rarer and account for <1% of infections.8–10
Until recently, the standard of care for all genotypes was a combination therapy of pegylated interferon and ribavirin.11 Side effects are universal and include fatigue, fever, and nausea with additional psychiatric effects of insomnia, headaches, irritability, and depression.12 Since 2011, there has been a steady stream of direct-acting antivirals (DAAs) that have been licensed for treatment. These new drugs, which inhibit viral NS5B RNA polymerase, NS3 protease, or NS5A phosphoprotein, are used alone or in combination with the existing interferon treatment and have delivered significantly higher sustained viral response rates, improved genotype coverage, and reduced treatment times.13,14 Side effects have been reduced, and interferon-free regimens are now possible.
Major downsides of the current therapies (including the DAAs) are side effects, high cost, and the late treatment of the disease, which commonly presents after years of chronic infection. Therefore, many patients already have irreversible liver damage and possibly liver cancer. Treatment rates are low, and, with an aging population, the incidence of cirrhosis and cancer is expected to triple by 2030 in developed countries.15 Resistance to the DAAs may become an increasing problem.16 Perhaps the largest barrier to treatment is the high cost. The older interferon treatment was expensive and already out of the reach of developing nations. Some less expensive treatments may become available to poorer nations when patents expire. However, the latest combination therapies may be beyond the reach of most countries, resulting in some controversy especially as production costs are relatively low.17
Despite ground-breaking improvements in treatment, the disease burden is set to increase both in poorer and in more affluent countries. Therefore, there is a pressing need for a prophylactic vaccine to provide protection from this disease and offer a potential route for eradication.
Virus structure and entry
HCV is a positive-strand RNA virus that belongs to the hepacivirus genus within the larger family of Flaviviridae that includes Dengue and Yellow fever viruses. The virus RNA genome is translated as a polyprotein that is cleaved to form three structural and seven nonstructural proteins (Figure 1A). The viral nucleocapsid assembles from the core protein and contains the viral RNA genome. The nucleocapsid itself is surrounded by the virus envelope that contains covalently linked complexes of the transmembrane proteins E1 and E2.18 The surfaces of E1 and E2 are highly glycosylated containing 4 and 9–11 N-linked sites, respectively. E2 also contains four putative O-linked glycosylation sites (Figure 1B).19
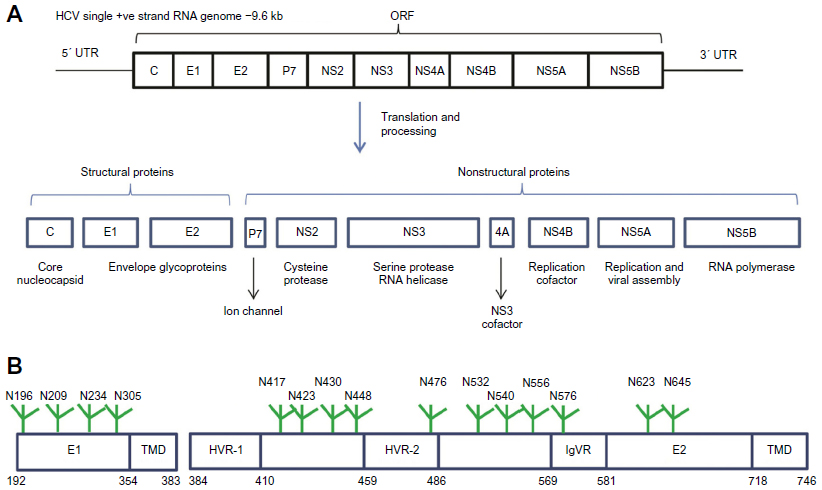 | Figure 1 HCV genome organization, polyprotein processing, and glycoprotein structure. |
The virus infects hepatocytes through a multistep process regulated by several intracellular signaling events.20 Virus particles are associated with lipoproteins, apolipoprotein E (ApoE), high-density lipoprotein, and low-density lipoprotein to form lipoviral particles. The entry process requires four essential receptors and coreceptors: scavenger receptor BI (SR-BI), CD81, and two tight junction proteins, claudin-1 and occludin. SR-BI is the first receptor to interact, initially with ApoE on the lipoviral particle and subsequently more directly with E2. The virus is taken into the cell via clathrin-mediated endocytosis and fuses with the endosomal membrane prior to uncoating and release of the viral genome.21 Until recently, E2 was believed to be a class II fusion protein, similar to those of other flaviviruses, and to mediate this fusion event. However, recent crystal structure data showed that E2 does not adopt the extended conformation of class II fusion proteins but has a compact, globular structure.22,23 The N terminus of E1 also has an unexpected fold and is unlikely to mediate class II fusion.24 The molecular detail of the roles of E1 and E2 in the fusion event is therefore unclear and will likely involve a novel entry mechanism.
Protective immune responses to HCV
Humoral responses
After initial infection by HCV, virus can be detected in serum after 1–3 weeks, but neither HCV-specific T-cells nor antibodies are detectable for 1–2 months. This delay in adaptive responses may contribute to the chronicity of HCV.25–27 It is widely accepted that cellular responses can resolve acute infection but the role of neutralizing antibodies (nAbs) in this regard is less clear.28 Several studies have shown that individuals can clear HCV infection without an effective humoral response, showing that nAbs are not absolutely required. Nevertheless, nAbs appear to be important in the control of chronic infection and prevention of reinfection.26,29–33 In one example, depletion of serum antibody (Ab) resulted in higher viremia and worsening of the disease and restoration of Ab levels resulted in disease improvement.34
Recent work has provided stronger evidence for nAbs in the resolution of acute infection. The early generation of high-titer broadly neutralizing Ab (bnAb) is associated with clearance of acute infection, whereas the later appearance of low-titer nAb with limited cross-reactivity is found in chronic disease.33,35–38 The role of Abs in protection from infection by HCV has also been demonstrated by passive immunization in animal models and in human beings.39–41 In human beings, this was observed in an incident involving a commercial intravenous immunoglobulin (IVIG) product prepared from pooled human plasma and used to treat primary immunodeficiency. After the introduction of screening by testing for HCV antibodies, one batch of the IVIG, from which units containing anti-HCV had been excluded, transmitted HCV to patients.42 The product was unfortunately contaminated with HCV, and the exclusion of plasma containing anti-HCV made it more infectious than earlier batches of IVIG made from unscreened plasma. Due to the protection provided by passive immunization, clinical trials are now proceeding to test the efficacy of HCV nAbs to prevent reinfection in liver transplant patients.43
Cellular responses
As with the humoral response, cellular responses to infection are delayed, and resolution of acute infection only occurs after the appearance of HCV-specific T-cells expressing IFN in the liver.44–47 The infection is met with a broad and strong helper T-cell (CD4+) and cytolytic T-cell (CD8+) response, which weakens and narrows in chronic disease.48,49 HCV-specific helper T-cells are difficult to detect in chronic disease, and the reason for their loss is poorly understood.50–52 CD8+ cells are also lost early in viral persistence, although this may be an adaptive response to prevent further tissue damage.53 Pathways leading to the loss or exhaustion of HCV-specific CD8+ cells may involve several factors, including the loss of CD4+ cells, viral mutation and chronic antigen stimulation, and also the appearance of regulatory T-cells.28
The requirement for both CD4+ and CD8+ cells in the resolution of infection has been demonstrated in an animal model where chimpanzees that previously cleared the infection were reinfected after the removal of one subset or the other.54,55 Cytotoxic T-cell activity (T-cell perforin expression and hepatocyte apoptosis) can also be correlated with disease resolution in the chimpanzee model, implying a role for this cell type.56
In summary, spontaneous resolution of infection is associated with an early adaptive immune response in the form of high-titer nAb and also a broad, strong, and sustained T-cell response where both helper and cytotoxic T-cells are absolutely required.
Vaccine approaches
Depending on race, age, genetics, and disease status, 20%–40% of individuals, after initial infection, eradicate the disease over a period of weeks or months.2,57 This has provided optimism that prophylactic vaccination to harness natural immunity could also be effective. Overall results using therapeutic vaccines have been disappointing, with only small reductions in viral loads, and therefore a prophylactic vaccine may be a more achievable target. A widely accepted goal of HCV treatment is the prevention of chronicity of infection, and this may be easier to achieve in uninfected subjects prophylactically than reverse it in infected patients with therapeutic vaccination. A confusing term in this regard is sterilizing immunity, which is commonly associated with prophylactic vaccination. Sterilizing immunity may be impossible to achieve with HCV, and more likely, a period of infection (or reinfection after future exposures) will precede viral clearance after prophylactic vaccination, similar to the scenario documented for patients who spontaneously resolve the disease.
So far, the development of a pan-genotypic vaccine to HCV has been challenging, mainly due to the genetic diversity of the virus. However, a vaccine that protected against a single common genotype would be a major advance. Another genetic problem for vaccine design is the high viral replication and mutation rate.58 The virus exists in the body as a constantly evolving quasispecies or vast swarm of closely related but distinct structures. This diversity presents particular problems for the humoral immune response and production of nAbs, which are the basis of all successful viral vaccines to date. HCV has also evolved a variety of strategies to evade antibody-mediated neutralization such as hypervariable regions (HVRs) within the envelope glycoproteins that act as immunological decoys (Table 1).28,59–68 How then can the immune system or a vaccine elicit a broadly neutralizing response to such an elusive target? One solution for vaccine development may be to focus on conserved epitopes. This strategy has also recently been adopted in vaccine design for human immunodeficiency virus (HIV) to overcome similar challenges of viral diversity and variability.69,70 Some rare bnAbs against HIV have been found after patient screening that are highly divergent from germline variable sequences, presumably having undergone extensive affinity maturation, and it has been suggested that novel vaccination strategies will be required to elicit them.71 This may also be the case for HCV, and one strategy that has been predicted to yield results is sequential vaccination with different but related antigens to more precisely direct the immune system.72 Advances of in silico modeling will greatly aid the design of novel immunogens to mimic chosen epitopes.73 Additionally, next-generation sequencing means detailed analysis of the immune responses to vaccines is possible, and this will enable researchers to track the affinity maturation of particular B-cell linages that could elicit bnAb.
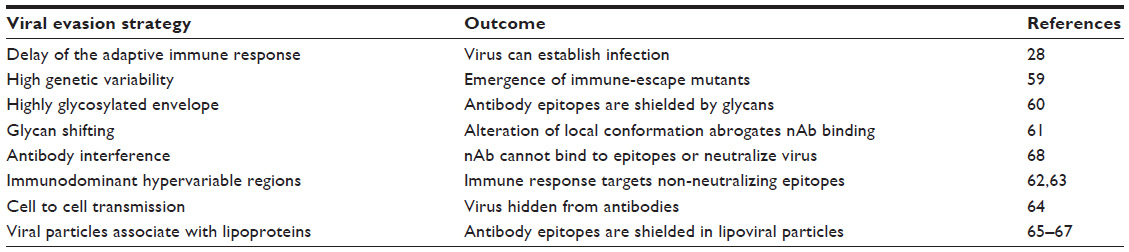 | Table 1 Viral evasion strategies to avoid antibody-mediated neutralization |
Prophylactic B-cell vaccines
Early vaccine research on HCV began exploiting the potential of the envelope glycoproteins E1 and E2. This approach produced some promising results in animal models.74 Substantial evidence of this approach comes from a meta-analysis of vaccine studies in chimpanzees, which concluded that structural proteins were better than nonstructural proteins at eliciting a protective response. This implies that nAbs are important to clear infection.75
Immunization of healthy volunteers with recombinant E1E2 heterodimer induced cross-reactive nAb responses to genotypes 1a, 1b, and 2a, but development of this vaccine is on hold because of the difficulty of producing intact recombinant heterodimer.76,77 The difficulty of producing recombinant protein can be overcome by heterologously expressing envelope glycoproteins in a viral vector or virus-like particle (VLP).78 Although the whole molecule approach shows some promise, it may fail due to same viral evasion strategies that contribute to chronic infection (Table 1). An appropriately modified E2 protein, for example, without HVRs, may avoid viral evasion strategies and ultimately be a more successful candidate.79 However, folding issues may limit the use of singly expressed or modified E2 proteins.80 Some recent research in chimpanzees failed to generate a nAb response using E2 lacking the HVR-1 region.81 The same work also demonstrated that only E1-vaccinated animals and not E2-vaccinated animals cleared HCV infection. In a Phase I clinical trial using a similar strategy, recombinant E1 generated low-to-moderate Ab responses alongside a clear T-helper 1 response in nearly all participants.82
In summary, the use of whole glycoproteins can generate bnAb responses, although there have been mixed results using recombinant singly expressed E1 and E2.
Prophylactic T-cell vaccines
Most HCV vaccines have been designed as therapeutic strategies and have focused on nonstructural epitopes to generate cellular (T-cell) responses. However, there are several promising prophylactic T-cell vaccine candidates that have progressed to clinical trials.83
One recent approach used adenoviral particles expressing the NS3–NS5 region of genotype 1b to generate long-lasting T-cell responses in healthy volunteers.84 This novel strategy used the adenoviral vectors human adenovirus 6 (Ad6) and chimpanzee adenovirus 3 (ChAd3), both containing DNA encoding the same region of the HCV genome. The immune system was primed with an initial inoculation of Ad6, and then boosted with several inoculations of ChAd3. These particular adenoviruses were chosen based on rare serotypes to reduce antivector immune responses, although such responses may have limited boosting in this study. The T-cell responses were predominately cytotoxic although helper T-cells were also present. T-cell responses were triggered by peptides from genotypes 1b, 1a, and to a lesser extent 3a, demonstrating a potentially wide treatment profile. Importantly, responses were also strong in a standard enzyme-linked immunospot (ELISPOT) assay measuring IFNγ (>1,000 SFC/106 peripheral blood mononuclear cells). A later study trial using ChAd3 as the priming vector and modified vaccinia Ankara (MVA) as the boosting vector had arguably an even better T-cell response profile.85 As previously, long-term memory T-cells were present but also higher CD4+ cell levels and even higher CD8+ cell responses. This was possibly due to lower immune responses to the MVA virus particles used for boosting. In addition, the authors carried out a more in-depth study of T-cell subsets to show a vaccination profile similar to that seen in other successful viral vaccines or in natural immunity.86,87 The next step is to discover if these responses are protective against infection. Phase I and Phase II trials to assess safety and efficacy are due for completion in 2016 (Table 2).88 The same strategy is also being tested in a Phase I/II trial as a therapy for HCV-infected patients, and it will be interesting to compare efficacy of these very promising viral vector stratagems.89 Two other viral vector vaccine strategies using adenoviruses are also currently being developed.90–92 In summary, the above vaccine strategies using viral vectors have great potential. However, formulating a vaccine with two different components may be expensive, and repeated boosting may not be practical in the developing world and for some patient groups.
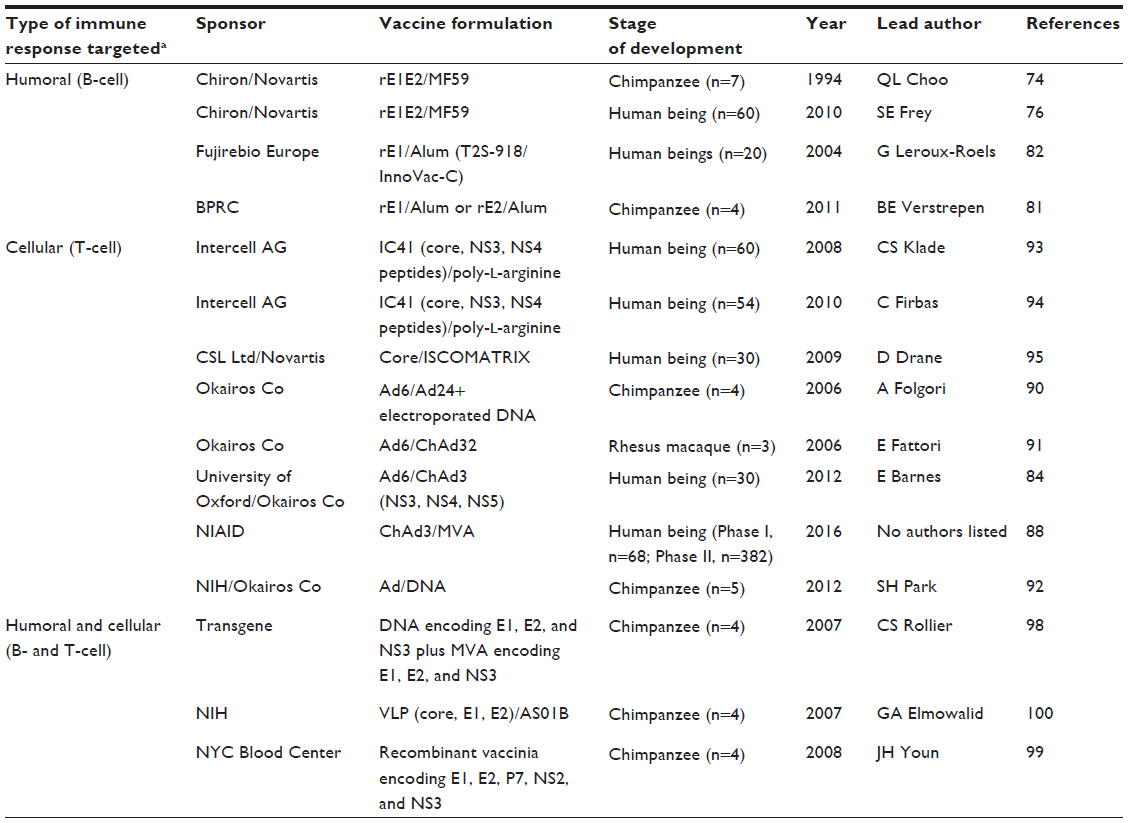 | Table 2 Current prophylactic vaccine candidates |
Another vaccine candidate being developed for both prophylaxis and therapy uses synthetic peptides derived from the core, NS3, and NS4 proteins that have been chosen to stimulate both cytotoxic and helper T-cells.93,94 Participants for this modality of treatment will occasionally have to be human leukocyte antigen (HLA) matched to the peptides used for vaccination. HLA classes that bind to the peptides present in a vaccine can be predetermined experimentally and by computer analysis. This should generate optimal immune responses in selected patients for a particular set of peptides. Volunteers for this study were previously-treated HCV patients who possessed the HLA-A 0201 allele, which is present in 50% of Caucasians. The results from two clinical trials in healthy volunteers showed that response was correlated with higher peptide doses and the use of adjuvants (poly l-arginine). However, the strength of the responses was very weak when compared to viral vector and VLP systems and there is no information on long-term memory responses. The company aims to develop this vaccine with a broader range of epitope targets and different adjuvants. This should help to increase the strength and breadth of T-cell responses achieved by this vaccine approach.
A third vaccine candidate designed to elicit protective cellular responses against the core protein has also reached clinical trials. This strategy involves injecting the core protein, expressed in yeast, with a novel proprietary adjuvant composed of saponin, phospholipid, and cholesterol that forms spherical micelle-like structures. In a Phase I trial, nearly all participants developed antibodies to the core protein, but T-cell responses were only present in two volunteers (25%) receiving the highest dose of vaccine.95 These trial results were disappointing given previous promising results in a Rhesus macaque model.96 This approach, which is also being considered as a therapy for chronically infected patients, will have to achieve high T-cell responder rates. Further trials are planned with higher doses of vaccine.
Prophylactic B- and T-cell combined vaccines
For a successful outcome, B-cell vaccines that contain recombinant protein or peptide from E1 and/or E2 may have to be combined with a T-cell vaccine and heterologously expressed. This would increase the possibility of a strong, broad, and sustained humoral and cellular response.97 Another option is for the vaccine to contain both B- and T-cell epitopes. Three vaccines that contain both B- and T-cell HCV epitopes have been tested in the chimpanzee. Two are based on strains of vaccinia virus and a third on a VLP (Table 2).98–100
A prime and boost regimen using DNA and MVA encoding E1E2 and NS3 has been developed. However, T-cell responses were transient and three of four animals were not protected from challenge with HCV.98
An HCV-based VLP vaccine encoding core, E1, and E2 has been developed by the National Institute of Health (USA). A high HCV-specific T-cell response was induced in four animals that were protected against subsequent challenge with a homologous strain.100 A lower dose to weight ratio in this study compared to previous mouse vaccinations may explain the lack of a humoral response. The high T-cell responses in this study show promise that this versatile vector may be developed into a successful vaccine approach.101
A lack of humoral response was also a characteristic of a vaccinia virus vaccine that encoded structural and nonstructural proteins.99 This vaccine, developed by the New York City Blood Center, induced a moderate T-cell response in four animals, and there was 100% protection from chronic disease after challenge with the same genotype. Some cross-genotypic protection was also found after combined challenge with six genotypes.
In summary, results from B- and T-cell combined vaccines have been mixed. In general, they have elicited good cellular responses and poor humoral responses.
New targets for future HCV vaccines
NAbs and key epitopes for vaccine design
Recent crystal structures of E2 peptides and of truncated E2 protein complexed to bnAb have provided a starting point to design immunogens that elicit epitope-specific antibodies. Some bnAbs bind to conserved linear epitopes, and these are of great interest because technically it is to easier to make an immunogen that represents a linear rather than a discontinuous epitope. However, nearly all the well-characterized bnAbs bind to discontinuous epitopes on E2 that are proposed to form part of the CD81 binding site.102 Extensive work has provided evidence for three main CD81 binding regions on the surface of E2 (regions 1–3 described later; Figure 2).68,103,104 BnAbs binding to these regions prevent E2 association with CD81 and presumably subsequent viral entry and fusion. Our understanding of the structure and location of neutralizing epitopes was greatly increased recently when two groups solved crystal structures of a truncated core of the extracellular domain of E2.22,23 The compact structure consists of a central β-sandwich flanked by front and back layers consisting of flexible loops and helices. To allow folding and crystallization, the N and C termini and also internal HVRs were omitted. Despite this, several areas of protein were disordered, including a key CD81 binding region (region 1). It has been proposed that a nontruncated E2, folded in the presence of E1, would adopt a slightly different structure with a different arrangement of disulfide bonds.105 Nevertheless, the structure shows that the proposed CD81 binding regions are juxtaposed on one face of the molecule (Figure 3). Notwithstanding these limitations, knowledge of the structure of E2 has rekindled interest in exploiting conserved neutralizing epitopes for vaccine design.
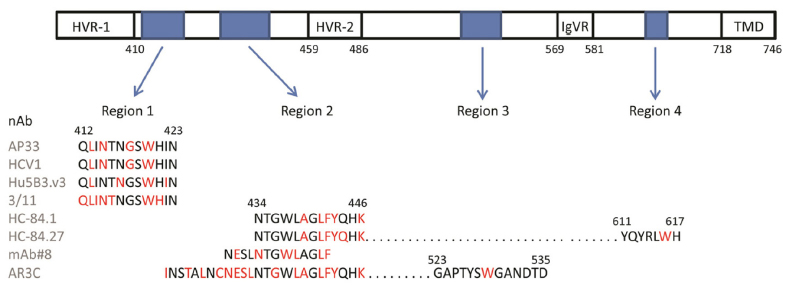 | Figure 2 A linear map of the E2 envelope glycoprotein, showing regions recognized by nAbs and implicated in binding to CD81. |
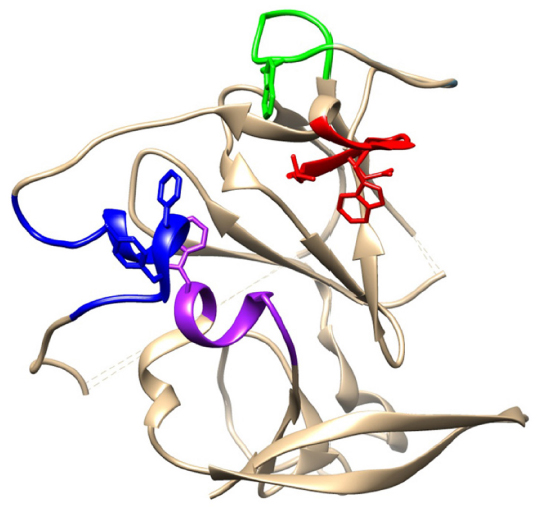 | Figure 3 Core structure of HCV E2 glycoprotein ectodomain, showing regions that are recognized by neutralizing antibodies and implicated in binding to CD81. |
Key epitopes for vaccine design
Region 1
The region between residues 412 and 423, immediately downstream of HVR-1, contains some highly conserved amino acid (aa) residues that are recognized as a series of overlapping epitopes by a number of monoclonal antibodies (Figure 2).68 One such antibody is the mouse monoclonal AP33, which inhibits infection in cell culture models of HCV and neutralizes virus of all genotypes in HCV pseudoparticle (HCVpp) assays.106,107 A humanized, affinity-matured version of AP33 (MRCT10.v362) has an approximately threefold higher neutralization potency on three tested genotypes.61 Therefore, this region is an exciting prospect for vaccine design.
The AP33 epitope has been characterized by alanine-scanning mutagenesis and X-ray crystallography (Figure 3).61,107–110 The major determinants of antibody recognition are residues Leu 413, Asn 415, Gly 418, and Trp 420. The latter is a contact residue for all bnAbs that recognize this region and is critical for binding of E2 to CD81.61,104,107–112 The structures of three antibody–peptide complexes with bnAbs AP33, HCV1, and hu5B3.v3 show this region forming a β-hairpin structure.61,108–111 A fourth antibody–peptide complex of the same region with bnAb 3/11 shows the peptide adopting a very different, extended conformation.112 A fifth complex reveals a composite structure comprising a looser turn and extended conformation.110 Therefore, it would appear that this region of E2 can adopt different conformations and is likely to be flexible. Indeed, this region is disordered in the crystal structure.23
Interestingly, this segment of E2 appears to have low antigenicity in HCV-infected patients and only 2.5% of patient sera tested positive to a peptide mimic.113 However, a vaccine based on this region could be much more antigenic and generate a stronger antibody response. Antibody-resistant mutants have been found by passaging HCV in the presence of bnAb AP33 in cell culture, which could indicate limitations of the protective effect of AP33-like antibodies in vivo.61,114 Encouragingly, another bnAb (HC-33) that is more resistant to immune escape has been isolated to this epitope.115 A combined immunogen based on the AP33 and HC-33 binding sites is an attractive prospect, as it raises the possibility of a vaccine that elicits a pan-genotypic bnAb response that is resistant to escape mutants.
Region 2
The region of E2 encompassed by aa residues 434–446 (Figures 2 and 3) was previously thought to elicit mainly antibodies that inhibited neutralization by bnAbs specific for region 1.68 However, the recent discovery of bnAbs that bind to this region has shown that, although some interference between binding and neutralization to either region is possible, there can also be additive or synergistic neutralization when bnAbs to both regions are present.116 The structure of this region has been determined both in the context of E2 core and as antibody-bound peptides.23,117,118 It consists of a long N-terminal loop and a short C-terminal helix of 1.5 turns. However, the angle and local conformation of the loop and helix vary in different structures and this has been interpreted to mean that region 2 can adopt different conformations, which may reflect different functional binding states of E2.117 Antibodies binding to this region can either be neutralizing or non-neutralizing depending on which conformation is bound. The short alpha helix contains the highly conserved aa residues Phe 442 and Tyr 443, which are implicated in binding to a hydrophobic pocket formed by the large extracellular loop on CD81.103,119,120 Phe 442 and Tyr 443 are also contact residues for several bnAbs, including HC-84.1, HC-84.27, and AR3C (Figure 2).23,118 The series of HC-84 antibodies that recognize this region have broad genotype specificity and some bind to linear epitopes.121 No immune-escape variants arose when the virus was passaged in the presence of the HC-84 series of bnAbs, showing that they are resistant to virus escape from neutralization.115 This region is another good target for an epitope-focused vaccine approach.
Region 3
This region, comprising aa residues 523–535, has been proposed as a major binding site for CD81 that is also recognized by several bnAbs, some of which are effective across all the common genotypes (Figures 2 and 3).68,122,123 It includes the highly conserved residues Tyr 527, Trp 529, Gly 530, and Asp 535 that have been identified by alanine-scanning mutagenesis as being critical for CD81 binding.104 Characterization of antibody recognition determinants using a variety of methods has provided evidence for contact with one or more of these highly conserved residues in a number of overlapping epitopes.102 However, recent structural data conflict with some of the previous mutagenesis findings. The structure of the AR3C-E2 complex shows that the bnAb AR3C does not contact residues Gly 530 and Asp 535, as indicated previously by alanine scanning, and that its major binding site is region 2 and not region 3 as previously reported.23,124 This may also be the case for other bnAbs such as CBH-5 that are predicted, on the basis of alanine scanning, to share the same recognition residues.122 It is likely that mutagenesis in this region changes the conformation of E2 and prevents antibody recognition at other sites. The major contact residue in this region for AR3C based on the crystal structure data is Trp 529, which, together with other proposed CD81 binding residues, would contribute to a hydrophobic interface (Figure 3). This segment of E2 appears to be relatively antigenic, and antibodies to it have been found in patient sera and in mice immunized with E2, although much of these data have been generated by alanine scanning and may not be reliable.124,125 This region also appears to be resistant to the emergence of escape variants and would be another good prospect for an epitope-based vaccine, although some uncertainty exists in the residues involved in antibody and CD81 binding.126
Region 4
Early mutagenesis studies highlighted a small region between aa residues 611 and 617 as a potential CD81 binding site.127 This finding has been supported by the characterization of the interaction sites of several bnAbs such as the HC-84 series and HC-11 (Figure 2).121 The tryptophan (W) present within this region (YPYRLWH) is a contact residue for some of the HC-84 series of bnAbs based on structural analysis and mutagenesis and could contribute to a hydrophobic interface for CD81 binding (Figure 3). This small segment of E2 is recognized by conformational antibodies, and any peptide-based vaccine would likely have to include other regions, described earlier, to allow antibody binding.
Other neutralizing epitopes
Two other regions have been described recently that are recognized by bnAb and which are not part of the CD81 binding site. These areas, termed antigenic regions 4 and 5 by Giang et al, are formed in the heterodimer by clusters of residues in a relatively unstudied part of E2 close to the membrane and perhaps also by residues in E1.123 The authors speculate that the epitopes are located at an interface between the two envelope proteins and are present only in the correctly folded E1E2 complex. Further characterization of these newly discovered epitopes is required to assess their prospects for vaccine design.
Future prospects
The potential of small conserved regions of E2 in a vaccine has been largely unexplored in human clinical studies. One approach could be immunization with peptides in combination with a suitable adjuvant. The peptide structure could be changed chemically, for example, by cyclization or possibly linked to a chemical or protein scaffold.128,129 Such changes of the natural structure could improve stability and antigenicity: a key issue with protein vaccination methods. A scaffold also offers the possibility of presenting multiple peptides in the correct orientation to mimic a discontinuous epitope in the native E2 structure. Such a strategy has been used to make a potential vaccine candidate for Respiratory Syncytial Virus.129 In this case computer modelling was used to design an immunogen that mimics the structure and orientation of an epitope bound by a well characterized nAb. A few animal studies have also used free peptides containing conserved epitopes of E2 to explore their potential to generate a humoral response. In one study, vaccination of mice with a T-cell helper epitope linked to peptide sequences generated nAb that inhibited entry of genotype 1a pseudoparticles into Huh-7 cells.130 In summary, the use of peptide antigens to mimic HCV structural epitopes has achieved mixed vaccination results. However, new technologies to improve stability, antigenicity, and orientation mean that they could still form part of a successful novel vaccination strategy. One approach would be to combine a peptide vaccine with another type of antigen such as a subunit vaccine with a prime and boost regimen to focus the immune system on particular epitopes.
The conserved epitopes could also be exploited using an anti-idiotype approach (Figure 4). In this strategy, a bnAb is used as the starting point to reverse-engineer a vaccine. By judicious screening, an antibody can be isolated whose variable regions mimic the shape of the original epitope. This antibody or a derived fragment of it is used as a vaccine. This novel vaccination approach has been used to treat cancer and is currently being developed for HCV.131–133
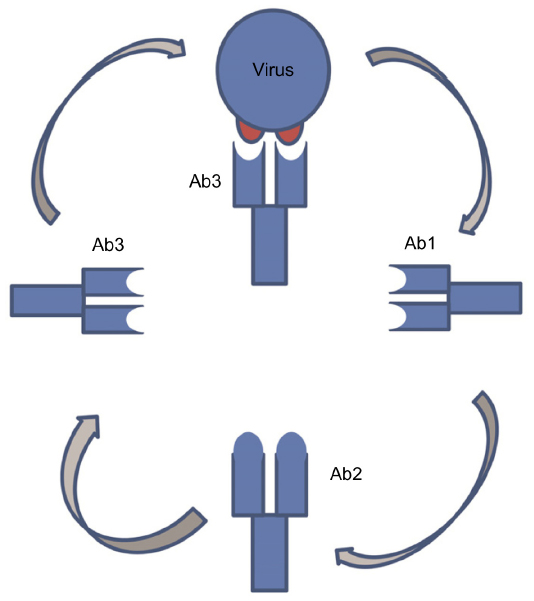 | Figure 4 Anti-idiotype vaccine approach. |
The epitopes could also be presented in a VLP or virus construct. This strategy has been used to express whole or truncated surface E1 and E2 in a vaccine context, but expression of the small conserved HCV epitopes in such constructs is unexplored. One platform that could be tested is a hepatitis B virus (HBV) VLP. Two different HBV VLPs are available, formed from either the core protein or the HBV surface antigen (HBVSAg). The HBV core protein that forms the capsid of the virus has proven to be a very flexible carrier of heterologous inserts. B- and T-cell epitopes have been inserted at the N and C termini as well as the major immunodominant region of the core protein in numerous vaccination experiments.134 Strong T-cell (helper and cytotoxic) responses are developed against the HBV core protein, and, given that cellular and humoral responses are both important in controlling HCV infection, the possibility of inserting HCV B- and T-cell epitopes into a HBV core protein is an intriguing one. The flexibility, high antigenicity, and compatibility with nearly any recombinant expression system make HBV core VLPs a very good candidate for an epitope-based vaccine for HCV. Although HBV core-based VLP vaccines have not progressed past the preclinical stage for HCV, two other HBV core-based vaccines have progressed to clinical testing for malaria and influenza A.135–137 Another possibility utilizing HBV is the insertion of HCV epitopes into the HBVSAg, which can also self-assemble with host lipids into VLPs without the nucleocapsid and is the basis of the successful worldwide HBV vaccine. In the field of HCV, this system has grounds for optimism as some recent work swapping the TM region of E1 and E2 with HBVSAg produced VLPs that induced high-titer nAb when used as a vaccine in an animal model.78,138 Many other VLP approaches using different platforms are also possible.101
A completely different approach would be the use of viral vectors. Viral vectors based on adenovirus or vaccinia could be used to express E2 epitopes in a standalone strategy or used in a prime-boost regimen when combined with E2 protein or peptides. A study in mice using Ad6 expressing full length and truncated E1E2 was combined in a prime and boost regimen with recombinant E1E2 protein to elicit strong cellular and bnAb responses. A similar approach using canary pox virus and the HIV surface protein gp120 has been the most successful HIV vaccine to date.139
Summary
Recent DAAs have revolutionized the therapeutic treatment of hepatitis C. However, the disease burden is still set to increase in developed and developing countries. A prophylactic vaccine could circumvent treatment issues such as late diagnosis, high costs, and provide an eradication route for this disease.
Immune correlates of protection deciphered from studying spontaneous resolvers and chronic patients have provided important clues to the potential attributes of a successful vaccine, although more work needs to be done to define this further.
The most advanced HCV vaccine to date involves the use of viral vectors with a prime and boost regimen. Notably, a similar strategy has led to the currently most successful HIV vaccine.
Recent crystal structures of the HCV envelope glycoprotein E2 have renewed interest in a rational vaccine approach to design immunogens that mimic conserved epitopes on E2.
Acknowledgment
The work in the authors’ laboratory is funded by the Medical Research Council, UK.
Disclosure
The authors report no conflicts of interest in this work.
References
Perz JF, Armstrong GL, Farrington LA, Hutin YJ, Bell BP. The contributions of hepatitis B virus and hepatitis C virus infections to cirrhosis and primary liver cancer worldwide. J Hepatol. 2006;45(4):529–538. | |
Micallef JM, Kaldor JM, Dore GJ. Spontaneous viral clearance following acute hepatitis C infection: a systematic review of longitudinal studies. J Viral Hepat. 2006;13(1):34–41. | |
Muhlberger N, Schwarzer R, Lettmeier B, Sroczynski G, Zeuzem S, Siebert U. HCV-related burden of disease in Europe: a systematic assessment of incidence, prevalence, morbidity, and mortality. BMC Public Health. 2009;9:34. | |
Yeung CY, Lee HC, Chan WT, Jiang CB, Chang SW, Chuang CK. Vertical transmission of hepatitis C virus: current knowledge and perspectives. World J Hepatol. 2014;6(9):643–651. | |
Garfein RS, Vlahov D, Galai N, Doherty MC, Nelson KE. Viral infections in short-term injection drug users: the prevalence of the hepatitis C, hepatitis B, human immunodeficiency, and human T-lymphotropic viruses. Am J Public Health. 1996;86(5):655–661. | |
van de Laar T, Pybus O, Bruisten S, et al. Evidence of a large, international network of HCV transmission in HIV-positive men who have sex with men. Gastroenterology. 2009;136(5):1609–1617. | |
Flamm SL, Parker RA, Chopra S. Risk factors associated with chronic hepatitis C virus infection: limited frequency of an unidentified source of transmission. Am J Gastroenterol. 1998;93(4):597–600. | |
Smith DB, Bukh J, Kuiken C, et al. Expanded classification of hepatitis C virus into 7 genotypes and 67 subtypes: updated criteria and genotype assignment web resource. Hepatology. 2014;59(1):318–327. | |
Sievert W, Altraif I, Razavi HA, et al. A systematic review of hepatitis C virus epidemiology in Asia, Australia and Egypt. Liver Int. 2011; 31(Suppl 2):61–80. | |
Messina JP, Humphreys I, Flaxman A, et al. Global distribution and prevalence of hepatitis C virus genotypes. Hepatology. 2015;61(1):77–87. | |
Manns MP, McHutchison JG, Gordon SC, et al. Peginterferon alfa-2b plus ribavirin compared with interferon alfa-2b plus ribavirin for initial treatment of chronic hepatitis C: a randomised trial. Lancet. 2001;358(9286):958–965. | |
Manns MP, Wedemeyer H, Cornberg M. Treating viral hepatitis C: efficacy, side effects, and complications. Gut. 2006;55(9):1350–1359. | |
Fox AN, Jacobson IM. Recent successes and noteworthy future prospects in the treatment of chronic hepatitis C. Clin Infect Dis. 2012; 55(Suppl 1):S16–S24. | |
Kim do Y, Ahn SH, Han KH. Emerging therapies for hepatitis C. Gut Liver. 2014;8(5):471–479. | |
Razavi H, Waked I, Sarrazin C, et al. The present and future disease burden of hepatitis C virus (HCV) infection with today’s treatment paradigm. J Viral Hepat. 2014;21(Suppl 1):34–59. | |
Welsch C, Jesudian A, Zeuzem S, Jacobson I. New direct-acting antiviral agents for the treatment of hepatitis C virus infection and perspectives. Gut. 2012;61(Suppl 1):i36–i46. | |
Hill A, Cooke G. Medicine. Hepatitis C can be cured globally, but at what cost? Science. 2014;345(6193):141–142. | |
Vieyres G, Thomas X, Descamps V, Duverlie G, Patel AH, Dubuisson J. Characterization of the envelope glycoproteins associated with infectious hepatitis C virus. J Virol. 2010;84(19):10159–10168. | |
Falkowska E, Kajumo F, Garcia E, Reinus J, Dragic T. Hepatitis C virus envelope glycoprotein E2 glycans modulate entry, CD81 binding, and neutralization. J Virol. 2007;81(15):8072–8079. | |
Zhu YZ, Qian XJ, Zhao P, Qi ZT. How hepatitis C virus invades hepatocytes: the mystery of viral entry. World J Gastroenterol. 2014; 20(13):3457–3467. | |
Sharma NR, Mateu G, Dreux M, Grakoui A, Cosset FL, Melikyan GB. Hepatitis C virus is primed by CD81 protein for low pH-dependent fusion. J Biol Chem. 2011;286(35):30361–30376. | |
Khan AG, Whidby J, Miller MT, et al. Structure of the core ectodomain of the hepatitis C virus envelope glycoprotein 2. Nature. 2014; 509(7500):381–384. | |
Kong L, Giang E, Nieusma T, et al. Hepatitis C virus E2 envelope glycoprotein core structure. Science. 2013;342(6162):1090–1094. | |
El Omari K, Iourin O, Kadlec J, et al. Unexpected structure for the N-terminal domain of hepatitis C virus envelope glycoprotein E1. Nat Commun. 2014;5:4874. doi: 10.1038/ncomms5874. | |
Courouce AM, Bouchardeau F, Girault A, Le Marrec N. Significance of NS3 and NS5 antigens in screening for HCV antibody. Lancet. 1994; 343(8901):853–854. | |
Thimme R, Oldach D, Chang KM, Steiger C, Ray SC, Chisari FV. Determinants of viral clearance and persistence during acute hepatitis C virus infection. J Exp Med. 2001;194(10):1395–1406. | |
Netski DM, Mosbruger T, Depla E, et al. Humoral immune response in acute hepatitis C virus infection. Clin Infect Dis. 2005;41(5):667–675. | |
Park SH, Rehermann B. Immune responses to HCV and other hepatitis viruses. Immunity. 2014;40(1):13–24. | |
Cooper S, Erickson AL, Adams EJ, et al. Analysis of a successful immune response against hepatitis C virus. Immunity. 1999;10(4):439–449. | |
Post JJ, Pan Y, Freeman AJ, et al; Hepatitis C Incidence and Transmission in Prisons Study (HITS) Group. Clearance of hepatitis C viremia associated with cellular immunity in the absence of seroconversion in the hepatitis C incidence and transmission in prisons study cohort. J Infect Dis. 2004;189(10):1846–1855. | |
Razvi S, Schneider L, Jonas MM, Cunningham-Rundles C. Outcome of intravenous immunoglobulin-transmitted hepatitis C virus infection in primary immunodeficiency. Clin Immunol. 2001;101(3):284–288. | |
Osburn WO, Fisher BE, Dowd KA, et al. Spontaneous control of primary hepatitis C virus infection and immunity against persistent reinfection. Gastroenterology. 2010;138(1):315–324. | |
Bjoro K, Froland SS, Yun Z, Samdal HH, Haaland T. Hepatitis C infection in patients with primary hypogammaglobulinemia after treatment with contaminated immune globulin. N Engl J Med. 1994; 331(24):1607–1611. | |
Ennishi D, Terui Y, Yokoyama M, et al. Monitoring serum hepatitis C virus (HCV) RNA in patients with HCV-infected CD20-positive B-cell lymphoma undergoing rituximab combination chemotherapy. Am J Hematol. 2008;83(1):59–62. | |
Raghuraman S, Park H, Osburn WO, Winkelstein E, Edlin BR, Rehermann B. Spontaneous clearance of chronic hepatitis C virus infection is associated with appearance of neutralizing antibodies and reversal of T-cell exhaustion. J Infect Dis. 2012;205(5):763–771. | |
Dowd KA, Netski DM, Wang XH, Cox AL, Ray SC. Selection pressure from neutralizing antibodies drives sequence evolution during acute infection with hepatitis C virus. Gastroenterology. 2009;136(7):2377–2386. | |
Pestka JM, Zeisel MB, Bläser E, et al. Rapid induction of virus-neutralizing antibodies and viral clearance in a single-source outbreak of hepatitis C. Proc Natl Acad Sci U S A. 2007;104(14):6025–6030. | |
Osburn WO, Snider AE, Wells BL, et al. Clearance of hepatitis C infection is associated with the early appearance of broad neutralizing antibody responses. Hepatology. 2014;59(6):2140–2151. | |
Farci P, Shimoda A, Wong D, et al. Prevention of hepatitis C virus infection in chimpanzees by hyperimmune serum against the hypervariable region 1 of the envelope 2 protein. Proc Natl Acad Sci U S A. 1996;93(26):15394–15399. | |
Morin TJ, Broering TJ, Leav BA, et al. Human monoclonal antibody HCV1 effectively prevents and treats HCV infection in chimpanzees. PLoS Pathog. 2012;8(8):e1002895. | |
Yu MY, Bartosch B, Zhang P, et al. Neutralizing antibodies to hepatitis C virus (HCV) in immune globulins derived from anti-HCV-positive plasma. Proc Natl Acad Sci U S A. 2004;101(20):7705–7710. | |
Bresee JS, Mast EE, Coleman PJ, et al. Hepatitis C virus infection associated with administration of intravenous immune globulin. A cohort study. JAMA. 1996;276(19):1563–1567. | |
MassBiologics. Open Label Study of the Efficacy and Safety of MBL-HCV1 in Combination With Oral Direct-Acting Antivirals in Patients Undergoing Liver Transplantation for Hepatitis C [Internet]; 2015 [cited January 21, 2015]. Available from: https://clinicaltrials.gov/ct2/show/NCT01532908. | |
Major ME, Dahari H, Mihalik K, et al. Hepatitis C virus kinetics and host responses associated with disease and outcome of infection in chimpanzees. Hepatology. 2004;39(6):1709–1720. | |
Thimme R, Bukh J, Spangenberg HC, et al. Viral and immunological determinants of hepatitis C virus clearance, persistence, and disease. Proc Natl Acad Sci U S A. 2002;99(24):15661–15668. | |
Dustin LB, Rice CM. Flying under the radar: the immunobiology of hepatitis C. Annu Rev Immunol. 2007;25:71–99. | |
Rehermann B. Pathogenesis of chronic viral hepatitis: differential roles of T cells and NK cells. Nat Med. 2013;19(7):859–868. | |
Day CL, Lauer GM, Robbins GK, et al. Broad specificity of virus-specific CD4+ T-helper-cell responses in resolved hepatitis C virus infection. J Virol. 2002;76(24):12584–12595. | |
Dustin LB, Cashman SB, Laidlaw SM. Immune control and failure in HCV infection – tipping the balance. J Leukoc Biol. 2014;96(4):535–548. | |
Callendret B, Walker C. A siege of hepatitis: immune boost for viral hepatitis. Nat Med. 2011;17(3):252–253. | |
Day CL, Seth NP, Lucas M, et al. Ex vivo analysis of human memory CD4 T cells specific for hepatitis C virus using MHC class II tetramers. J Clin Invest. 2003;112(6):831–842. | |
Gerlach JT, Diepolder HM, Jung MC, et al. Recurrence of hepatitis C virus after loss of virus-specific CD4(+) T-cell response in acute hepatitis C. Gastroenterology. 1999;117(4):933–941. | |
Wherry EJ. T cell exhaustion. Nat Immunol. 2011;12(6):492–499. | |
Grakoui A, Shoukry NH, Woollard DJ, et al. HCV persistence and immune evasion in the absence of memory T cell help. Science. 2003; 302(5645):659–662. | |
Shoukry NH, Grakoui A, Houghton M, et al. Memory CD8+ T cells are required for protection from persistent hepatitis C virus infection. J Exp Med. 2003;197(12):1645–1655. | |
Watanabe H, Wells F, Major ME. Clearance of hepatitis C in chimpanzees is associated with intrahepatic T-cell perforin expression during the late acute phase. J Viral Hepat. 2010;17(4):245–253. | |
Grebely J, Page K, Sacks-Davis R, et al; InC3 Study Group. The effects of female sex, viral genotype, and IL28B genotype on spontaneous clearance of acute hepatitis C virus infection. Hepatology. 2014;59(1):109–120. | |
Ribeiro RM, Li H, Wang S, et al. Quantifying the diversification of hepatitis C virus (HCV) during primary infection: estimates of the in vivo mutation rate. PLoS Pathog. 2012;8(8):e1002881. | |
von Hahn T, Yoon JC, Alter H, et al. Hepatitis C virus continuously escapes from neutralizing antibody and T-cell responses during chronic infection in vivo. Gastroenterology. 2007;132(2):667–678. | |
Helle F, Duverlie G, Dubuisson J. The hepatitis C virus glycan shield and evasion of the humoral immune response. Viruses. 2011;3(10):1909–1932. | |
Pantua H, Diao J, Ultsch M, et al. Glycan shifting on hepatitis C virus (HCV) E2 glycoprotein is a mechanism for escape from broadly neutralizing antibodies. J Mol Biol. 2013;425(11):1899–1914. | |
Bankwitz D, Steinmann E, Bitzegeio J, et al. Hepatitis C virus hypervariable region 1 modulates receptor interactions, conceals the CD81 binding site, and protects conserved neutralizing epitopes. J Virol. 2010;84(11):5751–5763. | |
Prentoe J, Jensen TB, Meuleman P, et al. Hypervariable region 1 differentially impacts viability of hepatitis C virus strains of genotypes 1 to 6 and impairs virus neutralization. J Virol. 2011;85(5):2224–2234. | |
Timpe JM, Stamataki Z, Jennings A, et al. Hepatitis C virus cell-cell transmission in hepatoma cells in the presence of neutralizing antibodies. Hepatology. 2008;47(1):17–24. | |
Dreux M, Pietschmann T, Granier C, et al. High density lipoprotein inhibits hepatitis C virus-neutralizing antibodies by stimulating cell entry via activation of the scavenger receptor BI. J Biol Chem. 2006; 281(27):18285–18295. | |
Bartosch B, Verney G, Dreux M, et al. An interplay between hypervariable region 1 of the hepatitis C virus E2 glycoprotein, the scavenger receptor BI, and high-density lipoprotein promotes both enhancement of infection and protection against neutralizing antibodies. J Virol. 2005;79(13):8217–8229. | |
Voisset C, Op de Beeck A, Horellou P, et al. High-density lipoproteins reduce the neutralizing effect of hepatitis C virus (HCV)-infected patient antibodies by promoting HCV entry. J Gen Virol. 2006;87(pt 9):2577–2581. | |
Sautto G, Tarr AW, Mancini N, Clementi M. Structural and antigenic definition of hepatitis C virus E2 glycoprotein epitopes targeted by monoclonal antibodies. Clin Dev Immunol. 2013;2013:450963. | |
Burton DR, Ahmed R, Barouch DH, et al. A blueprint for HIV vaccine discovery. Cell Host Microbe. 2012;12(4):396–407. | |
Ahmed FK, Clark BE, Burton DR, Pantophlet R. An engineered mutant of HIV-1 gp120 formulated with adjuvant Quil A promotes elicitation of antibody responses overlapping the CD4-binding site. Vaccine. 2012; 30(5):922–930. | |
Kwong PD, Mascola JR, Nabel GJ. Broadly neutralizing antibodies and the search for an HIV-1 vaccine: the end of the beginning. Nat Rev Immunol. 2013;13:693–701. | |
Wang S, Mata-Fink J, Kriegsman B, et al. Manipulating the selection forces during affinity maturation to generate cross-reactive HIV antibodies. Cell. 2015;160(4):785–797. | |
He L, Zhu J. Computational tools for epitope vaccine design and evaluation. Curr Opin Virol. 2015;11:103–112. | |
Choo QL, Kuo G, Ralston R, et al. Vaccination of chimpanzees against infection by the hepatitis C virus. Proc Natl Acad Sci U S A. 1994; 91(4):1294–1298. | |
Dahari H, Feinstone SM, Major ME. Meta-analysis of hepatitis C virus vaccine efficacy in chimpanzees indicates an importance for structural proteins. Gastroenterology. 2010;139(3):965–974. | |
Frey SE, Houghton M, Coates S, et al. Safety and immunogenicity of HCV E1E2 vaccine adjuvanted with MF59 administered to healthy adults. Vaccine. 2010;28(38):6367–6373. | |
Wong JA, Bhat R, Hockman D, et al. Recombinant hepatitis C virus envelope glycoprotein vaccine elicits antibodies targeting multiple epitopes on the envelope glycoproteins associated with broad cross-neutralization. J Virol. 2014;88(24):14278–14288. | |
Beaumont E, Patient R, Hourioux C, Dimier-Poisson I, Roingeard P. Chimeric hepatitis B virus/hepatitis C virus envelope proteins elicit broadly neutralizing antibodies and constitute a potential bivalent prophylactic vaccine. Hepatology. 2013;57(4):1303–1313. | |
McCaffrey K, Boo I, Poumbourios P, Drummer HE. Expression and characterization of a minimal hepatitis C virus glycoprotein E2 core domain that retains CD81 binding. J Virol. 2007;81(17):9584–9590. | |
McCaffrey K, Gouklani H, Boo I, Poumbourios P, Drummer HE. The variable regions of hepatitis C virus glycoprotein E2 have an essential structural role in glycoprotein assembly and virion infectivity. J Gen Virol. 2011;92(pt 1):112–121. | |
Verstrepen BE, Depla E, Rollier CS, et al. Clearance of genotype 1b hepatitis C virus in chimpanzees in the presence of vaccine-induced E1-neutralizing antibodies. J Infect Dis. 2011;204(6):837–844. | |
Leroux-Roels G, Depla E, Hulstaert F, et al. A candidate vaccine based on the hepatitis C E1 protein: tolerability and immunogenicity in healthy volunteers. Vaccine. 2004;22(23–24):3080–3086. | |
Roohvand F, Kossari N. Advances in hepatitis C virus vaccines, part two: advances in hepatitis C virus vaccine formulations and modalities. Expert Opin Ther Pat. 2012;22(4):391–415. | |
Barnes E, Folgori A, Capone S, et al. Novel adenovirus-based vaccines induce broad and sustained T cell responses to HCV in man. Sci Transl Med. 2012;4(115):115ra1. | |
Swadling L, Capone S, Antrobus RD, et al. A human vaccine strategy based on chimpanzee adenoviral and MVA vectors that primes, boosts, and sustains functional HCV-specific T cell memory. Sci Transl Med. 2014;6(261):261ra153. | |
Akondy RS, Monson ND, Miller JD, et al. The yellow fever virus vaccine induces a broad and polyfunctional human memory CD8+ T cell response. J Immunol. 2009;183(12):7919–7930. | |
Northfield JW, Loo CP, Barbour JD, et al. Human immunodeficiency virus type 1 (HIV-1)-specific CD8+ T(EMRA) cells in early infection are linked to control of HIV-1 viremia and predict the subsequent viral load set point. J Virol. 2007;81(11):5759–5765. | |
National Institute of Allergy and Infectious Diseases. Staged phase I/II hepatitis C prophylactic vaccine. In: ClinicalTrials.gov [Internet]. Bethesda, MD: National Library of Medicine (US); 2015 [cited January 6, 2015]. Available from: https://clinicaltrials.gov/show/NCT01436357. | |
ReiThera Srl. Study of a new MVA vaccine for hepatitis C virus. In: ClinicalTrials.gov [Internet]. Bethesda, MD: National Library of Medicine (US); 2015 [cited January 15, 2015]. Available from: https://clinicaltrials.gov/ct2/show/NCT01296451. | |
Folgori A, Capone S, Ruggeri L, et al. A T-cell HCV vaccine eliciting effective immunity against heterologous virus challenge in chimpanzees. Nat Med. 2006;12(2):190–197. | |
Fattori E, Zampaglione I, Arcuri M, et al. Efficient immunization of rhesus macaques with an HCV candidate vaccine by heterologous priming-boosting with novel adenoviral vectors based on different serotypes. Gene Ther. 2006;13(14):1088–1096. | |
Park SH, Shin EC, Capone S, et al. Successful vaccination induces multifunctional memory T-cell precursors associated with early control of hepatitis C virus. Gastroenterology. 2012;143(4):1048–60. e4. | |
Klade CS, Wedemeyer H, Berg T, et al. Therapeutic vaccination of chronic hepatitis C nonresponder patients with the peptide vaccine IC41. Gastroenterology. 2008;134(5):1385–1395. | |
Firbas C, Boehm T, Buerger V, et al. Immunogenicity and safety of different injection routes and schedules of IC41, a Hepatitis C virus (HCV) peptide vaccine. Vaccine. 2010;28(12):2397–2407. | |
Drane D, Maraskovsky E, Gibson R, et al. Priming of CD4+ and CD8+ T cell responses using a HCV core ISCOMATRIX vaccine: a phase I study in healthy volunteers. Hum Vaccin. 2009;5(3):151–157. | |
Polakos NK, Drane D, Cox J, et al. Characterization of hepatitis C virus core-specific immune responses primed in rhesus macaques by a nonclassical ISCOM vaccine. J Immunol. 2001;166(5):3589–3598. | |
Chmielewska AM, Naddeo M, Capone S, et al. Combined adenovirus vector and hepatitis C virus envelope protein prime-boost regimen elicits T cell and neutralizing antibody immune responses. J Virol. 2014;88(10):5502–5510. | |
Rollier CS, Paranhos-Baccala G, Verschoor EJ, et al. Vaccine-induced early control of hepatitis C virus infection in chimpanzees fails to impact on hepatic PD-1 and chronicity. Hepatology. 2007;45(3):602–613. | |
Youn JW, Hu YW, Tricoche N, et al. Evidence for protection against chronic hepatitis C virus infection in chimpanzees by immunization with replicating recombinant vaccinia virus. J Virol. 2008;82(21):10896–10905. | |
Elmowalid GA, Qiao M, Jeong SH, et al. Immunization with hepatitis C virus-like particles results in control of hepatitis C virus infection in chimpanzees. Proc Natl Acad Sci U S A. 2007;104(20):8427–8432. | |
Beaumont E, Roingeard P. Prospects for prophylactic hepatitis C vaccines based on virus-like particles. Hum Vaccin Immunother. 2013; 9(5):1112–1118. | |
Angus AG, Patel AH. Immunotherapeutic potential of neutralizing antibodies targeting conserved regions of the HCV envelope glycoprotein E2. Fut Microbiol. 2011;6(3):279–294. | |
Drummer HE, Boo I, Maerz AL, Poumbourios P. A conserved Gly436-Trp-Leu-Ala-Gly-Leu-Phe-Tyr motif in hepatitis C virus glycoprotein E2 is a determinant of CD81 binding and viral entry. J Virol. 2006;80(16):7844–7853. | |
Owsianka AM, Timms JM, Tarr AW, et al. Identification of conserved residues in the E2 envelope glycoprotein of the hepatitis C virus that are critical for CD81 binding. J Virol. 2006;80(17):8695–8704. | |
Castelli M, Clementi N, Sautto GA, et al. HCV E2 core structures and mAbs: something is still missing. Drug Discov Today. 2014;19(12):1964–1970. | |
Owsianka A, Tarr AW, Juttla VS, et al. Monoclonal antibody AP33 defines a broadly neutralizing epitope on the hepatitis C virus E2 envelope glycoprotein. J Virol. 2005;79(17):11095–11104. | |
Tarr AW, Owsianka AM, Timms JM, et al. Characterization of the hepatitis C virus E2 epitope defined by the broadly neutralizing monoclonal antibody AP33. Hepatology. 2006;43(3):592–601. | |
Potter JA, Owsianka AM, Jeffery N, et al. Toward a hepatitis C virus vaccine: the structural basis of hepatitis C virus neutralization by AP33, a broadly neutralizing antibody. J Virol. 2012;86(23):12923–12932. | |
Kong L, Giang E, Nieusma T, et al. Structure of hepatitis C virus envelope glycoprotein E2 antigenic site 412 to 423 in complex with antibody AP33. J Virol. 2012;86(23):13085–13088. | |
Li Y, Pierce BG, Wang Q, et al. Structural basis for penetration of the glycan shield of hepatitis c virus E2 glycoprotein by a broadly neutralizing human antibody. J Biol Chem. 2015; 290(16):10117–10125. | |
Kong L, Giang E, Robbins JB, et al. Structural basis of hepatitis C virus neutralization by broadly neutralizing antibody HCV1. Proc Natl Acad Sci U S A. 2012;109(24):9499–9504. | |
Meola A, Tarr AW, England P, et al. Structural flexibility of a conserved broadly neutralizing epitope in hepatitis C virus glycoprotein E2. J Virol. 2015;89:2170–2181. | |
Tarr AW, Owsianka AM, Jayaraj D, et al. Determination of the human antibody response to the epitope defined by the hepatitis C virus-neutralizing monoclonal antibody AP33. J Gen Virol. 2007; 88(11):2991–3001. | |
Dhillon S, Witteveldt J, Gatherer D, et al. Mutations within a conserved region of the hepatitis C virus E2 glycoprotein that influence virus-receptor interactions and sensitivity to neutralizing antibodies. J Virol. 2010;84(11):5494–5507. | |
Keck ZY, Angus AG, Wang W, et al. Non-random escape pathways from a broadly neutralizing human monoclonal antibody map to a highly conserved region on the hepatitis C virus E2 glycoprotein encompassing amino acids 412–423. PLoS Pathog. 2014;10(8):e1004297. | |
Keck Z, Wang W, Wang Y, et al. Cooperativity in virus neutralization by human monoclonal antibodies to two adjacent regions located at the amino terminus of hepatitis C virus E2 glycoprotein. J Virol. 2013;87(1):37–51. | |
Deng L, Ma L, Virata-Theimer ML, et al. Discrete conformations of epitope II on the hepatitis C virus E2 protein for antibody-mediated neutralization and nonneutralization. Proc Natl Acad Sci U S A. 2014; 111(29):10690–10695. | |
Krey T, Meola A, Keck ZY, Damier-Piolle L, Foung SK, Rey FA. Structural basis of HCV neutralization by human monoclonal antibodies resistant to viral neutralization escape. PLoS Pathog. 2013;9(5):e1003364. | |
Drummer HE, Wilson KA, Poumbourios P. Identification of the hepatitis C virus E2 glycoprotein binding site on the large extracellular loop of CD81. J Virol. 2002;76(21):11143–11147. | |
Harman C, Zhong L, Ma L, et al. A view of the E2-CD81 interface at the binding site of a neutralizing antibody against hepatitis C virus. J Virol. 2015;89(1):492–501. | |
Keck ZY, Xia J, Wang Y, et al. Human monoclonal antibodies to a novel cluster of conformational epitopes on HCV E2 with resistance to neutralization escape in a genotype 2a isolate. PLoS Pathog. 2012; 8(4):e1002653. | |
Owsianka AM, Tarr AW, Keck ZY, et al. Broadly neutralizing human monoclonal antibodies to the hepatitis C virus E2 glycoprotein. J Gen Virol. 2008;89(3):653–659. | |
Giang E, Dorner M, Prentoe JC, et al. Human broadly neutralizing antibodies to the envelope glycoprotein complex of hepatitis C virus. Proc Natl Acad Sci U S A. 2012;109(16):6205–6210. | |
Law M, Maruyama T, Lewis J, et al. Broadly neutralizing antibodies protect against hepatitis C virus quasispecies challenge. Nat Med. 2008;14(1):25–27. | |
Sabo MC, Luca VC, Prentoe J, et al. Neutralizing monoclonal antibodies against hepatitis C virus E2 protein bind discontinuous epitopes and inhibit infection at a postattachment step. J Virol. 2011;85(14):7005–7019. | |
Keck ZY, Saha A, Xia J, et al. Mapping a region of hepatitis C virus E2 that is responsible for escape from neutralizing antibodies and a core CD81-binding region that does not tolerate neutralization escape mutations. J Virol. 2011;85(20):10451–10463. | |
Roccasecca R, Ansuini H, Vitelli A, et al. Binding of the hepatitis C virus E2 glycoprotein to CD81 is strain specific and is modulated by a complex interplay between hypervariable regions 1 and 2. J Virol. 2003;77(3):1856–1867. | |
Werkhoven PR, van de Langemheen H, van der Wal S, Kruijtzer JAW, Liskamp RMJ. Versatile convergent synthesis of a three peptide loop containing protein mimic of whooping cough pertactin by successive Cu(I)-catalyzed azide alkyne cycloaddition on an orthogonal alkyne functionalized TAC-scaffold. J Pept Sci. 2014;20(4):235–239. | |
Correia BE, Bates JT, Loomis RJ, et al. Proof of principle for epitope-focused vaccine design. Nature. 2014;507(7491):201–206. | |
Torresi J, Fischer A, Grollo L, Zeng W, Drummer H, Jackson DC. Induction of neutralizing antibody responses to hepatitis C virus with synthetic peptide constructs incorporating both antibody and T-helper epitopes. Immunol Cell Biol. 2007;85(2):169–173. | |
Alfonso M, Díaz A, Hernández AM, et al. An anti-idiotype vaccine elicits a specific response to N-glycolyl sialic acid residues of glycoconjugates in melanoma patients. J Immunol. 2002;168(5):2523–2529. | |
Bendandi M. Idiotype vaccines for lymphoma: proof-of-principles and clinical trial failures. Nat Rev Cancer. 2009;9(9):675–681. | |
Owsianka A, Jeffery N, di Lorenzo C, Patel A. Towards a hepatitis C vaccine; isolation of an anti-idiotypic antibody that mimics a broadly neutralizing epitope on the E2 glycoprotein. In: 21st International Symposium on Hepatitis C virus and Related Viruses; Banff, Canada; 2014. | |
Pumpens P, Grens E. HBV core particles as a carrier for B cell/T cell epitopes. Intervirology. 2001;44(2–3):98–114. | |
Oliveira GA, Wetzel K, Calvo-Calle JM, et al. Safety and enhanced immunogenicity of a hepatitis B core particle Plasmodium falciparum malaria vaccine formulated in adjuvant Montanide ISA 720 in a phase I trial. Infect Immun. 2005;73(6):3587–3597. | |
Ibañez LI, Roose K, De Filette M, et al. M2e-displaying virus-like particles with associated RNA promote T helper 1 type adaptive immunity against influenza A. PLoS One. 2013;8(3):e59081. | |
Sanofi. Safety Study of Recombinant M2e Influenza-A Vaccine in Healthy Adults (FLU-A) [Internet]; 2015 [cited January 6, 2015]. Available from: https://clinicaltrial.gov/show/NCT00819013. | |
Netter HJ, Macnaughton TB, Woo WP, Tindle R, Gowans EJ. Antigenicity and immunogenicity of novel chimeric hepatitis B surface antigen particles with exposed hepatitis C virus epitopes. J Virol. 2001;75(5):2130–2141. | |
Vaccari M, Poonam P, Franchini G. Phase III HIV vaccine trial in Thailand: a step toward a protective vaccine for HIV. Expert Rev Vaccines. 2010;9(9):997–1005. | |
Pettersen EF, Goddard TD, Huang CC, et al. UCSF Chimera – a visualization system for exploratory research and analysis. J Comput Chem. 2004;25(13):1605–1612. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.