Back to Journals » Infection and Drug Resistance » Volume 13
Current and Future Perspectives on Isothermal Nucleic Acid Amplification Technologies for Diagnosing Infections
Authors Obande GA ![]() , Banga Singh KK
, Banga Singh KK
Received 19 October 2019
Accepted for publication 16 November 2019
Published 12 February 2020 Volume 2020:13 Pages 455—483
DOI https://doi.org/10.2147/IDR.S217571
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Sahil Khanna
Video abstract presented by Godwin Attah Obande.
Views: 4120
Godwin Attah Obande,1,2 Kirnpal Kaur Banga Singh1
1Department of Medical Microbiology & Parasitology, School of Medical Sciences, Health Campus, Universiti Sains Malaysia, Kelantan, Malaysia; 2Department of Microbiology, Faculty of Science, Federal University Lafia, Lafia, Nasarawa State, Nigeria
Correspondence: Kirnpal Kaur Banga Singh
Department of Medical Microbiology & Parasitology, School of Medical Sciences, Health Campus, Universiti Sains Malaysia, Kubang Kerian 16150, Kelantan, Malaysia
Tel +60 97676257
Fax +60 97676289
Email [email protected]
Abstract: Nucleic acid amplification technology (NAAT) has assumed a critical position in disease diagnosis in recent times and contributed significantly to healthcare. Application of these methods has resulted in a more sensitive, accurate and rapid diagnosis of infectious diseases than older traditional methods like culture-based identification. NAAT such as the polymerase chain reaction (PCR) is widely applied but seldom available to resource-limited settings. Isothermal amplification (IA) methods provide a rapid, sensitive, specific, simpler and less expensive procedure for detecting nucleic acid from samples. However, not all of these IA techniques find regular applications in infectious diseases diagnosis. Disease diagnosis and treatment could be improved, and the rapidly increasing problem of antimicrobial resistance reduced, with improvement, adaptation, and application of isothermal amplification methods in clinical settings, especially in developing countries. This review centres on some isothermal techniques that have found documented applications in infectious diseases diagnosis, highlighting their principles, development, strengths, setbacks and imminent potentials for use at points of care.
Keywords: amplification, diagnosis, disease, isothermal, polymerase chain reaction, resistance
Introduction
Infectious diseases are caused by viruses, fungi, bacteria, and parasites. They infect a lot of people and cause numerous deaths annually, especially in developing countries.1,2 Factors such as increasing mobility of humans, climate change, increase in the exchange of goods and services across different borders, and globalisation has aided infectious diseases transmission and dynamics across the world.3,4
Disease diagnosis plays critical roles in patient care. It helps with knowing the actual etiological agent in cases of infection, while also making it possible to identify infections arising from previously unknown, as well as new pathogens. With proper identification of the causal agent, effective therapy can be instituted and monitored, and efficient surveillance systems can be established to control disease spread. Although infectious disease patterns are changing rapidly, proper diagnosis is one way to monitor the changing trend continuously.5
Absence of proper, efficient and accurate diagnosis of infectious diseases portend severe problems for healthcare. Accurate and rapid diagnosis of a disease is crucial in determining what kind of treatment should be administered by health workers. Some avoidable deaths have occurred where health workers administered the wrong treatments as a result of the wrong diagnosis. In a study by Lu et al6 25.8% (32/124) avoidable deaths were recorded in the emergency department of a health facility over three years, seven of which were due to misdiagnosis and eight due to delayed diagnosis. In another multi-centre study of mortality associated with misdiagnosis, the researchers reported misdiagnosis in 11.6% (113/974) patients with 15% in-hospital mortality.7 In some other instances, it is not uncommon for treatment to commence later than is required for the most efficient clinical intervention as a result of inadequate testing facilities,8 delays in diagnosis of disease9 and slow diagnostic process.10 For instance, the use of stool culture in the diagnosis of Clostridium difficile diarrhoea takes 2–3 days to complete, thereby delaying commencement of treatment.10 When a patient gets treated with antibiotics without a proper and accurate diagnosis of causal agents involved, the outcome could be ineffective and antimicrobial resistance could result, leading to pathogens becoming more difficult to treat than they were previously. This problem results from the overuse of effective antimicrobial agents and drugs,11 which are becoming fewer by the day. Not less than half the population of adults and children who suffer upper respiratory tract infection are treated with antibiotics, even though the majority of such illnesses result from viral infections12–14 Antimicrobial resistance has recently become a serious problem, with increasing magnitude across the globe.11
From the simple microscope invented by Antonie van Leeuwenhoek to the present use of sophisticated technologies, identification of pathogens has evolved greatly. This evolution has been made possible by advancements in the study and understanding of different scientific fields, including Molecular Biology. Hence, the diagnosis of diseases can now be conducted from various sample types.5 Available automated systems used for biochemical testing such as VITEK 2 (bioMérieux, Marcy l’Etoile, France) and Analytical Profile Index 20 (bioMérieux, Marcy l’Etoile, France) report low sensitivity, as identification is phenotypic. The cost also makes it unaffordable in resource-constrained areas.15 Simple and rapid identification techniques have been established over time, which base identification on the molecular characteristics of microbes. NAAT is an essential molecular identification technique applied in many aspects of healthcare such as disease diagnosis and investigations into genetic disorders.16 These methods are sensitive, specific and require a shorter time to complete than the older culture methods. Polymerase chain reaction (PCR) which pioneered the revolution in molecular diagnosis, was developed in 1983 by Kary Mullis.17,18 Since then, various versions of PCR have been developed, such as quantitative, nested, real-time, multiplex, and immunocapture PCR. The advent of the PCR appeared to ease pathogen identification, but its cumbersome procedure, sophisticated equipment requirement, operation at varying temperatures and the extent of expertise needed for its operation has limited its usage.17,19 One advantage of PCR-based assays, however, is that sequencing tools can be used to analyse the resulting amplification products, availing much more information about the organism.20 Interestingly, molecular methods have been used to identify some organisms that cannot be cultured in the laboratory. For instance, the Hepatitis C virus (HCV) and Tropheryma whipplei were first detected using molecular methods. The advent of nucleotide sequencing and pyrosequencing have also assisted in both the identification of pathogens and understanding the genetic relatedness of pathogens.5 Real-time PCR has facilitated nucleic acid quantification through fluorescent-labelled probes and has been applied in the quantification of pathogens with reported accuracy, precision, ease of standardisation and rapidity. Despite this, real-time PCR still requires expensive instrumentation and special expertise to perform and is hence, not within reach of resource-constrained societies.21 Real-time PCR involves the use of techniques that requires trained personnel with adequate knowledge of its usage and interpretation and as such might not be easily applicable in all settings.17,19
Though many diagnostic methods now exist that facilitate more accurate diagnosis of disease agents, improvement of existing methods with capabilities for higher rapidity, accuracy, ease of use and less cost is still a crucial need. Point of care diagnosis of disease, which does not require sophisticated equipment or visiting the hospital environment, will enhance healthcare and surveillance if they are made simple enough to be used even with minimal training. This review takes a look at some techniques applied in infectious diseases diagnosis and highlights the important types of isothermal methods currently in use. Future directions of these simpler methods have been highlighted to encourage the development of simpler technologies, which will greatly aid healthcare delivery, especially in resource-constrained areas.
Isothermal Amplification Methods
One reason why nucleic acid amplification appears to be cumbersome is the need for initial extraction of the pathogen’s nucleic acid, amplification of the sequence of interest in the nucleic acid, and then analysis of products obtained,5 especially when the use of gel electrophoresis is involved. Less cumbersome and more straightforward non-PCR based isothermal techniques such as Nucleic Acid Sequence Based Amplification (NASBA),22 Sequence Mediated Amplification of RNA Technology (SMART),23 Strand Displacement Amplification (SDA),24 Loop-mediated Isothermal Amplification (LAMP)16 and most recently the Multiple Cross Displacement Amplification (MCDA)25 have been developed (Table 1). These methods operate at a uniform temperature without the need for variation during the process, therefore eliminating the use of thermocyclers. Also, real-time readings of amplification are possible with these methods, and amplified products detected by measuring turbidity or by visual inspection for colour change. This capability eliminates the need for gel electrophoresis. IA methods could become more popular than PCR soon, owing to their low energy requirement and simplicity which allows for their possible integration into simple, compact systems.26 IA methods differ from one another in features such as the number of primers and enzymes, the temperature of amplification and template types used. Some POC platforms based on IA have been developed with some already commercialised (Table 2).
 |  |  |
Table 1 Summary of Some Clinical Studies with IA Methods |
 |  |  |
Table 2 Commercial Diagnostic Kits, Platforms and Systems Using IA Techniques |
Loop-Mediated Isothermal Amplification (LAMP)
LAMP is an IA method first developed and reported in the year 2000.16 LAMP gained acceptance because it is highly specific, sensitive and rapid27 coupled with its isothermal nature, the use of high strand displacing DNA polymerase enzyme of Bacillus stearothermophilus (Bst), all facilitating amplification of minimal number of DNA into as much as 109 copies within one hour.16 LAMP assay requires four specific primers. These include the inner and outer primers that recognise a total of six different sequences of a DNA. Backward Inner Primer (BIP) and Forward Inner Primer (FIP) make up the inner primers. The assay also includes Forward Loop (LF) and Backward Loop (LB) primers for more rapid amplification (Figure 1). Similarly, Backward outer (B3) and Forward outer (F3) make up the outer primers. Sense and antisense sequences found on FIP (F2 and F1c) and BIP (B2 and B1c) interact with the initiation points for LAMP.16 The LAMP procedure consists of a single amplification period at between 60 to 65°C for 1 hr.
 |
Figure 1 Schematic illustration of LAMP. The reaction begins with annealing of the FIP and its extension by polymerase enzyme to form a new strand of DNA which is displaced by extension of F3 (non-cyclic phase). The new strand forms a loop at the 5ʹ end with compliment sequences at F1 and F1c. BIP also anneals, and a similar process as of FIP releases another new strand that forms a double stem-loop structure at both ends. A new double stem-loop DNA with a sequence complementary to the first strand and another stem-loop with double the length of the previous one is formed subsequently from self-primed extension by the DNA strand, and by annealing of FIP and BIP coupled with extensions and displacements. As products from subsequent steps get involved in the reaction, DNA structures of different sizes bearing inverted repeats on the same strand result with multiple loops formed as a result of alternately repeated sequences on the same strand. Reproduced from Notomi T, Okayama H, Masubuchi H, et al. Loop-mediated isothermal amplification of DNA. Nucleic Acids Res. 2000;28(12):E63–E63.16 |
Nucleic acid amplification by LAMP begins with the binding of the sense strand of FIP (F2) to the F2c of the target region, resulting in the synthesis of a complementary strand (Figure 1). This strand is displaced when F3 binds to F3c of the target sequence and initiates synthesis, resulting in a FIP-linked displaced strand with a loop-structured end. BIP initiates synthesis of another strand using the FIP-linked strand as a template. Displacement of the synthesised strand through B3-primed synthesis results in dumb-bell structured material which converts into a stem-loop by self-priming. The second stage utilises the stem-loop material in a series of repeated amplification cycles to yield a vast number of amplified strands of various stem lengths from the target sequence.16 Hence, amplification occurs by two types of repeated cycles around the formed loop: self-primed elongation at the 3ʹ end of the template via the stem-loop structure, and elongation occasioned by the inner primers around the loop.17
Detection of products is by gel electrophoresis, fluorescence detection or directly by turbidity due to magnesium pyrophosphate that accumulates as an end product of amplification. Intercalating fluorescent dyes like Pico and SYBR Green are applied to enhance direct visual detection of amplified products.28–30 Amplified products form bands which appear like a ladder on agarose gel electrophoresis.17,31
LAMP has the capacity for incorporation into assays for medical investigation, genetic testing, environmental testing and rapid testing of food products, especially in resource-constrained environments.17 LAMP has found application in detecting resistance genes such as blaKPC and blaNDM-1 in E. coli, Klebsiella pneumonia and Acinetobacter baumannii32 as well as identification of bacteria like E. coli,33 Mycobacteria,34 Vibrio parahemolyticus, Salmonella spp35 and A. baumannii.36 LAMP with a detection limit of 1ng has also been successfully used for rapid detection of Porphyromonas gingivalis and Aggregatibacter actinomycetemcomitans, both of which cause periodontitis in humans.37 LAMP assays for detecting yeasts,38 influenza viruses39 and Plasmodium have been developed.40 Different companies have also designed commercial LAMP kits (Table 2) for the detection of Salmonella,41–43 verotoxin-producing Escherichia coli44–46 and Campylobacter.47
Different studies have reported high specificity and sensitivity of LAMP assay. In a systematic review of 16 studies (6,979 samples) which evaluated the accuracy of LAMP in detecting Clostridium difficile disease in humans, LAMP was reported more sensitive and specific than anaerobic toxigenic culture (TC).48 TC (sensitivity of 90–100%, specificity of 98–100%) is one of the reference tests used for evaluation of novel testing methods for Clostridium difficile.49,50 Two different publications reported higher sensitivity of LAMP than PCR designed for Salmonella enterica with egg constituents unable to inhibit the assay.41,42
Researchers are now developing miniaturised versions of the LAMP assay. A reported study which used a commercial poly(methyl methacrylate) (PMMA) microchip with sample cross channel with a width of 100µm and depth of 30µm, achieved amplification, detection and analysis of a prostate-specific antigen gene using a single device.51 The study used LAMP for amplification, electrophoresis for detection of amplified products and analysis by visual observation, aided by SYBR Green I dye. It, therefore, follows that LAMP assay can be developed for use on a single device or chip wherever and whenever needed. Presently, commercial LAMP kits are available. Japan has approved a LAMP kit used for identifying and monitoring disease agents.52
LAMP is amenable to contamination quite easily, one reason being the number of pipetting steps and the use of multiple, specific primers. Performing LAMP, therefore, requires enormous care and strict adherence to aseptic techniques to prevent carryover contamination. Using DNA binding dyes for detection of amplified products preclude the opening of the reaction tubes post-amplification. Stains such as SYBR Green I, SYBR safe and calcein added to the reaction mixture before the amplification step, can aid visual detection of amplified products. Addition of Uracil-DNA-glycosylase (UDG-LAMP) before the amplification process has also been reported to prevent carryover contamination.53 In a two-step process, deoxyuridine triphosphate (dUTP) is added to all reaction mixtures for LAMP to enable incorporation of uracil to the resulting amplicons.48 In subsequent LAMP reactions, the addition of UDG to reaction mixtures ensures that uracil-containing LAMP amplicons from previous reactions are cleaved and degraded through hydrolysis at the phosphate backbone, leaving only the target DNA available for amplification. Heat is then used to deactivate the UDG enzyme. A second limitation of LAMP is the difficulty faced with multiplexing. The multiplex assay has not recorded as much success with LAMP as with PCR, due to the number of primers used.19 There is also the challenge of target selection and primer design. Appropriate targets could be missed by the software during the process, requiring manual intervention.54 False-positive amplification due to hybridisation by primers is also common.55 Hopefully, as the patent license (EP 1020534 B) for this technology expires in 2019, LAMP would experience more modifications. LAMP primers can be designed using software. Examples are the paid LAMP Designer (http://www.optigene.co.uk/lamp-designer/) by OptiGene Limited (England), and PrimerExplorer (Version 5, https://primerexplorer.jp/e/) provided free of charge by Eiken Chemical Company Limited (Japan).
Helicase-Dependent Amplification (HDA)
HDA was developed in 2004 and is a method that excludes the temperature cycling process necessary for the separation of double-stranded DNA (dsDNA). Instead, the DNA helicase enzyme separates the dsDNA. This separation facilitates primer annealing and elongation by polymerase enzyme.56 Helicase enzymes were found previously in E. coli in 1976.57,58 HDA is similar to the process of DNA replication in living organisms.26,59 The process begins when the helicase enzyme disrupts the hydrogen bonds of dsDNA and separates them using energy derived from ATP hydrolysis. The resulting single-stranded DNA (ssDNA) is covered by single-strand binding proteins (SSBs) which stabilise the strand and prevents reannealing between the two separated strands (Figure 2). Next, two primers bearing specific sequences anneal to the target regions on the ssDNA template. Extension by DNA polymerase enzyme takes place, leading to the addition of deoxynucleotide-triphosphates to produce two dsDNA. The resulting dsDNA undergo the same replication cycle, acting as templates. The first HDA assay was performed at 37°C and was able to amplify 103 copies of genomic DNA by over ten million folds, in addition to being able to directly amplifying DNA in human blood.56 An existing patent (EP 1539979 B) for this technology will remain valid until 2023.
 |
Figure 2 Helicase-dependent amplification. The process begins with strand separation by helicase, primer annealing, extension by polymerase and ends with two copies of the target. Adapted from Vincent M, Xu Y, Kong H. Helicase-dependent isothermal DNA amplification. EMBO Rep. 2004;5(8):795–800. Copyright © 2004 European Molecular Biology Organization.56 |
HDA has undergone some improvements over the times. An et al60 achieved amplification at 60-65°C by replacing the UvrD helicase used in the first reported assay56 with a thermostable combination of MutL homologue and UvrD helicase of Thermoanaerobacter tengcongensis. The resulting HDA assay was more specific and sensitive than the first reported assay.60 HDA was further enhanced by fusing helicase enzyme with DNA polymerase. The resulting enzyme named as hemilase, increased the length of amplified fragments from the initial 70-120bp to 2.3 kilobases in the improved HDA.61 Goldmeyer et al62 developed and reported the first thermophilic HDA (tHDA) to be coupled with reverse-transcription in a single tube, which was able to successfully amplify Ebola virus-armored RNA target by up to one million-fold within 10 mins, in addition to detecting as low as 20 Enterovirus particles per reaction.62 A helicase dependent OnChip method which was able to identify Staphylococcus aureus resistant to methicillin (MRSA) and Neisseria gonorrhoeae in single and duplex reactions respectively has also been developed.63 This was achieved by detecting the mecA gene of S. aureus and the pivNG gene of N. gonorrhoea. Huang et al64 designed an electricity-free, sensitive HDA assay which was successfully applied to detect Clostridium difficile from 8 diarrhoea patients stool samples, with a detection limit of 1.25 x 10−2 pg. The disposable low-cost platform consisted of a thermoplastic reaction chip made with commercial toe warmers, insulated with Styrofoam. The design was reportedly cheaper and less material-consuming than other traditional methods.64 Amplified products from HDA can be visualised in real-time by fluorescence and by using agarose gel electrophoresis.26,56
Some limitations of HDA include sample contamination, low reaction temperature, generation of non-specific amplification products due to primer mispairing and amplification inhibition.26,65 However, continuous improvement of HDA can surmount these problems. Primers for HDA can be designed using PrimerQuestSM® provided by Integrated DNA Technologies (Coralville, USA).64 A report also used the Primer3 program for reverse transcription HDA (RT-HDA) primer design.62 Two versions of this program are now available: Primer3Plus (http://primer3plus.com) and Primer3web (http://primer3.ut.ee/).
Signal-Mediated Amplification of RNA Technology (SMART)
The SMART technique which was first reported in 2001 by Wharam et al23 centres on amplified signals generated by the target sequence rather than direct amplification of target sequence and hence thermal cycling, or target sequence copying is not required.23 The method generates signals that are highly target-dependent, specific for different targets and can discriminate when there is a change in target-borne base pairs.66 The assay involves one oligonucleotide (single-stranded) probe for extending sequences and another that serves as the template. They both comprise a long end which anneals to the target and another shorter end with which they anneal to each other, only when the target sequence is present. When this happens, the two probes and the target form a three-way junction structure.23 Two different enzymes are involved in this method, namely T7 RNA polymerase and Bacillus stearothermophilus (Bst) DNA polymerase. A double-stranded promoter sequence of T7 RNA results when DNA polymerase synthesizes bases complementary to the single-strand (ss) of the opposite probe, thereby increasing the length of the extension probe (Figure 3). RNA signals are generated by T7 RNA polymerase only when the promoter sequence on the template probe becomes double-stranded (ds). RNA signal amplification occurs when the generated signal binds to another template oligonucleotide, from which DNA polymerase extension yields a double-stranded (ds) promoter. Additional rounds of RNA signal extension and transcription increases the generated signal, which can then be detected using a method known as enzyme-linked oligosorbent assay (ELOSA). An oligonucleotide linked with an enzyme and another labelled with biotin hybridises RNA signals in a sequence-specific manner to form a complex which is then captured in standard plate reader wells coated with streptavidin.23,67 The end detection system of the SMART method is by visualisation and measurement of the colour change that results in the well-contained solutions upon application of the substrate.67 Hence, this technique does not require gel electrophoresis and can simultaneously quantify multiple samples.
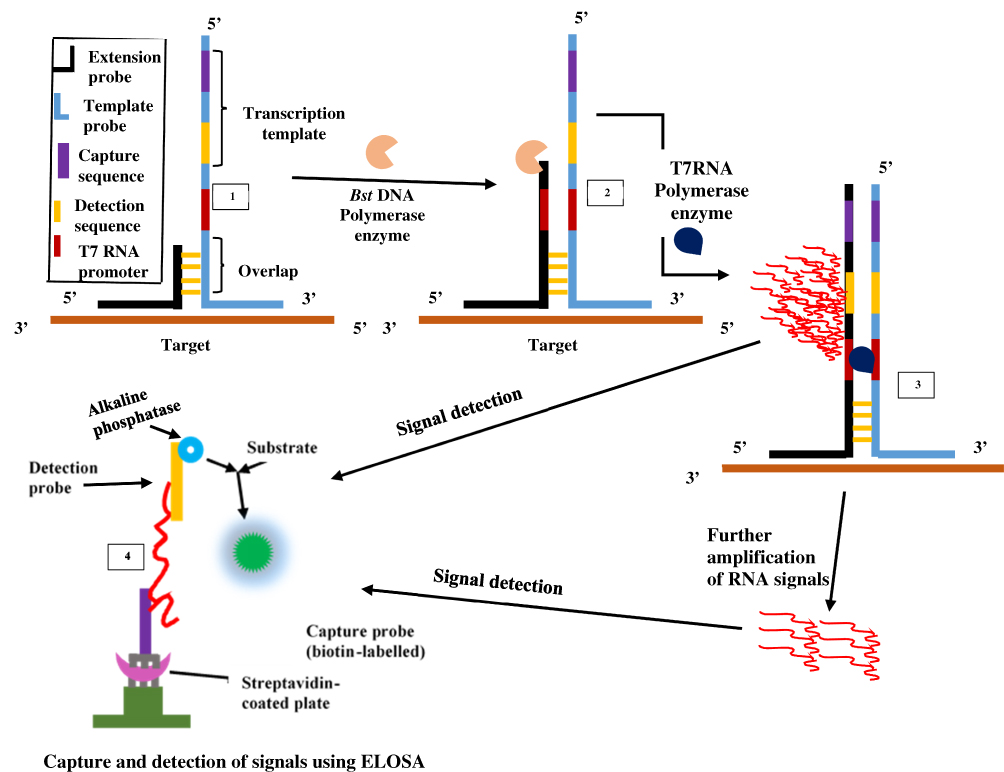 |
Figure 3 Signal-mediated amplification of RNA technology. A 3WJ forms; DNA polymerase extends the extension probe; T7 RNA polymerase promoter is formed, and T7 RNA polymerase generates signals (1st RNA). Signals can be transcribed by T7 RNA polymerase and detected by ELOSA (option 2) or further amplified to yield multiple 2nd RNA signals (option 1) which can then be detected. Adapted with permission from Springer Nature. Wharam SD, Hall MJ, Wilson WH. Detection of virus mRNA within infected host cells using an isothermal nucleic acid amplification assay: marine cyanophage gene expression within Synechococcus sp. Virol J. 2007;4(1):52.68 |
Although SMART is not as sensitive as PCR, it has been reported to be more tolerant to samples of various types like genomic DNA and total RNA both in E. coli, crude cultures and non-target DNA.67 This technology has found application in detecting phage viruses of marine Synechococcus;66 differentiation of virus-infected hosts from healthy hosts by detecting gene expression;68 and detection of coa (coagulase) and mecA genes of MRSA. Since the signals generated by this method is the same for all targets, the technique can be incorporated into microbiological detection of any target once the three-way junction structure for the target sequence can be designed.69 Patents for the SMART assay include EP-B-0, 666.927, AU672367 and WO99/37806.23 There is currently no reported demonstration in the published literature regarding multiplexing capability of SMART.
Nucleic Acid Sequence-Based Amplification (NASBA)
Unlike SMART, NASBA is a method that centres on actual amplification of ssRNA and DNA. It is most suitable for detecting RNA targets such as messenger RNA, ribosomal RNA and genomic RNA.67,70,71 It is a method related to transcription-mediated amplification72 and self-sustained sequence replication (3SR) developed by Guatelli et al.73 It was first introduced in 199122 and cannot be used for the amplification of double-stranded DNAs unless they are first denatured.59 NASBA requires only four to five cycles to achieve up to one million-fold amplification of RNA copies within 90 mins and does not require specialised equipment to perform.22,67 It works similarly to retroviral replication of RNA using complementary DNA intermediates (Figure 4). The process comprises three thermolabile enzymes, namely T7 DNA dependent RNA polymerase (DdRp), RNase H and avian myeloblastosis virus (AMV) reverse transcriptase, and requires two primers (P1 and P2). Two denaturation steps at 95°C are necessary to amplify a DNA target.71 Products amplified by the NASBA technique can be detected by gel electrophoresis,74 enzyme-linked gel assay (ELGA)75 and electro-chemiluminescence (ECL).76 NASBA-coupled molecular beacon for real-time detection of RNA77 has also been reported.
 |
Figure 4 Nucleic acid sequence-based amplification. P1 anneals to sense RNA, then extension by AMV reverse transcriptase enzyme follows, forming a hybrid of RNA and complementary DNA (cDNA). P2 binds to cDNA following RNA degradation by RNase H; extension by reverse transcriptase generates DNA with T7 RNA polymerase promoter sequence, which transcribes into multiple antisense RNA (-) copies by DNA dependent RNA polymerase. P2 anneals to resulting RNA (-) strands, undergoes extension and forms hybrids of RNA (-) and cDNA. RNA (-) is degraded again by RNase H while P1 binds to the new cDNA, followed by extension by reverse transcriptase to yield dsDNA bearing promoter sequence for T7 RNA polymerase and the cycle continues, generating multiple RNA copies. Adapted from Zanoli LM, Spoto G. Isothermal amplification methods for the detection of nucleic acids in microfluidic devices. Biosensors. 2012;3(1):18–43.59 |
NASBA is limited by the need for initial denaturation at a different temperature (65°C for RNA and 95°C for DNA) from the amplification reaction (41°C),59,71 and the need to add required thermolabile enzymes after the denaturation steps. Furthermore, amplification is efficient only with sequences that are 120–250 bp long. Despite these limitations, it is very suitable for diagnosis of RNA viruses and is less prone to contamination unlike PCR.78 Its low temperature of operation (41°C) is an advantage and speed of amplification (30 mins), coupled with the fact that unlike in PCR, primers used in the reaction does not significantly influence amplicon yield.
Different improvements and variants of NASBA have been developed with various applications. A commercial kit known as NucliSENS EasyQ System for HIV-1 RNA quantitation has been produced (Table 2). It was capable of detecting and quantifying HIV-1 virus RNA in clinical samples.79 The use of real-time NASBA (RT-NASBA) to detect Mycoplasma pneumoniae has been reported, though with low sensitivity than conventional NASBA.80 Other published applications of NASBA include quantitative detection (NASBA-QT) of HCV,81 Rhinovirus,82 Human papillomavirus,83 Human Cytomegalovirus in HIV-1 positive patients,84 Hepatitis A,74 Chlamydia trachomatis,85 Leishmania parasites,86 and Mycobacterium leprae.87 Selective amplification of RNA signals by NASBA is unaffected by comparable DNA sequence when present.71 Further improvements have steered establishment of duplex88–90 and multiplex88 NASBA for detecting various targets in non-clinical samples, in combination with different methods for detecting products.
NASBA, like other IA methods, has some limitations, a major one of which is the integrity of RNA recovered from samples for analysis. The labile nature of RNA could result in degradation and loss of viability in any event of delay between the time of collection, and assay performance.91 Also since the enzymes used are not thermostable, temperatures higher than 42°C can affect the enzymes and compromise the process. NASBA is also unlikely to efficiently amplify target sequences less than 100 bp and greater than 250 bp.71,92 Currently, there is no patent restriction on this IA method as the earlier patent has already expired.65
Recombinase Polymerase Amplification (RPA)
Initially, RPA was developed with a probe-based method for detecting products. It is a method that uses the enzymes recombinase and DNA polymerase together with DNA-binding proteins to achieve DNA amplification. The proteins used in RPA are involved in the process of synthesis, recombination and repair of DNA within living organisms. The first report of RPA used the large fragment of Bacillus subtilis Pol I and T4 uvsX as DNA polymerase and recombinase respectively. DNA sample pre-treatment or thermal denaturation step is not required in RPA because recombinase enzyme facilitates the separation of dsDNA.93 Recombinase acts as a catalyst that facilitates the binding of oligonucleotide primers to target DNA templates of matching sequences. RPA begins when a primer bound to recombinase scans for specific complementary sequence on the dsDNA and hybridises to it, following the separation of the double-strand by recombinase enzyme (Figure 5). Upon proper binding of the primer to template DNA, the recombinase enzyme disengages from the complex, leaving the 3ʹ end open for the DNA polymerase enzyme to attach. The displaced DNA strand is then coated by DNA-binding proteins while primer extension by DNA polymerase forms a new dsDNA which acts as a template for an additional cycle of amplification. Repeated rounds of this process generate rapid target amplification, producing several copies of DNA. Gel electrophoresis and real-time fluorescence are methods for detecting amplified products of RPA.93
 |
Figure 5 Recombinase polymerase amplification. Recombinase-bound primers scan for complementary sequence; recombinase enzyme separates the strands and SSB proteins prevent reannealing of separated strands; primers anneal, and extension by DNA polymerase takes place, leading to the formation of two dsDNA. Repetition of this cycle produces multiple copies of DNA. Adapted from Piepenburg O, Williams CH, Stemple DL, Armes NA. DNA detection using recombination proteins. PLoS Biol. 2006;4(7):e204–e204.93 |
The low-temperature requirement, coupled with simplicity of procedure and the ability of RPA to generate several copies of target DNA within a short time, makes it a suitable candidate method for incorporation into devices for disease diagnosis at points of care.91 Furthermore, RPA can be performed as a one-tube reaction with real-time detection, by including a fluorescent probe. Three kinds of fluorescent probes have been developed, namely the exo, nfo and fpg probes, which all contain a fluorophore and quencher.94 To aid primer design and solve problems associated with the process, a software known as Primer design for RPA (PrimedRPA) (https://omictools.com/primedrpa-tool) which can automate the process of RPA primer and exo probe design is now available.95 It is designed to identify conserved regions and filter off parts capable of causing a cross-reaction during RPA and is a Python-based package. Some systems have been developed in a bid to making RPA an applicable on-demand testing method. These include digital RPA known as the slipchip96,97 and microchip;98 multiplex RPA for bacteria namely Salmonella spp and Chronobacter spp,99 Listeria monocytogenes and Salmonella enterica serotype Enteritidis,100 Group B streptococci;101 parasites (Giardia, Cryptosporidium, and Entamoeba)102 and fungus (Botrytis cinerea, Pseudomonas syringae, and Fusarium oxysporum);103 mobile RPA in a suitcase which was named diagnostics-in-a-suitcase (Dias);104,105 microfluidic integrated RPA;106–109 and the one-step RPA assay.110 While a slip chip is microfluidic device lacking pumps and valves, a microchip contains pumps and valves that aid movement of sample and reaction reagents through channels linking reaction chambers. Although the initial patent (EP 0481065 B) over RPA has expired, patent rights still exist for some of the improvements it has experienced.
Some studies have reported the diagnosis of infectious diseases using RPA. Examples include in detecting and differentiating MRSA from MSSA, using multiplex RPA approach and lateral-flow strips.93 A lab-on-a-foil RPA system which consisted of a dry and liquid reagent microfluidic cartridge, a centrifugal analyser for incubation at 37°C and detection by real-time fluorescence, has been developed. The method successfully identified S. aureus mecA resistance gene.111 Francisella tularensis, which causes a zoonotic disease (tularemia) in both humans and animals has been successfully detected using a quantitative real-time RPA which showed a comparable result with PCR but was faster by 10 mins.112 Reverse transcription RPA (RT-RPA) has also been used to detect epidemic human Norovirus strain (GII.4 New Orleans).113 Yang et al114 used RPA with internal amplification control (RPA-IAC) to detect the toxR gene of Vibrio parahaemolyticus at a temperature of 37°C for 20 mins. Ahn and colleagues developed a paper chip single-step RPA for detecting foodborne pathogens.115 The method was able to simultaneously detect S. aureus, Shiga toxin-producing E. coli and Salmonella typhimurium at a low concentration (102cfu/mL) in spiked milk, at 37°C for 20 mins. An RPA coupled with lateral flow strips (RPA-LF) for detection of Coxiella burnetii was successful at specifically detecting and differentiating the target organism from other bacteria within 30 mins, combining RPA at 37°C and lateral flow at room temperature.116 The RPA-LF had a sensitivity of 10 and 7 copies per reaction for plasmid and genomic DNA, respectively. Similarly, an RPA lateral flow dipstick (RPA-LFD) method has also been successfully used to detect the aquatic cyprinid herpesvirus 2 (CyHV-2) which causes diseases in fishes, in a process that is 50 mins faster and sensitivity 100 times greater than PCR, and without cross-reaction with other aquatic viruses.117 Rohrman and Richards-Kortum118 also reported a plastic and paper-made proof-of-concept device that couples RPA with enzymatic amplification of HIV DNA at a sensitivity of 10 DNA copies per microliter and amplification time of 15 mins.
Rolling Circle Amplification (RCA)
RCA is an enzyme-based IA method. Its development was from the rolling circle replication that occurs naturally in many plasmids and viruses. It involves the amplification of short RNA or DNA primer from a circularised template DNA by RNA or DNA polymerase enzyme. The result of this amplification is a long ssDNA or ssRNA, which is composed of repeated sequences that are complementary to the dsDNA template and are adjacent to each other.119 The polymerase enzyme displaces the single strand of genetic material that results, while continuously amplifying the dsDNA template (Figure 6). This process continues until one or more factors such as depletion in nucleotide availability halt the process.91 RCA involves a DNA polymerase, a primer (RNA or DNA), a target (circular) and deoxynucleotide triphosphates (dNTPs). RCA uses enzymes such as Phi29, Bst, and Vent exo-DNA polymerases, but Phi29 DNA polymerase is most preferred.119 This is due to its high strand displacement, and ability to process very complex circular DNA templates such as those having organisational issues, multiple cross-overs and multiple branches. RCA products can be detected using gel electrophoresis120–122 or visually using fluorescence spectroscopy, microscopy and flow cytometry.123–127 Furthermore, incorporating nanoparticles such as gold nanoparticles (AuNPs), magnetic beads, and quantum dots into RCA products128–130 can aid the visualisation of products. Primers designed for PCR can be used for RCA.67
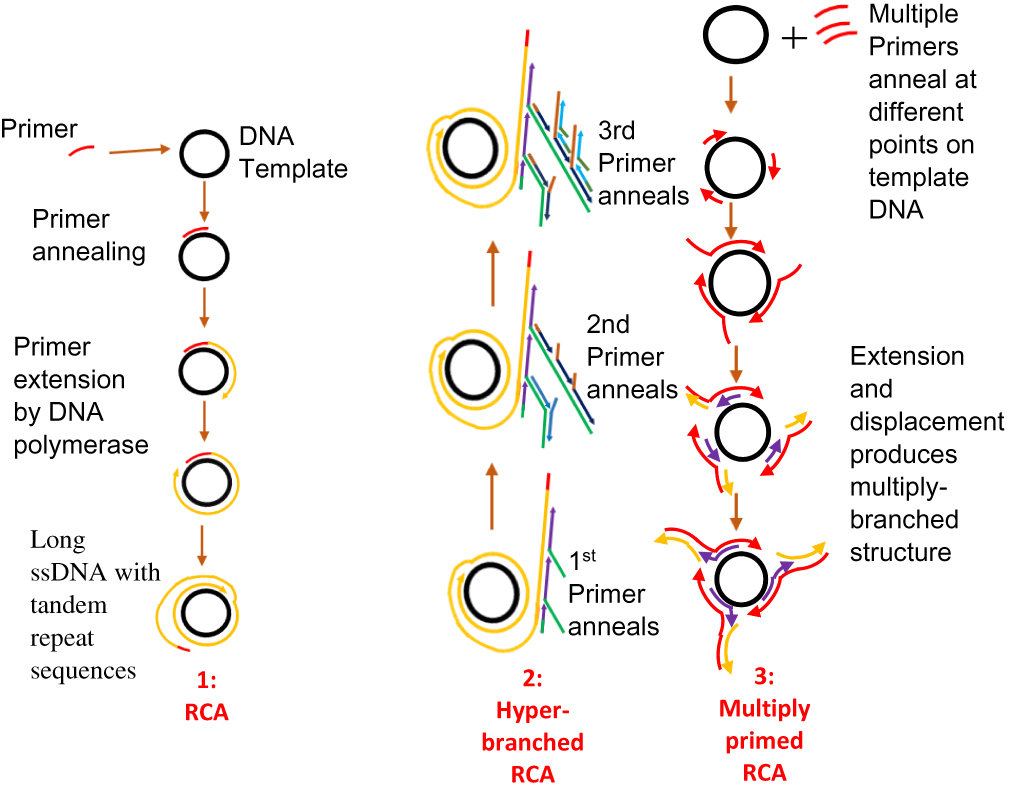 |
Figure 6 Rolling circle amplification. [1] Primer anneals to circular DNA which is continuously extended by polymerase enzyme to produce a displaced long ssDNA with tandem repeat sequences. Adapted with permission from Liu D, Daubendiek SL, Zillman MA, Ryan K, Kool ET. Rolling Circle DNA Synthesis: Small Circular Oligonucleotides as Efficient Templates for DNA Polymerases. J Am Chem Soc.1996;118(7):1587–1594. Copyright 1996 American Chemical Society.203 [2] Hyper-branched RCA in which more than one primer anneals to products of previous amplification or padlock probe is used to form a circular DNA. New strands arise from the extension of primers; as they are displaced, reverse primers anneal and are extended to generate more displaced strands, resulting in multiple copies of highly branched DNA. Adapted from Yan J, Zhou Y, Zheng AS, et al. Isothermal amplified detection of DNA and RNA. Mol. BioSyst. 2014;10:970-1003.70 [3] Multiprimed RCA or multiply primed RCA in which multiple primers anneal to different points on a circular DNA with simultaneous extension and displacement. Multiply-branched structures result. Adapted with permission from Dean FB, Nelson JR, Giesler TL, Lasken RS. Rapid amplification of plasmid and phage DNA using Phi29 DNA polymerase and multiply-primed rolling circle amplification. Genome Res. 2001;11(6):1095–1099.134 Copyright © 2001, Cold Spring Harbor Laboratory Press. |
Since its discovery in the mid-1990s, RCA has undergone some modifications.119 RCA initially utilised only circular ssDNA target as a template but not linear double-stranded targets.59 The design of oligonucleotides known as padlock probes,131 containing 3ʹ- and 5ʹ sequences that can bind with a target sequence with both ends have solved the problem. The probe becomes circularised when the ends are joined together by ligase. Long DNA sequences with tandem repeats can then be synthesised from this circularised padlock probe once a primer binds to it. A polymerase enzyme then amplifies the primer.70 The ligation process can be achieved either by using a template or without a template. Template mediated ligation can be achieved using the T4 DNA ligase while template-free ligation can be achieved using a special DNA ligase known as CircLigase.119,132,133
One of the attributes that set this method apart is the exponential amplification of target material using RCA. By a process known as multiprimed RCA or multiply-primed RCA, it is possible to apply more than one primer at a time to achieve amplification from multiple points on the same circular template. After primer annealing, DNA polymerase begins the extension of the primers with the successive displacement of newly synthesised strands. Primers further anneal to the displaced strands and elongation by DNA polymerase continues, resulting in the formation of a multiple-branched structure.119,134 A second method is known as the hyper-branched RCA (HRCA), in which a padlock probe is used at the start.135,136 Upon successful elongation of the primer by the polymerase enzyme, the resulting displaced linear products from the RCA are annealed to, by subsequent sets of reverse primers, which are further extended by DNA polymerase, and displaced once the end of an adjourning primer is reached. Multiple copies of highly branched DNA result from the repetition of the process with newly displaced single-stranded products. This RCA method is also known as ramification amplification (RAM)136 or cascade RCA.137
RCA possess some features that make it a suitable tool for some fields. For instance, it does not require temperature regulation as is done in PCR and could be performed in a liquid, on a solid, in vitro and in vivo. Also, detecting a single molecule using RCA is possible due to its adaptability either on a solid support or inside a cell such as in the identification of surface protein markers on tumour cells138 and measurement of protein levels in serum samples.139 Since RCA products contain repeated sequences, the technique can be customised or tailored towards desired sequences by manipulating the template. In addition to all these, amplification by this method is considered efficient, with multivalent capabilities.119
Although not much data appears to be available, RCA has been used in the diagnosis of some disease-causing agents. RCA designed with stem-loop primer has been used to detect HPV16 and HPV18 from carcinoma cells of the cervix, and cervical mucus exfoliated cell samples.128 Similarly, H5N1 influenza virus detection using hyperbranched RCA with a detection limit of 9fM has been reported.122 The authors reported that results obtained by RCA compared favourably to standard real-time PCR. Juul and colleagues140 reported a microfluidics RCA known as rolling-circle enhanced enzyme activity detection (REEAD) for detecting Plasmodium. In this protocol, topoisomerase I (pTopI) of the Plasmodium performs the required circularisation of the RCA substrate. Crude clinical samples could be detected using REEAD with high sensitivity.59 RCA assays can be completed with amplification of products ≤90 mins.122,135,141
As a limitation to this IA method, linear multimeric by-products can arise during ligation steps of the test.133 Inadequate binding sites for enzymes also hamper the effectiveness of ligation by enzymes, especially for small DNA.142 Long storage of RCA products results in non-specific cross-linking within and between component molecules, making mass production and storage of RCA systems a difficult venture. The problem of cross-linking between components can be minimized by fine-tuning parameters such as length of product formed, the sequence used and composition of the RCA assay. The use of a computer-aided application such as the cadnano to design and construct predictable DNA nanostructures has also been suggested to minimise non-specific interactions during RCA.119
Strand Displacement Amplification (SDA)
SDA is the IA method that gave rise to multiple displacement amplification and LAMP. It was first reported in 1992 by Walker et al24 and syndicates activity of an exonuclease-deficient DNA polymerase with a restriction endonuclease HincII and in combination with four different primers. The first two S1 and S2 primers contain restriction enzyme recognised sites at the 5ʹ end to the target complementary sequence, while the other two B1 and B2 primers are known as the bumper primers.
SDA begins with an initial denaturation step at 95°C to separate the double-stranded DNA to allow annealing between each strand and one each of the primers (Figure 7). The primers S1 and S2 are extended to produce a strand bearing restriction sites for endonuclease activity. These strands are each displaced by bumper primers B1 and B2 respectively, resulting in double-stranded DNAs and displaced single strands from S1 and S2 extensions. S1 and B1 extension, as well as S2 and B2, are done simultaneously by exo-klenow in the presence of dGTP, dCTP, TTP and thiol-modified dATP (dATPαS). In the following reactions, B2 and S2 anneal to S1 extension, while B1 and S1 anneal to S2 extension, followed by extension and displacement steps as in the previous step, giving rise to two fragments with hemiphosphorothioate HincII site on both ends and another pair bearing it only on one end. The newly synthesised DNA strands thus, incorporate the modified dATP and is hence, inactive to the endonuclease enzyme.
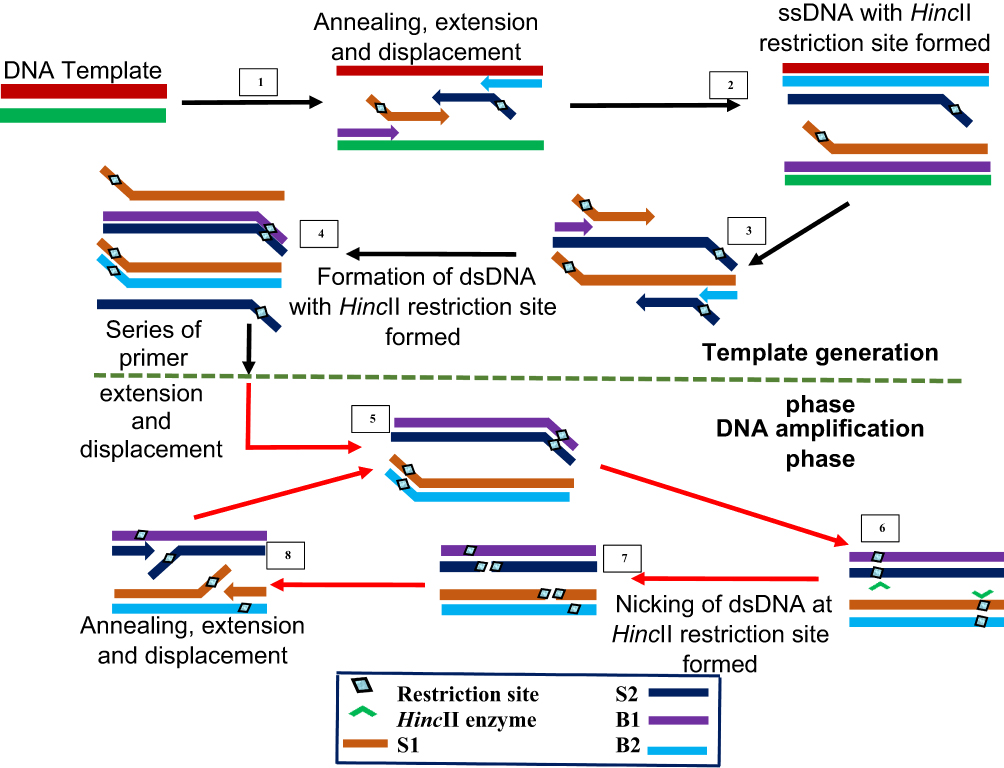 |
Figure 7 Strand displacement amplification. Denaturation of a dsDNA allows annealing of primers S1, S2, B1 and B2 which are extended simultaneously with the displacement of extended S1 and S2 strands bearing HincII restriction sites. When a dsDNA with both strands bearing HincII restriction sites is generated, the unmodified original strand extended from the primers is nicked by the enzyme, while the polymerase extension of the 3ʹ end of the newly synthesized strand results in the displacement of downstream DNA strands. The displaced strands then form the template for subsequent amplification which results in exponential amplification of the target. Adapted with permission from Walker TG. Empirical Aspects of Strand Displacement Amplification. Genome Res. 1993; 3:1–6.204 Copyright © 1993, Cold Spring Harbor Laboratory Press. |
This round of annealing and extension continues until a dsDNA with both strands bearing HincII restriction sites result. The strands are then nicked by the endonuclease enzyme, followed by polymerase extension of the strand at the nick, leading to multiple circles of nicking, extension and displacement. The endonuclease nicks the unmodified original strand extended from the primers, while the polymerase extension occurs at the 3ʹ end of the newly synthesised strand to displace DNA down the strand. The displaced strands then form the template for subsequent amplification which culminate to more rapid target amplification.24,143 SDA product detection is possible by methods such as gel electrophoresis,144–146 molecular beacons145,147 and fluorescence methods.148–150
SDA has been applied in the diagnosis of tuberculosis through reverse-transcriptase SDA.144 The technique which targeted alpha-antigen (85B protein) of Mycobacterium tuberculosis was successful at detecting and monitoring mRNA presence in patients undergoing tuberculosis treatment over 14 days, with an analytical sensitivity of 10 copies of target. Commercial kits for SDA (Table 2) are also available.
An example is the BD ProbeTecTM ET system designed for detecting Chlamydia trachomatis in urine145,151 and Neisseria gonorrhoea in urine.152 Clinical studies on patient urine and swab samples with the developed SDA-based assay reported a sensitivity of 100% and specificity of 100% for detection of C. trachomatis and N. gonorrhea nucleic acid145 while another reported a specificity of 99.2% and sensitivity of 99.3% for N. gonorrhoea using BD ProbeTecTM SDA system on urine samples, in which results were obtained in less than 5 hrs with better performance than culture method.152 Similarly, Van Der Pol et al153 reported sensitivity ranging from 80.5% to 98.5% when this assay was used to detect chlamydia and gonorrhoea infection in men and women using urine, cervical and urethral swabs. The reported BD ProbeTecTM ET system incorporates SDA with real-time fluorescence detection.
Despite the exponential nature of amplification by SDA, the method is limited by the inability to amplify base-pair sequences that are longer than 50 bp to 120 bp.67 There also appears to be very little known about the development of simple SDA for use at patient point of care and research purposes, probably due to the long-time required for running this method.91 A recent application of SDA has been in the detection of microRNAs.154 Dickinson and Company was assigned patent right to this IA technique in 1995, but the patent right now bears an expired status as at 2019.
Multiple Cross Displacement Amplification (MCDA)
MCDA is a new IA method that was first reported in 2015 by Wang et al25 It involves only one enzyme, a polymerase with strand displacement capability and is isothermal, operating at a single temperature throughout the reaction (between 60 – 68°C). It is also reported to be highly sensitive, specific and can be applied to rapid biological, environmental and clinical research. The reaction completes in less than 40 mins. Amplification by this method involves the use of 10 primers which are the amplification primers, cross primers and displacement primers. The primers include C1, C2, D1, D2, R1 and R2 which identify target sites C1s, C2s, D1s, D2s, R1s and R2s; F1 and F2 are the displacement primers, while the cross primers are CP1 and CP2 which respectively identify F1s, F2s, P1s and P2s. In the first step of the MCDA, CP1 anneals to P1s and extends, while the new strand is displaced by that synthesised from F1 extension (Figure 8). In step 2, C1, D1, F2, R1 and CP2 anneal to the displaced strand and are simultaneously extended to generate four different products, namely C1, R1, D1 and CP2. Extension of product D1 by primers C1 and CP1 results in a new product CP1/C1, while the C1 product forms an intramolecular stem-loop due to its complementary C1s sequence at its 3ʹ end and C1 sequence at its 5ʹ end. This stem-loop is then extended by CP1 to generate another product CP1/C1 of two strands which can reanneal to form loop structures stabilised by C1 and C1s base-pair double helix. These two strands can become templates for amplification by D1, C1 and CP1 to form products CP1/D1 and CP1/C1 respectively as in the first circle. In the second cycle of MCDA, the second product from step 2 (R1) undergoes a similar amplification process as C1 to yield products CP1/C1, CP1/D1, CP1/R1 and R1/R1s, respectively. The product CP2 from step 2 resulting from displacement by F2 primer can also form a stem-loop when extended, owing to complementary sequences on its 3ʹ and 5ʹ ends. Primers C2, D2 and R2 then anneal to this newly formed strand and a process similar to that of C1, D1 and R1 are repeated. The stem-loop structures formed over series of steps from these two cycles of amplification become templates in the exponential amplification cycle and undergo series of amplification and displacement reactions to yield multiple copies of the target DNA bearing inverted repeats.25
 |
Figure 8 Multiple cross displacement amplification. CP1 anneals to P1s to begin the reaction. Synthesis by F1 displaces the product formed by CP1, which is then annealed to by amplification primers D1, C1, R1, CP2 (cross primer) and F2 (displacement primer), leading to four different products (step 2). A new product CP1/D1 is formed by the extension of product D1 by C1 and CP1 (step 3), while the C1 product forms an intramolecular stem loop due to complementarity at its 3ʹ (C1s sequence) and 5ʹ (C1 sequence) ends. CP1 extends the stem loop, forming a new CP1/C1 product (step 4). These can then become templates from which amplification primers D1, C1 and CP1 generate products CP1/D1 and CP1/C1 respectively as before (Cycle 1). In cycle 2, product R1 undergoes a similar amplification process as C1 to yield products CP1/C1, CP1/D1, CP1/R1 and R1/R1s respectively. CP2 product displaced by F2 primer from step 2, is extended at the 3ʹ end to form a new double strand with stem loop structure (step 6). Primers C2, D2 and R2 then anneal to the newly formed strands to repeat similar form of amplification by C1, D1 and R1 (step 8). The two cycles generate several stem loop structures which become templates for exponential amplification to yield multiple copies of the target DNA with numerous inverted repeats. Reproduced with permission from Wang Y, Wang Y, Ma A-J, et al. Rapid and sensitive isothermal detection of nucleic-acid sequence by multiple cross displacement amplification. Sci Rep. 2015;5:11902.25 |
Products of MCDA amplification can be visualised by gel electrophoresis and colourimetric methods25,155 and also lateral flow biosensor,155–158 nanoparticles based biosensor155,159 and real-time turbidity.155 Primer design for MCDA uses software used for LAMP and PCR primer design which are PrimerExplorer (Eiken Chemical Company limited, Japan)and Primer Premier (Premier Biosoft International, USA).157
One major setback of this method is the generation of false-positive results due to the formation of primer-dimers and off-target hybrids, as well as problems of low sensitivity, high background and loss of signals that result when the primers and polymerase mix with triphosphates.155,160 Efforts made towards improving the method and solving these problems since its development, includes the development of self-avoiding molecular recognition system (SAMRS) assisted MCDA or SAMRS-MCDA. The SAMRS-MCDA system operates at 60°C and visualisation of products is by a lateral flow biosensor coupled with nanoparticles. Primers made up of self-avoiding molecular recognition system components, enables selective hybridisation to nucleotides apart from other SAMRS-containing sequences.155
MCDA and SAMRS-MCDA have been successfully used to detect disease-causing bacteria such as Shigella species,158 Vibrio parahaemolyticus,157 Listeria monocytogenes,160 Klebsiella pneumonia,159,161 Pseudomonas aeruginosa,155 S. aureus and MRSA by the mecA and nuc genes as targets.156 A multiplex MCDA was also successfully used to distinguish between MRSA, MSSA and non-S. aureus. There have been reports of impressive specificity and sensitivity for MCDA in the mentioned studies with detection of target organisms from samples such as sputum,159,161 blood,156 oyster,157 and faeces.158 With results from the mentioned studies comparable to established methods, MCDA could serve as a valuable on-the-spot diagnostic tool for rapidly detecting disease agents specifically in areas with resource constraints, giving its simplicity and cost-effectiveness.157–159 Wang et al25 have since applied for a patent of their novel methodology and sequences covered in their earlier work at the State Intellectual Property Office of the People’s Republic of China (application number CN201510280765.X).
Miniaturising IA for POC Use
An efficient testing system suitable for clinical application should be condensable into a simple point-of-care device with the most minimal technical expertise. A number of these IA techniques for which POC devices have been attempted are still at the proof-of-concept-stage. Such an ideal diagnostic method should be able to operate without sophisticated equipment, should combine preparation of the sample, amplification and product detection all in one, and still retain specificity, sensitivity, simplicity, robustness and cost-effectiveness.65
Microfluidic systems are those that utilise only a minimal volume of sample, usually in microlitres. This property makes contamination most unlikely and allows miniaturisation of equipment, thus making them very suitable for POC applications, especially in resource-constrained environments. They employ microchambers free of fluids and as such are simpler, energy-saving systems, not requiring heating and cooling systems, or fluid recycling.162 Valves, mixers and pumps can also be integrated, enabling the regulation of fluids and other device components. The flow of fluids in microfluidics can either be passive by diffusion or active by application of some external force that increases the diffusion rate. These have been used to achieve droplet-based and continuous flow technologies for controlling fluids in microfluidic devices.163 Microfluidic devices comprise of materials such as nitrocellulose, glass fibre, cellulose, paper and silicon, with paper and polymers being the most popular.65 A microfluidic device based on NASBA detected amplification using fluorescent measurements with a detection limit of 5 fmol/l has been reported.164 Paper-based devices have been used successfully in diagnostic assays based on colourimetric chemical detection and immunoassay of proteins, which probably informed the use of paper for IA devices. Kaarj et al165 used RT-LAMP based paper microfluidic chip for detection of Zika Virus from human urine and plasma samples. The device achieved a sensitivity as low as one copy/µL, and real-time quantification by smartphone imaging. Similarly, a paper-based RT-RPA microfluidic device designed by Magro et al166 for Ebola virus was tested on the field in Guinea, with reported 90% sensitivity to PCR, with multiplexing capability. The device used reagents freeze-dried on paper. Rohrman and Richards-Kortum reported an isothermal enzymatic amplification of DNA from HIV, using a lateral flow device with low-cost, light-weight for POC testing with DNA extracted from dried blood spots.118 A more recent report, however, described more efficient amplification when polyethersulfone and polycarbonate were used for LAMP and HDA, instead of cellulose paper, glass fibre, or nitrocellulose. Polyethersulfone though not widely applied in rapid diagnostic assays, proved to be a better material for rapid POC application with nucleic acid from Bordetella pertussis, Chlamydia trachomatis, Neisseria gonorrhoeae, and Influenza A H1N1.167
One major challenge of microfluidic devices is that of biomolecular interactions with the walls of microfluidic channels, which can reduce enzyme and primer concentrations, thereby resulting in reaction inhibition. However, the use of polyethersulfone may be more expensive for application than cellulose. Applying surface treatment (eg using polyethersulfone) to hydrophilic materials could solve the problem of non-specific adsorption of reaction components usually associated with surfaces.65 Surface coatings can come as permanent modifications such as using polyethene glycol (PEG) or blocking well surfaces with molecules such as bovine serum albumin. Problems also arise when micro-devices are poorly fabricated, leading to the presence of rough surfaces, junctions and sharp corners, all of which can reduce the concentration of biomaterials available for the reaction.162 Several methods available for fabrication of microfluidics include injection moulding, microthermoforming, hot embossing, casting, lithography and laser ablation.168 Hence, miniaturisation of IA into POC devices requires careful consideration of materials used in fabrication, as well as the method of fabrication to be used, in other to achieve efficient microfluidic devices.
Prospects of Isothermal Amplification
Given the increasing acceptance and application of isothermal methods for amplification of nucleic acids and detection of pathogenic microbes (Table 1), it is sufficient to project an isothermal-based diagnostic world soon. Notwithstanding the improvements recorded in the development and application of isothermal diagnostic assays to infectious disease diagnosis, some other methods are being established as fresh innovations or off-shoots of already existing technologies. Some of these include exponential amplification reaction (EXPAR),169 single chimeric primers isothermal amplification (SPIA),170 Isothermal and chimeric primer-initiated amplification of nucleic acids (ICAN),171 hairpin fluorescence probe assisted isothermal amplification172 and beacon-assisted detection amplification.173 Also, there appears to be the need for standardization of these methods as contrasting data exist regarding the performance of some of these methods, thereby making comparison difficult.91 The nearest future should witness more compact hand-held diagnostic technologies that operate on isothermal principles and still combine such simplicity with accuracy and rapidity. This advancement could positively alter the current state of healthcare in developing countries where the accurate diagnosis of diseases is still a huge problem while making diagnosis faster in developed countries. Winning the fight against the impact of diseases especially in resource-constrained societies requires simple, rapid, but yet sensitive diagnostic tests to be available for clinical application.
Conclusion
Rapid and accurate identification of disease agents is key to achieving good health and managing the ravaging effects of pathogens. Developing and improving already existing technologies is at the heart of this drive, and isothermal technologies hold a large stake, owing to characteristics such as simplicity, cost-effectiveness, robustness, sensitivity and specificity. In some cases, these methods have been reported to perform even better than PCR and other previously existing traditional diagnostic methods, which are more cumbersome and demanding in their application. Tailoring these methods to meet the massive and urgent diagnostic gap in resource-limited environments and developing countries could be easier to achieve than employing the use of culture-based methods and other more expensive and high skill-demanding techniques. Despite the huge potentials of these isothermal methods, some are yet to be commercialised or developed for clinical diagnosis at treatment points. Hence, it is important to develop hand-held technologies that will make isothermal diagnostic methods easily accessible and more suitable for use at point-of-care. For instance, improving the multiplexing ability and incorporating preparation of samples, purification steps and product detection into one simple step would increase the fortunes of isothermal methods in diagnostics. Isothermal methods with multiplexing capability would record higher application because it would translate to more efficient usage, arising from the liberty of processing multiple samples for multiple targets simultaneously. Thus, time, resources and energy will be saved and diagnosis-to-treatment time drastically reduced. Future improvements should also target rapid detection of resistance genes up to multiplexing levels, as most of the methods have only demonstrated identification of pathogens. Such an improvement would greatly strengthen the success of the fight against antimicrobial resistance and its attendant problems in disease treatment and management.
Acknowledgement
This review has the support of a Research University Grant (Geran Penyelidikan Bridging Universiti Sains Malaysia 304.PPSP.6316339). The funders did not influence review design, literature collection, preparation of the manuscript, analysis or decision to publish.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Tenover FC. The role for rapid molecular diagnostic tests for infectious diseases in precision medicine. Expert Rev Precis Med Drug Dev. 2018;3(1):69–77.
2. Al-Mulla NA, Taj-Aldeen SJ, El Shafie S, Janahi M, Al-Nasser AA, Chandra P. Bacterial bloodstream infections and antimicrobial susceptibility pattern in pediatric hematology/oncology patients after anticancer chemotherapy. Infect Drug Resist. 2014;7:289–299. doi:10.2147/IDR.S70486
3. Saker L, Lee K, Cannito B, Gilmore A, Campbell-Lendrum D Globalization and infectious diseases; a review of the linkages. Special Topics No 3 UNICEF/UNDP/World Bank/WHO. 2004:1–67.
4. Cheng Y-H, Lin Y-J, Chen S-C, et al. Assessing health burden risk and control effect on dengue fever infection in the southern region of Taiwan. Infect Drug Resist. 2018;11:1423–1435. doi:10.2147/IDR.S169820
5. Caliendo AM, Gilbert DN, Ginocchio CC, et al. Better tests, better care: improved diagnostics for infectious diseases. Clin Infect Dis. 2013;57 Suppl 3:S139–170. doi:10.1093/cid/cit578
6. Lu TC, Tsai CL, Lee CC, et al. Preventable deaths in patients admitted from emergency department. Emerg Med J. 2006;23(6):452–455. doi:10.1136/emj.2004.022319
7. Abe T, Tokuda Y, Shiraishi A, et al. In-hospital mortality associated with the misdiagnosis or unidentified site of infection at admission. Crit Care. 2019;23(1):202.
8. Ahmad RA, Mahendradhata Y, Utarini A, de Vlas SJ. Diagnostic delay amongst tuberculosis patients in Jogjakarta Province, Indonesia is related to the quality of services in DOTS facilities. Trop Med Int Health. 2011;16(4):412–423. doi:10.1111/tmi.2011.16.issue-4
9. Bojovic O, Medenica M, Zivkovic D, et al. Factors associated with patient and health system delays in diagnosis and treatment of tuberculosis in Montenegro, 2015-2016. PLoS One. 2018;13(3):e0193997. doi:10.1371/journal.pone.0193997
10. Frenz MB, McIntyre AS. Reducing delays in the diagnosis and treatment of Clostridium difficile diarrhoea. QJM. 2003;96(8):579–582. doi:10.1093/qjmed/hcg098
11. Elshafie S, Taj-Aldeen SJ. Emerging resistant serotypes of invasive Streptococcus pneumoniae. Infect Drug Resist. 2016;9:153–160. doi:10.2147/IDR.S102410
12. Hersh AL, Shapiro DJ, Pavia AT, Shah SS. Antibiotic prescribing in ambulatory pediatrics in the United States. Pediatrics. 2011;128(6):1053–1061.
13. Grijalva CG, Nuorti JP, Griffin MR. Antibiotic prescription rates for acute respiratory tract infections in US ambulatory settings. JAMA. 2009;302(7):758–766. doi:10.1001/jama.2009.1163
14. Roumie CL, Halasa NB, Grijalva CG, et al. Trends in antibiotic prescribing for adults in the United States–1995 to 2002. J Gen Intern Med. 2005;20(8):697–702. doi:10.1111/j.1525-1497.2005.0148.x
15. Soo PC, Tseng CC, Ling SR, et al. Rapid and sensitive detection of Acinetobacter baumannii using loop-mediated isothermal amplification. J Microbiol Methods. 2013;92(2):197–200. doi:10.1016/j.mimet.2012.11.020
16. Notomi T, Okayama H, Masubuchi H, et al. Loop-mediated isothermal amplification of DNA. Nucleic Acids Res. 2000;28(12):E63–E63. doi:10.1093/nar/28.12.e63
17. Notomi T, Mori Y, Tomita N, Kanda H. Loop-mediated isothermal amplification (LAMP): principle, features, and future prospects. J Microbiol. 2015;53(1):1–5. doi:10.1007/s12275-015-4656-9
18. Li J, Macdonald J, von Stetten F. Review: a comprehensive summary of a decade development of the recombinase polymerase amplification. Analyst. 2019;144(1):31–67.
19. Dhama K, Karthik K, Chakrabort S, et al. Loop-mediated isothermal amplification of DNA (LAMP): a new diagnostic tool lights the world of diagnosis of animal and human pathogens: a review. Pak J Biol Sci. 2014;17(2):151–166. doi:10.3923/pjbs.2014.151.166
20. Black EM, Lowings JP, Smith J, Heaton PR, McElhinney LM. A rapid RT-PCR method to differentiate six established genotypes of rabies and rabies-related viruses using TaqMan™ technology. J Virol Methods. 2002;105(1):25–35. doi:10.1016/S0166-0934(02)00062-9
21. Parida M, Sannarangaiah S, Dash PK, Rao PV, Morita K. Loop mediated isothermal amplification (LAMP): a new generation of innovative gene amplification technique; perspectives in clinical diagnosis of infectious diseases. Rev Med Virol. 2008;18(6):407–421. doi:10.1002/rmv.v18:6
22. Compton J. Nucleic acid sequence-based amplification. Nature. 1991;350:91. doi:10.1038/350091a0
23. Wharam SD, Marsh P, Lloyd JS, et al. Specific detection of DNA and RNA targets using a novel isothermal nucleic acid amplification assay based on the formation of a three-way junction structure. Nucleic Acids Res. 2001;29(11):e54–e54. doi:10.1093/nar/29.11.e54
24. Walker GT, Fraiser MS, Schram JL, Little MC, Nadeau JG, Malinowski DP. Strand displacement amplification—an isothermal, in vitro DNA amplification technique. Nucleic Acids Res. 1992;20(7):1691–1696. doi:10.1093/nar/20.7.1691
25. Wang Y, Wang Y, Ma A-J, et al. Rapid and sensitive isothermal detection of nucleic-acid sequence by multiple cross displacement amplification. Sci Rep. 2015;5:11902. doi:10.1038/srep11902
26. Barreda-García S, Miranda-Castro R, de-los-Santos-Álvarez N, Miranda-Ordieres AJ, Lobo-Castañón MJ. Helicase-dependent isothermal amplification: a novel tool in the development of molecular-based analytical systems for rapid pathogen detection. Anal Bioanal Chem. 2018;410(3):679–693. doi:10.1007/s00216-017-0620-3
27. Nurul Najian AB, Engku Nur Syafirah EA, Ismail N, Mohamed M, Yean CY. Development of multiplex loop mediated isothermal amplification (m-LAMP) label-based gold nanoparticles lateral flow dipstick biosensor for detection of pathogenic Leptospira. Anal Chim Acta. 2016;903:142–148. doi:10.1016/j.aca.2015.11.015
28. Mori Y, Kitao M, Tomita N, Notomi T. Real-time turbidimetry of LAMP reaction for quantifying template DNA. J Biochem Biophys Methods. 2004;59(2):145–157. doi:10.1016/j.jbbm.2003.12.005
29. Tomita N, Mori Y, Kanda H, Notomi T. Loop-mediated isothermal amplification (LAMP) of gene sequences and simple visual detection of products. Nat Protoc. 2008;3(5):877–882. doi:10.1038/nprot.2008.57
30. Mori Y, Nagamine K, Tomita N, Notomi T. Detection of loop-mediated isothermal amplification reaction by turbidity derived from magnesium pyrophosphate formation. Biochem Biophys Res Commun. 2001;289(1):150–154. doi:10.1006/bbrc.2001.5921
31. Abdullah J, Saffie N, Sjasri FAR, et al. Rapid detection of Salmonella Typhi by loop-mediated isothermal amplification (LAMP) method. Braz J Microbiol. 2014;45(4):1385–1391. doi:10.1590/S1517-83822014000400032
32. Solanki R, Vanjari L, Ede N, Gungi A, Soory A, Vemu L. Evaluation of LAMP assay using phenotypic tests and conventional PCR for detection of blaNDM-1 and blaKPC genes among carbapenem-resistant clinical Gram-negative isolates. J Med Microbiol. 2013;62(Pt 10):1540–1544. doi:10.1099/jmm.0.059907-0
33. Stratakos AC, Linton M, Millington S, Grant IR. A loop-mediated isothermal amplification method for rapid direct detection and differentiation of nonpathogenic and verocytotoxigenic Escherichia coli in beef and bovine faeces. J Appl Microbiol. 2017;122(3):817–828. doi:10.1111/jam.2017.122.issue-3
34. Iwamoto T, Sonobe T, Hayashi K. Loop-mediated isothermal amplification for direct detection of mycobacterium tuberculosis complex, M. avium, and M. intracellulare in sputum samples. J Clin Microbiol. 2003;41(6):2616–2622. doi:10.1128/JCM.41.6.2616-2622.2003
35. Liu N, Zou D, Dong D, et al. Development of a multiplex loop-mediated isothermal amplification method for the simultaneous detection of Salmonella spp. and Vibrio parahaemolyticus. Sci Rep. 2017;7:45601. doi:10.1038/srep45601
36. Wang Q, Zhou Y, Li S, et al. Real-time fluorescence loop mediated isothermal amplification for the detection of Acinetobacter baumannii. PLoS One. 2013;8(7):e66406. doi:10.1371/journal.pone.0066406
37. Hamzan NI, Fauzi FH, Taib H, Mohamad S. Simple and rapid detection of Porphyromonas gingivalis and Aggregatibacter actinomycetemcomitans by loop-mediated isothermal amplification assay. Bangladesh J Med Sci. 2018;17:3. doi:10.3329/bjms.v17i3.36995
38. Kasahara K, Ishikawa H, Sato S, Shimakawa Y, Watanabe K. Development of multiplex loop-mediated isothermal amplification assays to detect medically important yeasts in dairy products. FEMS Microbiol Lett. 2014;357(2):208–216. doi:10.1111/1574-6968.12512
39. Mahony J, Chong S, Bulir D, Ruyter A, Mwawasi K, Waltho D. Multiplex loop-mediated isothermal amplification (M-LAMP) assay for the detection of influenza A/H1, A/H3 and influenza B can provide a specimen-to-result diagnosis in 40 min with single genome copy sensitivity. J Clin Virol. 2013;58(1):127–131. doi:10.1016/j.jcv.2013.06.006
40. Lucchi NW, Demas A, Narayanan J, et al. Real-time fluorescence loop mediated isothermal amplification for the diagnosis of malaria. PLoS One. 2010;5(10):e13733.
41. Hara-Kudo Y, Yoshino M, Kojima T, Ikedo M. Loop-mediated isothermal amplification for the rapid detection of Salmonella. FEMS Microbiol Lett. 2005;253(1):155–161. doi:10.1016/j.femsle.2005.09.032
42. Ohtsuka K, Yanagawa K, Takatori K, Hara-Kudo Y. Detection of Salmonella enterica in naturally contaminated liquid eggs by loop-mediated isothermal amplification, and characterization of Salmonella isolates. Appl Environ Microbiol. 2005;71(11):6730. doi:10.1128/AEM.71.11.6730-6735.2005
43. Okamura M, Ohba Y, Kikuchi S, et al. Loop-mediated isothermal amplification for the rapid, sensitive, and specific detection of the O9 group of Salmonella in chickens. Vet Microbiol. 2008;132(1):197–204. doi:10.1016/j.vetmic.2008.04.029
44. Hara-Kudo Y, Konishi N, Ohtsuka K, et al. Detection of Verotoxigenic Escherichia coli O157 and O26 in food by plating methods and LAMP method: a collaborative study. Int J Food Microbiol. 2008;122(1–2):156–161. doi:10.1016/j.ijfoodmicro.2007.11.078
45. Yano A, Ishimaru R, Hujikata R. Rapid and sensitive detection of heat-labile I and heat-stable I enterotoxin genes of enterotoxigenic Escherichia coli by loop-mediated isothermal amplification. J Microbiol Methods. 2007;68(2):414–420. doi:10.1016/j.mimet.2006.09.024
46. Hill J, Beriwal S, Chandra I, et al. Loop-mediated isothermal amplification assay for rapid detection of common strains of Escherichia coli. J Clin Microbiol. 2008;46(8):2800–2804. doi:10.1128/JCM.00152-08
47. Yamazaki W, Taguchi M, Ishibashi M, et al. Development and evaluation of a loop-mediated isothermal amplification assay for rapid and simple detection of Campylobacter jejuni and Campylobacter coli. J Med Microbiol. 2008;57(4):444–451. doi:10.1099/jmm.0.47688-0
48. Lloyd A, Pasupuleti V, Thota P, et al. Accuracy of loop-mediated isothermal amplification for the diagnosis of Clostridium difficile infection: a systematic review. Diagn Microbiol Infect Dis. 2015;82(1):4–10. doi:10.1016/j.diagmicrobio.2015.02.007
49. Massey V, Gregson DB, Chagla AH, Storey M, John MA, Hussain Z. Clinical usefulness of components of the Triage immunoassay, enzyme immunoassay for toxins A and B, and cytotoxin B tissue culture assay for the diagnosis of Clostridium difficile diarrhea. Am J Clin Pathol. 2003;119(1):45–49. doi:10.1309/U8ATL52Q60XYAVX6
50. Poutanen SM, Simor AE. Clostridium difficile-associated diarrhea in adults. CMAJ. 2004;171(1):51–58. doi:10.1503/cmaj.1031189
51. Hataoka Y, Zhang L, Mori Y, Tomita N, Notomi T, Baba Y. Analysis of specific gene by integration of isothermal amplification and electrophoresis on poly(methyl methacrylate) microchips. Anal Chem. 2004;76(13):3689–3693. doi:10.1021/ac035032u
52. Mori Y, Notomi T. Loop-mediated isothermal amplification (LAMP): a rapid, accurate, and cost-effective diagnostic method for infectious diseases. J Infect Chemother. 2009;15(2):62–69. doi:10.1007/s10156-009-0669-9
53. Hsieh K, Mage PL, Csordas AT, Eisenstein M, Soh HT. Simultaneous elimination of carryover contamination and detection of DNA with uracil-DNA-glycosylase-supplemented loop-mediated isothermal amplification (UDG-LAMP). Chem Commun (Camb). 2014;50(28):3747–3749. doi:10.1039/c4cc00540f
54. Wong YP, Othman S, Lau YL, Radu S, Chee HY. Loop-mediated isothermal amplification (LAMP): a versatile technique for detection of micro-organisms. J Appl Microbiol. 2018;124(3):626–643. doi:10.1111/jam.13647
55. Watts MR, James G, Sultana Y, et al. A loop-mediated isothermal amplification (LAMP) assay for Strongyloides stercoralis in stool that uses a visual detection method with SYTO-82 fluorescent dye. Am J Trop Med Hyg. 2014;90(2):306–311. doi:10.4269/ajtmh.13-0583
56. Vincent M, Xu Y, Kong H. Helicase-dependent isothermal DNA amplification. EMBO Rep. 2004;5(8):795–800. doi:10.1038/sj.embor.7400200
57. Abdel-Monem M, Hoffmann-Berling H. Enzymic unwinding of DNA. Eur J Biochem. 1976;65(2):431–440. doi:10.1111/j.1432-1033.1976.tb10358.x
58. Abdel-monem M, Dürwald H, Hoffmann-Berling H. Enzymic Unwinding of DNA. Eur J Biochem. 1976;65(2):441–449. doi:10.1111/j.1432-1033.1976.tb10359.x
59. Zanoli LM, Spoto G. Isothermal amplification methods for the detection of nucleic acids in microfluidic devices. Biosensors. 2012;3(1):18–43. doi:10.3390/bios3010018
60. An L, Tang W, Ranalli TA, Kim H-J, Wytiaz J, Kong H. Characterization of a thermostable UvrD helicase and its participation in helicase-dependent amplification. J Biol Chem. 2005;280(32):28952–28958. doi:10.1074/jbc.M503096200
61. Motré A, Li Y, Kong H. Enhancing helicase-dependent amplification by fusing the helicase with the DNA polymerase. Gene. 2008;420(1):17–22. doi:10.1016/j.gene.2008.04.017
62. Goldmeyer J, Kong H, Tang W. Development of a novel one-tube isothermal reverse transcription thermophilic helicase-dependent amplification platform for rapid RNA detection. J Mol Diagn. 2007;9(5):639–644. doi:10.2353/jmoldx.2007.070012
63. Andresen D, von Nickisch-rosenegk M, Bier FF. Helicase dependent OnChip-amplification and its use in multiplex pathogen detection. Clin Chim Acta. 2009;403(1):244–248.
64. Huang S, Do J, Mahalanabis M, et al. Low cost extraction and isothermal amplification of DNA for infectious diarrhea diagnosis. PLoS One. 2013;8(3):e60059.
65. Maffert P, Reverchon S, Nasser W, Rozand C, Abaibou H. New nucleic acid testing devices to diagnose infectious diseases in resource-limited settings. Eur J Clin Microbiol Infect Dis. 2017;36(10):1717–1731.
66. Hall MJ, Wharam SD, Weston A, Cardy DLN, Wilson WH. Use of signal-mediated amplification of RNA technology (SMART) to detect marine cyanophage DNA. BioTechniques. 2002;32(3):604–611.
67. Li J, Macdonald J. Advances in isothermal amplification: novel strategies inspired by biological processes. Biosens Bioelectron. 2015;64:196–211. doi:10.1016/j.bios.2014.08.069
68. Wharam SD, Hall MJ, Wilson WH. Detection of virus mRNA within infected host cells using an isothermal nucleic acid amplification assay: marine cyanophage gene expression within Synechococcus sp. Virol J. 2007;4(1):52. doi:10.1186/1743-422X-4-52
69. Levi K, Bailey C, Bennett A, Marsh P, Cardy DLN, Towner KJ. Evaluation of an isothermal signal amplification method for rapid detection of methicillin-resistant staphylococcus aureus from patient-screening swabs. J Clin Microbiol. 2003;41(7):3187.
70. Yan L, Zhou J, Zheng Y, et al. Isothermal amplified detection of DNA and RNA. Mol Biosyst. 2014;10(5):970–1003. doi:10.1039/c3mb70304e
71. Deiman B, Van Aarle P, Sillekens P. Characteristics and applications of Nucleic Acid Sequence-Based Amplification (NASBA). Appl Biochem Biotechnol Part B Mol Biotechnol. 2002;20(2):163–179.
72. Gill P, Ghaemi A. Nucleic acid isothermal amplification technologies—a review. Nucleosides Nucleotides Nucleic Acids. 2008;27(3):224–243. doi:10.1080/15257770701845204
73. Guatelli JC, Whitfield KM, Kwoh DY, Barringer KJ, Richman DD, Gingeras TR. Isothermal, in vitro amplification of nucleic acids by a multienzyme reaction modeled after retroviral replication. Proc Natl Acad Sci U S A. 1990;87:1874–1878. doi:10.1073/pnas.87.5.1874
74. Jean J, Blais B, Darveau A, Fliss I. Detection of hepatitis a virus by the nucleic acid sequence-based amplification technique and comparison with reverse transcription-PCR. Appl Environ Microbiol. 2001;67(12):5593. doi:10.1128/AEM.67.12.5593-5600.2001
75. Samuelson A, Westmoreland D, Eccles R, Fox JD. Development and application of a new method for amplification and detection of human rhinovirus RNA. J Virol Methods. 1998;71(2):197–209. doi:10.1016/S0166-0934(98)00006-8
76. van Gemen B, van Beuningen R, Nabbe A, et al. A one-tube quantitative HIV-1 RNA NASBA nucleic acid amplification assay using electrochemiluminescent (ECL) labelled probes. J Virol Methods. 1994;49(2):157–167. doi:10.1016/0166-0934(94)90040-X
77. Leone G, van Gemen B, Schoen CD, van Schijndel H, Kramer FR. Molecular beacon probes combined with amplification by NASBA enable homogeneous, real-time detection of RNA. Nucleic Acids Res. 1998;26(9):2150–2155. doi:10.1093/nar/26.9.2150
78. Loens K, Ursi D, Goossens H, Ieven M. Nucleic acid sequence-based amplification. In: Walker JM, Rapley R, editors. Medical Biomethods Handbook. Totowa, NJ: Humana Press; 2005:273–291.
79. Ginocchio CC, Kemper M, Stellrecht KA, Witt DJ. Multicenter evaluation of the performance characteristics of the nuclisens HIV-1 QT assay used for quantitation of human immunodeficiency virus type 1 RNA. J Clin Microbiol. 2003;41(1):164. doi:10.1128/JCM.41.1.164-173.2003
80. Loens K, Ieven M, Ursi D, et al. Detection of Mycoplasma pneumoniae by real-time nucleic acid sequence-based amplification. J Clin Microbiol. 2003;41(9):4448–4450. doi:10.1128/JCM.41.9.4448-4450.2003
81. Damen M, Sillekens P, Cuypers HTM, Frantzen I, Melsert R. Characterization of the quantitative HCV NASBA assay. J Virol Methods. 1999;82(1):45–54. doi:10.1016/S0166-0934(99)00079-8
82. Loens K, Ieven M, Ursi D, et al. Improved detection of rhinoviruses by nucleic acid sequence-based amplification after nucleotide sequence determination of the 5ʹ noncoding regions of additional rhinovirus strains. J Clin Microbiol. 2003;41(5):1971–1976. doi:10.1128/JCM.41.5.1971-1976.2003
83. Zappacosta R, Sablone F, Pansa L, Buca D, Buca D, Analytic RS. Diagnostic performances of human papillomavirus E6/E7 mRNA test on up-to 11-year-old liquid-based cervical samples. A biobank-based longitudinal study. Int J Mol Sci. 2017;18(7):1480.
84. Zhang F, Tetali S, Wang XP, Kaplan MH, Cromme FV, Ginocchio CC. Detection of human cytomegalovirus pp67 late gene transcripts in cerebrospinal fluid of human immunodeficiency virus type 1-infected patients by nucleic acid sequence-based amplification. J Clin Microbiol. 2000;38(5):1920.
85. Morré SA, Sillekens PT, Jacobs MV, et al. Monitoring of Chlamydia trachomatis infections after antibiotic treatment using RNA detection by nucleic acid sequence based amplification. MP. 1998;51(3):149–154. doi:10.1136/mp.51.3.149
86. van der Meide WF, Schoone GJ, Faber WR, et al. Quantitative nucleic acid sequence-based assay as a new molecular tool for detection and quantification of Leishmania parasites in skin biopsy samples. J Clin Microbiol. 2005;43(11):5560–5566. doi:10.1128/JCM.43.11.5560-5566.2005
87. van der Vliet GM, Cho SN, Kampirapap K, et al. Use of NASBA RNA amplification for detection of Mycobacterium leprae in skin biopsies from untreated and treated leprosy patients. Int J Leprosy Mycobacterial Dis. 1996;64(4):396–403.
88. van Deursen PB, Gunther AW, van Riel CC, et al. A novel quantitative multiplex NASBA method: application to measuring tissue factor and CD14 mRNA levels in human monocytes. Nucleic Acids Res. 1999;27:17. doi:10.1093/nar/27.17.e15-i
89. Jean J, Blais B, Darveau A, Fliss I. Simultaneous detection and identification of hepatitis A virus and rotavirus by multiplex nucleic acid sequence-based amplification (NASBA) and microtiter plate hybridization system. J Virol Methods. 2002;105(1):123–132. doi:10.1016/s0166-0934(02)00096-4
90. Klerks MM, Leone GOM, Verbeek M, van den Heuvel JFJM, Schoen CD. Development of a multiplex AmpliDet RNA for the simultaneous detection of potato leafroll virus and potato virus Y in potato tubers. J Virol Methods. 2001;93(1):115–125. doi:10.1016/S0166-0934(01)00258-0
91. Craw P, Balachandran W. Isothermal nucleic acid amplification technologies for point-of-care diagnostics: a critical review. Lab Chip. 2012;12(14):2469–2486. doi:10.1039/c2lc40100b
92. Loens K, Ursi D, Ieven M, et al. Detection of mycoplasma pneumoniae in spiked clinical samples by nucleic acid sequence-based amplification. J Clin Microbiol. 2002;40(4):1339–1345. doi:10.1128/JCM.40.4.1339-1345.2002
93. Piepenburg O, Williams CH, Stemple DL, Armes NA. DNA detection using recombination proteins. PLoS Biol. 2006;4(7):e204–e204. doi:10.1371/journal.pbio.0040204
94. Moore MD, Jaykus L-A. Recombinase polymerase amplification: a promising point-of-care detection method for enteric viruses. Future Virol. 2017;12(8):421–429. doi:10.2217/fvl-2017-0034
95. Higgins M, Ravenhall M, Ward D, et al. PrimedRPA: primer design for recombinase polymerase amplification assays. Bioinformatics. 2018;35(4):682–684.
96. Shen F, Davydova EK, Du W, Kreutz JE, Piepenburg O, Ismagilov RF. Digital isothermal quantification of nucleic acids via simultaneous chemical initiation of recombinase polymerase amplification reactions on SlipChip. Anal Chem. 2011;83(9):3533–3540. doi:10.1021/ac200247e
97. Tsaloglou MN, Watson RJ, Rushworth CM, et al. Real-time microfluidic recombinase polymerase amplification for the toxin B gene of clostridium difficile on a SlipChip platform. Analyst. 2015;140(1):258–264. doi:10.1039/C4AN01683A
98. Yeh EC, Fu CC, Hu L, Thakur R, Feng J, Lee LP. Self-powered integrated microfluidic point-of-care low-cost enabling (SIMPLE) chip. Sci Adv. 2017;3(3):e1501645. doi:10.1126/sciadv.1501645
99. Santiago-Felipe S, Tortajada-Genaro LA, Morais S, Puchades R, Maquieira A. Isothermal DNA amplification strategies for duplex microorganism detection. Food Chem. 2015;174:509–515. doi:10.1016/j.foodchem.2014.11.080
100. Liu HB, Du XJ, Zang YX, Li P, Wang S. SERS-based lateral flow strip biosensor for simultaneous detection of listeria monocytogenes and salmonella enterica serotype enteritidis. J Agric Food Chem. 2017;65(47):10290–10299. doi:10.1021/acs.jafc.7b03957
101. Daher RK, Stewart G, Boissinot M, Bergeron MG. Isothermal recombinase polymerase amplification assay applied to the detection of group B streptococci in vaginal/anal samples. Clin Chem. 2014;60(4):660–666. doi:10.1373/clinchem.2013.213504
102. Crannell Z, Castellanos-Gonzalez A, Nair G, Mejia R, White AC, Richards-Kortum R. Multiplexed recombinase polymerase amplification assay to detect intestinal protozoa. Anal Chem. 2016;88(3):1610–1616. doi:10.1021/acs.analchem.5b03267
103. Lau HY, Wang Y, Wee EJH, Botella JR, Trau M. Field demonstration of a multiplexed point-of-care diagnostic platform for plant pathogens. Anal Chem. 2016;88(16):8074–8081. doi:10.1021/acs.analchem.6b01551
104. Abd El Wahed A, Patel P, Faye O, et al. Recombinase Polymerase Amplification Assay for Rapid Diagnostics of Dengue Infection. PLoS One. 2015;10(6):e0129682. doi:10.1371/journal.pone.0129682
105. Abd El Wahed A, Weidmann M, Hufert FT. Diagnostics-in-a-suitcase: development of a portable and rapid assay for the detection of the emerging avian influenza A (H7N9) virus. J Clin Virol. 2015;69:16–21. doi:10.1016/j.jcv.2015.05.004
106. Renner LD, Zan J, Hu LI, et al. Detection of ESKAPE bacterial pathogens at the point of care using isothermal DNA-based assays in a portable degas-actuated microfluidic diagnostic assay platform. Appl Environ Microbiol. 2017;83:4. doi:10.1128/AEM.02449-16
107. Branavan M, Mackay RE, Craw P, et al. Modular development of a prototype point of care molecular diagnostic platform for sexually transmitted infections. Med Eng Phys. 2016;38(8):741–748. doi:10.1016/j.medengphy.2016.04.022
108. Hu C, Kalsi S, Zeimpekis I, et al. Ultra-fast electronic detection of antimicrobial resistance genes using isothermal amplification and Thin Film Transistor sensors. Biosens Bioelectron. 2017;96:281–287. doi:10.1016/j.bios.2017.05.016
109. Kim TH, Park J, Kim CJ, Cho YK. Fully integrated lab-on-a-disc for nucleic acid analysis of food-borne pathogens. Anal Chem. 2014;86(8):3841–3848. doi:10.1021/ac403971h
110. Choi G, Jung JH, Park BH, et al. A centrifugal direct recombinase polymerase amplification (direct-RPA) microdevice for multiplex and real-time identification of food poisoning bacteria. Lab Chip. 2016;16(12):2309–2316. doi:10.1039/C6LC00329J
111. Lutz S, Weber P, Focke M, et al. Microfluidic lab-on-a-foil for nucleic acid analysis based on isothermal recombinase polymerase amplification (RPA). Lab Chip. 2010;10(7):887–893. doi:10.1039/b921140c
112. Euler M, Wang Y, Otto P, et al. Recombinase polymerase amplification assay for rapid detection of Francisella tularensis. J Clin Microbiol. 2012;50(7):2234.
113. Moore MD, Jaykus L-A. Development of a recombinase polymerase amplification assay for detection of epidemic human noroviruses. Sci Rep. 2017;7:40244. doi:10.1038/srep40244
114. Yang H-L, Wei S, Gooneratne R, et al. Development of a recombinase polymerase amplification assay for Vibrio parahaemolyticus detection with an internal amplification control. Can J Microbiol. 2018;64(4):223–230. doi:10.1139/cjm-2017-0504
115. Ahn H, Batule BS, Seok Y, Kim M-G. Single-step recombinase polymerase amplification assay based on a paper chip for simultaneous detection of multiple foodborne pathogens. Anal Chem. 2018;90(17):10211–10216. doi:10.1021/acs.analchem.8b01309
116. Qi Y, Yin Q, Shao Y, et al. Rapid and visual detection of coxiella burnetii using recombinase polymerase amplification combined with lateral flow strips. Biomed Res Int. 2018;2018:10.
117. Wang H, Sun M, Xu D, et al. Rapid visual detection of cyprinid herpesvirus 2 by recombinase polymerase amplification combined with a lateral flow dipstick. J Fish Dis. 2018;41(8):1201–1206.
118. Rohrman BA, Richards-Kortum RR. A paper and plastic device for performing recombinase polymerase amplification of HIV DNA. Lab Chip. 2012;12(17):3082–3088. doi:10.1039/c2lc40423k
119. Ali MM, Li F, Zhang Z, et al. Rolling circle amplification: a versatile tool for chemical biology, materials science and medicine. Chem Soc Rev. 2014;43(10):3324–3341.
120. Ali MM, Li Y. Colorimetric sensing by using allosteric-DNAzyme-coupled rolling circle amplification and a peptide nucleic acid–organic dye probe. Angew Chem Int Ed. 2009;48(19):3512–3515.
121. Zhao W, Gao Y, Kandadai SA, Brook MA, Li Y. DNA polymerization on gold nanoparticles through rolling circle amplification: towards novel scaffolds for three-dimensional periodic nanoassemblies. Angew Chem. 2006;45(15):2409–2413. doi:10.1002/anie.200600061
122. Hamidi SV, Ghourchian H, Tavoosidana G. Real-time detection of H5N1 influenza virus through hyperbranched rolling circle amplification. Analyst. 2015;140(5):1502–1509. doi:10.1039/c4an01954g
123. Ali MM, Su S, Filipe CDM, Pelton R, Li Y, Enzymatic manipulations of DNA oligonucleotides on microgel: towards development of DNA–microgel bioassays. Chem Commun. 2007;43:4459–4461. doi:10.1039/b709817k
124. Linck L, Reiß E, Bier F, Resch-Genger U. Direct labeling rolling circle amplification as a straightforward signal amplification technique for biodetection formats. Anal Methods. 2012;4(5):1215–1220.
125. Göransson J, Ke R, Nong RY, et al. Rapid identification of bio-molecules applied for detection of biosecurity agents using rolling circle amplification. PLoS One. 2012;7(2):e31068.
126. Sato K, Ishii R, Sasaki N, Sato K, Nilsson M. Bead-based padlock rolling circle amplification for single DNA molecule counting. Anal Biochem. 2013;437(1):43–45.
127. Ebai T, Souza de Oliveira FM, Löf L, et al. Analytically sensitive protein detection in microtiter plates by proximity ligation with rolling circle amplification. Clin Chem. 2017;63(9):1497. doi:10.1373/clinchem.2017.271833
128. Zhang B, Wang Q, Wu J, Chen Y, Wang J. Detection of nucleic acids with a novel stem-loop primer rolling circle amplification technique. Reports. 2018;64(2):69–80.
129. Schweitzer B, Wiltshire S, Lambert J, et al. Immunoassays with rolling circle DNA amplification: a versatile platform for ultrasensitive antigen detection. Proc Natl Acad Sci. 2000;97(18):10113. doi:10.1073/pnas.170237197
130. Cheng W, Yan F, Ding L, Ju H, Yin Y. Cascade signal amplification strategy for subattomolar protein detection by rolling circle amplification and quantum dots tagging. Anal Chem. 2010;82(8):3337–3342. doi:10.1021/ac100144g
131. Nilsson M, Malmgren H, Samiotaki M, Kwiatkowski M, Chowdhary BP, Landegren U. Padlock probes: circularizing oligonucleotides for localized DNA detection. Science (New York, NY). 1994;265(5181):2085–2088. doi:10.1126/science.7522346
132. Polidoros AN, Pasentsis K, Tsaftaris AS. Rolling circle amplification-RACE: a method for simultaneous isolation of 5ʹ and 3ʹ cDNA ends from amplified cDNA templates. Biotechniques. 2006;41(1):35–36, 38, 40 passim. doi:10.2144/000112205
133. Rubin E, Rumney S, Wang S, Kool ET. Convergent DNA synthesis: a non-enzymatic dimerization approach to circular oligodeoxynucleotides. Nucleic Acids Res. 1995;23(17):3547–3553. doi:10.1093/nar/23.17.3547
134. Dean FB, Nelson JR, Giesler TL, Lasken RS. Rapid amplification of plasmid and phage DNA using Phi29 DNA polymerase and multiply-primed rolling circle amplification. Genome Res. 2001;11(6):1095–1099. doi:10.1101/gr.180501
135. Lizardi PM, Huang X, Zhu Z, Bray-Ward P, Thomas DC, Ward DC. Mutation detection and single-molecule counting using isothermal rolling-circle amplification. Nat Genet. 1998;19(3):225–232. doi:10.1038/898
136. Zhang DY, Brandwein M, Hsuih T, Li HB. Ramification amplification: a novel isothermal DNA amplification method. Mole Diagn. 2001;6(2):141–150. doi:10.2165/00066982-200106020-00010
137. Thomas DC, Nardone GA, Randall SK. Amplification of padlock probes for DNA diagnostics by cascade rolling circle amplification or the polymerase chain reaction. Arch Pathol Lab Med. 1999;123(12):1170–1176. doi:10.1043/1543-2165-123.20.1170
138. Konry T, Smolina I, Yarmush JM, Irimia D, Yarmush ML. Ultrasensitive detection of low-abundance surface-marker protein using isothermal rolling circle amplification in a microfluidic nanoliter platform. Small. 2011;7(3):395–400. doi:10.1002/smll.201001620
139. Zhou H, Bouwman K, Schotanus M, et al. Two-color, rolling-circle amplification on antibody microarrays for sensitive, multiplexed serum-protein measurements. Genome Biol. 2004;5(4):R28. doi:10.1186/gb-2004-5-4-r28
140. Juul S, Nielsen CJ, Labouriau R, et al. Droplet microfluidics platform for highly sensitive and quantitative detection of malaria-causing plasmodium parasites based on enzyme activity measurement. ACS Nano. 2012;6(12):10676–10683. doi:10.1021/nn3038594
141. Ibarra-Meneses AV, Cruz I, Chicharro C, et al. Evaluation of fluorimetry and direct visualization to interpret results of a loop-mediated isothermal amplification kit to detect Leishmania DNA. Parasit Vectors. 2018;11(1):250. doi:10.1186/s13071-018-2836-2
142. Kuhn H, Demidov VV, Frank-Kamenetskii MD. Rolling-circle amplification under topological constraints. Nucleic Acids Res. 2002;30(2):574–580. doi:10.1093/nar/30.2.574
143. Walker GT, Little MC, Nadeau JG, Shank DD. Isothermal in vitro amplification of DNA by a restriction enzyme/DNA polymerase system. Proc Natl Acad Sci. 1992;89(1):392. doi:10.1073/pnas.89.1.392
144. Hellyer TJ, DesJardin LE, Teixeira L, Perkins MD, Cave MD, Eisenach KD. Detection of viable mycobacterium tuberculosis by reverse transcriptase-strand displacement amplification of mRNA. J Clin Microbiol. 1999;37(3):518–523.
145. Little MC, Andrews J, Moore R, et al. Strand displacement amplification and homogeneous real-time detection incorporated in a second-generation DNA probe system, BDProbeTecET. Clin Chem. 1999;45(6):
146. Chen Q, Bian Z, Chen M, et al. Real-time monitoring of the strand displacement amplification (SDA) of human cytomegalovirus by a new SDA-piezoelectric DNA sensor system. Biosens Bioelectron. 2009;24(12):3412–3418. doi:10.1016/j.bios.2009.06.012
147. Dong H, Zhang J, Ju H, et al. Highly sensitive multiple microRNA detection based on fluorescence quenching of graphene oxide and isothermal strand-displacement polymerase reaction. Anal Chem. 2012;84(10):4587–4593. doi:10.1021/ac300721u
148. Zhang -Z-Z, Zhang C-Y. Highly sensitive detection of protein with aptamer-based target-triggering two-stage amplification. Anal Chem. 2012;84(3):1623–1629. doi:10.1021/ac2029002
149. Cui W, Wang L, Jiang W. A dual amplification fluorescent strategy for sensitive detection of DNA methyltransferase activity based on strand displacement amplification and DNAzyme amplification. Biosens Bioelectron. 2016;77:650–655. doi:10.1016/j.bios.2015.10.040
150. Yan X, Tang M, Yang J, et al. A one-step fluorescent biosensing strategy for highly sensitive detection of HIV-related DNA based on strand displacement amplification and DNAzymes. RSC Adv. 2018;8(55):31710–31716. doi:10.1039/C8RA06480F
151. Verkooyen RP, Noordhoek GT, Klapper PE, et al. Reliability of nucleic acid amplification methods for detection of Chlamydia trachomatis in urine: results of the first international collaborative quality control study among 96 laboratories. J Clin Microbiol. 2003;41(7):3013. doi:10.1128/JCM.41.7.3013-3016.2003
152. Akduman D, Ehret JM, Messina K, Ragsdale S, Judson FN. Evaluation of a strand displacement amplification assay (BD ProbeTec-SDA) for detection of Neisseria gonorrhoeae in urine specimens. J Clin Microbiol. 2002;40(1):281. doi:10.1128/JCM.40.1.281-283.2002
153. Van Der Pol B, Ferrero DV, Buck-Barrington L, et al. Multicenter evaluation of the BDProbeTec ET System for detection of Chlamydia trachomatis and Neisseria gonorrhoeae in urine specimens, female endocervical swabs, and male urethral swabs. J Clin Microbiol. 2001;39(3):1008–1016. doi:10.1128/JCM.39.3.1008-1016.2001
154. Shi C, Liu Q, Ma C, Zhong W. Exponential strand-displacement amplification for detection of MicroRNAs. Anal Chem. 2014;86(1):336–339. doi:10.1021/ac4038043
155. Wang Y, Wang Y, Wang H, Xu J, Ye C. A label-free technique for accurate detection of nucleic acid-based self-avoiding molecular recognition systems supplemented multiple cross-displacement amplification and nanoparticles based biosensor. Artif Cells Nanomed Biotechnol. 2018;46(8):1671–1684. doi:10.1080/21691401.2017.1389748
156. Wang Y, Yan W, Fu S, et al. Multiple cross displacement amplification coupled with nanoparticles-based lateral flow biosensor for detection of staphylococcus aureus and identification of methicillin-resistant S. aureus. Front Microbiol. 2018;9:907. doi:10.3389/fmicb.2018.00907
157. Wang Y, Li H, Li D, et al. Multiple cross displacement amplification combined with gold nanoparticle-based lateral flow biosensor for detection of vibrio parahaemolyticus. Front Microbiol. 2016;7:2047.
158. Wang Y, Wang Y, Xu J, Ye C. Development of multiple cross displacement amplification label-based gold nanoparticles lateral flow biosensor for detection of Shigella spp. Front Microbiol. 2016;2016(7):1834.
159. Wang Y, Yan W, Wang Y, Xu J, Ye C. Rapid, sensitive and reliable detection of Klebsiella pneumoniae by label-free multiple cross displacement amplification coupled with nanoparticles-based biosensor. J Microbiol Methods. 2018;149:80–88. doi:10.1016/j.mimet.2018.05.003
160. Wang Y, Li H, Wang Y, et al. Development of multiple cross displacement amplification label-based gold nanoparticles lateral flow biosensor for detection of Listeria monocytogenes. Int J Nanomedicine. 2017;12:473–486. doi:10.2147/IJN.S123625
161. Niu L, Zhao F, Chen J, et al. Isothermal amplification and rapid detection of Klebsiella pneumoniae based on the multiple cross displacement amplification (MCDA) and gold nanoparticle lateral flow biosensor (LFB). PLoS One. 2018;13(10):e0204332. doi:10.1371/journal.pone.0204332
162. Asiello PJ, Baeumner AJ. Miniaturized isothermal nucleic acid amplification, a review. Lab Chip. 2011;11(8):1420–1430. doi:10.1039/c0lc00666a
163. Zanoli LM, Spoto G. Isothermal amplification methods for the detection of nucleic acids in microfluidic devices. Biosensors. 2013;3(1):18. doi:10.3390/bios3010018
164. Esch MB, Locascio LE, Tarlov MJ, Durst RA. Detection of viable Cryptosporidium parvum using DNA-modified liposomes in a microfluidic chip. Anal Chem. 2001;73(13):2952–2958. doi:10.1021/ac001508n
165. Kaarj K, Akarapipad P, Yoon J-Y. Simpler, faster, and sensitive zika virus assay using smartphone detection of loop-mediated isothermal amplification on paper microfluidic chips. Sci Rep. 2018;8(1):12438. doi:10.1038/s41598-018-30797-9
166. Magro L, Jacquelin B, Escadafal C, et al. Paper-based RNA detection and multiplexed analysis for Ebola virus diagnostics. Sci Rep. 2017;7(1):1347. doi:10.1038/s41598-017-00758-9
167. Linnes JC, Rodriguez NM, Liu L, Klapperich CM. Polyethersulfone improves isothermal nucleic acid amplification compared to current paper-based diagnostics. Biomed Microdevices. 2016;18(2):30. doi:10.1007/s10544-016-0057-z
168. Becker H, Gartner C. Polymer microfabrication technologies for microfluidic systems. Anal Bioanal Chem. 2008;390(1):89–111. doi:10.1007/s00216-007-1692-2
169. Van Ness J, Van Ness LK, Galas DJ. Isothermal reactions for the amplification of oligonucleotides. Proc Natl Acad Sci. 2003;100(8):4504. doi:10.1073/pnas.0730811100
170. Kurn N, Chen P, Heath JD, Kopf-Sill A, Stephens KM, Wang S. Novel isothermal, linear nucleic acid amplification systems for highly multiplexed applications. Clin Chem. 2005;51(10):1973. doi:10.1373/clinchem.2005.053694
171. Shimada M, Hino F, Sagawa H, Mukai H, Asada K, Kato I. [Development of the detection system for Mycobacterium tuberculosis DNA by using the isothermal DNA amplification method ICAN]. Rinsho Byori. 2002;50(5):528–532.
172. Ma C, Liu S, Shi C. Ultrasensitive detection of microRNAs based on hairpin fluorescence probe assisted isothermal amplification. Biosens Bioelectron. 2014;58:57–60. doi:10.1016/j.bios.2014.02.034
173. Connolly AR, Trau M. Rapid DNA detection by beacon-assisted detection amplification. Nat Protoc. 2011;6:772. doi:10.1038/nprot.2011.326
174. Norén T, Unemo M, Magnusson C, Eiserman M, Matussek A. Evaluation of the rapid loop-mediated isothermal amplification assay illumigene for diagnosis of clostridium difficile in an outbreak situation. APMIS. 2014;122(2):155–160. doi:10.1111/apm.12121
175. Lalande V, Barrault L, Wadel S, Eckert C, Petit J-C, Barbut F. Evaluation of a loop-mediated isothermal amplification assay for diagnosis of clostridium difficile infections. J Clin Microbiol. 2011;49(7):2714. doi:10.1128/JCM.01835-10
176. Chow WH, McCloskey C, Tong Y, et al. Application of isothermal helicase-dependent amplification with a disposable detection device in a simple sensitive stool test for toxigenic Clostridium difficile. J Mol Diagn. 2008;10(5):452–458. doi:10.2353/jmoldx.2008.080008
177. Gaydos CA, Schwebke J, Dombrowski J, et al. Clinical performance of the Solana(R) point-of-care trichomonas assay from clinician-collected vaginal swabs and urine specimens from symptomatic and asymptomatic women. Expert Rev Mol Diagn. 2017;17(3):303–306. doi:10.1080/14737159.2017.1282823
178. Uphoff TS, Buchan BW, Ledeboer NA, Granato PA, Daly JA, Marti TN. Multicenter evaluation of the solana group a streptococcus assay: comparison with culture. J Clin Microbiol. 2016;54(9):2388–2390. doi:10.1128/JCM.01268-16
179. Goldmeyer J, Li H, McCormac M, et al. Identification of Staphylococcus aureus and determination of methicillin resistance directly from positive blood cultures by isothermal amplification and a disposable detection device. J Clin Microbiol. 2008;46(4):1534. doi:10.1128/JCM.02234-07
180. Moosavian M, Seyed-Mohammadi S, Saki M, et al. Loop-mediated isothermal amplification for detection of Legionella pneumophila in respiratory specimens of hospitalized patients in Ahvaz, southwest Iran. Infect Drug Resist. 2019;12:529–534. doi:10.2147/IDR.S198099
181. Couturier B, Schlaberg R, Konzak C, Nicholes J, Law C, She RC. tcdA as a diagnostic target in a loop‐mediated amplification assay for detecting toxigenic clostridium difficile. J Clin Lab Anal. 2013;27(3):171–176. doi:10.1002/jcla.2013.27.issue-3
182. Cheaveau J, Nguyen H, Chow B, et al. Clinical validation of a commercial LAMP test for ruling out malaria in returning travelers: a prospective diagnostic trial. Open Forum Infect Dis. 2018;5(11):ofy260–ofy260. doi:10.1093/ofid/ofy209.172
183. Abirami N, Nidaullah H, Chuah L-O, et al. Evaluation of commercial loop-mediated isothermal amplification based kit and ready-to-use plating system for detection of Salmonella in naturally contaminated poultry and their processing environment. Food Control. 2016;70:74–78. doi:10.1016/j.foodcont.2016.05.035
184. Lim HSY, Zheng Q, Miks-Krajnik M, Turner M, Yuk H-G. Evaluation of commercial kit based on loop-mediated isothermal amplification for rapid detection of low levels of uninjured and injured salmonella on duck meat, bean sprouts, and fishballs in Singapore. J Food Prot. 2015;78(6):1203–1207. doi:10.4315/0362-028X.JFP-14-535
185. Tegegne B, Getie S, Lemma W, Mohon AN, Pillai DR. Performance of loop-mediated isothermal amplification (LAMP) for the diagnosis of malaria among malaria suspected pregnant women in Northwest Ethiopia. Malar J. 2017;16(1):34. doi:10.1186/s12936-017-1692-4
186. Nguyen VAT, Nguyen HV, Dinh TV, et al. Evaluation of Loopamp™MTBC detection kit for diagnosis of pulmonary tuberculosis at a peripheral laboratory in a high burden setting. Diagn Microbiol Infect Dis. 2018;90(3):190–195. doi:10.1016/j.diagmicrobio.2017.11.009
187. Matthias K, Sven E, Ulrich V, et al. Value of the eazyplex® CSF direct assay in rapid diagnosis of invasive meningococcal disease – case report. Acta Microbiol Immunol Hung. 2018;65(3):309–315. doi:10.1556/030.65.2018.021
188. Findlay J, Hopkins KL, Meunier D, Woodford N. Evaluation of three commercial assays for rapid detection of genes encoding clinically relevant carbapenemases in cultured bacteria. J Antimicrob Chemother. 2015;70(5):1338–1342. doi:10.1093/jac/dku571
189. Hinic V, Ziegler J, Straub C, Goldenberger D, Frei R. Extended-spectrum beta-lactamase (ESBL) detection directly from urine samples with the rapid isothermal amplification-based eazyplex(R) SuperBug CRE assay: proof of concept. J Microbiol Methods. 2015;119:203–205. doi:10.1016/j.mimet.2015.10.015
190. Rodel J, Bohnert JA, Stoll S, et al. Evaluation of loop-mediated isothermal amplification for the rapid identification of bacteria and resistance determinants in positive blood cultures. Eur J Clin Microbiol Infect Dis. 2017;36(6):1033–1040. doi:10.1007/s10096-016-2888-1
191. Mourez T, Delaugerre C, Vray M, Lemee V, Simon F, Plantier JC. Comparison of the bioMerieux NucliSENS EasyQ HIV-1 v2.0-HIV-1 RNA quantification assay versus abbott realtime HIV-1 and Roche Cobas TaqMan HIV-1 v2.0 on current epidemic HIV-1 variants. J Clin Virol. 2015;71:76–81. doi:10.1016/j.jcv.2015.08.007
192. Van Der Pol B, Hook EWI, Williams JA, Smith B, Taylor SN. Performance of the BD CTQx and GCQx amplified assays on the BD viper LT compared with the BD viper XTR system. Sex Transm Dis. 2015;42(9):521–523. doi:10.1097/OLQ.0000000000000313
193. Van Der Pol B, Williams JA, Fuller D, Taylor SN, Hook EW
194. Nye MB, Osiecki J, Lewinski M, et al. Detection of Chlamydia trachomatis and Neisseria gonorrhoeae with the cobas CT/NG v2.0 test: performance compared with the BD probeTec CT Qx and GC Qx amplified DNA and aptima AC2 assays. Sex Transm Infect. 2019;95(2):87. doi:10.1136/sextrans-2018-053545
195. Chevaliez S, Dubernet F, Dauvillier C, Hezode C, Pawlotsky JM. The new aptima HCV quant dx real-time TMA assay accurately quantifies hepatitis C virus genotype 1-6 RNA. J Clin Virol. 2017;91:5–11. doi:10.1016/j.jcv.2017.03.020
196. Bruni MP, Freitas da Silveira M, Stauffert D, et al. Aptima Trichomonas vaginalis assay elucidates significant underdiagnosis of trichomoniasis among women in Brazil according to an observational study. Sex Transm Infect. 2019;95(2):129. doi:10.1136/sextrans-2018-053567
197. Cook DA, Smith LW, Law J, et al. Aptima HPV assay versus hybrid capture® 2 HPV test for primary cervical cancer screening in the HPV FOCAL trial. J Clin Virol. 2017;87:23–29. doi:10.1016/j.jcv.2016.12.004
198. Ferrieri P, Nelson K, Thonen-Kerr E, Arbefeville S. Prospective evaluation of xpert xpress strep a automated PCR assay vs solana group a streptococcal NAAT vs conventional throat culture. Am J Clin Pathol. 2018;150(suppl_1):S157–S157.
199. Kurosaki Y, Magassouba NF, Oloniniyi OK, et al. Development and evaluation of Reverse Transcription-Loop-Mediated Isothermal Amplification (RT-LAMP) assay coupled with a portable device for rapid diagnosis of ebola virus disease in guinea. PLoS Negl Trop Dis. 2016;10(2):e0004472–e0004472. doi:10.1371/journal.pntd.0004472
200. Quoc NB, Phuong NDN, Chau NNB, Linh DTP. Closed tube loop-mediated isothermal amplification assay for rapid detection of hepatitis B virus in human blood. Heliyon. 2018;4(3):e00561. doi:10.1016/j.heliyon.2018.e00561
201. Craw P, Mackay RE, Naveenathayalan A, et al. A simple, low-cost platform for real-time isothermal nucleic acid amplification. Sensors (Basel). 2015;15(9):23418–23430. doi:10.3390/s150923418
202. Wang Y, Zhang B, Sun Y, Liu Y, Gu Y. Loop-mediated isothermal amplification on crude DNA as a point-of-care test for the diagnosis of mycoplasma-related vaginitis during early pregnancy. Lab Med. 2017;49(1):59–67. doi:10.1093/labmed/lmx063
203. Liu D, Daubendiek SL, Zillman MA, Ryan K, Kool ET. Rolling Circle DNA Synthesis: Small Circular Oligonucleotides as Efficient Templates for DNA Polymerases. J Am Chem Soc.1996;118(7):1587–1594. doi:10.1021/ja952786k
204. Walker G T.. Empirical Aspects of Strand Displacement Amplification. Genome Research. 1993; 3:1-6. doi:10.1101/gr.3.1.1
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.