Back to Journals » OncoTargets and Therapy » Volume 8
Current and emerging treatment options for hairy cell leukemia
Authors López-Rubio M, Garcia-Marco JA
Received 13 April 2015
Accepted for publication 13 July 2015
Published 19 August 2015 Volume 2015:8 Pages 2147—2156
DOI https://doi.org/10.2147/OTT.S70316
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Daniele Santini
Montserrat López-Rubio,1 Jose Antonio Garcia-Marco2
1Department of Hematology, Hospital Universitario Príncipe de Asturias, Alcalá de Henares, 2Department of Hematology, Hospital Universitario Puerta de Hierro Majadahonda, Majadahonda, Madrid, Spain
Abstract: Hairy cell leukemia (HCL) is a lymphoproliferative B-cell disorder characterized by pancytopenia, splenomegaly, and characteristic cytoplasmic hairy projections. Precise diagnosis is essential in order to differentiate classic forms from HCL variants, such as the HCL-variant and VH4-34 molecular variant, which are more resistant to available treatments. The current standard of care is treatment with purine analogs (PAs), such as cladribine or pentostatin, which provide a high rate of long-lasting clinical remissions. Nevertheless, ~30%–40% of the patients relapse, and moreover, some of these are difficult-to-treat refractory cases. The use of the monoclonal antibody rituximab in combination with PA appears to produce even higher responses, and it is often employed to minimize or eliminate residual disease. Currently, research in the field of HCL is focused on identifying novel therapeutic targets and potential agents that are safe and can universally cure the disease. The discovery of the BRAF mutation and progress in understanding the biology of the disease has enabled the scientific community to explore new therapeutic targets. Ongoing clinical trials are assessing various treatment strategies such as the combination of PA and anti-CD20 monoclonal antibodies, recombinant immunotoxins targeting CD22, BRAF inhibitors, and B-cell receptor signal inhibitors.
Keywords: hairy cell leukemia, purine analogs, rituximab, immunotoxins, vemurafenib, ibrutinib
Introduction
Hairy cell leukemia (HCL) is defined, according to the World Health Organization classification, as a mature peripheral B-cell neoplasm that accounts for 2%–3% of all adult leukemias.1 It is characterized by infiltration of the bone marrow, liver, and spleen by malignant B cells with hair-like cytoplasmic projections and an indolent course. HCL is more frequent in males, with an overall male to female ratio of 4:1, and the median age at onset is 52 years.2 Hairy cells have a characteristic immunophenotyping profile showing positivity for CD11c, CD25, and CD103 markers, in addition to the B-cell antigens CD20 and CD22. However, recurrent chromosomal translocations have not been identified. Recently, the BRAF V600E mutation has been identified activating the MEK-ERK pathway in patients with classic HCL (HCL-c).3 This finding has implications for the pathogenesis, diagnosis, and targeted therapy.
Precise diagnosis and detailed workup are essential because the clinical profile of HCL can closely mimic that of other chronic B-cell lymphoproliferative disorders that are treated differently. Variants of HCL, such as HCL-variant (HCL-v) and VH4-34 molecular variants, have a different immunophenotype and specific VH gene usage and are more resistant to available treatments. BRAF mutations are absent in both variants.4
Before the introduction of purine analogs (PAs), splenectomy and treatment with interferon alpha led to clinical and hematological responses; however, these were rarely complete and median survival was only 4 years.5–7 Front-line treatment of HCL is currently based on the purine nucleoside analogs pentostatin or cladribine. Both agents confer high and durable response rates, but 30%–40% of patients will eventually relapse 5–10 years after their first treatment.8,9 In this disease, the major challenge is the treatment of patients with multiple clinical relapses as well as those with HCL variants that are refractory to standard PA treatments.
This article provides an update of the treatment options that are currently available and reviews the results of clinical trials with novel molecules that may change the future therapeutic landscape of this rare disease.
Current standard of care
The main indications for treatment are symptomatic cytopenias or painful splenomegaly. If a patient is asymptomatic and cytopenias are minimal, it is reasonable to adopt a watch-and-wait policy. It should be noted that the risk of opportunistic infections in patients with monocytopenia, with or without neutropenia, is high. Therefore, even asymptomatic patients may be considered for early treatment.
First-line treatment
PA therapy as primary treatment
Since the effect of the PAs pentostatin and cladribine were discovered in HCL patients, treatment with these drugs currently remains the standard of care. Both agents induce complete remission (CR) in a high proportion of patients (>80%), and most studies demonstrate a median disease-free survival of >10 years.8–11 Nevertheless, pentostatin and cladribine have not been compared in large, randomized trials and most of the available response and toxicity data derive from published retrospective series. A long-term follow-up study by Else et al demonstrated no difference in outcome between the two agents.9 The overall response rates (ORRs) were 96%–100%, CR around 80%, and 10-year overall survival was between 85% and 100%, without any statistically significant differences. Relapse rates were similar for both groups (44% for pentostatin and 38% for cladribine). In another retrospective study, 107 patients treated with pentostatin or cladribine were evaluated and showed CR of 92% in pentostatin-treated patients and 88% in cladribine-treated patients, with an ORR of 100% in both groups. Minimal residual disease (MRD) was positive in 52% and 47% (P=0.445), respectively. Of those treated, 51% and 25% relapsed (P=0.016), and the median treatment-free interval (TFI) was 95 months for pentostatin versus 144 months for cladribine, without any significant differences. Considering the two groups, the data showed TFI of 170 months versus 44 months for patients with CR versus partial remission (PR).12
Doses and PA treatment schedules were recently published in the revised British guidelines for the diagnosis and management of HCL,13 which are included in Table 1. Different dosing and schedules have been used with similar results. Robak et al showed that cladribine could be infused over 2 hours instead of 24 hours, with 19 out of 23 patients achieving CR. The same study demonstrated that there was no statistically significant difference between CR rates in the group receiving the medication over 5 days versus 7 days, with the 5-day arm presenting less infectious complications.14 Cladribine was administered at doses of 0.15 mg/kg weekly for 6 weeks. Initial results suggested similar efficacy with decreased immunosuppression and infections.15 However, a larger follow-up trial that included 138 patients showed that this schedule did not significantly reduce toxicities.16 Furthermore, daily subcutaneous administration of cladribine over 7 consecutive days has been studied in 73 patients. The efficacy and toxicity were comparable to studies with intravenous cladribine and the plasma concentrations of subcutaneous cladribine were similar to those obtained using the intravenous route.17 Using a 5-day subcutaneous dosing regimen, von Rohr et al treated 62 HCL patients with cladribine 0.14 mg/kg/d. The CR rate achieved was 76% with a response rate of 97%.18
 | Table 1 Purine analog treatment schedules in HCL |
Multiple studies have demonstrated the type of response to PA as an important prognostic factor, with patients who only achieved a partial response faring significantly worse than those achieving CR.9,10,19 Therefore, achieving CR is an important treatment goal. Bone marrow assessment after cell count recovery (4–6 months after cladribine therapy or following eight to nine courses of pentostatin) is recommended, and a second course of PA therapy should be administrated if patients do not enter CR.
Monitoring MRD
Although both PAs are highly effective and lead to excellent overall survival, it appears that neither of them is curative since the majority of patients have evidence of MRD despite being in CR. Several authors have studied the presence of MRD using immunohistochemical, flow cytometry, or polymerase chain reaction techniques20–23 that detected MRD in 40%–60% of patients who had received PAs. Flow cytometry is clearly superior to other techniques, but the prognostic implication of detectable MRD after therapy for HCL may be confounded by several factors: variability in the sensitivity of various techniques used to assess MRD, nonuniformity of the criteria used to define MRD, variability in timing of MRD assessments after therapy, and the limited numbers of patients in reported studies.
However, the preponderance of the data suggests that persistence of MRD after therapy with nucleoside analogs is predictive of eventual disease recurrence. Tallman et al24 and Wheaton et al25 studied MRD using immunohistochemical techniques in patients treated with PAs. The prevalence of MRD detected after cladribine (13%) was lower than that after pentostatin treatment (26%) but did not differ significantly. However, the 4-year relapse-free survival rate (55% versus 88%) was significantly lower if MRD was detected.
In a retrospective study by López-Rubio et al12 MRD positivity (analyzed by immunophenotyping) was detected in 49% of 107 HCL patients treated with PAs, and significant differences were found between the two treatments. The estimated TFI by Kaplan–Meier analysis was 97 months in MRD+ patients, while the median TFI in MRD patients was not reached (P<0.059).
A study recently published by Garnache Ottou et al26 confirmed the utility of detecting MRD using the results of flow cytometry techniques in order to identify patients at a high risk of relapse.
Chemoimmunotherapy: rituximab with cladribine or pentostatin
The rationale for adding rituximab to PAs was based on the efficacy and safety of this combination in patients with relapsed disease,27 as well as the significant synergy of this combination observed in other lymphoproliferative disorders. Ravandi et al28 reported a Phase II clinical trial in 31 previously untreated patients with HCL. Cladribine was administered intravenously at 5.6 mg/m2 over 2 h/d for 5 consecutive days. Approximately 4 weeks after initiating cladribine, eight weekly doses of rituximab (375 mg/m2 intravenously) were administered. A CR rate of 100% was reported and, after a median follow-up of 25 months, the median CR, PFS, and overall survivals had not been reached. Patients achieved MRD-negative status after completion of treatment with rituximab, as demonstrated by flow cytometry and consensus polymerase chain reaction. Despite a significant decline in the number of CD4+ and CD8+ T cells, as well as serum immunoglobulin levels, no increase was observed in the incidence of opportunistic infections. A longer follow-up study may provide further information regarding the importance of achieving MRD-negative CR in long-term outcomes.
Cervetti et al29 analyzed the results of ten patients who received four cycles of rituximab after administration of cladribine. Before starting anti-CD20 antibody therapy, two patients were in CR, six in PR, and two showed no significant response to cladribine. All cases resulted to be IgH positive. Eight out of ten patients (four in PR, two in CR, and two unresponsive after 2-chlorodeoxyadenosine) were evaluable for response. Two months after the end of anti-CD20 therapy, all evaluated patients presented complete hematological remission. Moreover, rituximab increased the percentage of molecular remission up to 100% 1-year after the end of treatment. Interestingly, in all cases but one, including those that were persistently polymerase chain reaction-positive, semiquantitative molecular analyses showed MRD levels lower than those found before rituximab administration. The results not only confirm the therapeutic effect of rituximab but also show its relevance in eradicating MRD in HCL.
In a subsequent extended follow-up study, Cervetti et al30 analyzed 27 HCL patients treated with anti-CD20 after pretreatment with cladribine. Patients who demonstrated persistent MRD or detectable clinical disease were treated with rituximab (375 mg/m2 once a week for four doses). Hematological, immunological, and molecular analyses were repeated for 2 months, 6 months, and 12 months after the end of anti-CD20 treatment. The overall hematological response rate was 100% (CR 89%, PR 11%) 2 months after the last rituximab infusion. Molecular analysis revealed a progressive increment in the number of molecular remissions, with an overall molecular response rate of 70%. PFS was significantly affected both by the quality of response to rituximab (2-year PFS 50% for patients achieving PR versus 94% for cases in CR) (P<0.001) and by the molecular status (30% in cases MRD-positive versus 100% for patients MRD-negative; P<0.001).
Two ongoing studies are evaluating therapy with rituximab and PAs: one of them being an MD Anderson Cancer Center-supported “Phase II Study of 2CDA followed by rituximab in HCL” (NCT00412594), which is in recruitment phase; and the other, the National Cancer Institute-supported “Cladribine with simultaneous or delayed rituximab to treat HCL” (NCT00923013), with recruitment finished. The results of the two studies will provide useful data in the initial management of patients.
Role of interferon alpha and splenectomy
The role of interferon alpha is limited to patients presenting with severe pancytopenia and those with a pressing need for cell count recovery. A regimen of 3 mega-units three times a week will gradually improve blood counts and facilitate the subsequent use of either nucleoside analog.31
The principal indication for splenectomy is the finding of a very significant splenomegaly in the presence of low-level bone marrow infiltration.
Figure 1 presents a proposed algorithm for first-line treatment in HCL. This strategy is based on the expert opinion adopted by the Spanish CLL group. The aim is to eradicate MRD in patients with clinical response in order to improve the TFI. In these cases, the number of rituximab doses required to achieve a deeper response is not clear. Nevertheless, the cost and side effects of rituximab have to be taken into account, especially with respect to the addition of immunosuppression. This is the rationale behind the administration of four doses of rituximab and further evaluation of MRD status before consolidation with four additional doses if MRD+ is detected.
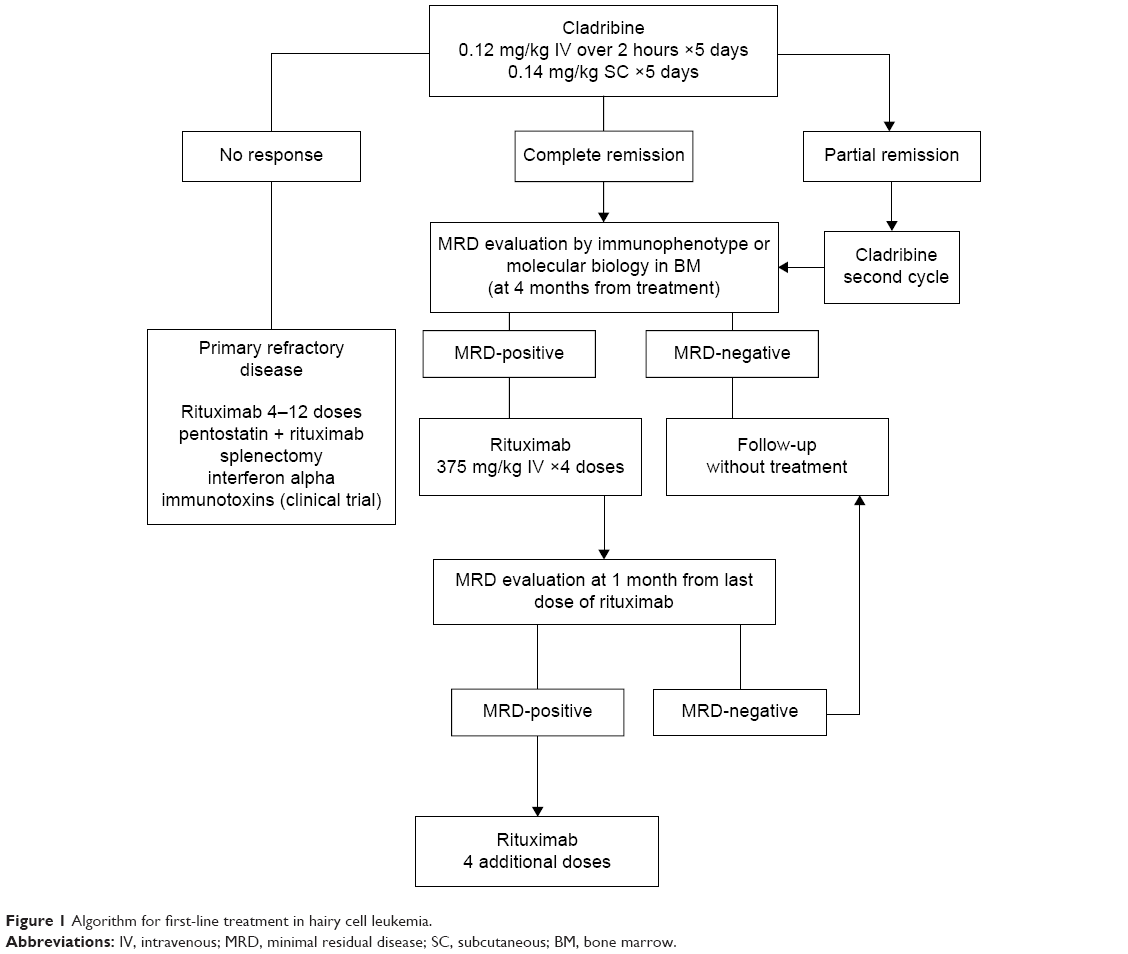 | Figure 1 Algorithm for first-line treatment in hairy cell leukemia. |
Treatment at relapse and refractory disease
Although relapsed and refractory diseases are vastly different entities, the majority of studies include both subsets when evaluating treatment options.
Patients with relapsed disease can be given additional courses of pentostatin or cladribine, although the response rate and duration tend to decrease with each subsequent course.9,32–34 The choice of agent at relapse may depend on the duration of first remission: if short (<2 years), the alternative agent should be used; if longer (>2 years), the patient should be retreated using the same agent. The combination of pentostatin or cladribine with rituximab is another therapeutic option.35 In a retrospective study published by Else et al36 18 patients with relapsed disease were treated with pentostatin or cladribine in combination with rituximab, showing a CR rate of 89%. Of the 13 patients who were evaluated for MRD, all were negative and responses were sustained at a median follow-up of 36 months with an estimated relapse rate of ~7%–11% at 3 years. Both agents were well tolerated in the long term with lymphocytopenia being the main concern.
Fludarabine or bendamustine combinations with rituximab have recently been explored in two small series with promising results,27,37 and an ongoing study (NCT01059786) is also comparing bendamustine with pentostatin in combination with rituximab in the multiply relapsed setting.
In relapsed patients, treatment with interferon should be considered in the absence of any other available alternative.38,39
Figure 2 shows a proposed algorithm for second-line treatment in HCL. Patients relapsing 2 years after initial treatment are treated with the same PA plus eight doses of rituximab; while those relapsing within the first 2 years after initial treatment should receive an alternative PA plus eight doses of rituximab. In the case of a second relapse, different treatment options are listed.
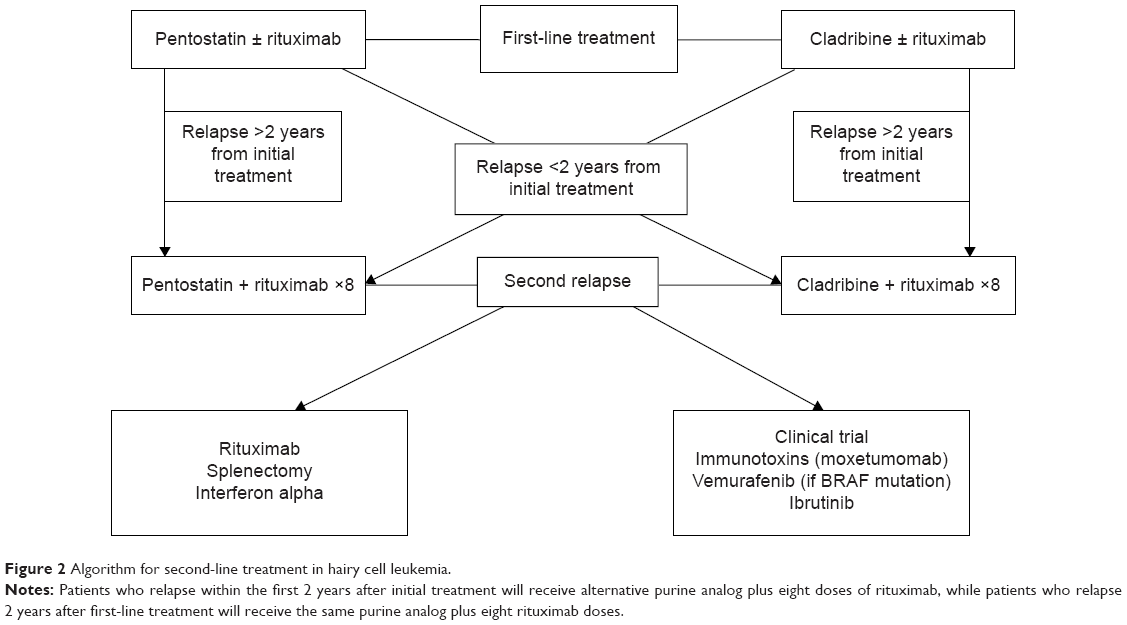 | Figure 2 Algorithm for second-line treatment in hairy cell leukemia. |
Recently, a subgroup of patients has been described that does not respond to first-line PA or subsequent treatments. These patients are characterized by the presence of leukocytosis, bulky splenomegaly, unmutated IGHV status, and p53 dysfunction, which generally confer resistance to PAs and poor prognosis.40 In cases with these clinical features, molecular analysis for p53 dysfunction (deletion/mutation) and IGHV mutational status are recommended to establish a different treatment approach in the context of clinical trials or the use of new agents based on molecular findings.
HCL Variant
HCL-v is considered to be unrelated to HCL and, according to the 2008 World Health Organization classification, is now separately categorized as an unclassifiable splenic B-cell lymphoma/leukemia together with the splenic diffuse red pulp variant of B-cell lymphoma. However, it is important in the differential diagnosis of HCL due to the different results of treatment.
HCL-v differs from HCL in its morphology, immunophenotype, and molecular characteristics. As in HCL, the cells in most HCL-v cases are villous and large. However, the cells in HCL-v have a distinct nucleolus and round nucleus resembling B-cell prolymphocytic leukemia. The immunophenotype of HCL-v cells differs from that of HCL in that, as a rule, CD25 and HC2 are not expressed. CD103 is expressed infrequently, and CD11c is nearly always positive. Moreover, patients with HCL-v have wild-type BRAF.4,41 HCL-v responds differently to standard HCL treatment, being generally resistant to interferon alpha and rarely achieving CR with either pentostatin or cladribine.
In the largest series published to date (n=58), splenectomy resulted in good PR in two-thirds of patients.42 Very rarely, patients may achieve CR after three or four courses of cladribine. Various case reports demonstrated the usefulness of rituximab either alone or after splenectomy in the treatment of HCL-v patients.43–45 In a recent study published by Kreitman et al46 ten HCL-v patients received cladribine 0.15 mg/kg on days 1–5, with eight weekly doses of rituximab 375 mg/m2 beginning on day 1. After 6 months, nine out of ten patients achieved CR, and eight remained free of MRD after a follow-up of 12–48 (median 27) months. No dose-limiting toxicities were observed when combining cladribine and rituximab. Cytopenias in CRs resolved within 7–211 (median 34) days without major infections. The authors concluded that although cladribine alone lacks effectiveness for early or relapsed HCL-v, cladribine with immediate rituximab achieves CRs without MRD and its administration is feasible.
Emerging treatment options in HCL
As a result of improved understanding of the pathobiology of HCL, new therapeutic targets have been identified (summarized in Table 2). Agents targeting these molecular pathways are under investigation, and some of them have demonstrated significant activity in relapsed patients. Ongoing and planned clinical trials are assessing several treatment strategies, such as the combination of PAs and various anti-CD20 monoclonal antibodies, recombinant immunotoxins targeting CD22 (eg, moxetumomab pasudotox),47,48 BRAF inhibitors, such as vemurafenib,49,50 and B-cell receptor (BCR) signaling inhibitors (eg, Bruton’s tyrosine kinase inhibitor ibrutinib). In Table 3, we show preliminary results and toxic effects of novel therapeutic agents in HCL.
 | Table 2 Novel therapeutic agents and their mechanism of action |
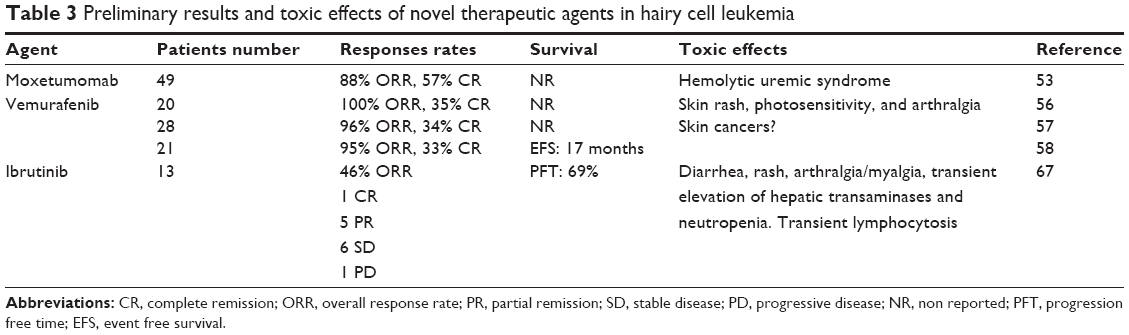 | Table 3 Preliminary results and toxic effects of novel therapeutic agents in hairy cell leukemia |
Immunotoxins
These hybrid agents contain a monoclonal antibody that identifies and binds to a specific cell target (CD25 or CD22 in the case of HCL) and the truncated toxin (pseudomona or diphtheria exotoxin), which is released inside the cell to block protein synthesis. A few clinical trials from the National Cancer Institute have studied the efficacy of recombinant immunotoxins against CD22 (BL22 and moxetumomab pasudotox) and CD25 (LMB-2) in patients with refractory HCL. Since CD25 is not universally overexpressed in HCL, the focus has been diverted to CD22, which is uniformly overexpressed in HCL.
Kreitman et al51 detailed the use of recombinant immunotoxin (BL22) in a Phase I study of patients resistant to first-line cladribine. The study included 16 HCL patients with CD22 expression and three had variant diseases. Of the patients treated, eleven achieved a CR and two attained a PR. Three patients ultimately relapsed (two with HCL-v) but achieved a second CR after retreatment. The side effects included dose-limiting cytokine release syndrome. Additionally, hemolytic uremic syndrome developed in two patients and both were resolved with plasmapheresis. No hematological toxicity was observed. In a Phase II clinical trial, immunotoxin BL22 was tested in 36 patients with relapsed and refractory HCL.52 After one cycle (40 μg/kg every other day, three doses), the CR rate was 25% and the ORR was 50%, with CR improving to 47% and ORR to 72% after retreatment. The median follow-up of this study was 26 months, with six patients relapsing and four of the 17 patients with a CR relapsed. ORR was impacted by spleen size. The regimen was well tolerated with mostly grade 1 and 2 hypoalbuminemia and elevated liver function tests. Three patients developed hemolytic uremic syndrome but none required plasmapheresis.
In order to enhance the efficacy and safety of BL22, the binding affinity of the immunotoxin to CD22 was improved by identifying a mutant (with three different amino acids) in the complementarity-determining region 3 of the monoclonal antibody–variable heavy chain. This immunotoxin is known as moxetumomab pasudotox, or HA22. An update of the Phase I dose escalation trial of moxetumomab pasudotox in 49 patients with relapsed and refractory HCL showed a CR rate of 57% and an ORR of 88%.53 Among patients who received a high dose (50 μg/kg every other day, three doses; n=33), 64% achieved CR and 13 out of 21 achieved MRD-negative CR, which was maintained after a median of 32 months. Two patients developed grade 2 hemolytic uremic syndrome, which was reversible without specific treatment. Those who had previously undergone splenectomy achieved PR. Patients with the lowest HCL burden appeared to have a better chance of a durable CR. A National Cancer Institute-supported study of moxetumomab pasudotox (NCT01829711) is ongoing in patients who are refractory or have relapsed at least twice after standard therapy.
BRAF and MAPK pathway inhibitors
The BRAF (V600E) mutation is a molecular hallmark of HCL-c. The BRAF gene is a member of the serine/threonine protein kinase family. Its product, the B-raf protein, is part of the signal transduction protein kinase family, which is critical to cell division and differentiation. BRAF mutations provide Ras-independent activation of the MAPK pathway, causing hyperactivation of ERK and thereby promoting the growth, survival, and differentiation of HCL cells. BRAF V600E mutations also occur in other cancers, such as malignant melanoma and papillary thyroid cancer. Clinical trials of BRAF inhibitors to treat HCL are motivated by results from the use of BRAF inhibitors to treat metastatic melanoma.
Vemurafenib is an oral agent that inhibits the thymidine kinase enzyme and specifically targets cells containing BRAF (V600E) mutations. Anecdotal case reports have demonstrated the activity of vemurafenib in patients with relapsed HCL.49,50,54 Preliminary results of a Phase II trial of vemurafenib in five patients with relapsed HCL have been reported.55 At the 2014 American Society of Hematology annual meeting, Park et al presented the data of 20 patients treated with vemurafenib 960 mg twice daily for 3 months.56 Patients with partial or complete response with detectable MRD were allowed to receive vemurafenib for up to three additional months. Twenty patients were evaluable for toxicity and 17 for disease response with a median follow-up of 10 months. Out of 20 patients, the ORR was 100%. Six patients achieved CR (four MRD- and two MRD+) and eleven achieved PR with very minimal disease. Tiacci et al57 reported the results of an Italian Phase II Clinical trial evaluating the efficacy and safety of vemurafenib in HCL patients who were refractory to or had relapsed after PAs. The study included 28 patients: six refractory to first-line treatment with PA and 21 who relapsed early and/or repeatedly after PA. Vemurafenib was administered orally on an outpatient basis at a dose of 960 mg twice daily for a median of 16 weeks and was generally well tolerated. The ORR was 96%, 34.6% CR and 61.4% PR, obtained after a median of 8 weeks and 9 weeks, respectively. Retreatment with vemurafenib was able to induce remissions in patients relapsing after a CR, but was less effective in patients relapsing after a PR. Sascha et al58 presented a European multicenter experience of 21 patients treated with vemurafenib. Patients had received a median of 3 (range 0–12) previous treatment lines. Vemurafenib was started at a dose of 240 mg bid in 18 patients and continued at this dose in 14 patients. In the remaining patients, doses were escalated between 480 mg and 960 mg with median duration of 90 (range 55–167) days. Blood counts improved in 20 patients who met response criteria. The median times to neutrophil and platelet count recovery and improvement of anemia were 39 days, 28 days, and 67 days, respectively. Seven patients achieved a CR and 13 patients achieved a PR. Patients who received >240 mg bid (n=8) did not achieve significantly more CRs than patients receiving 240 mg. The median observation time was 12 months (range 3–31 months) and median event-free survival (retreatment or death) was 17 months for all patients. Seven patients were retreated at relapse after a median of 10 months after stopping vemurafenib. All patients again demonstrated a response to vemurafenib.
The main toxic effects of vemurafenib are skin rash, photosensitivity, and arthralgia. Extrapolating the clinical data from studies of BRAF inhibitors in melanoma, some of the concerns with the use of vemurafenib in HCL are the development of skin cancers, relapse of HCL owing to activation of the MEK pathway, and development of resistance to vemurafenib.59,60 As a result, other agents, such as dabrafenib, are now being studied. Another strategy is to combine BRAF inhibitors with MEK inhibitors such as trametinib. In patients with melanoma, it has been shown that PFS durations were better after treatment with a combination of dabrafenib and trametinib than with dabrafenib alone,61 results that have been validated in in vitro studies of HCL cells.62 Patients with HCL-v and VH4-34 variants have mutations in the MAPK pathway; therefore, this combination strategy might be useful for patients with HCL-v or VH4-34, which generally do not respond well to PAs. However, the long-term impact of this strategy is currently unclear, and several reports are emerging of resistance to these BRAF inhibitors.59
Ibrutinib
BCR is a transmembrane receptor complex consisting of an extracellular portion-surface immunoglobulin receptor. This receptor consists of two heavy and two light chains, which bind to the antigen, and a cytoplasmic portion comprising a heterodimer of CD79a and CD79b. Activation of BCR results in the stimulation of various intracellular signaling pathways involving kinases, such as SYK, BTK, and LYN, thus stimulating lymphoid cell growth. In recent years, the development of BCR signaling kinase inhibitors has brought a paradigm shift in the therapeutic landscape of lymphoid malignancies. Currently, ibrutinib is the only BTK inhibitor that is commercially available. Ibrutinib has shown excellent results in chronic lymphocytic leukemia and mantle cell lymphoma.63,64
Sivina et al65 explored the expression and function of the BCR-associated kinase BTK and its inhibitor ibrutinib in HCL, demonstrating that BTK protein expression is present in HCL cells and that low ibrutinib concentrations induce full BTK target occupancy in HCL cells. Treatment with ibrutinib inhibited BCR downstream signaling and the proliferation and metabolism of HCL cells, suggesting that ibrutinib has a direct effect on HCL cell survival and growth. All this justifies the development of BCR-associated kinase inhibitors, such as ibrutinib, in patients with HCL.
A Phase II clinical trial (NCT01841723) is currently evaluating the use of ibrutinib in patients with relapsed HCL. Preliminary data presented at the American Society of Clinical Oncology (ASCO) suggested that this agent is well tolerated in HCL.66 These data were updated at the 20th Congress of European Hematology Association67 with the following results: 13 patients (two with HCL-v and eleven with relapsed HCL-c) received continuous oral ibrutinib (420 mg daily) in 28-day cycles. One MRD-negative complete response (HCL-c) and five partial responses have been observed (ORR 46%). Four additional patients (30%) with stable disease have experienced clinical benefit that does not meet criteria for PR and continue on treatment. At a median follow-up of 14.5 months, nine patients (69%) remain progression free on treatment, three patients (one with HCL-v and two with HCL-c) have progressed, and one patient (HCL-c) discontinued in cycle 8 after failing to resolve baseline neutropenia. The most frequent (>20%) grade 3/4 adverse events included lymphopenia (37%), hypophosphatemia (30%), neutropenia (23%), and infection (23%). Common grade 1/2 adverse events included myalgias (61%), headache (38%), dizziness (38%), diarrhea (38%), arthralgias (30%), rash (30%), and fatigue (30%). Other hematologic adverse events included grade 1/2 anemia (38%) and grade 1/2 thrombocytopenia (38%). Redistribution lymphocytosis (peaking at day 8) occurred in both HCL-v patients and in one HCL-c patient with circulating disease at baseline.
Expert commentary
HCL therapy with PAs has improved treatment outcomes, with long-term remissions and a life expectancy, which is not significantly different from that of a healthy matched population. However, long-term follow-up studies have shown that relapse-free survival curves do not reach a plateau with PAs and late relapses can occur; therefore, the treatment for relapsed and refractory disease remains a challenge. In patients with HCL, due to its marked efficacy, brief treatment duration and favorable toxicity profile, our first-line treatment is cladribine, which is followed by a second cycle if CR is not achieved. MRD should be monitored and we recommend four to eight doses of rituximab to try to achieve complete MRD-negative remission. It is expected that the results of ongoing studies will provide information regarding the effectiveness of adding rituximab to first-line treatment. Upon relapse after a long remission, and especially in patients treated with cladribine, we recommend an analysis of the mutational status of IGHV genes and a search for VH4-34 gene usage, together with an analysis of TP53 mutations. If negative, patients may be retreated with a second course of cladribine or pentostatin with rituximab. However, a change of the PA is recommended in patients who have only had a short remission. In patients in second relapse, refractoy disease, positivity for TP53 or BRAF V600E mutations or VH4-34 gene usage, treatment with targeted immunotoxins, BRAF inhibitors, either alone or in combination with MEK inhibitors, and BCR signaling inhibitors, such as ibrutinib, have provided new approaches. Positive Phase II data of moxetumomab pasudotox, an anti-CD22 immunotoxin, indicate that immunotoxin therapy can achieve durable MRD-negative CRs in patients with refractory disease. Vemurafenib and ibrutinib are currently the most promising agents undergoing clinical trials. The relative lack of serious side effects, such as myelotoxicity, and oral administration are the major advantages of these agents over conventional PAs.
Acknowledgments
The authors wish to thank Martin Hadley-Adams for reviewing the English language.
Disclosure
The authors report no conflicts of interest in this work.
References
Campo E, Swerdlow SH, Harris NL, Pileri S, Stein H, Jaffe ES. The 2008 WHO classification of lymphoid neoplasms and beyond: evolving concepts and practical applications. Blood. 2011;117(19):5019–5032. | ||
Cannon T, Mobarek D, Wegge J, Tabbara IA. Hairy cell leukemia: current concepts. Cancer Invest. 2008;26(8):860–865. | ||
Tiacci E, Trifonov V, Schiavoni G, et al. BRAF mutations in hairy-cell leukemia. N Engl J Med. 2011;364(24):2305–2315. | ||
Xi L, Arons E, Navarro W, et al. Both variant and IGHV4-34-expressing hairy cell leukemia lack the BRAF V600E mutation. Blood. 2012;119(14):3330–3332. | ||
Berman E, Heller G, Kempin S, Gee T, Tran LL, Clarkson B. Incidence of response and long-term follow-up in patients with hairy cell leukemia treated with recombinant interferon alfa-2a. Blood. 1990;75(4):839–845. | ||
Grever M, Kopecky K, Foucar MK, et al. Randomized comparison of pentostatin versus interferon alfa-2a in previously untreated patients with hairy cell leukemia: an intergroup study. J Clin Oncol. 1995;13(4):974–982. | ||
Federico M, Frassoldati A, Lamparelli T, et al. Long-term results of alpha interferon as initial therapy and splenectomy as consolidation therapy in patients with hairy cell leukemia. Final report from the Italian cooperative group for HCL. Ann Oncol. 1994;5(8):725–731. | ||
Chadha P, Rademaker AW, Mendiratta P, et al. Treatment of hairy cell leukemia with 2-chlorodeoxyadenosine (2-CdA): long-term follow-up of the Northwestern University experience. Blood. 2005;106(1):241–246. | ||
Else M, Dearden CE, Matutes E, et al. Long-term follow-up of 233 patients with hairy cell leukaemia, treated initially with pentostatin or cladribine, at a median of 16 years from diagnosis. Br J Haematol. 2009;145(6):733–740. | ||
Maloisel F, Benboubker L, Gardembas M, et al. Long-term outcome with pentostatin treatment in hairy cell leukemia patients. A French retrospective study of 238 patients. Leukemia. 2003;17(1):45–51. | ||
Zinzani PL, Tani M, Marchi E, et al. Long-term follow-up of front-line treatment of hairy cell leukemia with 2-chlorodeoxyadenosine. Haematologica. 2004;89(3):309–313. | ||
López-Rubio M, Da Silva C, Loscertales J, et al. Hairy cell leukemia treated initially with purine analogs: a retrospective study of 107 patients from the Spanish cooperative group on chronic lymphocytic leukemia (GELLC). Leuk Lymphoma. 2014;55(5):1007–1012. | ||
Jones G, Parry-Jones N, Wilkins B, Else M, Catovsky D. Revised guidelines for the diagnosis and management of hairy cell leukaemia and hairy cell leukaemia variant*. Br J Haematol. 2012;156(2):186–195. | ||
Robak T, Błasińska-Morawiec M, Krykowski E, et al. 2-chlorodeoxyadenosine (2-CdA) in 2-hour versus 24-hour intravenous infusion in the treatment of patients with hairy cell leukemia. Leuk Lymphoma. 1996;22(1–2):107–111. | ||
Lauria F, Bocchia M, Marotta G, Raspadori D, Zinzani PL, Rondelli D. Weekly administration of 2-chlorodeoxyadenosine in patients with hairy-cell leukemia: a new treatment schedule effective and safer in preventing infectious complications. Blood. 1997;89(5):1838–1839. | ||
Robak T, Jamroziak K, Gora-Tybor J, et al. Cladribine in a weekly versus daily schedule for untreated active hairy cell leukemia: final report from the polish adult leukemia group (PALG) of a prospective, randomized, multicenter trial. Blood. 2007;109(9):3672–3675. | ||
Juliusson G, Heldal D, Hippe E, et al. Subcutaneous injections of 2-chlorodeoxyadenosine for symptomatic hairy cell leukemia. J Clin Oncol. 1995;13(4):989–995. | ||
von Rohr A, Schmitz SF, Tichelli A, et al; Swiss Group for Clinical Cancer Research (SAKK), Bern, Switzerland. Treatment of hairy cell leukemia with cladribine (2-chlorodeoxyadenosine) by subcutaneous bolus injection: a phase II study. Ann Oncol. 2002;13(10):1641–1649. | ||
Dearden CE, Else M, Catovsky D. Long-term results for pentostatin and cladribine treatment of hairy cell leukemia. Leuk Lymphoma. 2011;52(suppl 2):21–24. | ||
Ellison DJ, Sharpe RW, Robbins BA, et al. Immunomorphologic analysis of bone marrow biopsies after treatment with 2-chlorodeoxyadenosine for hairy cell leukemia. Blood. 1994;84(12):4310–4315. | ||
Matutes E, Meeus P, McLennan K, Catovsky D. The significance of minimal residual disease in hairy cell leukaemia treated with deoxycoformycin: a long-term follow-up study. Br J Haematol. 1997;98(2):375–383. | ||
Bengio R, Narbaitz MI, Sarmiento MA, Palacios MF, Scolnik MP. Comparative analysis of immunophenotypic methods for the assessment of minimal residual disease in hairy cell leukemia. Haematologica. 2000;85(11):1227–1229. | ||
Sausville JE, Salloum RG, Sorbara L, et al. Minimal residual disease detection in hairy cell leukemia. Comparison of flow cytometric immunophenotyping with clonal analysis using consensus primer polymerase chain reaction for the heavy chain gene. Am J Clin Pathol. 2003;119(2):213–217. | ||
Tallman MS, Hakimian D, Kopecky KJ, et al. Minimal residual disease in patients with hairy cell leukemia in complete remission treated with 2-chlorodeoxyadenosine or 2-deoxycoformycin and prediction of early relapse. Clin Cancer Res. 1999;5(7):1665–1670. | ||
Wheaton S, Tallman MS, Hakimian D, Peterson L. Minimal residual disease may predict bone marrow relapse in patients with hairy cell leukemia treated with 2-chlorodeoxyadenosine. Blood. 1996;87(4):1556–1560. | ||
Garnache Ottou F, Chandesris MO, Lhermitte L, et al. Peripheral blood 8 colour flow cytometry monitoring of hairy cell leukaemia allows detection of high-risk patients. Br J Haematol. 2014;166(1):50–59. | ||
Gerrie AS, Zypchen LN, Connors JM. Fludarabine and rituximab for relapsed or refractory hairy cell leukemia. Blood. 2012;119(9):1988–1991. | ||
Ravandi F, O’Brien S, Jorgensen J, et al. Phase 2 study of cladribine followed by rituximab in patients with hairy cell leukemia. Blood. 2011;118(14):3818–3823. | ||
Cervetti G, Galimberti S, Andreazzoli F, et al. Rituximab as treatment for minimal residual disease in hairy cell leukaemia. Eur J Haematol. 2004;73(6):412–417. | ||
Cervetti G, Galimberti S, Andreazzoli F, et al. Rituximab as treatment for minimal residual disease in hairy cell leukaemia: extended follow-up. Br J Haematol. 2008;143(2):296–298. | ||
Habermann TM, Andersen JW, Cassileth PA, Bennett JM, Oken MM. Sequential administration of recombinant interferon alpha and deoxycoformycin in the treatment of hairy cell leukaemia. Br J Haematol. 1992;80(4):466–471. | ||
Saven A, Burian C, Koziol JA, Piro LD. Long-term follow-up of patients with hairy cell leukemia after cladribine treatment. Blood. 1998;92(6):1918–1926. | ||
Goodman GR, Burian C, Koziol JA, Saven A. Extended follow-up of patients with hairy cell leukemia after treatment with cladribine. J Clin Oncol. 2003;21(5):891–896. | ||
Jehn U, Bartl R, Dietzfelbinger H, Haferlach T, Heinemann V. An update: 12-year follow-up of patients with hairy cell leukemia following treatment with 2-chlorodeoxyadenosine. Leukemia. 2004;18(9):1476–1481. | ||
Grever MR. How I treat hairy cell leukemia. Blood. 2010;115(1):21–28. | ||
Else M, Dearden CE, Matutes E, et al. Rituximab with pentostatin or cladribine: an effective combination treatment for hairy cell leukemia after disease recurrence. Leuk Lymphoma. 2011;52(suppl 2):75–78. | ||
Burotto M, Stetler-Stevenson M, Arons E, Zhou H, Wilson W, Kreitman RJ. Bendamustine and rituximab in relapsed and refractory hairy cell leukemia. Clin Cancer Res. 2013;19(22):6313–6321. | ||
Benz R, Stussi G, Fehr J. Interferon as an alternative to purine analogues in the treatment of hairy cell leukaemia. Br J Haematol. 2010;148(4):664–665. | ||
Seymour JF, Estey EH, Keating MJ, Kurzrock R. Response to interferon-alpha in patients with hairy cell leukemia relapsing after treatment with 2-chlorodeoxyadenosine. Leukemia. 1995;9(5):929–932. | ||
Forconi F, Sozzi E, Cencini E, et al. Hairy cell leukemias with unmutated IGHV genes define the minor subset refractory to single-agent cladribine and with more aggressive behavior. Blood. 2009;114(21):4696–4702. | ||
Tiacci E, Schiavoni G, Forconi F, et al. Simple genetic diagnosis of hairy cell leukemia by sensitive detection of the BRAF-V600E mutation. Blood. 2012;119(1):192–195. | ||
Matutes E, Wotherspoon A, Brito-Babapulle V, Catovsky D. The natural history and clinico-pathological features of the variant form of hairy cell leukemia. Leukemia. 2001;15(1):184–186. | ||
Narat S, Gandla J, Dogan A, Mehta A. Successful treatment of hairy cell leukemia variant with rituximab. Leuk Lymphoma. 2005;46(8):1229–1232. | ||
Quach H, Januszewicz H, Westerman D. Complete remission of hairy cell leukemia variant (HCL-v) complicated by red cell aplasia post treatment with rituximab. Haematologica. 2005;90(suppl):ECR26. | ||
Yoshida T, Mihara K, Sugihara S, et al. Splenectomy followed by administration of rituximab is useful to treat a patient with hairy cell leukemia-variant. Ann Hematol. 2013;92(5):711–713. | ||
Kreitman RJ, Wilson W, Calvo KR, et al. Cladribine with immediate rituximab for the treatment of patients with variant hairy cell leukemia. Clin Cancer Res. 2013;19(24):6873–6881. | ||
Kreitman RJ, Wilson WH, Bergeron K, et al. Efficacy of the anti-CD22 recombinant immunotoxin BL22 in chemotherapy-resistant hairy-cell leukemia. N Engl J Med. 2001;345(4):241–247. | ||
Kreitman RJ, Squires DR, Stetler-Stevenson M, et al. Phase I trial of recombinant immunotoxin RFB4(dsFv)-PE38 (BL22) in patients with B-cell malignancies. J Clin Oncol. 2005;23(27):6719–6729. | ||
Dietrich S, Glimm H, Andrulis M, von KC, Ho AD, Zenz T. BRAF inhibition in refractory hairy-cell leukemia. N Engl J Med. 2012;366(21):2038–2040. | ||
Peyrade F, Re D, Ginet C, et al. Low-dose vemurafenib induces complete remission in a case of hairy-cell leukemia with a V600E mutation. Haematologica. 2013;98(2):e20–e22. | ||
Kreitman RJ, Wilson WH, White JD, et al. Phase I trial of recombinant immunotoxin anti-Tac(Fv)-PE38 (LMB-2) in patients with hematologic malignancies. J Clin Oncol. 2000;18(8):1622–1636. | ||
Kreitman RJ, Stetler-Stevenson M, Margulies I, et al. Phase II trial of recombinant immunotoxin RFB4(dsFv)-PE38 (BL22) in patients with hairy cell leukemia. J Clin Oncol. 2009;27(18):2983–2990. | ||
Arons E, Stetler-Stevenson M, Wilson WH, FitzGerald DJP, Pastan I. Pharmacokinetic analysis of response in hairy cell leukemia treated by anti-CD22 recombinant immunotoxin moxetumomab pasudotox. Blood. 2013;122(21):2871. | ||
Follows GA, Sims H, Bloxham DM, et al. Rapid response of biallelic BRAF V600E mutated hairy cell leukaemia to low dose vemurafenib. Br J Haematol. 2013;161(1):150–153. | ||
Park JH, Chung SS, Chung YR, et al. Phase II trial of the BRAF inhibitor, vemurafenib, in patients with braf mutant relapsed or refractory hairy cell leukemia. Blood. 2013;122(21):2876. | ||
Park JH, Chung SS, Chung YR, et al. Vemurafenib has potent antitumor activity in patients with relapsed/refractory BRAF mutant hairy cell leukemia. Blood. 2014;124:24. | ||
Tiacci E, De Caroli L, Zinzani PL, et al. Efficacy and safety of the BRAF inhibitor vemurafenib in hairy cell leukemia patients refractory to or relapsed after purine analogs: a phase-2 Italian clinical trial. Blood. 2014;124:150. | ||
Sascha D, Pircher A, Andrulis M, et al. BRAF inhibition in hairy cell leukemia: multicentre experience of 21 patients treated with vemurafenib. Blood. 2014;124:3634. | ||
Lito P, Rosen N, Solit DB. Tumor adaptation and resistance to RAF inhibitors. Nat Med. 2013;19(11):1401–1409. | ||
Lacouture ME, Duvic M, Hauschild A, et al. Analysis of dermatologic events in vemurafenib-treated patients with melanoma. Oncologist. 2013;18(3):314–322. | ||
Awad MM, Sullivan RJ. Dabrafenib in combination with trametinib for the treatment of metastatic melanoma. Expert Rev Clin Pharmacol. 2015;8(1):25–33. | ||
Pettirossi V, Santi A, Imperi E, et al. BRAF inhibitors reverse the unique molecular signature and phenotype of hairy cell leukemia and exert potent anti-leukemic activity. Blood. 2014;125(8):1207–1216. | ||
Badar T, Burger JA, Wierda WG, O’Brien S. Ibrutinib: a paradigm shift in management of CLL. Expert Rev Hematol. 2014;7(6):705–717. | ||
Herrera AF, Jacobsen ED. Ibrutinib for the treatment of mantle cell lymphoma. Clin Cancer Res. 2014;20(21):5365–5371. | ||
Sivina M, Kreitman RJ, Arons E, Ravandi F, Burger JA. The Bruton tyrosine kinase inhibitor ibrutinib (PCI-32765) blocks hairy cell leukaemia survival, proliferation and B cell receptor signalling: a new therapeutic approach. Br J Haematol. 2014;166(2):177–188. | ||
Jones JA, Andritsos L, Lucas D, Lozanski G, Hutchinson T. Preliminary safety and efficacy of the Bruton’s tyrosine kinase (BTK) inhibitor ibrutinib (IBR) in patients (pts) with hairy cell leukemia (HCL). J Clin Oncol. 2014;32:7063. | ||
Jones JA, Andritsos L, Ravandi F, et al. Safety and efficacy of the Bruton tyrosine kinase inhibitor Ibrutinib in patients with hairy cell leukemia: interim results of a Phase II study. Abstract S790. Presented at EHA20. June 2015. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.