Back to Journals » OncoTargets and Therapy » Volume 14
Current and Emerging Therapeutic Options for Hairy Cell Leukemia Variant
Authors Liu Q, Harris N, Epperla N ![]() , Andritsos LA
, Andritsos LA ![]()
Received 1 December 2020
Accepted for publication 6 February 2021
Published 9 March 2021 Volume 2021:14 Pages 1797—1805
DOI https://doi.org/10.2147/OTT.S242247
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Prof. Dr. Gaetano Romano
Qiuying Liu,1 Nicholas Harris,1 Narendranath Epperla,2 Leslie A Andritsos1,3
1Department of Internal Medicine, University of New Mexico, Albuquerque, NM, USA; 2Division of Hematology, The Ohio State University Comprehensive Cancer Center, Columbus, OH, USA; 3University of New Mexico Comprehensive Cancer Center, Albuquerque, NM, USA
Correspondence: Leslie A Andritsos
University of New Mexico, 1201 Camino de Salud, Albuquerque, NM, 87102, USA
Tel +1-505-925-0405
Fax +1-505-925-0408
Email [email protected]
Abstract: Hairy cell leukemia variant (HCL-v) is a rare B-cell lymphoproliferative disorder with distinct immunophenotypic and molecular characteristics when compared to classical hairy cell leukemia (HCL-c). In contrast to the enormous progress in therapeutic options for HCL-c, HCL-v remains a therapeutic challenge due to inferior outcomes with standard chemoimmunotherapy and BCR signaling pathway inhibitors, and due to the fact that HCL-v has limited molecular therapeutic targets. In addition, because of the rarity of the disease, there is a paucity of later phase studies or multicenter trials to guide treatment decisions. In this article, we briefly review the diagnostic criteria and clinical characteristics of HCL-v and present a comprehensive overview of current therapeutic options in HCL-v.
Keywords: salvage therapy, rare lymphoid malignancies, HCL-v, HCL-c
Introduction
The hairy cell leukemias (HCLs) are a rare group of hematological malignancies initially described in 1958 by Bouroncle and colleagues.1 First felt to represent one disease entity, these mature B-cell lymphoproliferative disorders were initially termed leukemic reticuloendotheliosis and characterized by distinctive malignant cells with slightly indented nuclei and circumferential cytoplasmic projections.2,3 Additional refinements in diagnostic capabilities ultimately revealed that classical HCL (HCL-c) and variant HCL (HCL-v) are biologically distinct entities and in 2008 the World Health Organization (WHO) reclassified the disorders into separate categories of lymphoproliferative disorders.4 While these diseases may have similar clinical presentations, responses to therapy and outcomes remain divergent.2,5 Despite marked improvements in outcomes of HCL-c,3,6 patients with HCL-v continue to have inferior responses to therapies and a significantly lower survival rate.
HCL-v, first described by Cawley et al,7 occurs at an incidence of 0.03 per 100,000 persons per year and constitutes around 0.4% of all chronic lymphoid malignancies.8–10 There is a male predominance with a male to female ratio of 6:1. HCL-v is a disease of the elderly, with a median age of 71 years.11 Patient usually present with splenomegaly, leukocytosis without monocytopenia, and a hypercellular bone marrow that can be easily aspirated.2,12 Patients tend to develop cytopenias as a result of hypersplenism rather than bone marrow failure.9,13 As noted above, while there may be a number of commonalities between the initial presentations of HCL-c and HCL-v, the clinical course of HCL-v is more aggressive,10 with a medial overall survival (OS) of 9 years11,14 in contrast to HCL-c in which patients may not experience a decline in OS due to improvements in therapies.15,16
Because of the differences between clinical outcomes of the HCLs, it is of paramount importance that a correct diagnosis is assigned early in the disease course. Table 1 compares the clinical and laboratory features commonly seen in the two disease entities.6,17,18 The immunophenotype of neoplastic cells in HCL-v is notable for lack of expression of CD25, CD123 and CD200, which are seen in almost all cases of HCL-c.2,11 The bone marrow in HCL-v is typically aspirable, unlike the “dry tap” encountered on bone marrow aspiration in HCL-c patients.12 Mutation in the V600E serine/threonine kinase BRAF oncogene is almost universally detected in all HLC-c cases, which has therapeutic and prognostic implications.6 While BRAF V600E mutations have not been documented in HCL-v, patients may harbor mutations of MAP2K1 and/or exhibit increased usage of IGHV4-34,11,19 both of which have been associated with inferior outcomes. In addition, while routine karyotyping, interphase cytogenetics, and mutational analyses are not commonly performed in the HCL-v, these may reveal deletions or mutations of TP53 in HCL-v in up to 38% of cases, which may have clinical and therapeutic implications.11,20
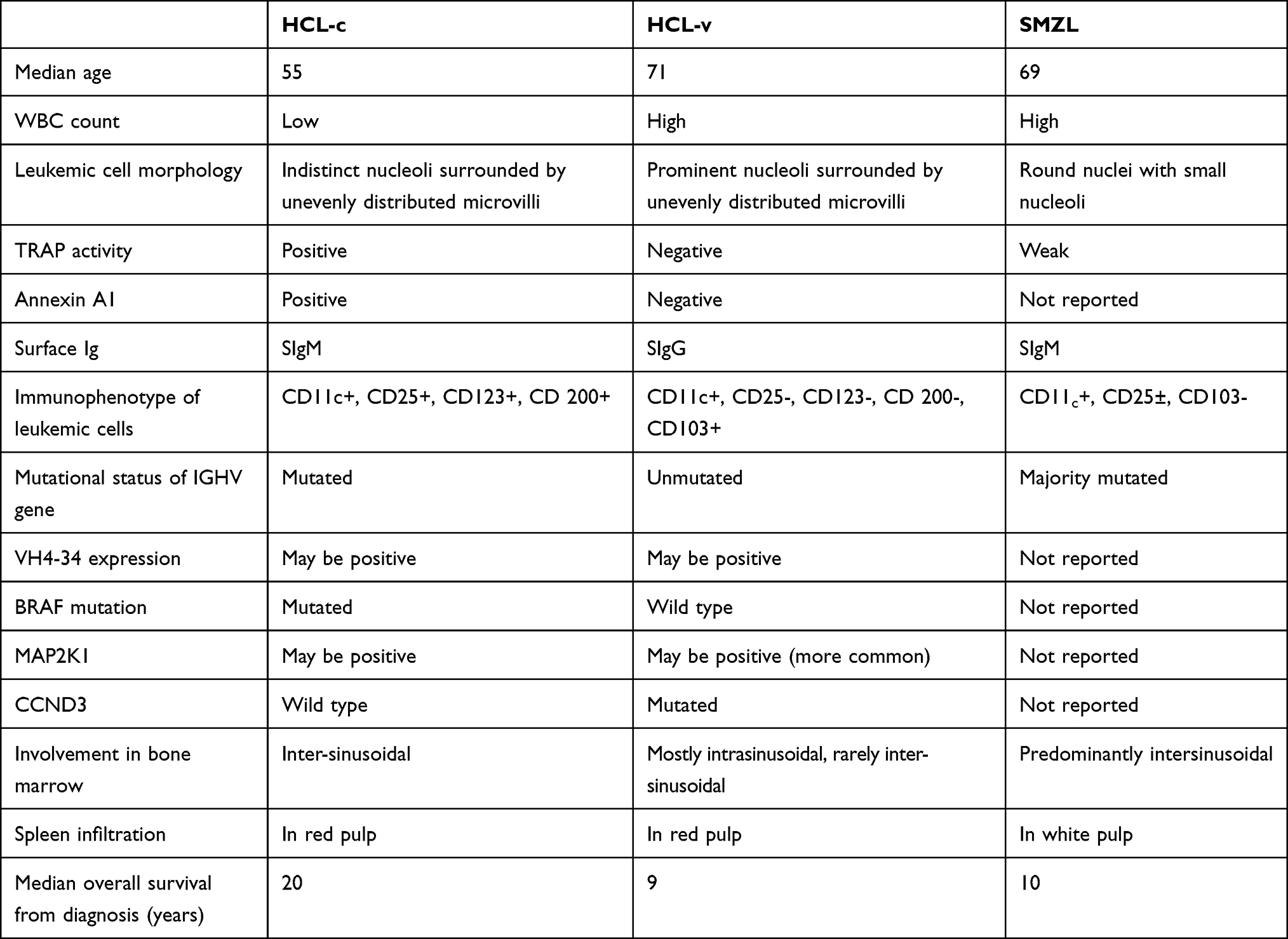 |
Table 1 Comparison of Clinical and Laboratory Characteristics of Classical Hairy Cell Leukemia (HCL-c) and Hairy Cell Leukemia Variant (HCL-v) |
It is of note that therapies that are highly effective in HCL-c will usually not be as successful when treating HCL-v11,21 and choice of therapy is a major challenge in this disease. In addition, there are currently no formal recommendations regarding timing of treatment initiation and therapeutic decision-making in HCL-v, although recent guidelines published for the management of HCL may be useful in determining when to initiate treatment and how to measure responses.22
Assessment of Response
Response assessments following completion of therapy are essential because they provide guidance regarding the future clinical course. These evaluations include complete blood count evaluation, physical examination including assessment of spleen and liver size and nodal involvement if applicable, and a bone marrow biopsy to determine percentage of residual bone marrow involvement if any. In addition, patients who had adenopathy or organomegaly prior to starting therapy should have repeat imaging.22 The timing of bone marrow biopsy varies based on the therapy received. Usually, a bone marrow biopsy is performed 4 to 6 months after receiving cladribine22,23 as the bone marrow may require many months before recovery after treatment with a purine analog. A complete response (CR) is defined as near normalization of peripheral blood counts, regression of splenomegaly and an absence of morphologic evidence of HCL on both peripheral blood smear and the bone marrow examination.23,24 With the help of immunohistochemical stains, CR can be further classified into those with and without evidence of minimal residual disease (MRD). MRD is defined as the presence of suspicious cells in the bone marrow biopsy by morphology or immunohistochemistry.25 A partial response (PR) is achieved when there has been near normalization of the peripheral blood counts with a minimum of 50% improvement in organomegaly and bone marrow biopsy infiltration.22–24
Current Therapy Options
Surgery and Radiation
Splenectomy
In a study performed by Matutes et al in 2001, 74% (13 out of 19) of patients with HCL-v achieved a hematological response after splenectomy,9 with a median duration of response of 4 years. Some authors also suggest that previous splenectomy may improve the response to alkylator-based chemotherapy and purine nucleoside analogs in patients with HCL-v.26 Hence, splenectomy is a potential therapeutic option in HCL-v, either as a first-line or as subsequent treatment, as it may correct cytopenias and removes a significant bulk of the disease. Patients who undergo splenectomy will need to receive immunizations prior to surgery to protect against encapsulated organisms.
Splenic irradiation is a palliative treatment for hypersplenism and splenic pain in patients with lymphoproliferative or myeloproliferative disorders.27 It is most commonly used for chronic lymphocytic leukemia and is well tolerated.28 Roughly 85–90% of patients achieve resolution of symptomatic splenomegaly that lasted 6 to 12 months.28,29 Hence, splenic irradiation may be a useful alternative to splenectomy for elderly patients or those who are not surgical candidates. In a case report by Sgarabotto et al, CR was achieved with splenic radiotherapy alone in a 79-year-old patient with HCL-v.30 Sasaki et al reported a patient with relapsed HCL-v and treated with splenic irradiation followed by alemtuzumab. Although the patient did not achieve CR/PR with irradiation alone, it attenuated splenomegaly (spleen size reduced from 12 to 4 cm below the left costal margin) and reduced the number of circulating leukemic cells.31 These case reports suggest that splenic irradiation may be a potential alternative to splenectomy for symptomatic elderly patients with high surgical risks and limited treatment options.
Interferon
Interferon alpha (IFN-α) was one of the first agents developed for treatment of HCL. However, the clinical trial that identified rapid hematological responses in patients with HCL predated the WHO reclassification which separated HCL-c and HCL-v into different disease categories, indicating that there were very likely both subtypes of patients in this study.4 Small studies reporting outcomes specifically in HCL-v showed less encouraging results, with one study showing that 14% (2/14 patients) of HCL-v patients who received IFN-α achieved transient partial responses,9 with another study finding no objective responses (no responses in 7 HCL-v patients treated with IFN).8 These variable/poor responses to IFN-α in HCL-v may be due to a low number of IFN-α receptors, which are more abundant in HCL-c.32 Hence, IFN-α should be reserved for patients with HCL-c.
Purine Nucleoside Analogs
The purine nucleoside analogs, cladribine and pentostatin, are the drugs of choice for the up-front treatment of HCL-c with exceptional response rates and long OS even when used as a single agents.33,34 However, their effects as single agents in HCL-v are suboptimal. In a study of HCL-v patients (n=6) treated with 5 days of cladribine, the overall response rate was 33% (2/6 achieved partial responses) and the responses lasted for 60+ and 29+ months, respectively.35 This suboptimal response is echoed by data from other retrospective studies showing the majority of HCL-v patients only achieve transient PR and rarely CR.8,9,26,36 Similar results have been documented with pentostatin with 54% of patients achieving PR with a median response duration of about 15 months.9 Based on these results, patients with HCL-v should no longer receive single-agent purine nucleoside analogs, even for up-front treatment.
Monoclonal Antibodies
Rituximab
Rituximab is a chimeric anti-CD20 monoclonal antibody that has been used in CD20-expressing B-cell malignancies.33,37 There have been several case reports demonstrating efficacy of rituximab either alone or as consolidation therapy following splenectomy in the treatment of HCL-v. Narat et al reported a case of a 53-year-old man with refractory HCL-v who was treated with weekly rituximab for 4 weeks, after which he achieved a CR.38 At the time of publication, the patient had been in CR for more than 19 months. Similar findings were reported by other case reports suggesting that rituximab is a promising therapy for patients with HCL-V.31,39
Alemtuzumab
Alemtuzumab is a humanized igG1 anti CD52 antibody that binds to cell membrane of normal and malignant lymphocyte and is effective in the treatment of lymphoid malignancies.13 CD52 expression was reported in 92–100% of the malignant cells in classical HCL and HCL-v.40 However, the efficacy of this agent is only limited to a few case reports. Telek et al reported a case of a 58-year-old male with HCL-v who received weekly alemtuzumab for 12 weeks.41 Hematologic remission was achieved after 8 weeks of treatment. Another study reported a patient with refractory HCL-v who received alemtuzumab after pretreatment with splenic irradiation.31 Although splenic irradiation reduced degree of splenomegaly and number of circulating leukemic cells (from 229.0 to 63.6 x 10(9)/L), subsequent treatment with alemtuzumab eliminated leukemic cells from the peripheral blood by day 12 of treatment and splenomegaly was resolved. In vitro studies showed that alemtuzumab induced leukemic cell death, which confirmed the activity of alemtuzumab in this patient. Given the paucity of data, it is unclear if alemtuzumab is effective in HCL-v. Additionally, alemtuzumab has limited clinical availability due to changes in utilization for hematological malignancies and may not be available in all areas.
Emerging Therapy Options
Antibody-Toxin Conjugates
BL22
An immunotoxin is a fusion of a bacterial toxin with a monoclonal antibody directed against a specific cell surface target.33 Both classical and variant HCL are strongly positive for CD22,5,42–44 an adhesion molecule expressed exclusively on B cells.45 BL22 is a recombinant immunotoxin containing a truncated Pseudomonas exotoxin and variable domains from anti-CD22,13 and was a precursor to moxetumomab pasudotox. BL22 is currently not available for use in the treatment of HCL or HCL-v but was tested in HCL-v. In a Phase 1 trial, 3 patients with HCL-v were treated with BL22 and achieved CR without MRD.42,46 This protocol called for 2 cycles of consolidation therapy regardless of the presence or absence of MRD. Two patients relapsed within a year, but CR was achieved with re-treatment with BL22. In this trial, BL22 associated toxicities included reversible hemolytic uremic syndrome and a dose limiting cytokine release syndrome categorized by fever, hypotension, and arthralgia, however these toxicities were not noted in the three patients with HCL-v.
Moxetumomab Pasudotox
More recently, a higher affinity version of BL22, termed HA22, or moxetumomab pasudotox was developed.5 Upon binding to CD22, HA22 is internalized, inhibiting protein translation and promoting apoptosis.47 In the phase 1 trial of moxetumomab pasudotox in HCL, two patients with HCL-v were enrolled48 but the responses were not specifically described. In a multi-center, Phase 3 trial, three patients with HCL-v and high disease burden were enrolled. Neither of these patients achieved a CR with moxetumomab pasudotox.49 However, it was shown to be quite effective for relapsed/refractory HCL-c with a complete response rate of 30%. Among complete responders, 85% achieved MRD negativity by immunohistochemistry.49 Based on these results, moxetumomab pasudotox was approved by the FDA in 2018 for the treatment of patients with relapsed/refractory HCL who have received at least 2 prior systemic therapies including at least one purine nucleoside analog.47 However, given the limited data, it is unclear whether this agent is effective in HCL-v.
Chemo-Immunotherapy
Building on the success of chemotherapies and immunotherapies as single agents in HCL and other B-cell malignancies, combination chemoimmunotherapy has been studied in HCL-v. Chow et al reported that rituximab can sensitize malignant B cells to chemotherapies like cladribine.50 In one study, 5 patients with HCL-v received 5 days of cladribine followed by 8 weekly doses of rituximab.51 All 5 patients achieved CR, and 2 patients remained in CR for a median of 13.5 months. Similarly, another study treated 10 HCL-v patients with concurrent cladribine and rituximab52 (5 days of cladribine 0.15 mg/kg and weekly rituximab 375mg/m2 for 8 weeks). At 6 months, 9 of 10 patients had achieved a CR. MRD was assessed by examining the presence of suspicious cells in bone marrow biopsy by immunohistochemistry or the presence of leukemic cells (defined as CD22 positive cells) in marrow aspirate or blood by flow cytometry. Eight out of the 9 patients were MRD negative at a median of 27 months. No dose-limiting toxicities were observed. Visentin et al also reported 3 patients with HCL-v who were treated with 4 cycles of bendamustine with rituximab.53 All 3 patients were elderly and were able to complete the regimen with tolerable toxicities. All 3 patients achieved a CR and remained in CR at 19 months of follow up. These reports suggest that chemoimmunotherapy produces superior responses versus single-agent therapy and should be considered in fit patients.
BCR Singling Pathway Inhibitors
BTK Inhibitors
Ibrutinib is an oral small-molecule inhibitor of bruton’s tyrosine kinase (BTK), which is uniformly expressed on HCL cells.33 It is currently approved for use in the treatment of B-cell lymphoproliferative disorders (including mantle cell lymphoma, marginal zone lymphoma and chronic lymphocytic leukemia) and acute graft versus host disease following hematopoietic cell transplantation (HCT). BTK plays a crucial role in B-cell receptor signaling and its inhibition halts HCL cell proliferation and cell survival in a dose-dependent manner. It is hypothesized that ibrutinib inhibits CXCR4 signaling, which is highly expressed in HCL cells. In addition, ibrutinib down-regulates the activation of MAPK/ERK pathway, which ultimately reduces HCL cell proliferation and survival.54 There is currently an ongoing single-arm, multicenter Phase 2 trial evaluating ibrutinib as a single agent in both classical HCL and HCL-V.55 At the time, the interim results were published, 3 out of 11 patients with HCL-V had achieved a partial response.56 However, 2 patients ultimately discontinued ibrutinib due to toxicities. There have also been several case reports of use of ibrutinib in patients with HCL-V. Bohn et al reported an 82-year-old man with multiple medical comorbidities who was diagnosed with HCL-V at age 77.57 He was initially treated with bendamustine plus with rituximab, however had to discontinue therapy due to development of a type IV hypersensitivity reaction to bendamustine. He next received cladribine followed by ofatumumab with no objective response. Finally, he was treated with ibrutinib as off-label salvage therapy and achieved >50% decrease in splenomegaly and resolution of lymphocytosis.57 In addition, Jain et al reported a 79-year-old diagnosed with CLL and HCL-V who responded well to ibrutinib and venetoclax.58 These early results of ibrutinib in HCL-V remain promising and further studies evaluating refinements of BTK inhibition including combination regimens, or risk stratified approaches targeting patients with deletions or mutations of TP53, may improve overall responses.
MAP2K Inhibitors
Activating MAP2K mutations are seen in up to half of the patients with HCL-v19,59 and MEK inhibitors are therefore attractive potential therapeutic options. Trametinib is an inhibitor of MEK which reversibly binds to MEK1 and MEK2, preventing downstream phosphorylation of ERK, in turn decreasing cellular proliferation and survival. It is currently approved by the FDA for the treatment of BRAF p.V600E mutant melanoma. However, to date no prospective trials have been published evaluating its use in HCL-v. Our group60 reported a 52-year-old man with relapsed/refractory HCL-v who had previously received multiple lines of therapy including cladribine, BL22, pentostatin/rituximab, splenectomy, single-agent rituximab, ibrutinib, bendamustine/rituximab and allogeneic transplantation from a matched unrelated donor. His disease relapsed on day +350 post-transplant, at which time sequencing of his peripheral blood revealed a somatic MAP2K1 p.K57N mutation that constitutively activates MEK. The patient was started on trametinib 2mg by mouth daily as a single agent and achieved a PR, suggesting a potential role for trametinib in the subgroup of patients who harbor mutations in MAP2K1. To this end, there is currently a phase 2 trial evaluating the MEK inhibitor binimetinib in patients with refractory classical HCL and HCL-v.61
CAR-T Therapy and Hematopoietic Cell Transplantation
Hematopoietic Cell Transplantation
Given the success of HCT in other hematological malignancies, the question arises whether this approach would be effective in HCL-v. Likewise, given the fact that HCL-v expresses CD19 this disease may be potentially approached with CD19 CAR-T. However, there is limited data available regarding the usefulness of either autologous or allogeneic transplantation or CAR-T. With respect to autologous transplantation, this is generally only effective in patients with chemotherapy-sensitive disease and is not curative in chronic lymphoproliferative disorders. In one report by Goldaniga et al,62 a patient with aggressive relapsed HCL-v underwent autologous transplantation using a conditioning regimen of melphalan 200 mg/m2 and thiotepa 15 mg/kg. This patient achieved both clinical and molecular CR but only maintained his remission for 16 months. By contrast, allogeneic transplantation may be a potentially curative treatment option given the graft-versus tumor effect with demonstrated efficacy in indolent lymphomas such as follicular lymphoma (MD Anderson data for reference). However, limited data using this approach is available. Busemann et al63 reported a 60-year-old man with HCL-v with refractory disease who underwent matched unrelated donor allogeneic transplantation using a conditioning regimen of treosulfan (30 g/m2), fludarabine (150 mg/m2) and anti-thymocyte-globulin (380 mg). This patient achieved a clinical and molecular remission lasting for 3.5 years. Given the unlikely long-term benefit of autologous transplantation this cannot be recommended at this time; allogeneic transplantation may have a role in select cases but should be performed as part of a clinical trial. Finally, with respect to CAR-T, it is likely that some patients with HCL-v were enrolled in the initial clinical trials and those results are eagerly anticipated. Again, this approach would not be recommended outside the context of a clinical trial given the potential toxicities and unknown potential benefit.
Conclusion
HCL-v is a rare lymphoproliferative disorder with a high incidence of poor risk molecular and cytogenetic features. In contrast to HCL-c, little progress has been made in improving therapeutic options for HCL-v. Table 2 presents a list of therapies reported in HCL-v. For fit patients, we recommend initial therapy with chemo-immunotherapy as this has been shown to provide the highest overall response rate. In addition, splenectomy may provide significant clinical benefit in patients with massive splenomegaly. For patients who require salvage therapy, limited options are available and we would encourage clinical trial participation whenever possible. Outside of a clinical trial, BCR signaling pathway inhibitors, MEK inhibition in patients with MAP2K1 mutations, and anti-CD20 as a single agent in select patients may provide some benefit. There remain a number of unexplored treatment approaches in HCL-v including targeted therapy combinations, BH3-mimetics, and immunotherapeutic approaches such as CAR-T and bispecific antibodies. HCL-v is clearly a disease in need of improved therapeutic options and future trials will likely utilize multi-agent approaches and a molecular-based treatment assignment.
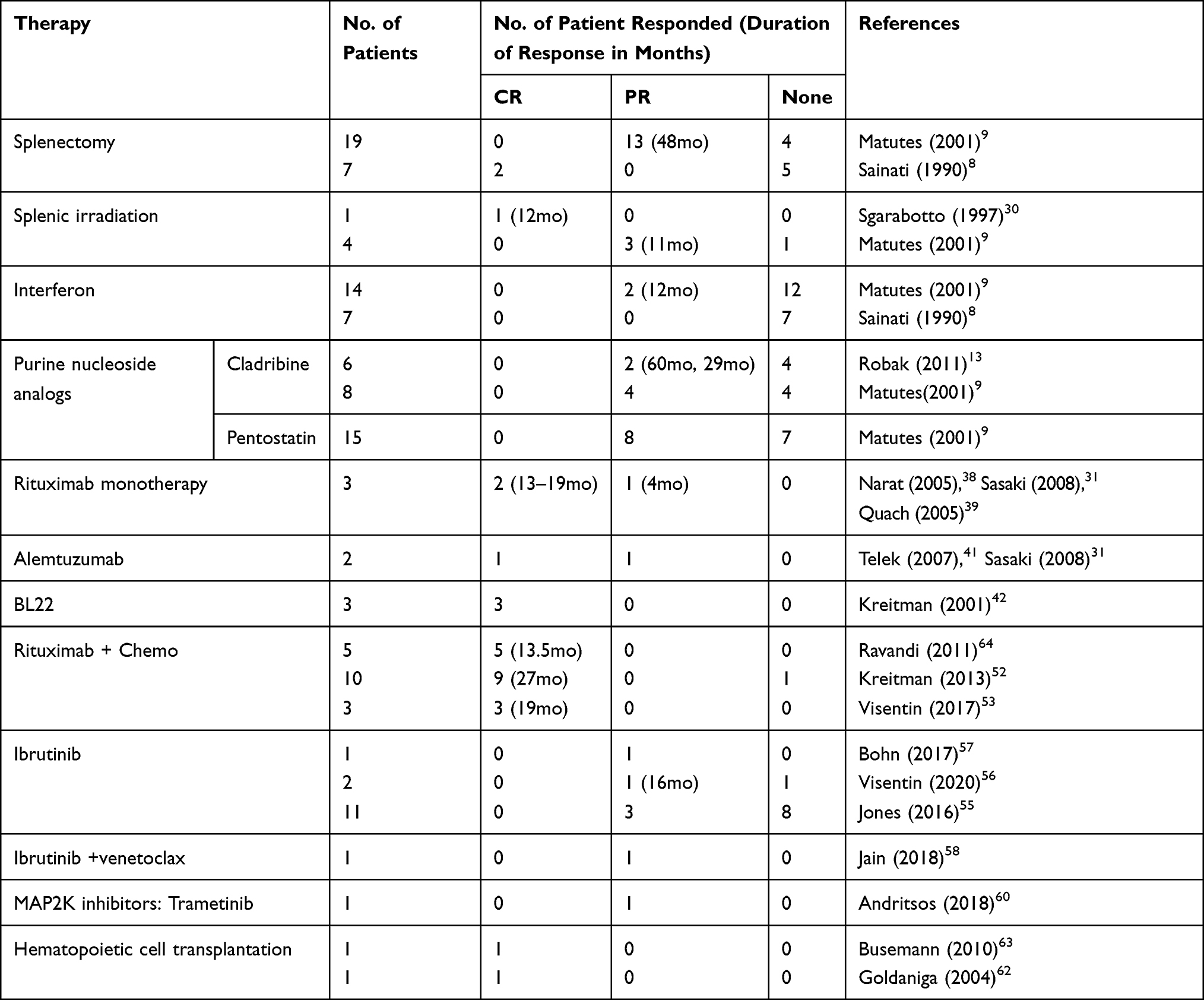 |
Table 2 Therapies Used in Hairy Cell Leukemia Variant (HCL-v) |
Disclosure
QSL and NH report no conflicts of interest. NE is on Speaker’s Bureau for Verastem Oncology and Beigene; received honoraria from Pharmacyclics. LA provides consultation services for Innate Pharma. LAalso receives research funding from the Hairy Cell Leukemia Foundation. The authors report no other conflicts of interest in this work.
References
1. Bouroncle BA, Wiseman BK, Doan CA. Leukemic reticuloendotheliosis. Blood. 1958;13(7):609–630. doi:10.1182/blood.V13.7.609.609
2. Sarvaria A, Topp Z, Saven A. Current therapy and new directions in the treatment of hairy cell leukemia: a review. JAMA Oncol. 2016;2(1):123–129. doi:10.1001/jamaoncol.2015.4134
3. Grever MR, Blachly JS, Andritsos LA. Hairy cell leukemia: update on molecular profiling and therapeutic advances. Blood Rev. 2014;28(5):197–203. doi:10.1016/j.blre.2014.06.003
4. Swerdlow SH, Campo E, Pileri SA, et al. The 2016 revision of the World Health Organization classification of lymphoid neoplasms. Blood. 2016;127(20):2375–2390. doi:10.1182/blood-2016-01-643569
5. Kreitman RJ. Hairy cell leukemia: present and future directions. Leuk Lymphoma. 2019;60(12):2869–2879. doi:10.1080/10428194.2019.1608536
6. Shao H, Calvo KR, Grönborg M, et al. Distinguishing hairy cell leukemia variant from hairy cell leukemia: development and validation of diagnostic criteria. Leuk Res. 2013;37(4):401–409. doi:10.1016/j.leukres.2012.11.021
7. Cawley JC, Burns GF, Hayhoe FG. A chronic lymphoproliferative disorder with distinctive features: a distinct variant of hairy-cell leukaemia. Leuk Res. 1980;4(6):547–559. doi:10.1016/0145-2126(80)90066-1
8. Sainati L, Matutes E, Mulligan S, et al. A variant form of hairy cell leukemia resistant to alpha-interferon: clinical and phenotypic characteristics of 17 patients. Blood. 1990;76(1):157–162. doi:10.1182/blood.V76.1.157.157
9. Matutes E, Wotherspoon A, Brito-Babapulle V, Catovsky D. The natural history and clinico-pathological features of the variant form of hairy cell leukemia. Leukemia. 2001;15(1):184–186. doi:10.1038/sj.leu.2401999
10. Wiber M, Maitre E, Cornet E, Salaün V, Naguib D, Troussard X. Variant form of hairy cell leukemia. Clin Case Rep. 2019;7(6):1161–1166. doi:10.1002/ccr3.2176
11. Matutes E, Martínez-Trillos A, Campo E. Hairy cell leukaemia-variant: disease features and treatment. Best Pract Res Clin Haematol. 2015;28(4):253–263. doi:10.1016/j.beha.2015.09.002
12. Wotherspoon A, Attygalle A, Mendes LS. Bone marrow and splenic histology in hairy cell leukaemia. Best Pract Res Clin Haematol. 2015;28(4):200–207. doi:10.1016/j.beha.2015.10.019
13. Robak T. Hairy-cell leukemia variant: recent view on diagnosis, biology and treatment. Cancer Treat Rev. 2011;37(1):3–10. doi:10.1016/j.ctrv.2010.05.003
14. Chandran R, Gardiner SK, Smith SD, Spurgeon SE. Improved survival in hairy cell leukaemia over three decades: a SEER database analysis of prognostic factors. Br J Haematol. 2013;163(3):407–409. doi:10.1111/bjh.12490
15. Benz R, Arn K, Andres M, et al. Prospective long-term follow-up after first-line subcutaneous cladribine in hairy cell leukemia: a SAKK trial. Blood Adv. 2020;4(15):3699–3707. doi:10.1182/bloodadvances.2020002160
16. Paillassa J, Cornet E, Noel S, et al. Analysis of a cohort of 279 patients with hairy-cell leukemia (HCL): 10 years of follow-up. Blood Cancer J. 2020;10(5):62. doi:10.1038/s41408-020-0328-z
17. Maitre E, Cornet E, Troussard X. Hairy cell leukemia: 2020 update on diagnosis, risk stratification, and treatment. Am J Hematol. 2019;94(12):1413–1422. doi:10.1002/ajh.25653
18. Robak T. Management of hairy cell leukemia variant. Leuk Lymphoma. 2011;52(Suppl 2):53–56. doi:10.3109/10428194.2011.566392
19. Mason EF, Brown RD, Szeto DP, et al. Detection of activating MAP2K1 mutations in atypical hairy cell leukemia and hairy cell leukemia variant. Leuk Lymphoma. 2017;58(1):233–236. doi:10.1080/10428194.2016.1185786
20. Angelova EA, Medeiros LJ, Wang W, et al. Clinicopathologic and molecular features in hairy cell leukemia-variant: single institutional experience. Mod Pathol. 2018;31(11):1717–1732. doi:10.1038/s41379-018-0093-8
21. Ahmed S, Rai KR. Interferon in the treatment of hairy-cell leukemia. Best Pract Res Clin Haematol. 2003;16(1):69–81. doi:10.1016/s1521-6926(02)00084-1
22. Grever MR, Abdel-Wahab O, Andritsos LA, et al. Consensus guidelines for the diagnosis and management of patients with classic hairy cell leukemia. Blood. 2017;129(5):553–560. doi:10.1182/blood-2016-01-689422
23. Cornet E, Delmer A, Feugier P, et al. Recommendations of the SFH (French society of haematology) for the diagnosis, treatment and follow-up of hairy cell leukaemia. Ann Hematol. 2014;93(12):1977–1983. doi:10.1007/s00277-014-2140-y
24. Jones G, Parry-Jones N, Wilkins B, Else M, Catovsky D. Revised guidelines for the diagnosis and management of hairy cell leukaemia and hairy cell leukaemia variant*. Br J Haematol. 2012;156(2):186–195. doi:10.1111/j.1365-2141.2011.08931.x
25. Tallman MS, Hakimian D, Kopecky KJ, et al. Minimal residual disease in patients with hairy cell leukemia in complete remission treated with 2-chlorodeoxyadenosine or 2-deoxycoformycin and prediction of early relapse. Clin Cancer Res. 1999;5(7):1665–1670.
26. Dunphy CH, Petruska PJ. Atypical prolymphocytic variant of hairy-cell leukemia: case report and review of the literature. Am J Hematol. 1996;53(2):121–125. doi:10.1002/(sici)1096-8652(199610)53:2<121::aid-ajh11>3.0.co;2-i
27. Paulino AC, Reddy SP. Splenic irradiation in the palliation of patients with lymphoproliferative and myeloproliferative disorders. Am J Hosp Palliat Care. 1996;13(6):32–35. doi:10.1177/104990919601300613
28. Zaorsky NG, Williams GR, Barta SK, et al. Splenic irradiation for splenomegaly: a systematic review. Cancer Treat Rev. 2017;53:47–52. doi:10.1016/j.ctrv.2016.11.016
29. Lavrenkov K, Krepel-Volsky S, Levi I, Ariad S. Low dose palliative radiotherapy for splenomegaly in hematologic disorders. Leuk Lymphoma. 2012;53(3):430–434. doi:10.3109/10428194.2011.614708
30. Sgarabotto D, Vianello F, Radossi P, et al. Remission in hairy cell leukemia-variant following splenic radiotherapy alone. Leuk Lymphoma. 1997;26(3–4):395–398. doi:10.3109/10428199709051790
31. Sasaki M, Sugimoto K, Mori T, Karasawa K, Oshimi K. Effective treatment of a refractory hairy cell leukemia variant with splenic pre-irradiation and alemtuzumab. Acta Haematol. 2008;119(1):48–53. doi:10.1159/000115785
32. Dadmarz R, Evans T, Secher D, Marshall N, Cawley JC. Hairy cells possess more interferon receptors than other lymphoid cell types. Leukemia. 1987;1(4):357–361.
33. King AC, Kabel CC, Pappacena JJ, Stump SE, Daley RJ. No loose ends: a review of the pharmacotherapy of hairy cell and hairy cell leukemia variant. Ann Pharmacother. 2019;53(9):922–932. doi:10.1177/1060028019836775
34. López-Rubio M, Garcia-Marco JA. Current and emerging treatment options for hairy cell leukemia. Onco Targets Ther. 2015;8:2147–2156. doi:10.2147/ott.s70316
35. Robak T, Błasińska-Morawiec M, Błoński J, et al. 2-Chlorodeoxyadenosine (cladribine) in the treatment of hairy cell leukemia and hairy cell leukemia variant: 7-year experience in Poland. Eur J Haematol. 1999;62(1):49–56. doi:10.1111/j.1600-0609.1999.tb01114.x
36. Palomera L, Domingo JM, Sola C, Azaceta G, Calvo MT, Gutierrez M. Cladribine (2-chlorodeoxyadenosine) therapy in hairy cell leukemia variant. A report of three cases. Haematologica. 2002;87(1):107–108.
37. Nieva J, Bethel K, Saven A. Phase 2 study of rituximab in the treatment of cladribine-failed patients with hairy cell leukemia. Blood. 2003;102(3):810–813. doi:10.1182/blood-2003-01-0014
38. Narat S, Gandla J, Dogan A, Mehta A. Successful treatment of hairy cell leukemia variant with rituximab. Leuk Lymphoma. 2005;46(8):1229–1232. doi:10.1080/10428190500083433
39. Quach H, Januszewicz H, Westerman D. Complete remission of hairy cell leukemia variant (HCL-v) complicated by red cell aplasia post treatment with rituximab. Haematologica. 2005;90:Ecr26.
40. Quigley MM, Bethel KJ, Sharpe RW, Saven A. CD52 expression in hairy cell leukemia. Am J Hematol. 2003;74(4):227–230. doi:10.1002/ajh.10428
41. Telek B, Batár P, Udvardy M. Successful alemtuzumab treatment of a patient with atypical hairy cell leukaemia variant. Orv Hetil. 2007;148(38):1805–1807. doi:10.1556/oh.2007.28169
42. Kreitman RJ, Wilson WH, Bergeron K, et al. Efficacy of the anti-CD22 recombinant immunotoxin BL22 in chemotherapy-resistant hairy-cell leukemia. N Engl J Med. 2001;345(4):241–247. doi:10.1056/nejm200107263450402
43. Kreitman RJ, Pastan I. Immunobiological treatments of hairy-cell leukaemia. Best Pract Res Clin Haematol. 2003;16(1):117–133. doi:10.1016/s1521-6926(03)00003-3
44. Kreitman RJ, Pastan I. Development of recombinant immunotoxins for hairy cell leukemia. Biomolecules. 2020;10(8):8. doi:10.3390/biom10081140
45. Clark EA. CD22, a B cell-specific receptor, mediates adhesion and signal transduction. J Immunol. 1993;150(11):4715–4718.
46. Kreitman RJ, Squires DR, Stetler-Stevenson M, et al. Phase I trial of recombinant immunotoxin RFB4(dsFv)-PE38 (BL22) in patients with B-cell malignancies. J Clin Oncol. 2005;23(27):6719–6729. doi:10.1200/jco.2005.11.437
47. Janus A, Robak T. Moxetumomab pasudotox for the treatment of hairy cell leukemia. Expert Opin Biol Ther. 2019;19(6):501–508. doi:10.1080/14712598.2019.1614558
48. Kreitman RJ, Tallman MS, Robak T, et al. Minimal residual hairy cell leukemia eradication with moxetumomab pasudotox: phase 1 results and long-term follow-up. Blood. 2018;131(21):2331–2334. doi:10.1182/blood-2017-09-803072
49. Kreitman RJ, Dearden C, Zinzani PL, et al. Moxetumomab pasudotox in relapsed/refractory hairy cell leukemia. Leukemia. 2018;32(8):1768–1777. doi:10.1038/s41375-018-0210-1
50. Chow KU, Sommerlad WD, Boehrer S, et al. Anti-CD20 antibody (IDEC-C2B8, rituximab) enhances efficacy of cytotoxic drugs on neoplastic lymphocytes in vitro: role of cytokines, complement, and caspases. Haematologica. 2002;87(1):33–43.
51. Ravandi F. Chemo-immunotherapy for hairy cell leukemia. Leuk Lymphoma. 2011;52(Suppl 2):72–74. doi:10.3109/10428194.2011.565096
52. Kreitman RJ, Wilson W, Calvo KR, et al. Cladribine with immediate rituximab for the treatment of patients with variant hairy cell leukemia. Clin Cancer Res. 2013;19(24):6873–6881. doi:10.1158/1078-0432.ccr-13-1752
53. Visentin A, Imbergamo S, Frezzato F, et al. Bendamustine plus rituximab is an effective first-line treatment in hairy cell leukemia variant: a report of three cases. Oncotarget. 2017;8(66):110727–110731. doi:10.18632/oncotarget.21304
54. Sivina M, Kreitman RJ, Arons E, Ravandi F, Burger JA. The bruton tyrosine kinase inhibitor ibrutinib (PCI-32765) blocks hairy cell leukaemia survival, proliferation and B cell receptor signalling: a new therapeutic approach. Br J Haematol. 2014;166(2):177–188. doi:10.1111/bjh.12867
55. Jones J, Andritsos L, Kreitman RJ, et al. Efficacy and safety of the bruton tyrosine kinase inhibitor ibrutinib in patients with hairy cell leukemia: stage 1 results of a phase 2 study. Blood. 2016;128(22):1215. doi:10.1182/blood.V128.22.1215.1215
56. Visentin A, Imbergamo S, Trimarco V, et al. Ibrutinib in relapsed hairy cell leukemia variant: a case report and review of the literature. Hematol Oncol. 2020;38(5):823–826. doi:10.1002/hon.2810
57. Bohn JP, Wanner D, Steurer M. Ibrutinib for relapsed refractory hairy cell leukemia variant. Leuk Lymphoma. 2017;58(5):1224–1226. doi:10.1080/10428194.2016.1239262
58. Jain P, Kanagal-Shamanna R, Konoplev S, Zuo Z, Estrov Z. Biclonal IGHV-4-34 hairy cell leukemia variant and CLL – successful treatment with ibrutinib and venetoclax. Am J Hematol. 2018;93(12):1568–1569. doi:10.1002/ajh.25264
59. Parry-Jones N, Joshi A, Forconi F, Dearden C. Guideline for diagnosis and management of hairy cell leukaemia (HCL) and hairy cell variant (HCL-V). Br J Haematol. 2020;191(5):730–737. doi:10.1111/bjh.17055
60. Andritsos LA, Grieselhuber NR, Anghelina M. Trametinib for the treatment of IGHV4-34, MAP2K1-mutant variant hairy cell leukemia. Leuk Lymphoma. 2018;59(4):1008–1011. doi:10.1080/10428194.2017.1365853
61. Binimetinib for people with relapsed/refractory BRAF wild type hairy cell leukemia and variant; 2020. Available from: https://clinicaltrials.gov/ct2/show/NCT04322383.
62. Goldaniga M, Guffanti A, Gianelli U, Magni M, Lambertenghi Deliliers G, Baldini L. Clinical and molecular complete remission in a case of variant hairy cell leukemia treated with DHAP followed by high-dose chemotherapy plus rituximab. Haematologica. 2004;89(11):Ecr41.
63. Busemann C, Schüler F, Krüger W, et al. Late extramedullary relapse after allogeneic transplantation in a case of variant hairy cell leukaemia. Bone Marrow Transplant. 2010;45(6):1117–1118. doi:10.1038/bmt.2009.293
64. Ravandi F, O’Brien S, Jorgensen J, et al. Phase 2 study of cladribine followed by rituximab in patients with hairy cell leukemia. Blood. 2011;118(14):3818–3823. doi:10.1182/blood-2011-04-351502
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.