Back to Journals » Cancer Management and Research » Volume 14
Current and Emerging Therapeutic Approaches for Extracranial Malignant Rhabdoid Tumors
Authors Nemes K, Johann PD, Tüchert S, Melchior P, Vokuhl C, Siebert R, Furtwängler R ![]() , Frühwald MC
, Frühwald MC ![]()
Received 15 September 2021
Accepted for publication 11 January 2022
Published 9 February 2022 Volume 2022:14 Pages 479—498
DOI https://doi.org/10.2147/CMAR.S289544
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Seema Singh
Karolina Nemes,1 Pascal D Johann,1,2 Stefanie Tüchert,3 Patrick Melchior,4 Christian Vokuhl,5 Reiner Siebert,6 Rhoikos Furtwängler,7 Michael C Frühwald1
1Paediatrics and Adolescent Medicine, Swabian Children’s Cancer Center, University Medical Center Augsburg, Augsburg, Germany; 2Division of Pediatric Neurooncology, German Cancer Consortium (DKTK), German Cancer Research Center (DKFZ), Heidelberg, Germany; 3Department of Diagnostic and Interventional Radiology, University Hospital Augsburg, Augsburg, Germany; 4Department of Radiation Oncology, University of Saarland, Homburg, Germany; 5Section of Pediatric Pathology, Department of Pathology, University Hospital Bonn, Bonn, Germany; 6Institute of Human Genetics, Ulm University & Ulm University Medical Center, Ulm, Germany; 7Department of Pediatric Hematology and Oncology, University of Saarland, Homburg, Germany
Correspondence: Michael C Frühwald
EU-RHAB Registry, Swabian Children’s Cancer Center, Paediatrics and Adolescent Medicine, University Medical Center Augsburg, Stenglinstr. 2, Augsburg, 86156, Germany
, Tel +49 821 400 9342
, Fax +49 821 400 179201
, Email [email protected]
Abstract: Extracranial malignant rhabdoid tumors (extracranial MRT) are rare, highly aggressive malignancies affecting mainly infants and children younger than 3 years. Common anatomic sites comprise the kidneys (RTK – rhabdoid tumor of kidney) and other soft tissues (eMRT – extracranial, extrarenal malignant rhabdoid tumor). The genetic origin of these diseases is linked to biallelic pathogenic variants in the genes SMARCB1, or rarely SMARCA4, encoding subunits of the SWI/SNF chromatin-remodeling complex. Even if extracranial MRT seem to be quite homogeneous, recent epigenome analyses reveal a certain degree of epigenetic heterogeneity. Use of intensified therapies has modestly improved survival for extracranial MRT. Patients at standard risk profit from conventional therapies; most high-risk patients still experience a dismal course and often therapy resistance. Discoveries of clinical and molecular hallmarks and the exploration of experimental therapeutic approaches open exciting perspectives for clinical and molecularly stratified experimental treatment approaches. To ultimately improve the outcome of patients with extracranial MRTs, they need to be characterized and stratified clinically and molecularly. High-risk patients need novel therapeutic approaches including selective experimental agents in phase I/II clinical trials.
Keywords: extracranial malignant rhabdoid tumors, eMRT, RTK, experimental therapy, immunotherapy
Introduction
Epidemiology
Malignant rhabdoid tumors (MRT) are rare, aggressive malignancies arising predominantly in infants and young children <3 years. As a separate entity, they were initially described in 1978 as morphologically distinct from Wilms tumors.1 The term rhabdoid was conceived to account for the histological resemblance of MRT cells to rhabdomyoblasts.2 MRT are commonly located in the central nervous system (~65%) (ATRT – atypical teratoid/rhabdoid tumor), but also extracranial (~35%) in the kidneys (RTK – RT of the kidney) and other soft tissues (eMRT – extracranial, extrarenal malignant RT) (eg, liver, neck, thorax, retroperitoneum, pelvis) (Figure 1A).3,4 The 5-year overall- and disease-free survival for extracranial MRT are significantly better than for ATRT, due to a higher percentage of gross total resection (GTR), radiotherapy (RTx) and older age at diagnosis.5,6
 |
Figure 1 Localization of extracranial MRT (EU-RHAB registry data). (A) Anatomical localization of patients with extracranial MRT (n=185) registered between 2007 and 2020. The most common localizations of primary tumors are highlighted bold. (B) Distribution of patients with extracranial MRT (n=185) by age and localization registered between 2004 and 2020. * Patients with ATRT and RTK or eMRT. (C) Distribution of patients with eMRT (n=118) by age and primary site registered between 2004 and 2020. *Other sites: bladder (n=2), heart (n=1), skin (n=2), pancreas (n=1), adrenal gland (n=1), clavicle (n=1), brachial plexus (n=1). |
Within the UK and Germany, the age-standardized annual incidence rates of eMRT are 5–5.7 per million in the first year of life and decrease to 0.1–0.2 at age 5 years.3,4 According to the comprehensive database of the International Incidence of Childhood Cancer study (IICC) including 14 world regions, and five ethnic groups in the US, 327 cases of RTK were reported in children aged 0 to 14 years between 2001 and 2010, representing age standardized incidence rates of 0.2 per million, amounting to 2% of renal tumors, respectively.7 Figure 1B and C present the distribution of patients with extracranial MRT (n = 185) by age and primary site registered within the EU-RHAB registry between 2004 and 2020.
While initial clinical reports of extracranial MRTs distinguished renal (RTK) from extrarenal sites (eMRT), extracranial MRTs seem genetically to be quite homogeneous; nevertheless, there are certain differences between the two tumor types: RTK tend to present earlier in life, usually within the first year8,9 (median age range 10.6 to 13 months) compared to eMRT with a median age of 16.8.10 RTK is furthermore characterized by an early onset of local and distant metastases. As many as 10% to 15% of patients with RTK present synchronous ATRT at diagnosis, and in many cases exhibit pathogenic germline variants in SMARCB1 (or rarely SMARCA4).4
Genetics and Molecular Subgroups
Essentially, all MRT are genetically characterized by biallelic loss of function mutations in SMARCB1, a classic tumor suppressor gene encoding BAF47 (also called INI1)11,12 or rarely in SMARCA413–15 in chromosome 22q11.23 and 19p13.2, respectively. SMARCB1 encodes a core subunit, SMARCA4 the catalytic subunit of the SWI/SNF chromatin-remodeling complex. SMARCB1 is suggested to be the primary gene associated with MRT development.16
Despite a certain phenotypic as well as epigenetic heterogeneity (localization, response to therapy, survival) no other recurrent genetic alterations apart from SMARCB1 (SMARCA4) mutations have been identified. This is in accordance with experimental studies that suggest epigenetic mechanisms as the key drivers of cancers resulting from SMARCB1 loss.17 While the significance of epigenetic mechanisms for the intracranial counterpart, ATRT, is well established5 and while DNA methylation profiling is an important asset in the diagnostics of childhood CNS tumors, its role is less clear, but currently actively investigated in sarcomas and associated neoplasias.18–20
Cooperative studies demonstrated that ATRT comprise three molecular subgroups with distinct epigenomic, transcriptional, clinico-pathologic, and therapeutic features.19,21–26 Employing differential gene expression analyses, Chun et al demonstrated two distinct molecular subgroups in extracranial MRT (subgroups 1 and 2), which exhibit ATRT- and RTK-like gene expression profiles. Within subgroup 1, significantly overexpressed genes were linked to BMP signaling and differentiation. In subgroup 2, the most significantly overexpressed genes were linked to cell adhesion and migration, WNT signaling and differentiation.19
In an integrative analysis of genomic, transcriptomic and epigenomic profiles of 301 MRT, five DNA methylation subgroups wereassociated with anatomic sites, SMARCB1 mutation patterns, gene expression pathways, DNA methylation pathway enrichment and immune cell infiltration: Group 1 – “ATRT-MYC-like”, Group 2 – “ATRT-TYR-like”, Group 3 – “RTK-like”, Group 4 – “Extrarenal MRT-like”, Group 5 – “ATRT-SHH-like”).23 Notably, the expression subgroup 2 largely corresponded to Group 3 (“RTK-like”), while there was no clear equivalent for subgroup 1.
Group 1 – “ATRT-MYC-like”, 3 – “RTK-like” and 4 – “Extrarenal MRT-like” overexpressed the HOX- and other homeobox-containing genes, involved in mesodermal development. The Group 2 – “ATRT-TYR-like and Group 5 – “ATRT-SHH-like” demonstrated increased expression of genes involved in neural or neural crest development. These results suggest that extracranial MRT share molecular features with ATRT-MYC.23 Another feature – distinguishing Group 2 – “ATRT-TYR-like and Group 5 – “ATRT-SHH-like” from most other pediatric brain tumors is genome wide hypermethylation (Figure 2).
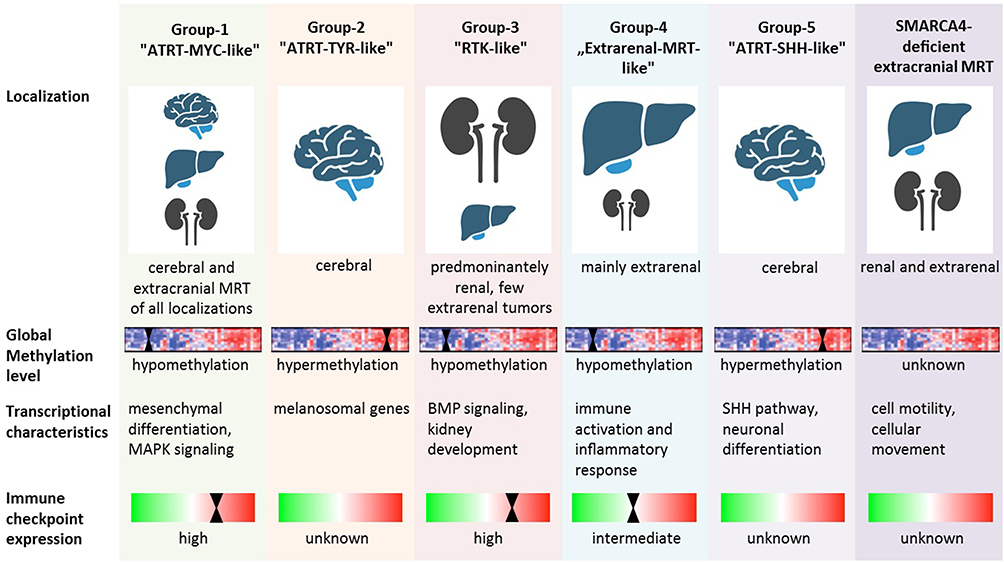 |
Figure 2 Overview of molecular features of the different extracranial MRT subgroups (based on Chun et al23 and Andrianteranagna et al28). |
Thus far, it remains speculative whether this feature has therapeutic implications: A study by Brocks et al described an induction of cryptic transcription start sites (and thus putative neoantigens) following exposure to demethylating agents.27 Whether this characteristic may also be found in a priori hypomethylated tumors remains to be studied.
Information on the specific molecular characteristics of SMARCA4-deficient extracranial MRT has been sparse until very recently. A recent study shed light on the specific transcriptomic and methylation characteristics of these entities.28 While SMARCB1-deleted extracranial MRT clustered together with ATRT-MYC, the SMARCA4-deficient counterparts tended to form a separate cluster. Along a similar line, the transcriptomic characteristics of SMARCA4-deficient extracranial MRT differed from SMARCB1-deficient extracranial MRT. The molecular characteristics of the different extracranial MRT subgroups are depicted in Figure 2.
Recent in vivo and in vitro studies using tyrosine kinase inhibitors (dasatinib, nilotinib) demonstrated inhibition of ATRT-MYC cells and were correlated with upregulation of PDGFRB.22 Alimova et al showed that in group 2 “ATRT-TYR-like” the MYC oncogene is involved in tumorigenesis, representing unique promoter occupancy. MYC inhibition by genetic and chemical agents in vitro as well as in vivo repressed tumorigenesis in ATRT cells.29
Germline Mutation
Approximately 25–30% of patients exhibit apathogenic germline mutation in SMARCB1 (rhabdoid tumor predisposition syndrome (RTPS 1)) or SMARCA4 (RTPS 2). Individuals with RTPS typically present prior to their first birthday frequently with synchronous, multifocal tumors and extensive disease.30 Germline pathogenic mutations in SMARCB1 associated with RTPS1 occur in most cases de novo and pedigrees with transmission across generations are rare. In contrast, germline pathogenic mutations of SMARCA4 associated with RTPS2 are inherited from a parent in more than 50%,13,14,31 suggesting incomplete penetrance. Nevertheless, our current understanding of potential (germline) mosaicism as well as of factors influencing penetrance of RTPS and related disorders remain poorly understood.
Current Conventional Therapeutic Approaches in Extracranial MRT
Due to the rarity of extracranial MRT and the lack of controlled clinical trials, large data sets of uniformly treated patients are exceptional. Most information on therapeutic success comes from retrospective studies (Table 1). Currently, there is no established standard treatment approach for extracranial MRT, and most individuals are treated on intensive multimodal regimens, combining early surgical resection of primary tumor (if feasible GTR), chemotherapy including intensive multidrug regimens and local radiotherapy to all sites of disease involvement or high dose chemotherapy (HDCT) followed by autologous stem-cell rescue. Certain cytostatics such as anthracyclines, alkylating, platinating agents and vinca alkaloids are employed in all current approaches.6,8–10,32–34
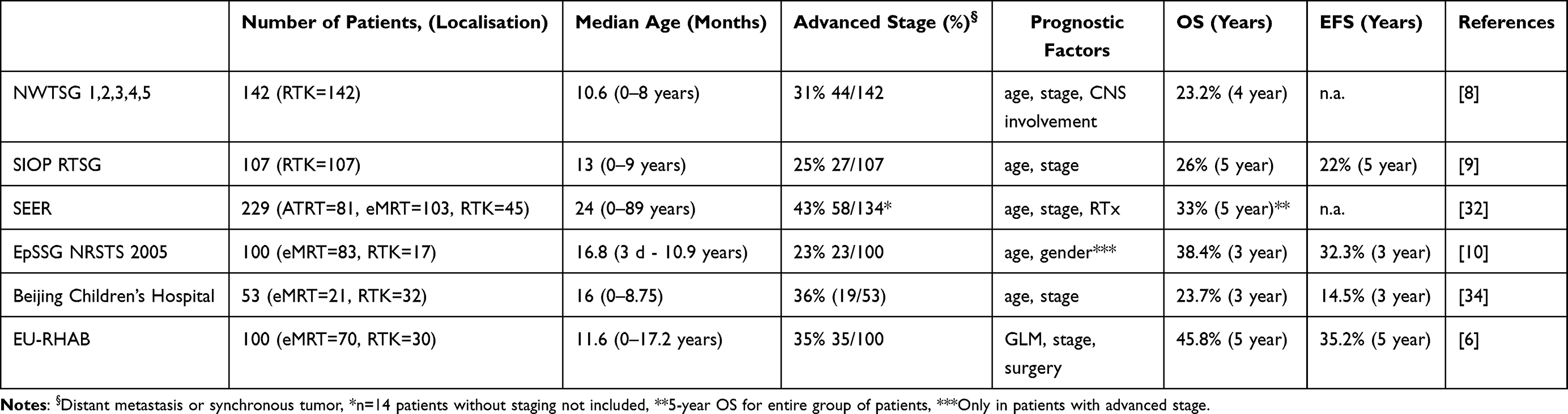 |
Table 1 Summary of Retrospective and Prospective Trials/Registries of Extracranial MRT |
The National Wilms Tumor Study Group Trial (NWTSG) registered 142 patients with renal rhabdoid tumors between 1969 and 2002. The effect of treatment was problematic to interpret as patients had not been treated uniformly. Initial analyses did not demonstrate any improvement compared to historical cohorts. The 4-year overall survival was only 23.2%. Age at diagnosis was a highly significant prognostic factor (p < 0.001). Patients diagnosed beyond 2 years of age, demonstrated a 41.1% (95% CI: 16–51%) 4-year overall survival rate. Higher stage, specifically the presence of a CNS lesion, was predictive of poor prognosis. Patients with stages I and II demonstrated significantly superior 4-year overall survival rates with 41.8% compared to outcome of patients with stage III/IV/V (15.9%).8
In the SIOP series, 107 patients with renal rhabdoid tumors were registered and treated between 1993 and 2005. The intention of the SIOP trials was to apply preoperative chemotherapy which consisted of weekly vincristine and 2-weekly actinomycin D (VA) for a period of 4 weeks for stages I–III tumors and vincristine, actinomycin D and doxorubicin (VAD) for stage IV tumours for a period of 6 weeks. In case of stage V, treatment was started with VA, and if the response was not satisfactory, doxorubicin was added after 4 weeks. A total of 60 out of 107 patients with RTK received preoperative chemotherapy. The advantages of preoperative chemotherapy could not be analyzed in this study; in 38 patients with SIOP stages I, II or III, vincristine and actinomycin D were applied for 4 weeks and in 22 patients with stage IV vincristine, actinomycin D and doxorubicin was given for 6 weeks. Postoperative treatment was a four-drug regimen consisting of etoposide, carboplatin, ifosfamide/cyclophosphamide and epi/doxorubicin. Additional radiotherapy was given to patients with SIOP stage II, III and IV stage. Unfortunately, the 5-year overall- and event-free survival of 26% (95% CI: 18–37%) and 22% (95% CI: 14–33%) did not show any improvement over other historical controls. The most important prognostic factor was young age at diagnosis; patients younger than 12 months at diagnosis demonstrated a 9.6% (95% CI: 3.7–25%) 5-year event-free survival (EFS) compared to patients older than 24 months (39.5%, 95% CI: 24–65.2%). There was also a significant difference in 5-year EFS between stage I (50%, 95% CI: 18.8–100%) and other stage groups.9
In an analysis of the Surveillance, Epidemiology, and End Results (SEER) program, 229 patients (ATRT = 81, eMRT = 103, RTK = 45) with malignant rhabdoid tumors of any anatomical region were included (1986 and 2005). The 5-year overall survival for the whole cohort was 33 ± 3.4%. Univariate and multivariate analyses disclosed that age (>2 years) at diagnosis, localized stage, and use of radiotherapy were significantly associated with improved survival. Multivariate analyses demonstrated that age (less than 2 years at diagnosis; hazard ratio (HR) – 1.79), distant metastasis (HR – 4.56) and absence of radiotherapy (HR – 1.89) were independent predictors of inferior survival.32
In the European Paediatric Soft Tissue Sarcoma Study Group (EpSSG) trial on Non-Rhabdomyosarcoma Soft Tissue Sarcoma 2005 (EpSSG NRSTS 2005) a total of 100 patients (eMRT=83, RTK=17) were prospectively registered and treated (2005–2014). The 3-year OS and EFS were 38.4% (95% CI: 28.8–47.9%) and 32.3% (95% CI: 23.2–41.6%), respectively. Older age (> 1 year) at diagnosis was the only significant prognostic factor on univariate analysis. The 4-year OS and EFS of patients <1 year at diagnosis were 20.1% (95% CI: 7.9–36.3%) and 17.2% (95% CI: 6.3–32.7%). Multivariate analyses disclosed that age at diagnosis (<1 year) (HR – 2.6) and gender (males) (only in advanced stage) (HR – 2.9) were independent prognostic factors for inferior survival.10
In a retrospective single-institution experience of the Beijing Children’s Hospital, 53 non-uniformly treated patients with extracranial MRT (eMRT = 21, RTK = 32) were included between 2007 and 2017. The 3-year OS- and EFS were poor (23.7% and 14.5%, respectively). Age (<1 year) and advanced stage at diagnosis were significantly associated with inferior survival.34
From 2009 until 2018, a total of 100 patients with extracranial MRT (n = 70 eMRT, n = 30 RTK) were included into the EU-RHAB registry. The 5-year OS and EFS estimates for the whole cohort were 45.8 ± 5.4% and 35.2 ± 5.1%, respectively. On univariate analyses, age at diagnosis (≥12 months), localized stage, absence of synchronous tumors, absence of a germline mutation (GLM), gross total resection (GTR), radiotherapy and achievement of a complete remission (CR) were significantly associated with favorable outcomes. In a multivariate, stepwise Cox regression model, presence of a GLM, advanced stage and lack of a GTR remained independent prognostic factors. Two risk groups are distinguishable: 1) patients at standard risk (SR) with localized disease (M0), gross total resection (GTR+) and without proof of a germline mutation (GLM-) demonstrated significantly superior 5-year OS rates (72.2 ± 9.9%), compared to those of a 2) high-risk group (HR) with one of the features; distant metastasis (M+) and/or incomplete resection (GTR-) and/or GLM+ (32.5 ± 6.2%).6
High-Dose-Chemotherapy (HDCT)
The role of high-dose-chemotherapy (HDCT) followed by autologous stem-cell rescue in intracranial rhabdoid tumors (ATRT) has been evaluated repeatedly.35,36 Its role in the treatment of extracranial MRT remains ill defined. Data of 251 patients diagnosed with nephroblastoma included in three consecutive SIOP/GPOH studies demonstrated a potential survival benefit of HDCT for certain relapse situations.37 Venkatramani et al described a benefit of HDCT on survival in patients with extracranial MRT, however only 4/10 initially intended patients received HDCT.33 In a retrospective analysis of all 58 patients with RTK (1991 to 2014) treated according to SIOP9/GPO, SIOP93-01/GPOH, SIOP2001/GPOH, and the European Rhabdoid Tumor Registry, comparable outcome with and without HDCT was detected.38 Similarly, no survival benefit for patients with extracranial MRT treated with HDCT according EU-RHAB protocol was recently reported in the EU-RHAB registry (n = 100).6
Role of Radiotherapy
Based on currently available data, the exact role of the timing, target volumes and optimal doses of radiotherapy (RTx) has not been established on an evidence-based level. The NWTS series suggests a role of RTx in the local control of RTK. The 4-year OS was 28.5% in patients treated with RTx compared to patients without (12%), however following adjustment of data for age and stage, significant benefits of RTx disappeared.8 The SEER series indicated improved survival rates for patients treated by RTx. In a multivariate model RTx was an independent prognostic factor for survival (p = 0.0006), however only 23% of patients <3 years received RTx.32 The EpSSG study did not confirm a significant benefit of RTx. This could have been confounded by age and/or stage.10 In fact, RTx was administered to older patients (reluctance to apply RTx to very young children) and those with advanced stages. In EU-RHAB patients treated by RTx (according to protocol-defined dose and volume), survived significantly longer compared to those without RTx (5-year OS 56.6 ± 6.9% vs 22.5 ± 7.7%). Nevertheless, the benefits of RTx in patients with SIOP stage I or IRS I await further definition. Within the EU-RHAB registry, none of the non-irradiated patients with stages IRS I or SIOP I relapsed.6 Melchior et al analyzed 58 patients with RTK treated by multimodality strategies. None of the five non-irradiated patients with local stage I relapsed.39
Role of Anatomical Location: EMRT versus RTK
Anatomical localization of extracranial rhabdoid tumors seems to have an impact on survival. The 5-year OS for RTK is in general considered to be inferior compared to eMRT (36.5% versus 50.1%).6 The poorer survival of patients with RTK may possibly be related to clinical (see above) and anatomical characteristics. RTK patients are often younger, present with synchronous tumors at diagnosis and in advanced stages with consecutive survival rates of only 20–25%. However, patients with SIOP stage I have an excellent survival.6,40 A distinct chemotherapy resistance characterizes RTK. Young age often delays radiotherapy. In addition, early recurrence of disease is more frequent, which may be attributable to a higher rate of GLM in RTKs.6,40 In fact, 33% of all completely characterized patients with RTK demonstrated a GLM contrasting with 16% in eMRT. Although eMRT often present with large primary tumors (> T3), patients demonstrated mostly a favorable response to induction chemotherapy commonly followed by local radiotherapy (Figure 3).
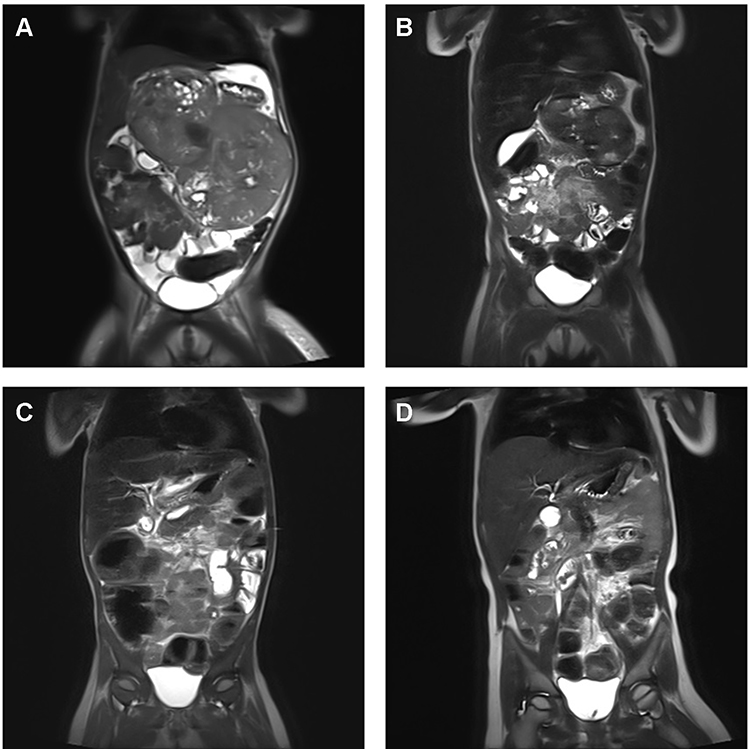 |
Figure 3 Response to standardized chemotherapy in a patient with extensive primary eMRT – Imaging results. (A) Diagnostic imaging at 27 months of age with inoperable, abdominal eMRT (14x14x9.8 cm), without distant metastasis, without germline mutation. (B) MRI before confirmed pathological diagnosis of eMRT, one course I2-VAd (ifosfamide, vincristine, Adriamycin) according CWS VAIA protocol, and after confirmed diagnosis, two courses ICE (ifosfamide, cyclophosphamide, etoposide) according EU-RHAB were given. After three courses of chemotherapy, tumor regression (5.6x8.8x4.2 cm), and stable disease < 25% was detected. (C) Imaging following chemotherapy according EU-RHAB was continued with one course of DOX (doxorubicin) and VCA (vincristine, cyclophosphamide, actinomycin D). Eventually the tumor was resected subtotally, and stable disease < 10% was achieved. (D) Following resection radiotherapy of the tumor bed (for abdomen up to 36 Gy) with boost to celiac trunk (up to 45 Gy) was performed, and therapy was completed with one course of VCA (vincristine, cyclophosphamide, actinomycin D), IC (ifosfamide, cyclophosphamide), VC (vincristine, cyclophosphamide) and DOX (doxorubicin) was given. The patient achieved complete remission, and is alive at 32 months following diagnosis. |
Potential Targeted Therapeutic Approaches to Extracranial MRT
Until very recently, preclinical investigations have mainly focused on the specific interrogation of SMARCB1-related biology and dysfunctionality of the SWI/SNF complex, which may affect a whole spectrum of associated oncogenic signaling pathways (Table 2).41–67 SWI/SNF chromatin remodeling complexes exist in three distinct, final-form complexes: canonical BAF (cBAF), PBAF, and ncBAF. Inhibition of the BRD9 subunit of the SWI/SNF complex decreased MRT cell proliferation and thus presented potential new cancer-specific therapeutic targets.68,69
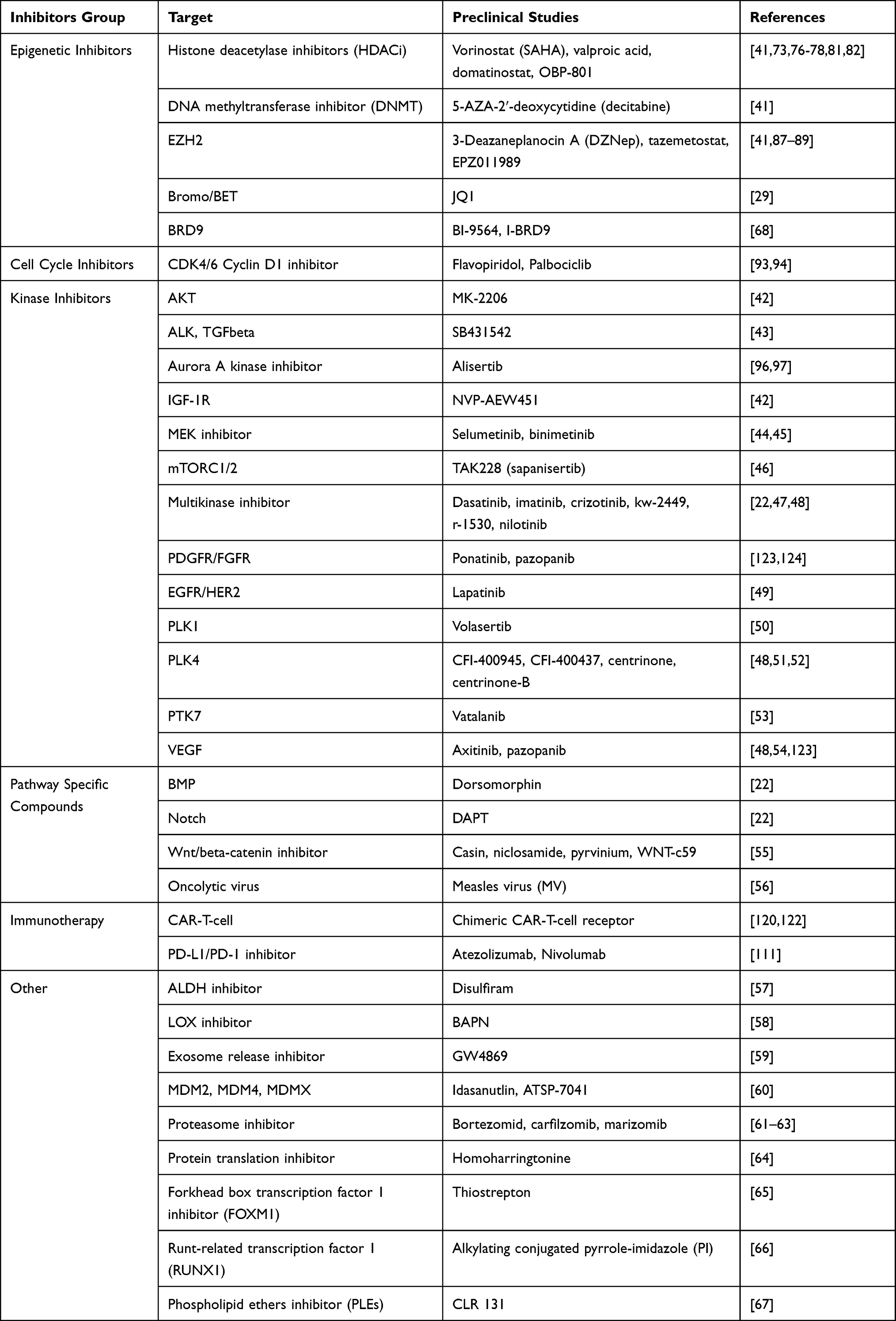 |
Table 2 Overview of Preclinical Studies on Pediatric MRT |
Targeting the Epigenome
Epigenetic modifications are tightly associated with the function of the SWI/SNF complex. Changes in the chromatin structure affect the modulation of gene expression.70,71 The inhibition of enzymatic activities involved in epigenetic regulation by enzymes such as HDAC (histone deacetylases), DNMT (DNA methyltransferases) or EZH2 (enhancer of zeste homolog 2) is the subject of active clinical trials.
HDAC (Histone Deacetylases) Inhibitors
Targeting histone acetylation is an attractive tool in the treatment of MRT as several HDAC are overexpressed in primary tumors and cell lines.72 Preclinical studies in MRT demonstrated synergistic effects of the HDAC inhibitor (HDCAi) vorinostat (SAHA) in combination with fenretinide, tamoxifen and doxorubicin in inducing cell cycle arrest and apoptosis.73 Low-dose panobinostat (LBH589) caused growth arrest, intra-tumor ossification and lineage maturation of malignant rhabdoid tumor cells in vitro and in vivo.74 Custers et al discovered that combined HDAC and mTOR inhibition mimics MRT differentiation.75 Recently, a small molecule epigenetic modulator known as domatinostat (4SC-202), inhibiting both class I HDACs and Lysine Demethylase (LSD1) was investigated, and cytotoxic and cytostatic effects on malignant rhabdoid tumor cells were detected.76 The histone deacetylase inhibitor, OBP-801 induced apoptosis in malignant rhabdoid tumor cells by epigenetically releasing the silencing of NOXA, a key mediator of MRT apoptosis.77 HDACi potentiate the antiproliferative effects of radiotherapy on MRT in vivo and in vitro.78,79 The HDAC inhibitor vorinostat was tolerated well when used alone or in combination with bortezomib or temozolomide in clinical trials including MRTs (NCT01076530, NCT00217412).80,81 Valproic acid was evaluated in a Phase I trial in children with refractory, solid tumors including CNS tumors; however, only one patient with ATRT was included, and one confirmed partial response (glioblastoma multiforme) and only one minor response (brainstem glioma) were observed.82
DNMT (DNA Methyltransferases) Inhibitors
The DNMT inhibitors, 5-azacytidine and 5-aza-2ʹdeoxycytidine (decitabine) suppress tumor growth by blocking the cell cycle, inducing apoptosis and promoting cellular differentiation.83 Decitabine in combination with doxorubicin and cyclophosphamide has shown promising effects in phase I trials in children with neuroblastoma and other solid tumors exclusive of MRT.84 An ongoing phase I trial employs decitabine and pembrolizumab (PD-1 monoclonal antibody) in patients with relapsed, refractory or progressive lymphomas and solid tumors including extracranial MRT (NCT03445858).
EZH2 (Enhancer of Zeste Homolog 2) Inhibitors
The antagonistic relationship between SWI/SNF and the polycomb repressive complex 2 (PRC2) plays a critical role in gene transcription.85,86 EZH2 (enhancer of zeste homolog 2) is a core enzymatic subunit of PRC2. Preclinical studies demonstrated that the EZH2 inhibitor, DZNep (3-deazaneplanocin A) inhibited cell growth in MRT in vitro and strongly potentiated the effects of ionizing radiation on ATRT cells.87 Tazemetostat (EPZ-6438) is a selective orally bioavailable inhibitor of EZH2ʹs enzymatic activity. In EZH2-mutant xenografts tazemetostat caused dose-dependent growth inhibition and reduction in H3K27me3 levels in malignant rhabdoid tumors.88 Furthermore, EPZ011989 in combination with standard of care reagents significantly improved time to event in MRT, although this effect was observed in only a minority of the combination experiments.89 In this first-in-human trial, tazemetostat disclosed a favorable safety profile and antitumor activity in patients with refractory B-cell lymphoma and advanced solid tumors.90 Preliminary data indicate that tazemetostat is generally well tolerated in children, and displays promising antitumor activity in ATRT.91 In an recently closed phase I/II trial, tazemetostat was applied to children with relapsed, refractory or progressive solid tumors including extracranial MRT (NCT02601937, NCT03155620, NCT03213665).
Tyrosine Kinase Inhibitors Targeting the CDK4/CDK6/Cyclin D1/RB Pathway
Biallelic inactivation of SMARCB1 in MRT cells increases the expression of cyclin D1, and upregulation of D-type cyclin-dependent kinases 4 and 6 (CDK4/6) promotes activation of the transcription factor E2F resulting in cell cycle progression.92 Non-specific CDK inhibitors such as flavopiridol combined with tamoxifen affected cyclin D1 and inhibited malignant rhabdoid tumor cell growth in vitro.93 Palbociclib (PD0332991) and ribociclib (LEE011) are both orally bioavailable selective cyclin-dependent kinase 4/6 inhibitors. In vitro palbociclib combined with radiotherapy promoted apoptosis in ATRT cells, in vivo a combination of both inhibited tumor growth.94
Recently, a phase I trial of the CDK4/6 inhibitor ribociclib (NCT017747876) in MRT, neuroblastomas and other CDK4/6-amplified malignancies demonstrated acceptable safety and favorable pharmacokinetics in children. Fifteen patients with MRT (n = 13 ATRT, n = 2 eMRT) received ribociclib, and two patients with ATRT achieved prolonged disease stabilization.95 In ongoing phase I/II studies, abemaciclib (NCT02644460, NCT04238819) and palbociclib (NCT03526250, NCT03709680) are applied to children with relapsed, refractory or progressive solid tumors including MRT.
Aurora Kinase A Inhibitors
Aurora kinase A (AURKA) is a direct downstream target of SMARCB1 and overexpressed in rhabdoid tumors. Targeting AURKA in malignant rhabdoid tumor cell lines in vitro and in xenografts demonstrated strong responses to the selective aurora kinase A inhibitor, alisertib (MLN-8237).96 Preclinical data moreover demonstrated that aurora kinase A inhibition enhanced radiation sensitivity of malignant rhabdoid tumor cell lines, making this compound an attractive agent for combination therapy.97 Alisertib (MLN-8237) is currently in clinical trials (phase I/II) for different tumor indications in adults and children including ATRT. Disease stabilization and/or regression of tumors was detected in four children with ATRT.98 In a Phase II trial of alisertib in children with recurrent/refractory solid tumors (n = 139), none of the four patients included with MRT (ATRT = 2, eMRT = 2), demonstrated a response; the response rate of alisertib as a single agent in the whole cohort was less than 5%.99 In a phase I trial of patients with advanced solid tumors (excluded MRT), addition of TAK-228 (mTOR inhibitor) to alisertib potentiated the antitumor activity of alisertib in vivo, resulting in increased cell death and apoptosis, the combination treatment was well tolerated.100 In a phase II trial of alisertib, as a single-agent in patients <22 years with recurrent or progressive malignant rhabdoid tumors (NCT02114229), the drug was well tolerated in children with recurrent ATRT. A third of the patients demonstrated disease stabilization for >6 months.101
Immunotherapy Approaches
Immunotherapy is an attractive anti-cancer strategy. This approach may particularly suit targeting diffuse, infiltrative tumors. Currently, FDA has approved immune modulators such as checkpoint inhibitors, chimeric antigen receptor T-cells (CAR T-cells), monoclonal antibodies and bispecific T-cell engagers (BiTEs) for use in children with cancer (Figure 4).102
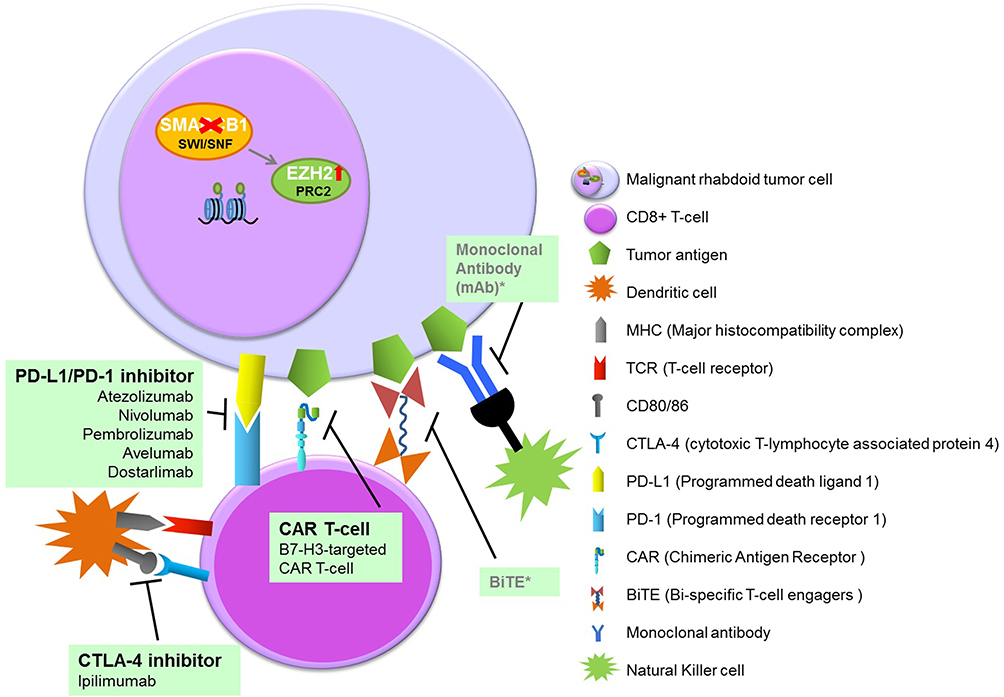 |
Figure 4 Overview of immunotherapy in pediatric MRT. Note: *For pediatric extracranial MRT not yet studied. |
Checkpoint Inhibitors
Success of immunotherapies based on immune checkpoint blockade is associated with the promotion of T-cell responses against tumor antigens. Currently, two classes of checkpoint inhibitors have been approved, those inhibiting the programmed death receptor 1 (PD-1) or its ligand (PD-L1) and inhibitors of the cytotoxic T-cell lymphocyte-associated protein 4 (CTLA-4).103
Recent studies describe a critical role for the composition of the tumor immune microenvironment (TIME) in tumor-immune interaction and in response to therapy.104,105 According to the effectiveness of tumor immune therapies, tumors are frequently described as “hot” or “cold”.106 Hot” tumors are frequently characterized by a high tumor mutation burden (TMB) in the coding genome, expression of PD-1 ligand, and by infiltration of cytotoxic lymphocytes expressing PD-1.107 Childhood tumors are generally considered “cold” due to low TMB,108 PD-1 expression and T-cell infiltrates.109 Recently, unique immune signatures for each ATRT subgroup were described, as well as for extracranial MRT. Despite the typical low TMB in MRT, a high rate of immune infiltration by a CD8+T subpopulation was detected, specifically in extracranial MRT and in ATRT-MYC.23 Grabovska et al described the TIME approach in more than 6000 pediatric tumors including MRTs and detected increased CD8+ T infiltration in extracranial MRT associated with inferior survival.110
In an experimental MRT model, checkpoint blockade therapy induced tumor-regression and immune response.111 A patient with eMRT treated with the anti-PD-L1 checkpoint inhibitor, atezolizumab demonstrated a transient objective response.112 In a phase I/II trial of the anti-PD-L1 compounds, atezolizumab and pembrolizumab in children and young adults with solid tumors, both compounds were well tolerated; however, responses were restricted, and observed in only a few rare PD-L1-positive tumor types.113,114 In a case report, partial response to conventional chemotherapy combined with atezolizumab was described in a high-grade metastatic tumor with rhabdoid features.115 The low tumor mutational burden in most pediatric cancers limits the number of neoantigens for immunotherapies. The routine use of checkpoint inhibitors in monotherapy has thus moved towards combinations with other targeted agents, conventional cytostatics as well as other modalities, such as surgery or radiotherapy.116 Inhibiting EZH2 in combination with immune checkpoint blockade has emerged as an attractive approach based on the immunologic effects seen in both regulatory T-cells and tumors, including increases in PD-L1 expression, in the setting of EZH2 inhibition. Wang et al demonstrated that disruption of EZH2 function in regulatory T-cells (Tregs) promotes potent cancer immunity.117 Goel et al described an increase in tumor immunogenicity due to suppression of the proliferation of Tregs following CDK4/6 inhibition. This provides a rationale for new combination regimens comprising CDK4/6 inhibitors and immunotherapies.118 PD-1 and CTLA-4 function on different levels of the immune response; combination therapy may enhance the response. In an ongoing trial PD-1/CTLA-4 signaling blockade in combination is tested in recurrent/refractory pediatric cancers, including extracranial MRT (NCT04416568).102
Current findings help to define future strategies for immune checkpoint inhibitors either by focusing research on specific disease subpopulations (eg, ATRT-MYC, extracranial MRT), or by providing the means to identify therapeutic combination partners that augment T-cell infiltration and proliferation in “immune cold” tumor microenvironments.119
CAR T-Cells
T-cells expressing chimeric CD-specific antigen receptors (CARs) target tumor-associated antigens directly on the tumor cell surface. However, many solid tumor antigens are expressed in lower levels on the surface of cancer cells, and that low-density antigen expression is insufficient for optimal CAR activation. B7-H3 (CD276) is a checkpoint molecule expressed highly on pediatric tumors. Immunotherapy employing B7-H3-targeted CAR T-cells are currently investigated clinically in children and adults with refractory extracranial solid tumors.120,121 Theruvath et al discovered that ATRTs express high levels of B7-H3, and B7-H3 CAR T-cells were highly active in ATRTs in vitro and in vivo in a xenograft murine model.122
Monoclonal antibodies bind to a specific tumor surface antigen and activate NK cells and macrophages via Fc receptor binding. The currently best studied monoclonal antibody for pediatric solid tumors is the anti-GD2, dinutuximab, FDA approved for neuroblastoma. However monoclonal antibodies targeting other pediatric solid tumors have been less successful.102 BiTEs (bi-specific T-cell engagers) are synthetic molecules that connect and activate T-cells with tumor-specific antigens. This leads to T-cell activation and subsequent cytolysis of the tumor. BiTEs for pediatric solid tumors are just beginning to be explored. One example is a phase I trial with anti-GD2 BiTE in neuroblastoma and osteosarcoma (NCT02173093).
Future Directions
Even though survival rates for patients with extracranial MRT demonstrated little improvement in recent years, multimodal treatment resulted in a remarkable survival benefit for a substantial part of patients with standard risk profiles.6 Albeit another group of patients (patients with high-risk factors) certainly need innovative therapeutic options. In the near future, it will be extremely important to implement careful patient selection for trial stratification. Recently, two risk groups have been identified:6
1) Standard risk group characterized by localized disease, gross total resection and without a germline mutation demonstrated significantly superior 5-year OS 72.2% compared to
2) High-risk group presenting with one of the features distant metastasis and/or incomplete resection and/or germline mutation (5-year OS 32.5%).
Standard risk patients may be treated with conventional chemotherapy approaches such as the EU-RHAB or EpSSG approaches, to avoid the uncertainties of experimental drug trials. For this group, it is imperative to minimize treatment toxicity among others by optimizing the sequence of modalities, the intensity of treatment, the doses and use of advanced techniques of radiotherapy. Current technical innovations in radiotherapy and changes in concepts using highly conformal radiotherapy fields, limit damage to surrounding normal tissue and improve the therapeutic utility of modern techniques, especially in very young children. However, for patients with SIOP or IRS stage I omission of RTx may be an option. More data and pooling of international series are urgently needed.
Patients with high-risk factors may benefit from inclusion into phase I trials using mechanism specific, epigenetic approaches (eg, HDAC-, DNMT- or EZH2-inhibitors) but also as target specific (eg, CDK4/6 inhibitor, ribociclib) frontline therapy. Preliminary results of phase I/II trials of EZH2 inhibitors demonstrated (NCT02601937, NCT03155620, NCT03213665) promising antitumor activity in ATRT.91 A number of promising targeted agents are currently in early phase clinical trials for patients with extracranial rhabdoid tumors, including inhibitors of HDACs, vorinostat (NCT04308330) and CUDC-907 (HDAC + PI3K inhibitor) (NCT02909777), CDK4/6 inhibitors, abemaciclib (NCT02644460, NCT04238819) and palbociclib (NCT03709680, NCT03526250), and the inhibitor of aurora kinase A, alisertib (NCT02114229) (Table 3).
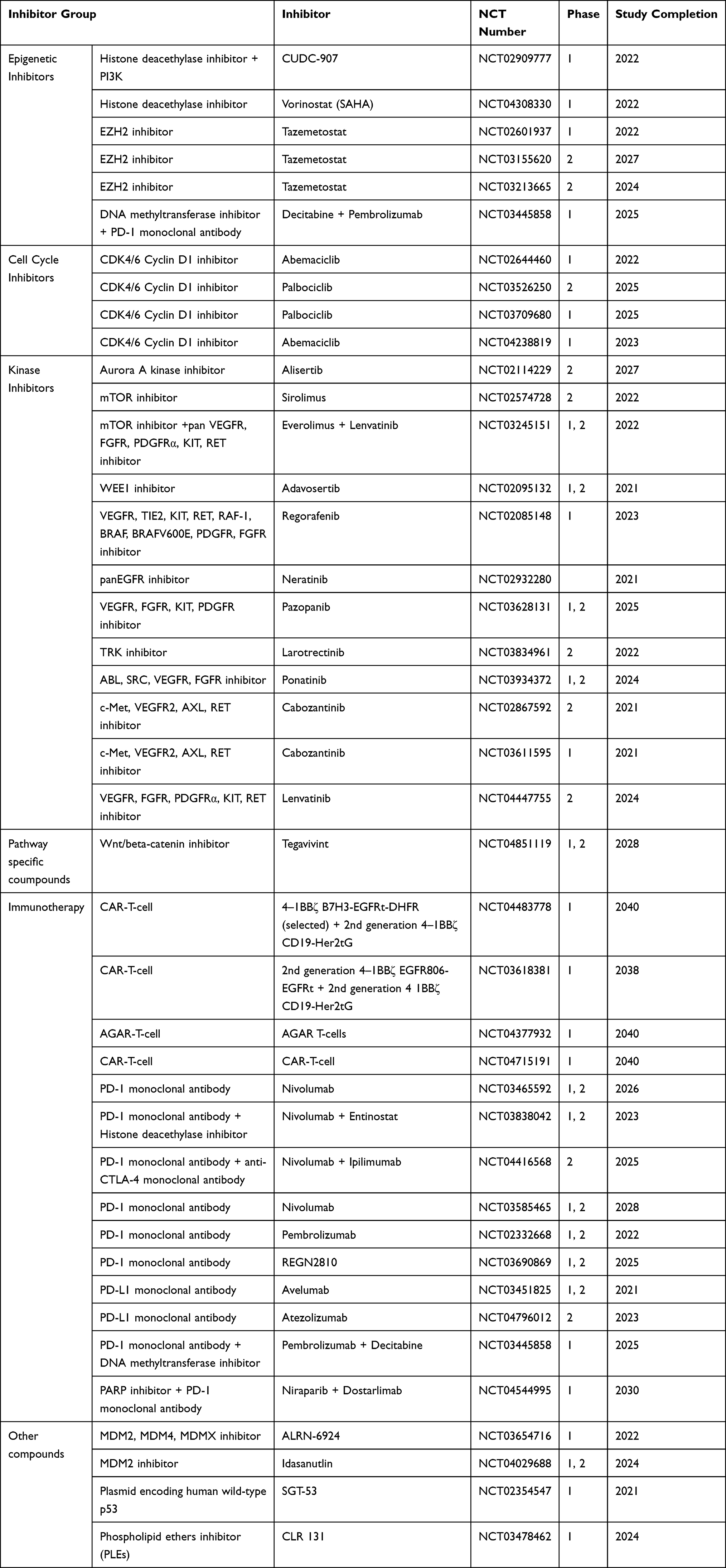 |
Table 3 Overview of Ongoing Experimental Clinical Trials in Pediatric Extracranial MRT |
Tyrosine kinase inhibitors consistently demonstrated antitumor activity in malignant rhabdoid tumors,22,123,124 multi-kinase inhibitors as single agents or in combination with other agents are involved in 10 phase I/II ongoing studies for extracranial MRT (Table 3). Immunotherapy, especially checkpoint blockade has become a major focus of preclinical and clinical investigations in high risk extracranial RT.23,111,113,114 Currently, 14 ongoing clinical trials including immunotherapies enrolling patients with extracranial MRT are listed at clinicaltrials.gov (Table 3).
Due to the modest number of patients with extracranial MRT, for the most promising targeted agents, it may be advisable, after careful in vitro and in vivo studies, to initiate limited trials with small patient numbers and smart biostatistical approaches. Thus, a selection of compounds that deserve going on to phase II combination trials for upfront treatment may be rapidly detected. On the other side, many basket phase I/II studies recruit only a very small number of MRT patients among a broader patient group. Thus, recruiting sufficient numbers of extracranial MRT patients in a stratified fashion according to novel risk factors and biomarkers (such as molecular subgroups), in collaborative, international consortia is imperative.
Discovery of heterogeneity in extracranial MRT especially on an epigenetic level has enormous potential for future risk and treatment stratification. Together with integrated risk stratification, precise risk-adapted treatment options will be developed for this very young population. It is imperative to characterize the relevance of germline disposition syndromes (RTPS), ie, whether the mono-allelic germline mutation in the SWI/SNF complex has an impact on patient pharmacogenomics or immune response. An improved understanding of the genotype-phenotype correlation and its consequences for therapy is of rather high importance.
Acknowledgments
MCF is supported by the “Deutsche Kinderkrebsstiftung” DKS 202.10, by “Deutsche Forschungsgemeinschaft” DFG FR 1516/4-1 and by DKH 70113981. RS received grant support for infrastructure by the KinderKrebsInitiative Buchholz/Holm-Seppensen and by DKH 70114040. PDJ is supported by the Else-Kroener-Fresenius Stiftung. We thank P. Neumayer and S. Breitmoser for expert assistance.
Disclosure
Reiner Siebert has received speaker’s honoraries from Astra Zeneca and Roche. None of the other authors declares any conflict of interest.
References
1. Beckwith JB, Palmer NF. Histopathology and prognosis of Wilms tumors: results from the First National Wilms’ Tumor Study. Cancer. 1978;41(5):1937–1948. doi:10.1002/1097-0142(197805)41:5<1937::aid-cncr2820410538>3.0.co;2-u
2. Judkins AR, Mauger J, Ht A, Rorke LB, Biegel JA. Immunohistochemical analysis of hSNF5/INI1 in pediatric CNS neoplasms. Am J Surg Pathol. 2004;28(5):644–650. doi:10.1097/00000478-200405000-00013
3. Cancer statistics reports for the Germany. Available from: http://www.kinderkrebsregister.de/dkkr/ergebnisse/jahresberichte/jahresbericht-2019.html.
4. Brennan B, Stiller C, Bourdeaut F. Extracranial rhabdoid tumours: what we have learned so far and future directions. Lancet Oncol. 2013;14(8):e329–e336. doi:10.1016/S1470-2045(13)70088-3
5. Frühwald MC, Hasselblatt M, Nemes K, et al. Age and DNA methylation subgroup as potential independent risk factors for treatment stratification in children with atypical teratoid/rhabdoid tumors. Neuro Oncol. 2020;22(7):1006–1017. doi:10.1093/neuonc/noz244
6. Nemes K, Bens S, Kachanov D, et al. Clinical and genetic risk factors define two risk groups of extracranial malignant rhabdoid tumours (eMRT/RTK). Eur J Cancer. 2021;142:112–122. doi:10.1016/j.ejca.2020.10.004
7. Nakata K, Colombet M, Stiller CA, Pritchard-Jones K, Steliarova-Foucher E. Incidence of childhood renal tumours: an international population-based study. Int J Cancer. 2020;147(12):3313–3327. doi:10.1002/ijc.33147
8. Tomlinson GE, Breslow NE, Dome J, et al. Rhabdoid tumor of the kidney in the National Wilms’ Tumor Study: age at diagnosis as a prognostic factor. J Clin Oncol. 2005;23(30):7641–7645. doi:10.1200/JCO.2004.00.8110
9. van den Heuvel-eibrink MM, van Tinteren H, Rehorst H. Malignant rhabdoid tumours of the kidney (MRTKs), registered on recent SIOP protocols from 1993 to 2005: a report of the SIOP renal tumour study group. Pediatr Blood Cancer. 2011;56(5):733–737. doi:10.1002/pbc.22922
10. Brennan B, De Salvo GL, Orbach D, et al. Outcome of extracranial malignant rhabdoid tumours in children registered in the European Paediatric Soft Tissue Sarcoma Study Group Non-Rhabdomyosarcoma Soft Tissue Sarcoma 2005 Study-EpSSG NRSTS 2005. Eur J Cancer. 2016;60:69–82. doi:10.1016/j.ejca.2016.02.027
11. Hasselblatt M, Isken S, Linge A, et al. High-resolution genomic analysis suggests the absence of recurrent genomic alterations other than SMARCB1 aberrations in atypical teratoid/rhabdoid tumors. Genes Chromosomes Cancer. 2013;52(2):185–190. doi:10.1002/gcc.22018
12. Kieran MW, Roberts CW, Chi SN, et al. Absence of oncogenic canonical pathway mutations in aggressive pediatric rhabdoid tumors. Pediatr Blood Cancer. 2012;59(7):1155–1157. doi:10.1002/pbc.24315
13. Hasselblatt M, Nagel I, Oyen F, et al. SMARCA4-mutated atypical teratoid/rhabdoid tumors are associated with inherited germline alterations and poor prognosis. Acta Neuropathol. 2014;128(3):453–456. doi:10.1007/s00401-014-1323-x
14. Schneppenheim R, Fruhwald MC, Gesk S, et al. Germline nonsense mutation and somatic inactivation of SMARCA4/BRG1 in a family with rhabdoid tumor predisposition syndrome. Am J Hum Genet. 2010;86(2):279–284. doi:10.1016/j.ajhg.2010.01.013
15. Holsten T, Bens S, Oyen F, et al. Germline variants in SMARCB1 and other members of the BAF chromatin-remodeling complex across human disease entities: a meta-analysis. Eur J Hum Genet. 2018;26(8):1083–1093. doi:10.1038/s41431-018-0143-1
16. Wilson BG, Roberts CW. SWI/SNF nucleosome remodellers and cancer. Nat Rev Cancer. 2011;11(7):481–492. doi:10.1038/nrc3068
17. Tegeder I, Thiel K, Erkek S, et al. Functional relevance of genes predicted to be affected by epigenetic alterations in atypical teratoid/rhabdoid tumors. J Neurooncol. 2019;141(1):43–55. doi:10.1007/s11060-018-03018-6
18. Koelsche C, Stichel D, Griewank KG, et al. Genome-wide methylation profiling and copy number analysis in atypical fibroxanthomas and pleomorphic dermal sarcomas indicate a similar molecular phenotype. Clin Sarcoma Res. 2019;9:2. doi:10.1186/s13569-019-0113-6
19. Chun HE, Lim EL, Heravi-Moussavi A, et al. Genome-wide profiles of extra-cranial malignant rhabdoid tumors reveal heterogeneity and dysregulated developmental pathways. Cancer Cell. 2016;29(3):394–406. doi:10.1016/j.ccell.2016.02.009
20. Koelsche C, Schrimpf D, Stichel D, Sill M, Sahm F. Sarcoma classification by DNA methylation profiling. Nat Commun. 2021;12(1):498. doi:10.1038/s41467-020-20603-4
21. Johann PD, Erkek S, Zapatka M, et al. Atypical teratoid/rhabdoid tumors are comprised of three epigenetic subgroups with distinct enhancer landscapes. Cancer Cell. 2016;29(3):379–393. doi:10.1016/j.ccell.2016.02.001
22. Torchia J, Golbourn B, Feng S, et al. Integrated (epi)-genomic analyses identify subgroup-specific therapeutic targets in CNS rhabdoid tumors. Cancer Cell. 2016;30(6):891–908. doi:10.1016/j.ccell.2016.11.003
23. Chun HE, Johann PD, Milne K, et al. Identification and analyses of extra-cranial and cranial rhabdoid tumor molecular subgroups reveal tumors with cytotoxic T cell infiltration. Cell Rep. 2019;29(8):2338–2354.e7. doi:10.1016/j.celrep.2019.10.013
24. Birks DK, Donson AM, Patel PR, et al. High expression of BMP pathway genes distinguishes a subset of atypical teratoid/rhabdoid tumors associated with shorter survival. Neuro Oncol. 2011;13(12):1296–1307. doi:10.1093/neuonc/nor140
25. Birks DK, Donson AM, Patel PR, et al. Pediatric rhabdoid tumors of kidney and brain show many differences in gene expression but share dysregulation of cell cycle and epigenetic effector genes. Pediatr Blood Cancer. 2013;60(7):1095–1102. doi:10.1002/pbc.24481
26. Ho B, Johann PD, Grabovska Y, et al. Molecular subgrouping of atypical teratoid/rhabdoid tumors-a reinvestigation and current consensus. Neuro Oncol. 2020;22(5):613–624. doi:10.1093/neuonc/noz235
27. Brocks D, Schmidt CR, Daskalakis M, et al. DNMT and HDAC inhibitors induce cryptic transcription start sites encoded in long terminal repeats. Nat Genet. 2017;49(11):1661. doi:10.1038/ng.3889
28. Andrianteranagna M, Cyrta J, Masliah-Planchon J, et al. SMARCA4-deficient rhabdoid tumours show intermediate molecular features between SMARCB1-deficient rhabdoid tumours and small cell carcinomas of the ovary, hypercalcaemic type. J Pathol. 2021;255(1):1–15. doi:10.1002/path.5705
29. Alimova I, Pierce A, Danis E, et al. Inhibition of MYC attenuates tumor cell self-renewal and promotes senescence in SMARCB1-deficient Group 2 atypical teratoid rhabdoid tumors to suppress tumor growth in vivo. Int J Cancer. 2019;144(8):1983–1995. doi:10.1002/ijc.31873
30. Bourdeaut F, Lequin D, Brugières L, et al. Frequent hSNF5/INI1 germline mutations in patients with rhabdoid tumor. Clin Cancer Res. 2011;17(1):31–38. doi:10.1158/1078-0432.CCR-10-1795
31. Nemes K, Bens S, Bourdeaut F, et al. Rhabdoid tumor predisposition syndrome. Adam MP, Ardinger HH, Pagon RA, et al. editors. GeneReviews(®). Seattle: University of Washington, Seattle Copyright © 1993–2021, University of Washington, Seattle. GeneReviews is a registered trademark of the University of Washington, Seattle; 1993.
32. Sultan I, Qaddoumi I, Rodríguez-Galindo C, Nassan AA, Ghandour K, Al-Hussaini M. Age, stage, and radiotherapy, but not primary tumor site, affects the outcome of patients with malignant rhabdoid tumors. Pediatr Blood Cancer. 2010;54(1):35–40. doi:10.1002/pbc.22285
33. Venkatramani R, Shoureshi P, Malvar J, Zhou S, Mascarenhas L. High dose alkylator therapy for extracranial malignant rhabdoid tumors in children. Pediatr Blood Cancer. 2014;61(8):1357–1361. doi:10.1002/pbc.25093
34. Cheng H, Yang S, Cai S. Clinical and prognostic characteristics of 53 cases of extracranial malignant rhabdoid tumor in children. A single-institute experience from 2007 to 2017. Oncologist. 2019;24(7):e551–e558. doi:10.1634/theoncologist.2018-0416
35. Benesch M, Bartelheim K, Fleischhack G, et al. High-dose chemotherapy (HDCT) with auto-SCT in children with atypical teratoid/rhabdoid tumors (AT/RT): a report from the European Rhabdoid Registry (EU-RHAB). Bone Marrow Transplant. 2014;49(3):370–375. doi:10.1038/bmt.2013.208
36. Hoffman LM, Richardson EA, Ho B, et al. Advancing biology-based therapeutic approaches for atypical teratoid rhabdoid tumors. Neuro Oncol. 2020;22(7):944–954. doi:10.1093/neuonc/noaa046
37. Furtwängler R, Nourkami N, Alkassar M, et al. Update on relapses in unilateral nephroblastoma registered in 3 consecutive SIOP/GPOH studies - A report from the GPOH-nephroblastoma study group. Klin Padiatr. 2011;223(3):113–119. doi:10.1055/s-0031-1275293
38. Furtwängler R, Kager L, Melchior P, et al. High-dose treatment for malignant rhabdoid tumor of the kidney: no evidence for improved survival-The Gesellschaft für Pädiatrische Onkologie und Hämatologie (GPOH) experience. Pediatr Blood Cancer. 2018;65(1):e26746. doi:10.1002/pbc.26746
39. Melchior P, Dzierma Y, Rübe C, et al. Local stage dependent necessity of radiation therapy in rhabdoid tumors of the kidney (RTK). Int J Radiat Oncol Biol Phys. 2020;108(3):667–675. doi:10.1016/j.ijrobp.2020.04.046
40. Walz AL, Fernandez CV, Geller JI. Novel therapy for pediatric and adolescent kidney cancer. Cancer Metastasis Rev. 2019;38(4):643–655. doi:10.1007/s10555-019-09822-4
41. Unland R, Borchardt C, Clemens D, Kool M, Dirksen U, Frühwald MC. Analysis of the antiproliferative effects of 3-deazaneoplanocin A in combination with standard anticancer agents in rhabdoid tumor cell lines. Anticancer Drugs. 2015;26(3):301–311. doi:10.1097/CAD.0000000000000181
42. Li T, Wang J, Liu P, et al. Insulin-like growth factor 2 axis supports the serum-independent growth of malignant rhabdoid tumor and is activated by microenvironment stress. Oncotarget. 2017;8(29):47269–47283. doi:10.18632/oncotarget.17617
43. Jeibmann A, Schulz J, Eikmeier K, et al. SMAD dependent signaling plays a detrimental role in a fly model of SMARCB1-deficiency and the biology of atypical teratoid/rhabdoid tumors. J Neurooncol. 2017;131(3):477–484. doi:10.1007/s11060-016-2326-3
44. Weingart MF, Roth JJ, Hutt-Cabezas M, et al. Disrupting LIN28 in atypical teratoid rhabdoid tumors reveals the importance of the mitogen activated protein kinase pathway as a therapeutic target. Oncotarget. 2015;6(5):3165–3177. doi:10.18632/oncotarget.3078
45. Shahab S, Rubens J, Kaur H, Sweeney H, Eberhart CG, Raabe EH. MEK inhibition suppresses growth of atypical teratoid/rhabdoid tumors. J Neuropathol Exp Neurol. 2020;79(7):746–753. doi:10.1093/jnen/nlaa042
46. Rubens JA, Wang SZ, Price A, et al. The TORC1/2 inhibitor TAK228 sensitizes atypical teratoid rhabdoid tumors to cisplatin-induced cytotoxicity. Neuro Oncol. 2017;19(10):1361–1371. doi:10.1093/neuonc/nox067
47. Oberlick EM, Rees MG, Seashore-Ludlow B, et al. Small-molecule and CRISPR screening converge to reveal receptor tyrosine kinase dependencies in pediatric rhabdoid tumors. Cell Rep. 2019;28(9):2331–2344.e8. doi:10.1016/j.celrep.2019.07.021
48. Suri A, Bailey AW, Tavares MT. Evaluation of protein kinase inhibitors with PLK4 cross-over potential in a pre-clinical model of cancer. Int J Mol Sci. 2019;20(9):2112. doi:10.3390/ijms20092112
49. Singh A, Lun X, Jayanthan A, et al. Profiling pathway-specific novel therapeutics in preclinical assessment for central nervous system atypical teratoid rhabdoid tumors (CNS ATRT): favorable activity of targeting EGFR-ErbB2 signaling with lapatinib. Mol Oncol. 2013;7(3):497–512. doi:10.1016/j.molonc.2013.01.001
50. Alimova I, Pierce AM, Harris P, et al. Targeting Polo-like kinase 1 in SMARCB1 deleted atypical teratoid rhabdoid tumor. Oncotarget. 2017;8(57):97290–97303. doi:10.18632/oncotarget.21932
51. Sredni ST, Bailey AW, Suri A, et al. Inhibition of polo-like kinase 4 (PLK4): a new therapeutic option for rhabdoid tumors and pediatric medulloblastoma. Oncotarget. 2017;8(67):111190–111212. doi:10.18632/oncotarget.22704
52. Sredni ST, Suzuki M, Yang JP, et al. A functional screening of the kinome identifies the Polo-like kinase 4 as a potential therapeutic target for malignant rhabdoid tumors, and possibly, other embryonal tumors of the brain. Pediatr Blood Cancer. 2017;64(11):e26551. doi:10.1002/pbc.26551
53. Messerli SM, Hoffman MM, Gnimpieba EZ, Bhardwaj RD. Therapeutic targeting of PTK7 is cytotoxic in atypical teratoid rhabdoid tumors. Mol Cancer Res. 2017;15(8):973–983. doi:10.1158/1541-7786.MCR-16-0432
54. Obaid H, Kannappan S, Gupta M, et al. In vitro investigation demonstrates IGFR/VEGFR receptor cross talk and potential of combined inhibition in pediatric central nervous system atypical teratoid rhabdoid tumors. Curr Cancer Drug Targets. 2020;20(4):295–305. doi:10.2174/1568009619666191111153049
55. Chakravadhanula M, Hampton CN, Chodavadia P, et al. Wnt pathway in atypical teratoid rhabdoid tumors. Neuro Oncol. 2015;17(4):526–535. doi:10.1093/neuonc/nou229
56. Studebaker AW, Hutzen B, Pierson CR, Shaffer TA, Raffel C, Jackson EM. Oncolytic measles virus efficacy in murine xenograft models of atypical teratoid rhabdoid tumors. Neuro Oncol. 2015;17(12):1568–1577. doi:10.1093/neuonc/nov058
57. Lee YE, Choi SA, Kwack PA, et al. Repositioning disulfiram as a radiosensitizer against atypical teratoid/rhabdoid tumor. Neuro Oncol. 2017;19(8):1079–1087. doi:10.1093/neuonc/now300
58. Golan H, Shukrun R, Caspi R, et al. In vivo expansion of cancer stemness affords novel cancer stem cell targets: malignant rhabdoid tumor as an example. Stem Cell Rep. 2018;11(3):795–810. doi:10.1016/j.stemcr.2018.07.010
59. Yang YP, Nguyen PNN, Ma HI, et al. Tumor mesenchymal stromal cells regulate cell migration of atypical teratoid rhabdoid tumor through exosome-mediated miR155/SMARCA4 pathway. Cancers (Basel). 2019;11(5):720. doi:10.3390/cancers11050720
60. Howard TP, Arnoff TE, Song MR, Giacomelli AO. MDM2 and MDM4 are therapeutic vulnerabilities in malignant rhabdoid tumors. Cancer Res. 2019;79(9):2404–2414. doi:10.1158/0008-5472.CAN-18-3066
61. Nakano Y, Takadera M, Miyazaki M, et al. Drug screening with a novel tumor-derived cell line identified alternative therapeutic options for patients with atypical teratoid/rhabdoid tumor. Hum Cell. 2021;34(1):271–278. doi:10.1007/s13577-020-00438-3
62. Morin A, Soane C, Pierce A, et al. Proteasome inhibition as a therapeutic approach in atypical teratoid/rhabdoid tumors. Neurooncol Adv. 2020;2(1):vdaa051. doi:10.1093/noajnl/vdaa051
63. Tran HM, Wu KS, Sung SY. Upregulation of protein synthesis and proteasome degradation confers sensitivity to proteasome inhibitor bortezomib in Myc-atypical teratoid/rhabdoid tumors. Cancers (Basel). 2020;12(3):752. doi:10.3390/cancers12030752
64. Howard TP, Oberlick EM, Rees MG. Rhabdoid tumors are sensitive to the protein-translation inhibitor homoharringtonine. Clin Cancer Res. 2020;26(18):4995–5006. doi:10.1158/1078-0432.CCR-19-2717
65. Shibui Y, Kohashi K, Tamaki A, et al. The forkhead box M1 (FOXM1) expression and antitumor effect of FOXM1 inhibition in malignant rhabdoid tumor. J Cancer Res Clin Oncol. 2021;147(5):1499–1518. doi:10.1007/s00432-020-03438-w
66. Daifu T, Mikami M, Hiramatsu H, Iwai A, Umeda K. Suppression of malignant rhabdoid tumors through Chb-M’-mediated RUNX1 inhibition. Pediatr Blood Cancer. 2020;68(2):e28789. doi:10.1002/pbc.28789
67. Marsh IR, Grudzinski J, Baiu DC, et al. Preclinical pharmacokinetics and dosimetry studies of (124)I/(131) I-CLR1404 for treatment of pediatric solid tumors in murine xenograft models. J Nucl Med. 2019;60(10):1414–1420. doi:10.2967/jnumed.118.225409
68. Krämer KF, Moreno N, Frühwald MC, Kerl K. BRD9 inhibition, alone or in combination with cytostatic compounds as a therapeutic approach in rhabdoid tumors. Int J Mol Sci. 2017;18(7):1537. doi:10.3390/ijms18071537
69. Michel BC, D’Avino AR, Cassel SH, et al. A non-canonical SWI/SNF complex is a synthetic lethal target in cancers driven by BAF complex perturbation. Nat Cell Biol. 2018;20(12):1410–1420. doi:10.1038/s41556-018-0221-1
70. Kim KH, Roberts CW. Mechanisms by which SMARCB1 loss drives rhabdoid tumor growth. Cancer Genet. 2014;207(9):365–372. doi:10.1016/j.cancergen.2014.04.004
71. Carugo A, Minelli R, Sapio L, et al. p53 is a master regulator of proteostasis in SMARCB1-deficient malignant rhabdoid tumors. Cancer Cell. 2019;35(2):204–220.e9. doi:10.1016/j.ccell.2019.01.006
72. Perla A, Fratini L, Cardoso PS, et al. Histone deacetylase inhibitors in pediatric brain cancers: biological activities and therapeutic potential. Front Cell Dev Biol. 2020;8:546. doi:10.3389/fcell.2020.00546
73. Kerl K, Ries D, Unland R, et al. The histone deacetylase inhibitor SAHA acts in synergism with fenretinide and doxorubicin to control growth of rhabdoid tumor cells. BMC Cancer. 2013;13:286. doi:10.1186/1471-2407-13-286
74. Muscat A, Popovski D, Jayasekara WS, et al. Low-dose histone deacetylase inhibitor treatment leads to tumor growth arrest and multi-lineage differentiation of malignant rhabdoid tumors. Clin Cancer Res. 2016;22(14):3560–3570. doi:10.1158/1078-0432.CCR-15-2260
75. Custers L, Khabirova E, Coorens THH. Somatic mutations and single-cell transcriptomes reveal the root of malignant rhabdoid tumours. Nat Commun. 2021;12(1):1407. doi:10.1038/s41467-021-21675-6
76. Hoffman MM, Zylla JS, Bhattacharya S, et al. Analysis of dual class I histone deacetylase and lysine demethylase inhibitor domatinostat (4SC-202) on growth and cellular and genomic landscape of atypical teratoid/rhabdoid. Cancers (Basel). 2020;12(3):756. doi:10.3390/cancers12030756
77. Sugimoto Y, Katsumi Y, Iehara T. The novel histone deacetylase inhibitor, OBP-801, induces apoptosis in rhabdoid tumors by releasing the silencing of NOXA. Mol Cancer Ther. 2020;19(10):1992–2000. doi:10.1158/1535-7163.MCT-20-0243
78. Knipstein JA, Birks DK, Donson AM, Alimova I, Foreman NK, Vibhakar R. Histone deacetylase inhibition decreases proliferation and potentiates the effect of ionizing radiation in atypical teratoid/rhabdoid tumor cells. Neuro Oncol. 2012;14(2):175–183. doi:10.1093/neuonc/nor208
79. Thiemann M, Oertel S, Ehemann V, et al. In vivo efficacy of the histone deacetylase inhibitor suberoylanilide hydroxamic acid in combination with radiotherapy in a malignant rhabdoid tumor mouse model. Radiat Oncol. 2012;7:52. doi:10.1186/1748-717X-7-52
80. Muscal JA, Thompson PA, Horton TM, et al. A phase I trial of vorinostat and bortezomib in children with refractory or recurrent solid tumors: a Children’s Oncology Group phase I consortium study (ADVL0916). Pediatr Blood Cancer. 2013;60(3):390–395. doi:10.1002/pbc.24271
81. Fouladi M, Park JR, Stewart CF, et al. Pediatric phase I trial and pharmacokinetic study of vorinostat: a Children’s Oncology Group phase I consortium report. J Clin Oncol. 2010;28(22):3623–3629. doi:10.1200/JCO.2009.25.9119
82. Su JM, Thompson P, Ou CN, et al. Phase 1 study of valproic acid in pediatric patients with refractory solid or CNS tumors: a children’s oncology group report. Clin Cancer Res. 2011;17(3):589–597. doi:10.1158/1078-0432.CCR-10-0738
83. Yan W, Herman JG, Guo M. Epigenome-based personalized medicine in human cancer. Epigenomics. 2016;8(1):119–133. doi:10.2217/epi.15.84
84. George RE, Lahti JM, Adamson PC, et al. Phase I study of decitabine with doxorubicin and cyclophosphamide in children with neuroblastoma and other solid tumors: a Children’s Oncology Group study. Pediatr Blood Cancer. 2010;55(4):629–638. doi:10.1002/pbc.22607
85. Margueron R, Reinberg D. The Polycomb complex PRC2 and its mark in life. Nature. 2011;469(7330):343–349. doi:10.1038/nature09784
86. Kim KH, Roberts CW. Targeting EZH2 in cancer. Nat Med. 2016;22(2):128–134. doi:10.1038/nm.4036
87. Alimova I, Birks DK, Harris PS, et al. Inhibition of EZH2 suppresses self-renewal and induces radiation sensitivity in atypical rhabdoid teratoid tumor cells. Neuro Oncol. 2013;15(2):149–160. doi:10.1093/neuonc/nos285
88. Kurmasheva RT, Sammons M, Favours E, et al. Initial testing (stage 1) of tazemetostat (EPZ-6438), a novel EZH2 inhibitor, by the pediatric preclinical testing program. Pediatr Blood Cancer. 2017;64(3):e26218. doi:10.1002/pbc.26218
89. Kurmasheva RT, Erickson SW, Earley E, Smith MA, Houghton PJ. In vivo evaluation of the EZH2 inhibitor (EPZ011989) alone or in combination with standard of care cytotoxic agents against pediatric malignant rhabdoid tumor preclinical models-A report from the Pediatric Preclinical Testing Consortium. Pediatr Blood Cancer. 2021;68(2):e28772. doi:10.1002/pbc.28772
90. Italiano A, Soria JC, Toulmonde M, et al. Tazemetostat, an EZH2 inhibitor, in relapsed or refractory B-cell non-Hodgkin lymphoma and advanced solid tumours: a first-in-human, open-label, phase 1 study. Lancet Oncol. 2018;19(5):649–659. doi:10.1016/S1470-2045(18)30145-1
91. Chi NS, Bourdeaut F, Laetsch TW et al. Phase 1 Study of Tazemetostat, an Enhancer of Zeste Homolog-2 Inhibitor, Pediatric Patients With Relapsed/Refractory Integrase Interacor 1-Negative Tumors. ASCO 2020.Available from: https://www.epizyme.com/wp-content/uploads/2021/06/EZH-102_ASCO-2020_Poster_Final.pdf.
92. Kohashi K, Oda Y. Oncogenic roles of SMARCB1/INI1 and its deficient tumors. Cancer Sci. 2017;108(4):547–552. doi:10.1111/cas.13173
93. Cimica V, Smith ME, Zhang Z, Mathur D, Mani S, Kalpana GV. Potent inhibition of rhabdoid tumor cells by combination of flavopiridol and 4OH-tamoxifen. BMC Cancer. 2010;10:634. doi:10.1186/1471-2407-10-634
94. Hashizume R, Zhang A, Mueller S, et al. Inhibition of DNA damage repair by the CDK4/6 inhibitor palbociclib delays irradiated intracranial atypical teratoid rhabdoid tumor and glioblastoma xenograft regrowth. Neuro Oncol. 2016;18(11):1519–1528. doi:10.1093/neuonc/now106.euro-oncology
95. Geoerger B, Bourdeaut F, DuBois SG, et al. A phase I study of the CDK4/6 inhibitor ribociclib (LEE011) in pediatric patients with malignant rhabdoid tumors, neuroblastoma, and other solid tumors. Clin Cancer Res. 2017;23(10):2433–2441. doi:10.1158/1078-0432.CCR-16-2898
96. Maris JM, Morton CL, Gorlick R, et al. Initial testing of the Aurora kinase A inhibitor MLN8237 by the Pediatric Preclinical Testing Program (PPTP). Pediatr Blood Cancer. 2010;55(1):26–34. doi:10.1002/pbc.22430
97. Venkataraman S, Alimova I, Tello T, et al. Targeting Aurora Kinase A enhances radiation sensitivity of atypical teratoid rhabdoid tumor cells. J Neurooncol. 2012;107(3):517–526. doi:10.1007/s11060-011-0795-y
98. Wetmore C, Boyett J, Li S, et al. Alisertib is active as single agent in recurrent atypical teratoid rhabdoid tumors in 4 children. Neuro Oncol. 2015;17(6):882–888. doi:10.1093/neuonc/nov017
99. Mossé YP, Fox E, Teachey DT. A phase II study of alisertib in children with recurrent/refractory solid tumors or leukemia: children’s oncology group phase I and pilot consortium (ADVL0921). Clin Cancer Res. 2019;25(11):3229–3238. doi:10.1158/1078-0432.CCR-18-2675
100. Davis SL, Ionkina AA, Bagby SM, Orth JD, Gittleman B. Preclinical and dose-finding phase I trial results of combined treatment with a TORC1/2 inhibitor (TAK-228) and Aurora A kinase inhibitor (alisertib) in solid tumors. Clin Cancer Res. 2020;26(17):4633–4642. doi:10.1158/1078-0432.CCR-19-3498
101. Upadhyaya S, Campagne O, Robinson GW. Phase II study of alisertib as a single agent in recurrent or progressive atypical teratoid rhabdoid tumors. J Clin Oncol. 2020;38(15_suppl):10542. doi:10.1200/JCO.2020.38.15_suppl.10542
102. Wedekind MF, Denton NL, Chen CY, Cripe TP. Pediatric cancer immunotherapy: opportunities and challenges. Paediatr Drugs. 2018;20(5):395–408. doi:10.1007/s40272-018-0297-x
103. Michot JM, Bigenwald C, Champiat S, et al. Immune-related adverse events with immune checkpoint blockade: a comprehensive review. Eur J Cancer. 2016;54:139–148. doi:10.1016/j.ejca.2015.11.016
104. Binnewies M, Roberts EW, Kersten K, et al. Understanding the tumor immune microenvironment (TIME) for effective therapy. Nat Med. 2018;24(5):541–550. doi:10.1038/s41591-018-0014-x
105. Hirata E, Sahai E. Tumor microenvironment and differential responses to therapy. Cold Spring Harb Perspect Med. 2017;7(7):a026781. doi:10.1101/cshperspect.a026781
106. Grabovska Y, Mackay A, O’Hare P, Crosier S, Finetti M. Pediatric pan-central nervous system tumor analysis of immune-cell infiltration identifies correlates of antitumor immunity. Nat Commun. 2020;11(1):4324. doi:10.1038/s41467-020-18070-y
107. Petralia F, Tignor N, Reva B, et al. Integrated proteogenomic characterization across major histological types of pediatric brain cancer. Cell. 2020;183(7):1962–1985.e31. doi:10.1016/j.cell.2020.10.044
108. Samstein RM, Lee CH, Shoushtari AN. Tumor mutational load predicts survival after immunotherapy across multiple cancer types. Nat Genet. 2019;51(2):202–206. doi:10.1038/s41588-018-0312-8
109. Gröbner SN, Worst BC, Weischenfeldt J, et al. The landscape of genomic alterations across childhood cancers. Nature. 2018;555(7696):321–327. doi:10.1038/nature25480
110. Yarmarkovich M, Maris JM. When cold is hot: immune checkpoint inhibition therapy for rhabdoid tumors. Cancer Cell. 2019;36(6):575–576. doi:10.1016/j.ccell.2019.11.006
111. Leruste A, Tosello J, Ramos RN, et al. Clonally expanded T cells reveal immunogenicity of rhabdoid tumors. Cancer Cell. 2019;36(6):597–612.e8. doi:10.1016/j.ccell.2019.10.008
112. Bourdeaut F, Thaku MD, Bergthold G, Karski E. ATRT-11. Marked response to Atezolizumab in a patient with rhabdoid tumor: a case study from the imatrix-atezolizumab trial. Neuro-Oncology. 2017;19(Suppl4):iv3. doi:10.1093/neuonc/nox083.010
113. Geoerger B, Zwaan CM, Marshall LV, et al. Atezolizumab for children and young adults with previously treated solid tumours, non-Hodgkin lymphoma, and Hodgkin lymphoma (iMATRIX): a multicentre phase 1–2 study. Lancet Oncol. 2020;21(1):134–144. doi:10.1016/S1470-2045(19)30693-X
114. Geoerger B, Kang HJ, Yalon-Oren M, et al. Pembrolizumab in paediatric patients with advanced melanoma or a PD-L1-positive, advanced, relapsed, or refractory solid tumour or lymphoma (KEYNOTE-051): interim analysis of an open-label, single-arm, phase 1–2 trial. Lancet Oncol. 2020;21(1):121–133. doi:10.1016/S1470-2045(19)30671-0
115. Hoppmann A, Williams AP, Coleman A, et al. Partial response to carboplatin, etoposide phosphate, and atezolizumab in a pediatric patient with high-grade metastatic tumor with rhabdoid and focal neuroendocrine features. Pediatr Blood Cancer. 2020;67(2):e28048. doi:10.1002/pbc.28048
116. Morrissey KM, Yuraszeck TM, Li CC, Zhang Y, Kasichayanula S. Immunotherapy and novel combinations in oncology: current landscape, challenges, and opportunities. Clin Transl Sci. 2016;9(2):89–104. doi:10.1111/cts.12391
117. Wang D, Quiros J, Mahuron K, et al. Targeting EZH2 reprograms intratumoral regulatory T cells to enhance cancer immunity. Cell Rep. 2018;23(11):3262–3274. doi:10.1016/j.celrep.2018.05.050
118. Goel S, DeCristo MJ, Watt AC, et al. CDK4/6 inhibition triggers anti-tumour immunity. Nature. 2017;548(7668):471–475. doi:10.1038/nature23465
119. Leruste A, Chauvin C, Pouponnot C, Bourdeaut F, Waterfall JJ. Immune responses in genomically simple SWI/SNF-deficient cancers. Nature. 2017;548(7668):471–475. doi:10.1038/nature23465
120. Majzner RG, Theruvath JL, Nellan A, et al. CAR T cells targeting B7-H3, a pan-cancer antigen, demonstrate potent preclinical activity against pediatric solid tumors and brain tumors. Clin Cancer Res. 2019;25(8):2560–2574. doi:10.1158/1078-0432.CCR-18-0432
121. Du H, Hirabayashi K, Ahn S, et al. Antitumor responses in the absence of toxicity in solid tumors by targeting B7-H3 via chimeric antigen receptor T cells. Cancer Cell. 2019;35(2):221–237.e8. doi:10.1016/j.ccell.2019.01.002
122. Theruvath J, Sotillo E, Mount CW, et al. Locoregionally administered B7-H3-targeted CAR T cells for treatment of atypical teratoid/rhabdoid tumors. Nat Med. 2020;26(5):712–719. doi:10.1038/s41591-020-0821-8
123. Chauvin C, Leruste A, Tauziede-Espariat A, et al. High-throughput drug screening identifies pazopanib and clofilium tosylate as promising treatments for malignant rhabdoid tumors. Cell Rep. 2017;21(7):1737–1745. doi:10.1016/j.celrep.2017.10.076
124. Wong JP, Todd JR, Finetti MA, et al. Dual targeting of PDGFRα and FGFR1 displays synergistic efficacy in malignant rhabdoid tumors. Cell Rep. 2016;17(5):1265–1275. doi:10.1016/j.celrep.2016.10.005
 © 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.