Back to Journals » ImmunoTargets and Therapy » Volume 14
Current Advances and Challenges in CAR-T Therapy for Hematological and Solid Tumors
Authors Zhang G ![]() , Bai M, Du H, Yuan Y
, Bai M, Du H, Yuan Y ![]() , Wang Y
, Wang Y ![]() , Fan W, Zhu H
, Fan W, Zhu H ![]() , Wu D, He P
, Wu D, He P ![]() , Xue B
, Xue B ![]()
Received 5 February 2025
Accepted for publication 16 June 2025
Published 27 June 2025 Volume 2025:14 Pages 655—680
DOI https://doi.org/10.2147/ITT.S519616
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Professor Michael Shurin
Gengtian Zhang,1,* Mengyao Bai,1,* Hanzhi Du,2 Yue Yuan,2 Yidan Wang,2 Weijing Fan,3 Huachao Zhu,2 Di Wu,2 Pengcheng He,2 Busheng Xue2
1Health Science Center, Xi’an Jiaotong University, Xi’an, 710061, People’s Republic of China; 2Department of Hematology, The First Affiliated Hospital of Xi’an Jiaotong University, Xi’an, 710061, People’s Republic of China; 3Department of Radiation Oncology, The First Affiliated Hospital of Xi’an Jiaotong University, Xi’an, 710061, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Pengcheng He, Department of Hematology, The First Affiliated Hospital of Xi’an Jiaotong University, No. 277, Yanta District, Xi’an, 710061, People’s Republic of China, Email [email protected] Busheng Xue, Department of Hematology, The First Affiliated Hospital of Xi’an Jiaotong University, No. 277, Yanta District, Xi’an, 710061, People’s Republic of China, Email [email protected]
Introduction: Chimeric antigen receptor (CAR) T-cell therapy has revolutionized the treatment of refractory hematological malignancies, yet significant challenges persist in extending its success to solid tumors. This review aims to provide a comprehensive overview of the current landscape and future perspectives of CAR-T therapy in both hematological malignancies and solid tumors.
Methods: A thorough literature search was conducted to identify relevant preclinical and clinical studies, as well as review articles, focusing on CAR-T therapy in various hematological malignancies and solid tumors. The collected information was synthesized to discuss the current applications, challenges, and strategies for improving CAR-T therapy in these settings.
Results: CAR-T therapy has demonstrated impressive clinical outcomes in treating certain hematological malignancies, such as B-cell lymphoma, leukemia, and multiple myeloma. However, the efficacy of CAR-T cells in solid tumors has been limited due to various obstacles, including tumor heterogeneity, immunosuppressive microenvironment, and off-tumor toxicities. Strategies to overcome these challenges involve advanced CAR designs, combination therapies, and novel approaches to CAR-T cell manufacturing and engineering.
Conclusion: While CAR-T therapy has revolutionized the treatment of some hematological malignancies, significant hurdles remain in extending its success to solid tumors. Continued research efforts focusing on improving CAR-T cell efficacy, safety, and accessibility will be crucial in unlocking the full potential of this innovative immunotherapeutic approach across a broad spectrum of cancer types.
Keywords: CAR-T, hematological malignancies, solid tumors, translational advances, clinical challenges
Introduction
Hematological malignancies and solid tumors represent two major categories of cancer, each encompassing multiple specific types and treatment approaches. Hematological malignancies originate from blood-forming tissues, such as bone marrow or immune system cells, and include leukemia, lymphoma, and myeloma. Leukemia develops due to uncontrolled proliferation of immature white blood cells, which impairs the production and function of normal blood cells.1 It is classified into acute types (such as acute lymphocytic leukemia, acute myeloid leukemia) and chronic types (such as chronic lymphocytic leukemia, chronic myeloid leukemia). Lymphoma and multiple myeloma originate from lymphatic system and plasma cells respectively, leading to tumor-mass effects, bone destruction and abnormal antibodies.
Treatment modalities include chemotherapy, targeted therapy and novel precision medicine approaches,2,3 which either eliminate rapidly dividing cancer cells directly or target-specific signaling pathways to kill tumor cells.4,5 While immunotherapy eradicates cancer cells through terminating the inhibited immune system or importing engineered immune cells.
Solid tumors arise from a variety of bodily tissues including epithelial, connective, and nervous tissues, manifesting as abnormal growths or masses in organs. Solid tumors comprise a diverse group of cancers, including breast cancer, lung cancer, and various sarcomas. Treatment strategies often involve a multimodal approach, incorporating surgery, radiotherapy, chemotherapy, targeted therapy, and immunotherapy. Surgical resection is the primary means for local entity tumors. Radiotherapy can destroy cancer cells and is often used in combination with surgery or as a standalone treatment.6 Chemotherapy uses drugs to target cancer cells but has multiple side effects. Targeted therapy is more tolerable and has become a standard treatment regimen. Immunotherapy enhances the immune system’s ability to recognize and attack cancer cells, with checkpoint inhibitors demonstrating significant efficacy.7,8
Current applications of CAR-T therapy demonstrate notable efficacy in hematologic malignancies with expanding indications, significant potential in solid tumors and autoimmune diseases, and preliminary feasibility in HIV/AIDS treatment. Concurrent advancements include leveraging CRISPR technology to develop universal CAR-T (UCAR-T) products, aiming to reduce manufacturing costs and enhance therapeutic accessibility.
Beyond the limitations (antigen escape, tumor heterogeneity, immunosuppressive tumor microenvironment, T cell exhaustion and CAR-T cell associated toxicities) observed in both hematologic malignancies and solid tumors, CAR-T cell therapy faces critical challenges in manufacturing and safety, including inadequate standardization of production processes.
In summary, as a cutting-edge medical technology, CAR-T cell therapy has broad prospects and potential in the treatment of multiple cancers.1
The Development Journey of CAR
The concept of a chimeric T cell receptor was first introduced in 1987, which integrates the variable domains (VH/VL) derived from an antibody with the constant domains derived from a T cell receptor (TCR).9,10 In 1989, a chimeric T cell receptor (cTCR) composed of the variable heavy and light chains of the anti-2,4,6-trinitrophenyl (TNP) antibody Sp6 was introduced, which were fused with the constant regions of the α and β TCR chains, respectively.9,11 Then, scFvR emerged, which is a fusion of a single-chain variable fragment (scFv) with the intracellular signaling domains of CD3ζ or FcεRIγ. It boasts higher vector delivery efficiency and can independently mediate cell activation signals. The double-chain cTLR and single-chain scFvR are recognized as the prototypes of modern chimeric antigen receptors (CARs).9,12
The first-generation CAR exhibited good anticancer activity in mouse models (Figure 1),13–15 but the therapeutic outcomes were not satisfactory in patients with ovarian cancer and metastatic renal cell carcinoma. The first-generation CAR has a relatively simple structure, leading to a short duration of action in vivo and an inability to provide sustained antitumor effects.16,17 Compared to the first-generation CAR, the second-generation CAR features an additional costimulatory domain in the intracellular signaling transduction region. The costimulatory domain is crucial for T-cell activation and functional regulation,18 typically consisting of CD28, CD27,19 ICOS,20 4–1BB, among others, with CD28 and 4–1BB being the most common. The costimulatory signal enhances the proliferation of CAR-T cells and increases the secretion of TNF-α and IL-2.21 The third-generation CAR-T cells introduce a more complex CAR structure on the surface of T cells, with their intracellular signaling transduction region containing two costimulatory domains. Multiple costimulatory domains can activate more signaling pathways, significantly enhancing the proliferative capacity of T cells.22 In particular, the third-generation anti-CD30 CAR-T cells have demonstrated robust antitumor activity in tumor xenograft models and miner side effects or potential risks of tumorigenesis was observed.23 The fourth-generation CARs and the next-generation CARs adopt more sophisticated designs in their structures, incorporating molecules to endow CAR-T cells with additional characteristics and functions.24 These CARs can be classified into two types: cells utilizing secretory molecules (such as bispecific T cell engagers (BiTEs), bispecific antigens, pro-inflammatory cytokines, agonists, and blockers of T cell receptors) and cells utilizing membrane receptors (such as chemokine receptors and switch receptors). A novel bispecific CAR with a circular architecture that is capable of simultaneously targeting both CD19 and CD22. This design demonstrates enhanced therapeutic efficacy against B-cell malignancies.25 These advancements reduce the likelihood of uncontrollable inflammatory side effects and equip CAR-T cells with potent tumor-killing activity and the ability to regulate the immune system, enabling them to more effectively eliminate tumor cells. Furthermore, these improvements can induce CAR-T cells to more efficiently enter tumor sites and enhance their ability to infiltrate tumors.26
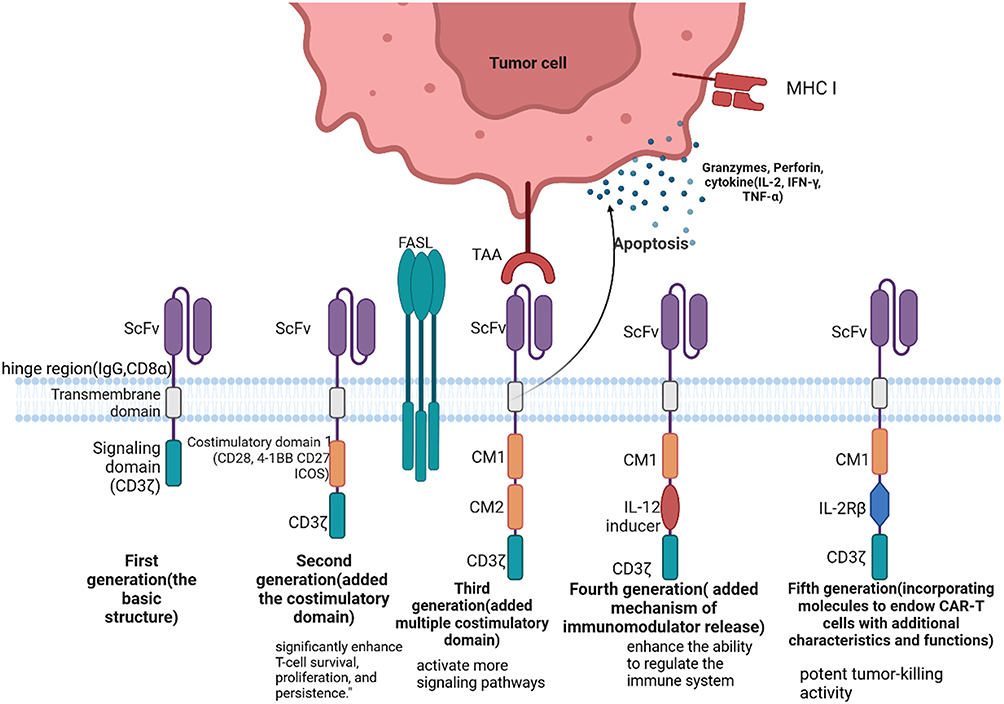 |
Figure 1 The development of CAR and CAR structure. scFv is Single-Chain Variable Fragment. CM is Chimeric Module. Created in BioRender. Bai, M. (2025) https://BioRender.com/6vdqf71. |
CAR Structure and Anti-Tumor Mechanisms
The Specific Structure of CAR
CARs feature a modular construct comprising four essential parts: an antigen recognition and binding domain, a hinge region, a transmembrane domain, and an intracellular signaling domain. Each of these components serves a unique purpose, and achieving an optimal CAR design involves numerous variations in the arrangement and composition of these protein domains.27
The antigen recognition and binding domain of CAR is specifically the single-chain variable fragment (scFv),28 which is composed of the light chain variable region (VL) and heavy chain variable region (VH) of a monoclonal antibody, connected by a polypeptide linker. It is located in the extracellular portion of CAR and serves as the foundation for CAR’s specific binding to tumor antigens, thereby endowing T cells with the ability to specifically recognize and bind to target antigens. The scFv is a small molecular fragment isolated from antibodies (like anti-GXM monoclonal antibody from hybridoma 18B729) that retains the specific binding capacity to antigens. The interaction between the two structural domains of VL and VH, as well as the relative position of the complementarity-determining regions (CDRs), both influence the affinity of CAR molecules for their target antigens.30 Additionally, the coding sequence of scFv determines its amino acid composition, which in turn affects its three-dimensional structure and binding capacity to target antigens.31 In the design of CAR, by selecting scFvs that exhibit sufficient affinity for tumor cell surface receptors while avoiding off-target effects targeting normal cells, the accuracy and safety of CAR can be ensured.
The hinge region, together with the antigen recognition and binding domain, constitutes the extracellular portion of CAR. Specifically, the hinge is a peptide segment that links the antigen recognition domain (typically scFv) and the transmembrane domain. It allows CAR to be more flexible extracellularly.32 Most hinge regions of CAR are derived from the hinge of IgG or the extracellular domain of CD8-α/CD28. By modifying the hinge region, there are certain benefits to CAR-T cells. For instance, the hinge-modified area can mitigate the cleavage of the extracellular portion of CAR, thereby enhancing the stability of CAR-T cells. Additionally, it can promote the expansion of CAR-T cells and improve the binding affinity between CAR and target antigens (such as CD70).33
The transmembrane domain (TM) is a crucial component of the structure and function of chimeric antigen receptor T-cells (CAR-T cells). It primarily serves as an anchoring structure that connects the extracellular domain and the intracellular domain, embedding the CAR into the cell membrane.34 It typically originates from transmembrane receptor proteins such as CD8-α, CD28, ICOS, or members of the tumor necrosis factor receptor superfamily (TNFRS). The primary function of the transmembrane domain is to conduct activation signals, transmitting signals received by the extracellular region of the CAR to the intracellular region, triggering the activation of T-cells. The choice of transmembrane domain influences the activation signals transmitted to the intracellular domain, thereby affecting the activation and function of CAR-T cells.34 Compared to cells carrying the CD8 TM or the 4–1BB TM, CAR-T cells with the ICOS TM exhibit enhanced proliferation and persistence in vivo.35
The intracellular signaling region of CARs primarily includes the signal transduction domain (such as CD3ζ) and costimulatory domains (such as CD28, 4–1BB). The signal transduction domain is responsible for conducting signals generated after antigen binding into T cells,30 triggering their activation and function. Costimulatory molecules can enhance T cells’ responsiveness to activation signals, and different combinations of costimulatory domains can affect the selectivity and function of CAR-T cells.36,37
Killing Mechanisms of CAR-T Cells
When CAR-T cells come into contact with tumor cells, the CARs on their surface specifically recognize and bind to the corresponding antigens on the surface of tumor cells. Once the CAR binds to the antigen on the tumor cell surface, the signaling pathway within the CAR-T cell is activated, and the immunoreceptor tyrosine-based activation motifs (ITAMs) within CD3ζ undergo phosphorylation. This triggers downstream signaling pathways such as MAPK and NF-κB, converting the externally recognized signal into an intracellular activation signal. Meanwhile, the costimulatory domain binds to its corresponding ligand, delivering costimulatory signals into the T cell to provide the secondary signal for T cell activation. This promotes sustained T cell proliferation and the release of cytokines. These cytokines not only enhance T cell infiltration into tumor tissues but also recruit other immune cells in the body (such as macrophages and other innate immune cells) to collectively eliminate tumor cells. Upon binding, a non-classical immune synapse (IS) forms on the surface of CAR-T cells.38 After the formation of the IS, CAR-T cells employ two primary mechanisms to eliminate tumor cells. CAR-T cells release perforin and granzymes that rapidly disrupt the tumor cell membrane and intracellular organelles, leading to their rapid death.38,39 The second is an indirect immune-mediated pathway, where CAR-T cells release cytokines such as interferon-γ (IFN-γ), which recruit and activate other immune cells to cooperatively attack tumor cells, resulting in the progressive destruction of tumor tissue.40 Additionally, upon IS formation, FasL on the surface of CAR-T cells engages with Fas receptors on the surface of tumor cells, triggering the Fas-mediated apoptotic pathway, which further contributes to tumor cell death.41,42
The Determinants of CAR Functionality
The affinity of the scFv for the target antigen is a fundamental determinant of CAR functionality. High-affinity scFv can more effectively recognize and bind to target antigens, thereby enhancing the recognition and killing ability of CAR-T cells towards tumor cells.43 For instance, high-affinity ROR1-CAR can more effectively recognize and bind to target antigens, demonstrating potent killing ability against tumor cells both in vitro and in vivo, and showing favorable safety and tolerability profiles in clinical trials.44 In certain situations, T cells that transiently express high-affinity CARs (such as through mRNA electroporation technology) can also maintain effective antitumor activity. This method helps to reduce the risk of long-term myeloid toxicity while maintaining the effectiveness of the treatment.45 The results can be supported when the antigen density on target cells is low. However, when the antigen density on target cells is high, increasing the affinity of CAR does not yield any benefits, even CARs with low affinity can make T cells more easily activated by high-density EGFR.46 This indicates that target antigen also has a certain impact on function of CAR. Additional research has found that integrating different binders such as FMC63 and SJ25C1 onto CARs can regulate the affinity of the CARs, enabling CAR-T cells to target tumor cells with high antigen density while avoiding recognition of healthy cells with low antigen density. This strategy can provide new insights and methodologies for optimizing CAR-T cell therapy.47 In addition, the charge density of CARs, particularly the positively charged patches (PCP) on their antigen-binding domains, exerts a significant influence on CAR functionality. The functional adaptability of CAR-T cells is intimately linked to the intensity of tonic signaling, which is modulated by the PCP on the surface of the CAR antigen-binding domain. Insufficient tonic signaling impairs the persistence of CAR-T cells, whereas excessive tonic signaling leads to CAR-T cell exhaustion.48,49 The functionality of CAR-T cells is also affected by their antigen-binding domains. CARs with different antigen-binding domains, such as 2H1-GXMR-CAR and 18B7-GXMR-CAR, exhibit distinct affinities, specificities, and binding kinetics towards the same antigen (GXM). These variations subsequently influence the recognition efficiency and targeting ability of CAR-T cells.43 Modification in the hinge region, transmembrane domain, and costimulatory domain also confer certain benefits to CAR expression.50 Studies have demonstrated that replacing the IgG4 molecule with the CD8α molecule as the hinge/transmembrane domain can improve GXMR-CAR expression. Additionally, the CD8-α-hinge/transmembrane domain plays a crucial role in robust signaling conduction by GXMR-CAR. The use of the 4–1BB molecule as a costimulatory domain can enhance the intensity of signaling.51 Another study has shown that CAR-T cells containing the CD8α hinge region and transmembrane domain exhibit lower levels of cytokine production and antigen-induced cell death (AICD) compared to CAR-T cells containing the CD28 hinge region and transmembrane domain.52
Comparison of CAR-T Cell Therapy and Traditional Treatments
For relapsed or refractory (R/R) large B-cell lymphoma (LBCL), the traditional second-line standard treatment (high-dose chemotherapy + autologous stem cell transplantation, ASCT) has limited efficacy. Studies have shown that CAR-T therapy demonstrates superior efficacy in patients who relapse or are refractory within 12 months, significantly improving the objective response rate (ORR), complete response (CR) rate, prolonging event-free survival (EFS) and progression-free survival (PFS), ultimately enhancing overall survival (OS), and exhibiting good safety.53 Gastric cancer has a poor prognosis and is prone to drug resistance with traditional therapies, while CAR-T therapy demonstrates stronger efficacy and manageable toxicity,54,55 and the insertion of a suicide gene can further enhance safety.56 Acute myeloid leukemia (AML) has high toxicity and is prone to drug resistance with traditional chemotherapy, whereas CAR-T (eg, AdCAR-T) offers more precise targeting.57 In multiple myeloma (MM), the high targeting specificity of CAR-T therapy against BCMA reduces damage to normal cells.58 Moreover, CAR-T therapy achieves long-term remission in B-cell malignancies that are refractory to conventional therapies.59 Traditional chemotherapy for mantle cell lymphoma (MCL) is gradually being replaced by targeted therapies (including CAR-T).60
Achievements of CAR-T Therapy in Hematological Malignancies
B Cell Malignancy
Currently, CD19 and B-cell maturation antigen (BCMA) are most common targets in CAR-T therapy, particularly in hematological malignancies. A series of studies focusing on anti-CD19 and anti-BCMA have demonstrated remarkable success in treating B cell malignancies.2 Anti-CD19 CAR-T cells have derived unprecedented success in patients with Relapsed/Refractory B cell malignancies. As a result, the FDA approved 6 kinds of CAR-T cell products on the clinical treatments for these patients.47,61 SJ25C1 is the first CD19 scFv proved to be feasible and efficient and it has been used to treat refractory acute lymphoblastic leukemia.47 To date, the anti-CD19 CAR-T cell products have derived impressive outcomes in treating B-ALL, a Phase I trial among children and young adult patients conducted by the NCI showed that 62% patients achieved a complete response (CR) and 90.3% of them were minimal residual disease (MRD)-negative. Furthermore, the clinical trial also demonstrated that consolidative alloHSCT after CAR-T treatment provided prolonged disease control, indicating CAR-T a perspective therapy before HSCT.62
Anti-CD19 CAR-T cells also achieved a rapid and durable response in the patients with non-Hodgkin’s Lymphoma (NHL) and it is gradually regarded as a standard treatment for R/R aggressive NHL after two or more lines of therapy.63
Except for CD19, CD20 is also a prospective target in treating B-cell lymphomas. CD20 is overexpressed in 90% B-cell lymphomas,64 and it is reported to be in higher phosphorylation in malignant B cells. Therefore, it is an ideal target in treating NHL and anti-CD20 CAR-T therapy has already achieved some results in clinical trials. However, some patients eventually relapsed after the therapy.65
Similar to CD19, CD22 is expressed in most B-ALL, making it a promising target for future therapies. Anti-CD22 CAR-T cell products may serve as effective treatments following the failure of anti-CD19 CAR-T therapies. A single-center, dose-escalation Phase I clinical trial involving 58 participants with ALL demonstrated that 70.2% of patients achieved a complete response (CR), with 87.5% of those being MRD-negative after infusion. Additionally, the CAR-T cells exhibited strong in vivo expansion during treatment. Another trial involving 21 participants who received anti-CD22 CAR-T cells found that 17 of them achieved a CR, with 9 being MRD-negative. Notably, an engineered 4–1BB costimulatory anti-CD22 CAR-T, whose scFv is constructed with VH and VL linked by a short amino acid sequence, was observed to form a significantly stronger immunological synapse and demonstrated enhanced cytotoxicity in a mouse model of ALL. This finding suggests a promising approach to enhance the efficacy of CAR-T therapy against ALL by modifying the CAR structure.66–69 Anti-CD22 CAR-T cell product may become an effective therapy after the failure of anti-CD19 CAR-T therapy.66–68
Other targets treating B cell malignancies, such as CD137 releasing chronic lymphocytic leukemia (CLL), CD30 on HL,70 CD23 and ROR1 treating B cell lymphomas,69,70 are also under investigation and have achieved some results as well.
CD7 Targeted CAR-T and Other Targets in Treating T Cell Malignancy
T cell malignancies are aggressive hematological cancers that often result in poor prognosis.71 Nelarabine-based treatment is commonly used for T-ALL and T-LBL, however, many patients experience relapse following this therapy,72–74 so more options are in urgent need. CD7 is expressed in over 95% T-ALL and T cell lymphoma, which makes it a promising target for CAR-T therapy (Table 1). However, due to the expression of CD7 in normal T cell, anti-CD7 CAR-T cell products may induce GvHD, fratricide and allo-rejection.75,76 Fortunately, CD7 does not play a crucial role in T cell development and function, suggesting that strategies such as deleting the CD7 gene or blocking its expression on the cell surface could help mitigate these risks.77,78 Hence, deleting the gene of CD7 or blocking its expression on cell surface might be feasible strategies to avoid fratricide and T cell aplasia.75–78 Furthermore, Zhang’s research indicates that compared to autologous anti-CD7 CAR-T therapy, patients treated with allogeneic products show a better prognosis and lower levels of CRS, ICANS and GvHD.79
 |
Table 1 Prospective Targets and Products for T Cell Malignancies and Clinical Trials Conducted Till Now |
Other observed targets, such as CD5, CD37, CCR4 and TRBC1 have also shown promising results in the treatment of T cell malignancies.84
Targeting BCMA as Well as Other Potential Targets in Treating Multiple Myeloma
Multiple myeloma (MM) is a plasma cell malignancy characterized by hypercalcemia, marrow failure and the overexpression of M-protein.85–87 Over the past few decades, proteasome inhibitors, which interfere with the intracellular machinery of misfolded protein disposal via inhibiting the ubiquitin-proteasome system,88 have proven effective in treating MM.89 Additionally, other approaches, such as immunomodulatory drugs and monoclonal antibodies, have also shown promise.90 Unfortunately, many patients suffer from R/R MM, highlighting the urgent need for more effective treatments.
BCMA, a member of tumor necrosis factor receptor (TNFR) superfamily,91,92 is highly expressed in malignant plasma cells compared to normal bone marrow-derived cells (BMMCs), making it an excellent target for CAR-T therapy.91 So far, the FDA has approved two anti-BCMA CAR-T products for treating R/R MM.87 Idecabtagene vicleucel (Ide-cel) is the first autologous targeted BCMA CAR-T cell products approved by FDA and EMA on the treatment of R/R MM.93 Ide-cel is a second-generation CAR-T cell product with 4–1BB as the costimulatory domain.94 A clinical trial involving 128 patients who were infused with Ide-cel after at least three prior treatment regimens that reported that 94 patients exhibited a response, with 42 achieving a complete response. CAR-T cells were detectable in vivo in 36% participants 12 months after infusion. However, these T cells appear to be ineffective in preventing recurrence.95 In another Phase III clinical trial, patients treated Ide-cel demonstrated a higher response rate compared to the standard treatment group (71% versus 42%), as well as complete response rate. Additionally, the progression-free survival of Ide-cel group was significantly longer than standard group (13.3 months compared to 4.4 months). Intriguingly, while the incidence of adverse events was higher in the Ide-cel group, the rate of infections did not exceed that of the standard treatment group.96 Although BCMA loss is rare, elevated levels of soluble BCMA are closely associated with recurrence following Ide-cel treatment.94 Alternatively, adding PI3K inhibitor bb007 during the ex vivo culture of Ide-cel may induce an elevation of memory-like T cells, potentially enhancing clinical responses.97 Cilta-cel, another anti-BCMA CAR-T therapy that utilizes two BCMA-targeting single domain antibodies to form its scFv, shows improved avidity and persistence.98,99 A clinical trial suggested that 97% of patients responded to treatment, with 65% achieving a complete response.98 Additionally, CARTITUDE-1, another clinical trial of Cilta-cel, showed a similar result: 7 out of 20 patients were MRD negative and ORR was 60%.99 Consequently, Cilta-cel achieved remarkable outcomes in the treatment patients with R/R MM.
In addition to viral vector CAR-T products, non-viral vector cell therapies are also under investigation. P-BCMA-101 is an anti-BCMA cell product using piggyBac™ (PB) DNA Modification System instead of a viral vector. This change brought a higher proportion of stem cell memory T phenotype, greater efficacy, and a lower incidence of CRS.100
Unfortunately, while BCMA-targeted CAR-T therapy demonstrates significant affinity and antitumor activity, some patients still experience relapse due to BCMA loss or down-regulated expression.101,102 G protein-coupled receptor, class C group 5 member D (GPRC5D) is found to be highly expressed in MM cells but shows low or no expression in normal B cells, T cells and NK cells through immunohistochemical analyses.101 In vivo experiment involving mice injected with OPM2-ffLuc cells followed by GPRC5D CAR-T cells indicated that GPRC5D CAR-T cells significantly increased survival. Notably, primary T cells modified with GPRC5D (109) scFv exhibited enhanced in vivo activity, providing a promising approach to screen CAR.102 MCARH109, which is composed of a humanized GPRC5D scFv, 4–1BB costimulatory domain and CD3ζ signaling domain, demonstrated superior antitumor activity in a clinical trial, with 71% of patients showing a response at all doses and 59% achieving a partial response or better. However, two patients experienced rare persistent cerebellar disorder, necessitating further research. Furthermore, the First Affiliated Hospital of Zhejiang University School of Medicine conducted a first-in-human, single-center, single-arm, Phase 1 trial with GRC5D CAR-T cells (OriCAR-017). All ten patients showed an overall response, four of whom achieved a very good partial response, with no neurological toxicity detected during the dose escalation stage.103
Other potential CAR-T targets, including CD38, CD138, SLAMF7, CD229 and integrin β7, are also being investigated for the treatment of MM.2,103
Achievements of CAR-T Therapy in Solid Tumors
CAR-T therapy in solid tumors presents a distinct set of challenges and opportunities. The investigation of CAR-T therapy in solid tumors such as neuroblastoma, glioblastoma, and various carcinomas is an active area of research, with both preclinical and clinical studies paving the way for potential advancements in cancer treatment.104
GD2 is the most extensively targeted antigen in CAR-T cell therapy for solid tumor like neuroblastoma (Table 2). A third-generation CAR-T cell product has demonstrated excellent response in a phase 1 trial involving patients with neuroblastoma and osteosarcoma; however, all patients eventually experienced disease progression.105 Transcriptomic analysis revealed that elevated levels of myeloid-derived suppressor cells (MDSC) may hinder CAR-T expansion. Furthermore, gene set enrichment analysis (GSEA) indicated that in patients with effective CAR-T expansion, non-classical monocytes significantly increased. These findings suggest that enhancing the effectiveness of anti-GD2 CAR-T cell product could be achieved through myeloid-based interventions.105 Del Bufalo et al developed a GD2-redirected CAR-T (GD2-CART01) utilizing CD28 and 4–1BB to treat neuroblastoma. They also incorporated the iCaspase9 gene as a safety switch to avoid mitigate potential toxicity. Following infusion, 9 out of 27 patients demonstrated a response, with an overall survival rate of 66% at three years among those who responded.106 Notably, CD8+ CAR-T cells predominated in the peripheral blood, and no exhausting markers were detected.107 In another study, a fourth-generation CAR-T (4SCAR-T) exhibited significant anti-tumor activity against glioblastoma. MRI scans demonstrated tumor size reduction in four patients following CAR-T cell infusion. Researchers found that there was an increase in CD8+T cells within tumor tissue compared to pre-infusion levels. Interestingly, counts of CD163+ M2 macrophage decreased in tumor specimens’ post-infusion, indicating that 4SCAR-T may be reprogramed the immunosuppressive tumor environment mediated by M2 macrophages.108 Additionally, an epidermal growth factor receptor variant III (EGFRvIII)-directed CAR-T cell product demonstrated effective anti-glioblastoma activity. Following CART-EGFRvIII infusion, there was a significant increase in both the number and clonotypic of tumor-infiltrating T cells. The infusion product’s clone constituted a substantial proportion of the tumor-infiltrating T cells, as confirmed by RNAscope ISH analysis. Moreover, levels of immunosuppressive molecules such as PD-1 increased markedly compared to pre-infusion levels, offering new insights into the drug resistance mechanisms associated with CAR-T therapy.109
 |
Table 2 Prospective Targets and Products for Solid Tumors and Clinical Trials Conducted Till Now |
For breast cancer, particularly triple-negative breast cancer (TNBC), CAR-T cell therapy has achieved some outcomes as well. Zhao et al developed an AXL -targeted CAR-T cell that expresses IL-7 receptor (AXL-CAR-T.C7R).106 In vitro co-culture with AXL-positive TNBC cells demonstrated increased expression of activated molecules, enhanced cell proliferation, and elevated levels of cytokines release compared to AXL-CAR-T. These findings suggest that C7R can effectively activate CAR-T cells in vitro.106 Furthermore, researchers found that AXL-CAR-T.C7R group had a higher CD8:CD4 ratio, resulting in improved treatment efficacy. Additionally, the C7R group had an increased number of tumor-infiltrating T cells, contributing to a stronger antitumor response.106 Another study developed a MUC1-targeted CAR-T cell that incorporates CD28 and CD3ζ (MUC28z), demonstrating high antigen specificity and potent cytotoxicity. After co-culture with HCC70 cells, there was a significant increase in the release of IFN-γ and granzyme B. Notably, the additional of an IFN-γ antibody during co-culture and led to a decrease in tumor lysis ability, indicating that the cytotoxicity of MUC28z partially depended on IFN-γ.110 Moreover, in vivo xenografts models demonstrated that MUC28z remarkably restricted the growth of HCC70 tumors compared to the control. Researchers also observed another interesting phenomenon: CD11c expression was upregulated following the recognition of tMUC1 by MUC28z, suggesting the expansion of tMUC1-targeted CTL. Further experiments may focus on the elucidating the specific regulatory mechanisms involved.110
Claudin18.2 (CLDN18.2) is the most widely used targeted antigen in CAR-T therapy for gastric cancer (GC).111,126 Qi et al developed a second-generation CLDN18.2-targeted CAR-T therapy (CT041), which achieved remarkable results in clinical trials, with 83.3% of patients experiencing tumor regression after infusion.112 Intriguingly, they found that a lower ratio of CD45RA+/CCR7− T cells among the infused CAR-T cells may be associated with longer progression-free survival (PFS). Furthermore, CAR-T expansion in ascites may aid in eliminating diffuse peritoneal metastatic lesions.111 Another study introduced a bi-specific Trop2/PD-L1 CAR-T cell, which demonstrated strong antitumor activity against Trop2+/PD-L1+ BGC823 cells and resulted in more cytokines release.112 Zhang et al. Conducted experiments using NKG2D-targeted CAR-T cells in combination with DKK1 inhibitors. The inhibition of DKK1 significantly enhanced the antitumor activity and persistence of NKG2D CAR-T cells by up-regulating the expression of NKG2DL in gastric cancer cell lines and tumor tissues, while also reducing the numbers of myeloid-derived suppressor cells (MDSCs) M2 macrophages. Additionally, DKK1 inhibition can induce a “hot” tumor microenvironment and increase the number of tumor-infiltrating T cells, suggesting an enhanced antitumor activity.113 In addition to claudin18.2 and HER2, FOLR1 is expressed in over a third of gastric cancer patients, making it a promising target in treating advanced GC. A Korean team firstly constructed a FOLR1-targeted CAR-T cell product. Co-culture experiment showed an improved cytotoxicity and higher cytokines release. Furthermore, they also concealed that FOLR CAR-T lyses GC cells through granzyme B mostly and apoptotic proteins in GC cells increased after co-culture.114 A recent study focused on a mesothelin-targeted CAR-T cell with IL-15/IL-15Rα (CAR-ss-T) inserted to the extracellular region of CAR showed strong killing activity and cytokines production in vitro. Interestingly, the cytotoxic effect of CAR-ss-T was enhanced following GLIPR1 knockdown, which also inhibited GC cell proliferation and migration. In the follow-up studies, inserting the gene segment of GLIPR1 antibody into the CAR structure may be a promising approach.115
An engineered cell-surface GRP78 targeted CAR-T cell (Pep42-BBZ CAR-T) demonstrated potent cytotoxic against A549 and H1299 lung cancer cell lines in vitro.116 In addition, in vivo experiment indicated that CAR-T cells effectively infiltrated tumors and strongly inhibited the proliferation of cancer cells.116 Moto et al constructed a CD98 heavy chain targeted CAR-T using mAb R8H283 to treat non-small cell lung cancer (NSCLC), resulting in a reduced tumor burden in xenograft model. Intriguingly, R8H283 appeared to specifically target NSCLC cells exhibiting high levels of CD98hc, without affecting normal monocytes and lymphocytes. Further investigation is needed to determine whether this specific response is associated with the change in CD98hc glycosylation on the surface of NSCLC cell.117 In a ROR1-targeted CAR-T experiment, researchers constructed a 3D tumor model using the SISmuc platform and reported strong cytotoxic activity of ROR1-targeted CAR-T cells.118 Additionally, ligand-based CAR-T cell product has been shown to be safe and effective against NSCLC.119 A recent study reported a ligand-based anti-EGFR CAR-T for treating NSCLC, where in researchers engineered two EGFR-targeted CAR-T (EGF-CAR and TGFα-CAR) with piggyBac transposon system. Both CARs exhibited effective expansion, with naive or stem cell-like T cells comprising over 99% of the population, indicating a favorable memory phenotype. The EGF-CAR displayed robust cytotoxicity in co-culture experiments, and further research suggested that longer spacers may enhance the efficacy of anti-EGFR ligands. Furthermore, the study indicated that EGF-CAR co-expressed with TIM-3 showed improved expansion and antitumor activity, positioning TIM-3 as a potential target for research into immunosuppressive markers.119 Small cell lung cancer (SCLC) represents an aggressive subtype of lung cancer, often associated with poor prognosis and limited treatment options.120,121,127 Delta-like ligand 3 (DLL3) is the most widely studied target for SCLC. SCLC-targeted CAR-T cells presented a high efficacy with a low toxicity. Engineered T cells completely killed cancer cells even with low DLL3 density in vivo.127 AMG 119 is the first DLL3-targeted CAR-T therapy, exhibiting robust antitumor activity and low toxicity in clinical trial. Moreover, cyto-dynamics revealed a strong expansion and persistence of these cells in patients.120 Nie et al constructed a multi-chain chimeric antigen receptor targeting DLL3 (DLL3-DT CAR-T). They utilized DAP12 as the activation domain instead of CD3ζ and introduced TREM1 as a signaling chain linking the scFv. The DT CAR-T exhibited a larger proportion of Tcm and pathway analysis indicated that these cells were activated through the phosphorylation of ZAP70 and SYK, similar to NK cells and myeloid cells, which may contribute to their strong resistance against TME and enhanced cytotoxicity.121
Other solid tumors, such as liver metastases and colorectal cancer, are characterized by complex microenvironments and antigenic heterogeneity. In response, scientists have developed CAR-T cells targeting specific antigens like CEA,122 glypican-3,123 GUCY2C and TAG-72,124,125 each of which is demonstrated overexpressed in certain carcinoma subtypes.
In summary, the application of CAR-T therapy in solid tumors, including neuroblastoma, glioblastoma, and various carcinomas, is a rapidly evolving field marked by both promise and complexity. Preclinical studies have laid the ground for understanding of the mechanisms by which CAR-T cells can target tumor-specific antigens, providing valuable insights into potential therapeutic strategies. Clinical trials are critical in translating these findings to benefit patients, necessitating careful consideration of safety, efficacy, and the unique challenges posed by each tumor type. As research progresses, the adaptation and refinement of CAR-T therapy holds the potential to significantly impact the treatment landscape for patients with solid tumors, offering hope for improved outcomes in cancers that have traditionally been difficult to treat.
Limitations and Potential Strategies for Overcoming Challenges in Therapy for Hematological Malignancies and Solid Tumors
Despite significant advancements in cancer treatment, CAR-T therapy continues to encounter unique and formidable challenges, primarily due to the complex biological nature of these tumors.
Antigen Escape and Tumor Heterogeneity
Antigen escape refers to the phenomenon in which tumor cells downregulate or completely lose the expression of the antigens targeted by CAR-T cells. This evasion strategy enables tumor cells to survive under immune pressure exerted by the therapy, leading to relapse in many blood cancer patients following CAR-T infusion. A similar phenomenon has been observed in the treatment of solid tumors.128 For instance, downregulation of IL13Ra2 has been observed in CAR-T therapies targeting glioblastoma.129 To address antigen escape, one potential solution is to target multiple antigens simultaneously or to modify CAR-T cells to recognize more than one antigen. For example, a CRISPR/Cas9-engineered dual-targeted CAR-T that targets CD19 and CD22 has shown to provide a more robust and reliable mechanism for identifying and destroying cancer cells in the treatment of ALL.130 Preclinical and clinical studies have demonstrated enhanced efficacy and persistence with this approach while effectively mitigating CD19 escape.130 Furthermore, combining CAR-T therapy with BiTEs represents another promising strategy. Engineered CAR-T cells can secrete BiTEs, which enhance the anti-tumor response of CAR-T. CAR-T cells that secrete BiTEs exhibited superior cytotoxicity and persistence against glioblastoma. In addition, BiTEs selectively promote the expansion of TEM and help reverse T cell exhaustion. Given these encouraging outcomes, targeting multiple antigens and incorporating BiTEs not only enhances anti-tumor activity but also helps prevent antigen escape and relapse.131
Another significant challenge in cancer treatment is tumor heterogeneity.132 Solid tumors are composed of a diverse mixture of cell types, each potentially possessing different genetic and phenotypic characteristics. This heterogeneity extends to the expression of antigens on the surfaces of tumor cells that CAR-T therapies are designed to recognize. Consequently, the uniform expression of a single target antigen, which is prevalent in blood cancers, is rarely observed in solid tumors. The variability in antigen expression complicates the identification of a single or even a few target antigens that CAR-T cells can effectively target, making it one of the main factors associated with immune escape.132,133 An innate-like NKp30+CD8+CAR-T exhibited enhanced killing capacity against tumor heterogeneity. In experiments involving tumors with HER2 heterogeneity, this engineered CAR-T demonstrated strong efficacy in killing target cells, even in the absence of HER2 expression. Thus, NK-like T cells expressing CARs represent a promising approach to tackle tumor heterogeneity by leveraging a combination of innate and adaptive immunity.132
Immunosuppressive Tumor Microenvironment
The immunosuppressive microenvironment within solid tumors poses another substantial obstacle to effective therapy. This environment is rich in cells and molecules that actively suppress immune responses, creating a hostile setting for CAR-T cells attempting to infiltrate and eradicate tumor cells. Key factors, such as regulatory T-cells, MDSCs,134 and secreted cytokines like TGF-β and IL-10, can inhibit T-cell activity and proliferation. Additionally, physical barriers, including dense stroma and abnormal vasculature, hinder the infiltration and distribution of CAR-T cells throughout the tumor. This combination of immunosuppression and physical obstruction diminishes the effectiveness of CAR-T cells, severely limiting their therapeutic potential.134,135
One promising strategy is to overcome these challenges involves combining CAR-T cell therapy with checkpoint inhibitors. Checkpoint inhibitors are a class of drugs designed to block proteins that cancer cells use to evade immune responses.136 For instance, a mesothelin-targeted CAR-T cell therapy combined with PD-1 inhibitor pembrolizumab has been reported to reduce tumor size and enhance efficacy in treating malignant pleural disease. Superior persistence was also observed in clinical trials (NCT02414269), suggesting that anti-PD-1 therapies can restore the function of exhausted CAR-T cells.133 Further advancements include the work by Chen et al, who developed a bi-specific scFv that blocks the PD-1/PD-L1 pathway while targeting TREM2 within the TME to treat colorectal cancer. By blocking the PD-1 pathway, they effectively inhibited immunosuppressive action, and targeting TREM2 significantly reduced the proportion of M2 TAMs and MDSCs. This approach enhanced CAR-T cells infiltration and remodeled the immunosuppressive microenvironment, thereby strengthening the antitumor capacity.137 While immune checkpoint blockade (ICB) has shown effectiveness in solid tumors, it is also beneficial in hematological malignancies. For example, CRISPR-Cas9 mediated CTLA-4 deletion endowed potent killing capacity of CD19-CAR-T against leukemia by maintaining CAR expression on T cell surface, enhancing CAR-T cell proliferation, and relieving the inhibiting effect of CD28 costimulatory signaling. However, blocking PD-1 and CTLA-4 simultaneously did not yield the expected better efficacy which needed further research.138
Another promising approach involves genetically engineering of CAR-T cells to express cytokines or costimulatory molecules.61,139 Cytokines are small proteins that play a crucial role in cell signaling, effectively enhancing the immune system’s ability to mount a coordinated response against pathogens and cancer cells. By equipping CAR-T cells with the capacity to produce specific cytokines, we can improve their proliferation, survival, and cytotoxic functions. Likewise, the addition of costimulatory molecules provides extra activation signals to CAR-T cells, ensuring their sustained potency and persistence within the tumor microenvironment.139 For instance, interleukins such as IL-15 and IL-18 have been shown to boost T cell proliferation and cytotoxicity.140,141 IL-15 significantly enhances the antitumor activity and longevity of CAR-T cells by promoting sustained proliferation and increasing the frequency of TCM and stem-like cells. Notably, IL-15 secreted by engineered CAR-T cells has been reported to be more effective than soluble IL-15.140 In a study by Chmielewski et al, CAR-T cells redirected against CEA were engineered to release IL-18. This secreted IL-18 induced T-BethighFoxO1low phenotype in the engineered T cells, leading to an acute inflammatory response. IL-18 remarkably facilitated CAR-T cells expansion, while also remodeling the TME through by reducing suppressor cells, such as Treg and M2 macrophage. Unexpectedly, IL-18 significantly mitigated T cell exhaustion, suggesting enhanced persistence.141 In addition to interleukin, LIGHT, or tumor necrosis factor superfamily member 14 (TNFSF14) has also been shown to significantly improve antitumor activity of CAR-T cells. LIGHT enhances cytotoxicity not only by promoting CAR-T cells expansion but also by upregulating the expression of CCL19, which recruits normal DCs and NK cells, helping to reshape the immunosuppressive tumor environment. Furthermore, CAR-T cells expressing LIGHT demonstrated increased adhesion to vessel wall, leading to tumor vascular normalization and promotion of highly immune-infiltrating TLSs.142 Moreover, the combination of interleukin and chemokines targets the TME to reverse the immunosuppressive microenvironment. A GPC3-targeted CAR-T co-expressing IL-7 and CCL19 exhibited enhanced cytotoxicity and improved persistence. The GPC3-IL-7-CCL19-CAR-T remodeled TME and improved cytotoxicity against HCC by recruiting DCs and reducing the proportion of MDSCs, while also upregulating CD4+TEM subset levels, indicating its potential for clinical application in HCC treatment.143 In addition to cytokines, the incorporation of co-stimulatory molecules presents a promising strategy in CAR design. CD28 is critical for early antigen stimulation, while 4–1BB plays more significant in T cell expansion.144 Tandem co-stimulation with ICOS and OX40 has been shown to regulate the expression of anti-apoptotic molecules such as Bcl-2 and Bcl-xL, resulting in a superior cytotoxicity and enhanced persistence. Similar effects, such as reduced exhaustion biomarkers and increased TCM/TEM ratio, have been observed in ICOS-OX40-CD3ζ CAR-T cells.145 Taken together, the incorporation of cytokines and co-stimulatory molecules has demonstrated significant potential, suggesting that these strategies will play an increasingly important role in further CAR-T engineering.139,144
T Cell Exhaustion
Chronic antigen exposure and persistent mitogenic stimulation can induce an exhausted phenotype of T cells, characterized by diminished effector function, commonly referred to as T cell exhaustion.146–148 This phenomenon is a major contributor to cancer relapse, highlighting the urgent need for effective interventions. While targeting PD-1/PD-L1 pathway can enhance persistence of exhausted T cells, it does not reverse the epigenetic changes with exhaustion.149 Epigenetic reprogramming has the potential to mitigate T cell exhaustion, improve trafficking and infiltration, and promote a memory phenotype in T cells, ultimately leading to enhanced persistence and better prognosis.146–148,150,151 DNA methylation has been demonstrated to promote T cell exhaustion and inhibit anti-PD-1 immunotherapy.149 For instance, ALL patients treated with CAR-T therapy exhibited genome-wide DNA methylation changes, including TCF7 and LEF1, which are crucial for maintaining naïve and memory T cell state(Figure 2).150 Furthermore, DNA methyltransferase 3a (DNMT3a) has been identified as a factor that induces exhaustion in CD8+T cells. Consequently, the deletion of DNMT3a may represent an effective strategy to inhibit DNA methylation and prevent T cell exhaustion.147,152
 |
Figure 2 Preventing T cell exhaustion and enhancing cytotoxicity through metabolic reprogramming. Knockout of regnase-1, overexpressing GLUT1, CARs secreting IL10 and modifying CAR with 4–1BB strongly enhance oxidative phosphorylation, thereby leading to memory phenotype and intensive persistence to T cell exhaustion. Knockout of SIRT1, overexpressing GLUT1 and CD28 domain increase glycolysis and facilitating T cell differentiation. Knockout of RASA2 reinforces fatty acid oxygen, enhancing anti-PD-1 activity. Upregulating the expression of ADRB2 and IL10 inhibit PD-1 as well. Despite energy metabolism, T cell exhaustion can be decreased through the inhibiting SOCE signaling pathway and preventing endoplasmic reticulum stress. Knockout of regnase-1 and roquin-1 simultaneously activate NF-kB pathway, resulting in enhanced cytotoxicity. In addition, knockout of SIRT1 and knockdown of ADRB2 regulate the expression of transcription factors, increasing the level of GzmB and IFN-γ, leading to enhanced cytotoxicity as well. Created in BioRender. Su, S. (2025) https://BioRender.com/3s9395v. |
Decitabine, a DNA methyltransferase inhibitor approved for clinical use, has been shown to persistently degrade DNMT3a and enhance the effects of epigenetic reprogramming in CAR-T cells exposed to tumor antigen. Low-dose decitabine treatment leads to the downregulation of genes associated with exhaustion while promoting the upregulation of memory-associated transcription factors like TCF7, BCL6 and LEF1. As a result, decitabine increases the proportion of stem-like T cells, enhancing their tumor-homing ability and antitumor potential.153 The Demethylation of Runx3 promoter, alongside high levels of methylation in Runx3 gene, has been identified as key factors in decitabine-induced anti-PD1 activity. The reprogramming of DNA methylation at Runx3 alleviates T cell exhaustion and boosts the population of CD8+T cells, suggesting an enhanced antitumor capacity. Consequently, the combination of decitabine with anti-PD-1 therapy has been shown to significantly inhibit tumor growth and increased TILs, presenting a promising approach for future clinical treatments.154
Despite the role of DNA methylation in T cell exhaustion, antigen-independent tonic signaling from CARs can also contribute to this phenomenon through spontaneous receptor clustering.149 Transient inhibition of CAR signaling has been shown to reduce the phenotypic hallmarks of exhaustion and promote a memory-like phenotype in T cells. For instance, Weber et al modified a topically signaling CAR by incorporating a C-terminal destabilizing domain (DD), allowing for the transient inhibition of CAR activity. In addition, rested CARs displayed enhanced accessibility at the TCF7 locus, which is crucial for T cell memory. Similar results were observed in CAR-T cells combined with tyrosine kinase inhibitor dasatinib.155
Histone methyltransferase SUV39H1 plays a significant role in silencing genes associated with the differentiation of CD8+T cells into effector cells, as well as genes linked to stem cell expression programs, through the catalysis of H3K9me3 catalysis.149,156,157 Deletion of SUV39H1 increases chromatin accessibility of genes associated with stem/memory T cells and enhances mitochondrial fitness. Notably, advancements have been made in PSMA-targeted CAR-T therapies for treating prostate cancer.158 Moreover, SUV39H1 has been found to epigenetically regulate the reactivity of CD8+T cells when combined with anti-PD-1 therapy by downregulating inhibitory receptors. Further research may focus on the specific mechanisms by which inhibiting SUV39H1 promotes the efficacy of CAR-T cells in conjunction with ICI therapy.158,159 EZH1, a crucial inhibitory factor of hematopoietic multipotency in the early mammalian embryo,160 regulates chromatin accessibility through H3K27me3 trimethylation.161 Knockout of EZH1 can induce the differentiation of iPSCs (human induced pluripotent stem cells)-derived T cells into mature effector and memory T cells. CAR-T cells with EZH1 repression (EZ-T) demonstrated enhanced cytolysis activity and prolonged persistence through epigenetic reprogramming in mice model. Intriguingly, this increased persistence is mainly due to the enrichment of TCRαβ T cells.162 MEK1/2 inhibition (MEKi) significantly enhances the metabolic fitness of CD8+T cells and induces naïve phenotype. MEKi promotes metabolic fitness and mitochondrial mass through inhibiting the MAPK pathway, leading to increased levels of PGC1α and SIRT3, and enhancing fatty acid oxidation.163 Furthermore, MEKi also suppresses cyclin D1, delaying the cell cycle and thereby promoting metabolic fitness. Taken together, MEKi-induced high metabolism and FAO create favorable conditions for the development of a memory phenotype, attenuating T cell exhaustion and enhancing antitumor activity.163 Additionally, Tox is overexpressed in exhausted CD8+T cells and drives transcriptional programs associated with exhaustion.164,165 Constitutive expression of Stat5a in Texprog cells has been shown to reverse the epigenetic programs of exhaustion, antagonized Tox and converted exhausted T cells to effector/NK-like phenotype. Stat5a appears to play a direct role in regulating chromatin accessibility and transcriptional activity, thereby collaborating with PD-L1 blockade to enhance cytotoxicity and persistence.166
Moreover, the engineering of CAR-T cells can be advanced by optimizing their metabolic pathways to improve their survival, functionality, and resistance to the immunosuppressive signals prevalent in the tumor environment. Tumor cells often create a hostile metabolic landscape that depletes essential nutrients and generates suppressive metabolites. By fine-tuning these metabolic pathways, CAR-T cells can be rendered more resilient and effective in navigating these challenging conditions.167
The immunosuppressive TME is typically characterized by elevated levels of lactate, lipids and hypoxia, which can contribute to T cell exhaustion. Naive T cells usually experience a metabolic switch mediated by PI3K-Akt-mTOR pathway, relying on oxidative phosphorylation for their energy needs. In contrast, mature CD8+ effector T cells are more dependent on aerobic glycolysis.167,168 Glucose deprivation, often instigated by cancer cells, can lead to T cell exhaustion and dysfunction due to decreased glucose availability.169 Moreover, diminished glucose consumption can trigger the upregulation of inhibitory molecules, such as PD-1 and LAG-3 on the T cell surface.170
Additionally, Arginase and indoleamine-pyrrole 2,3-dioxygenase (IDO) secreted by MDSCs and TAMs selectively deplete arginine and tryptophan, further compromising the metabolic fitness and tumor-infiltration capabilities of CD8+T cells.168–171 Furthermore, glutamine deprivation has been shown to induce mitophagy, thereby impairing the cytotoxic capacity of TILs in HCC.172 Hypoxia can recruit Treg through the induction of chemokine such as CCL28, while simultaneously reducing the proportion of TCM and TEM. This dynamic ultimately inhibits CD8+T cell activity and promotes terminal differentiation state.170,173
The accumulation of metabolites and hazardous substances inhibits T cell activity also contributes to T cell exhaustion as well. Lactate has been shown to drive the conversion of pro-inflammatory T cells to Treg by upregulating Foxp3 and inhibiting the secreting of IL-17A.174 Elevated cholesterol levels in TME have been shown to regulate the expression of immune checkpoints through endoplasmic reticulum (ER) stress. The ER stress sensor XBP1 becomes activated, leading to the upregulation of PD-1 and 2B4, which are indicative of an exhausted T cell phenotype.175 Furthermore, high levels of unbound free fatty acid (FFA) have also been reported to impair antitumor activity in breast cancer.176,177 IFN-γ further enhances the expression of SLC1A5, which increases tryptophan uptake in tumor-repopulating cells (TRCs).178 IDO1 metabolizes tryptophan into kynurenine, which subsequently activates aryl hydrocarbon receptor (AhR). This activation leads to the upregulation of PD-1 via overexpression of Siglec-15, ultimately resulting in the exhaustion of CD8+T cells.179,180
Restoring T cell activity through metabolic reprogramming is an effective strategy. For instance, the 4–1BB domain has been shown to increase memory phenotype and promote mitochondrial aerobic metabolism, while CD28 activation of the PI3K-AKT increases glycolysis, thereby facilitating T cell differentiation.168,181 The knockout of SIRT1, along with enhanced degradation of FOXO1 by the proteasome, promotes glycolysis and increases granzyme B levels, indicating a significant enhancement in cytotoxicity and persistence in CD8+ memory T cells under resting conditions.180 Interestingly, inhibition of glycolysis can also induce the differentiation of T cells into memory phenotype.181,182 For instance, miR-143 has been reported to inhibit glucose transporter 1 (GLUT-1), thereby decreasing glucose uptake and glycolysis, which in turn increases the proportion of memory T cells,183 but overexpression of GLUT1 is observed to enhance the antitumor activity of CAR-T cells.178
Moreover, elevated levels of L-arginine can reprogram metabolism, converting glycolysis to oxidative phosphorylation and maintaining a higher proportion of TCM and TEM.184 As previously mentioned, abnormal lipid metabolism can also lead to T cell exhaustion. Fatostatin, is an inhibitor of SREBP2, not only reduces cholesterol levels in T cell, but also alleviates ER stress mediated by XBP1. Consequently, fatostatin effectively prevents T cell exhaustion and reduce Treg.185 In addition, inhibiting cholesterol esterification has been shown to enhance cytotoxic effects and increase the proportion of CD8+ T cells.186
Beyond cholesterol metabolism, fatty acid catabolism also plays a crucial role in regulating T cell function. Enhanced fatty acid oxygen in hypoglycemia and hypoxic TME is essential for maintaining normal function of CD8+ TILs. FAs can also be converted to acetyl-CoA, which participates in TCA cycle and glycolysis, contributing to energy production. Collectively, metabolic reprogramming drives increased FAO and enhances overall efficacy, including response to anti-PD-1 reaction.187 Moreover, an increase in extracellular potassium reprograms T cell function through autophagy decreases histone acetylation. These epigenetic alterations help maintain stem-like characteristics, suggesting enhanced persistence and antitumor activity.188 Furthermore, SOCE-inhibitor BTP-2 reduces intracellular calcium signaling via SOCE-Calcineurin-NFAT pathway, resulting in higher proliferation and lower exhaustion signals.189 Other promising strategies including targeting REGNASE-1 to improve mitochondria fitness, ablating RASA2,190 ADRB2,191 secreting IL10192 and briefly inhibiting mitochondrial pyruvate carrier (MPC) to promote memory phenotype have shown to be effective in treating various cancers.193,194 Interestingly, knockout REGNASE-1 and ROQUIN-1 simultaneously enhanced inflammatory potential and TCM phenotype, thereby increasing cytotoxicity.195
Taken together, targeting the epigenetic programs and metabolism of T cells represents a promising approach to prevent T cell exhaustion and enhance therapeutic efficacy. Further research should concentrate on elucidating the specific molecular mechanisms underlying reprogramming and explore the integration of epigenetic with metabolic strategies to overcome the immunosuppressive TME.
CAR-T Cell Associated Toxicities
Despite the remarkable achievements in CAR-T cell therapy, potential toxicities such as CRS, ICANS, cytopenia-related complications and on-target, off-tumor toxicities pose substantial challenges to its clinical application. These toxicities are often linked to factors such as high tumor burden, target antigen and CAR-T doses.196 Addressing and mitigating CAR-T-associated toxicities, along with enhancing the safety in CAR-T cell therapies, are urgent priorities in the field of immunotherapy. A comprehensive understanding of those challenges is essential for maximizing therapeutic benefits while minimizing adverse effects. Several strategies are being explored by researchers and clinicians to achieve these goals, focusing on genetic engineering advancements and optimizing treatment protocols.197
CRS is an immune inflammatory response typically characterized by extensive proliferation of CAR-T cells and a significant increase in serum cytokines following CAR-T cells infusion. Cytokines such as IFN-γ, TNF-α and GM-CSF activate monocyte/macrophage system, further exacerbating the inflammatory response(Figure 3).196,197 Clinical manifestations of CRS can range from mild flu-like symptoms, including fever and fatigue, to severe, life-threatening complications such as hypotension, hypoxia, and multiple-organ dysfunction.197
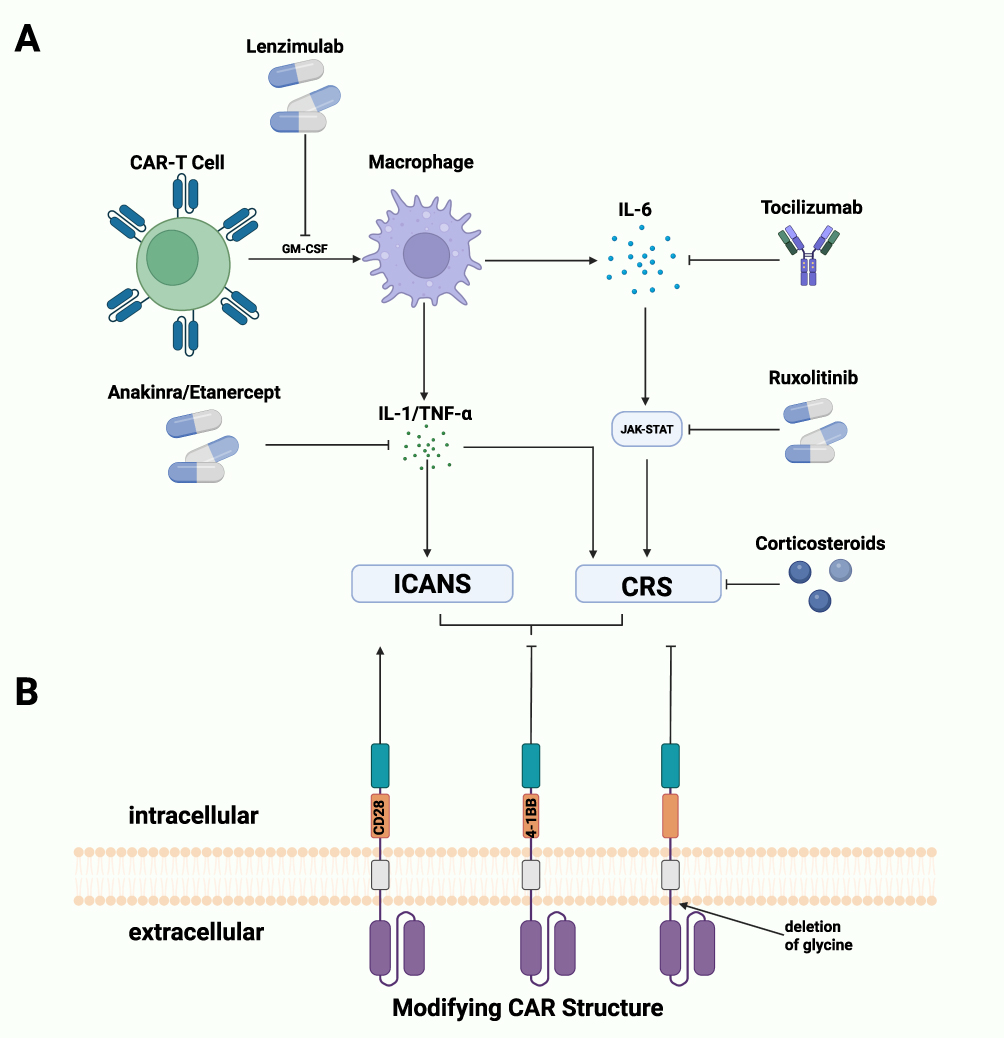 |
Figure 3 (A) Small molecules preventing CRS and ICANS. Unspecific medication corticosteroids strongly decrease the function and production of cytokines. Lenzimulab inhibits GM-CSF, avoiding the activation of macrophage. Tocilizumab along with Ruxolitinib block IL6-JAK-STAT signaling, strongly decrease inflammatory activity. Anakinra and etanercept inhibit IL-1 and TNF-α respectively, releasing CRS and ICANS mediated by these cytokines. (B) Modifying CAR structure to avoid adverse event. CD28 usually leads to higher toxicity while 4–1BB is considered safer. Deletion glycine of hinge domain decrease the level of pro-inflammatory cytokines, leading to a lower risk of adverse events. Created in BioRender. Su, S. (2025) https://BioRender.com/w6pe2ht. |
Another serious adverse event is ICANS. Patients with ICANS may experience symptoms such as fever, epilepsy, cerebral edema and myoclonus. Notably, ICANS often occurs concurrently or after CRS, suggesting a possible correlation between these two conditions.198 ICANS is characterized by elevated cytokine levels in cerebrospinal fluid (CSF) and disruptions to the blood–brain barrier with activated myeloid cells in central nervous system contributing to its development.198,199 IL-6 is generally regarded as a primary cytokine responsible for inducing CRS.200 As a result, the IL-6R-specific inhibitor tocilizumab has been approved by FDA for the treatment of CRS. Tocilizumab has been shown effectiveness in managing grade 2 CRS and gastric mucosa injury in a 72-year-old male patient with pancreatic cancer following anti-claudin18.2 CAR-T cells infusion.196,201 For severe cases of ICANS, intrathecal administration of tocilizumab may offer a viable treatment option.202 While tocilizumab has achieved excellent outcomes in the management of CRS, its effectiveness against ICANS is limited due to its poor penetration of the blood–brain barrier (BBB). Anakinra, a recombinant humanized IL-1 receptor antagonist, has been shown to effectively control fever, attenuated neurotoxicity, and decrease levels of inflammatory biomarkers like C reactive protein (CRP) in a single-center clinical trial, making it a promising candidate for treating refractory CRS following the failure of tocilizumab and corticosteroids.203 Moreover, the peak level of TNF-α has been associated with the severity of CRS, leading to the consideration of the TNF-α inhibitor etanercept as a new option for CRS management. Etanercept has demonstrated its ability to reduce TNF-α production without affecting CAR-T cell activity.204 Recent studies have indicated that monocytes and macrophages promote the development of CRS and ICANS, presenting a novel approach to manage these toxicities through targeting GM-CSF.205 Lenzimulab, a GM-CSF inhibitor, has been shown to nearly completely neutralize GM-CSF in vitro. Surprisingly, this neutralization enhanced CAR-T cell efficacy and persistence in a CART19 xenograft model. Furthermore, lenzimulab reduced the number of macrophages in brain, indicating a reduction in both CRS and neurotoxicity.206
Despite the availability of specific inhibitors, non-specific medication such as corticosteroids remain first-line treatment for ICANS and second-line options for severe CRS.207 Corticosteroids effectively inhibit the production and function of cytokines. Due to their potential immunosuppressive effects, corticosteroids are typically reserved for treating ICANS of grade 2 or higher. High dose of corticosteroids is generally believed to alleviate typical symptoms of associated with ICANS.207,208
Tyrosine kinase inhibitors, including ibrutinib and ruxolitinib, are promising agents for managing CAR-T cell-associated toxicities. IL-6 ultimately activates JAK-Stat pathway.209 Ruxolitinib, a JAK 1/2 inhibitor with broad anti-inflammatory activity, is commonly used to treat allergy reactions, immune responses and COVID-19. It has shown surprising efficacy in addressing steroid-refractory CRS without compromising the antitumor activity of CAR-T cells, suggesting its potential as an adjuvant therapy for CRS.210,211 Similarly, BTK inhibitor ibrutinib has demonstrated effectiveness against cytokine release syndrome, significantly preventing CRS in B cell malignancies following the infusion of CD19-targeting CAR-T cells.212 Moreover, ibrutinib has been found to enhance efficacy and the CR ratio among patients receiving CART-19 by promoting CAR-T cells activity. This suggests that the combination of CAR-T therapy and ibrutinib could be a promising approach in treating non-Hodgkin lymphoma.213,214
Another potential strategy for reducing toxicities involves modifying CAR structure to reduce toxicities. Previous studies have demonstrated that CD28 costimulatory domain is usually associated with higher risk of toxicity while 4–1BB domain is considered safer. Additionally, the deletion of glycine decreased the flexibility of CD8α hinge domain, leading to lower levels of pro-inflammatory cytokines and promoting safety.215 Moreover, managing CAR-T cell-associated toxicities extend beyond genetic engineering and pre-infusion treatments; effective post-treatment monitoring and management strategies are equally essential. Early detecting toxicities, particularly neurotoxicity and CRS, is crucial for timely intervention. Using standardized grading systems for adverse events assists clinicians in promptly adjusting treatment protocols.196
Tumor-associated antigens (TAA) are often co-expressed on both cancer cells and non-malignant tissues, which poses a challenge for CAR-T cells. This co-expression makes it difficult for CAR-T cells differentiate between tumor and normal organs, leading to inevitable tissue damage known as “On-Target, Off-tumor Toxicity” (OTOT).215 For instance, CD19 is expressed on both leukemia cells and brain mural cells, increasing the risk of neurotoxicity.216 CAR-T cells targeting CD19 and BCMA have been observed to attack normal B cells and plasma cells, resulting in hypogammaglobulinaemia.217 While OTOT is particularly prevalent in the treatment of solid tumors, it is also a concern in blood cancers. HER2-specific CAR-T therapy has yielded significant results in advanced biliary tract and pancreatic cancers; however, patients experiencing reversible severe upper gastrointestinal hemorrhage were detected following CAR-T cell infusion.218 Claudin18.2 (CLDN18.2) is another example that is co-expressed in both gastric cancer and normal gastric mucosal epithelium. As a result, CLDN18.2-targeted CAR-T cell infusion led to severe gastric mucosal damage in a phase 1 clinical trial.111 Similarly, low-dose of CAIX-targeted CAR-T cells used to treat metastatic renal cell carcinoma resulted in liver enzyme disturbances due to the CAIX expression in bile duct epithelium.219 Collectively, there is an urgent need for effective strategies to mitigate OTOT in the application of CAR-T. One potential strategy is to modify the affinity of scFv in the CAR structure. Reducing this affinity may decrease the likelihood of targeting normal cells that express physiologic levels of the antigen.43,47 Incorporating suicide gene technology into CAR-T represents a promising strategy for mitigating potential toxicities.2 This mechanism function as a safety switch, enabling clinicians to control the proliferation and activity of CAR-T cells when potentially severe side effects are arising. A common approach involves the use of herpes simplex virus thymidine kinase (HSV-TK) gene, which makes the modified CAR-T cells susceptible to antiviral drugs such as ganciclovir (GCV). Studies have shown that CAR-T cells expressing GM2 can be effectively diminished by GCV.220 Additionally, iCaspase9 serves as another suicide switch, with previous experiments demonstrating its effectiveness in managing CAR-T related side effects.107
Split CAR systems offer another approach to managing CAR-T cell activity, specifically addressing concerns related to overactivation and off-target effects. In these systems, the CAR is divided into two separate components, which require activation by different antigens to function effectively. This dual-requirement mechanism enhances specificity, as both antigens must be present for the CAR to activate.221 For instance, Halliwell et al developed a dual-positive CAR targeting ALK and B7H3 or GD2 using an “AND” structure. In this configuration, T cells are activated and can kill target cells only when ALK co-expresses with B7H3 or GD2 on tumor cells. This co-CAR strategy not only addresses the challenge of low ALK antigen density, thereby enhancing antitumor activity, but also helps to avoid “on-target, off-tumor toxicity” associated with high expression of GD2 or B7H3.222 Additionally, an IF-THEN gate integrating the SynNotch system has been reported to mitigate off-tumor toxicity. In this system, CD19 svFv SynNotch is co-expressed with “PDbody”, creating a dual CAR that targets both CD19 and PD-L1, the PDbody was engineered to bind to PD-L1 with a preference for a slightly lower pH, which is typical in the tumor microenvironment. To prevent the activation of CD19 SynNotch in normal B cells, pre-treatment with fludarabine and cyclophosphamide can be employed prior to infusion.223 Overall, split CAR systems reduce the risk of inadvertently targeting healthy cells that lack the specific antigen combination. By refining the specificity of CAR-T cell targeting, these systems can significantly decrease the incidence of adverse effects caused by non-specific or unwanted interactions, thus enhancing both safety and therapeutic outcomes.222,223
CAR-T Cell Therapy in Other Diseases
In autoimmune diseases such as immune-mediated necrotizing myopathy (IMNM), where traditional pharmacological methods have limited efficacy, CAR-T cell therapy has demonstrated significant therapeutic effects. Patients treated with BCMA-targeted CAR-T cells exhibited favorable safety effects.224
CAR-T cell therapies are also being explored for the treatment of systemic lupus erythematosus (SLE), with a notable focus on anti-CD19 CAR-T cells. These CD19 CAR-T cells have been shown to induce B cell depletion, leading to a rapid decline in anti-dsDNA antibodies disappeared rapidly.225 In a Phase 1 clinical trial, an engineered CAR-T cell that expresses two CARs targeting CD19 and BCMA demonstrated the ability to alleviate symptoms of SLE and relieve lupus nephritis (LN). Remarkably, the humoral immunity of patients was effectively reset, with only mild CRS observed, indicating a favorable safety profile.226
Conclusions and Future Expectations
CAR-T therapy has achieved breakthrough efficacy in treating hematological malignancies (eg, leukemia, lymphoma), providing patients with durable remission. However, its application in solid tumors remains limited by challenges such as antigen heterogeneity, TME, and T cell exhaustion. Future directions include: Developing multi-target CAR architectures to mitigate antigen escape risks; Enhancing CAR-T cell recognition and cytotoxicity through CRISPR-based strategies, such as precise knockout of IL-4 proteins or insertion of dual-target receptors (eg, CD19+CD22); Restoration of LAT activity; Engineering tunable safety switches via CRISPR to control treatment-related toxicities; Optimizing manufacturing protocols to improve therapeutic consistency; Reverse the exhaustion by introducing PD-1-CD28 chimeric genes; Adopting combination strategies with existing treatments, including synergy with immune checkpoint inhibitors, combination with tumor-targeting antibodies, integration with small-molecule drugs, coordinated use with oncolytic viruses, and concurrent application with radiotherapy.
The preparation time for universal CAR-T (UCAR-T) therapy has been markedly reduced, which may also lead to a significant reduction in treatment costs at scale. A number of clinical trials, including those conducted by our group (NCT06663046), are currently underway or will soon be initiated to evaluate the safety and effectiveness of this approach. Moreover, CAR-T cell therapeutic products capable of treating multiple cancer patients can be manufactured from cells obtained from a single healthy donor. Gene editing technology enables the knockout of TCR, HLA, or CD52 molecules on the surface of T cells in UCAR-T therapy, thereby eliminating graft-versus-host disease (GVHD) caused by UCAR-T cells against patients and host-versus-graft reactions (HVG) of patients against allogeneic UCAR-T cells. This facilitates large-scale production and reduces costs. While overcoming barriers in solid tumors remains critical, ongoing technological advancements will expand CAR-T’s therapeutic indications, ultimately improving global cancer treatment outcomes. This rapidly evolving field holds promise for further unleashing the immune system’s anti-cancer potential.
Acknowledgments
This study was supported by the National Natural Science Foundation of China (No. 82470226 and 82300255).
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Hu D, Yuan S, Zhong J, et al. Cellular senescence and hematological malignancies: from pathogenesis to therapeutics. Pharmacol Ther. 2021;223:107817. doi:10.1016/j.pharmthera.2021.107817
2. Zhang X, Zhu L, Zhang H, et al. CAR-T cell therapy in hematological malignancies: current opportunities and challenges. Front Immunol. 2022;13:927153. doi:10.3389/fimmu.2022.927153
3. Rosenquist R, Bernard E, Erkers T, et al. Novel precision medicine approaches and treatment strategies in hematological malignancies. J Intern Med. 2023;294(4):413–436. doi:10.1111/joim.13697
4. Cassetta L, Pollard JW. Targeting macrophages: therapeutic approaches in cancer. Nat Rev Drug Discov. 2018;17(12):887–904. doi:10.1038/nrd.2018.169
5. Li W, Wang F, Guo R, et al. Targeting macrophages in hematological malignancies: recent advances and future directions. J Hematol Oncol. 2022;15(1):110. doi:10.1186/s13045-022-01328-x
6. Beckers C, Pruschy M, Vetrugno I. Tumor hypoxia and radiotherapy: a major driver of resistance even for novel radiotherapy modalities. Semin Cancer Biol. 2024;98:19–30. doi:10.1016/j.semcancer.2023.11.006
7. Shin MH, Oh E, Kim Y, et al. Recent advances in CAR-based solid tumor immunotherapy. Cells. 2023;12(12):1606. doi:10.3390/cells12121606
8. Sivori S, Pende D, Quatrini L, et al. NK cells and ILCs in tumor immunotherapy. Mol Aspects Med. 2021;80:100870. doi:10.1016/j.mam.2020.100870
9. Mitra A, Barua A, Huang L, et al. From bench to bedside: the history and progress of CAR T cell therapy. Front Immunol. 2023;14:1188049. doi:10.3389/fimmu.2023.1188049
10. Kuwana Y, Asakura Y, Utsunomiya N, et al. Expression of chimeric receptor composed of immunoglobulin-derived V regions and T-cell receptor-derived C regions. Biochem Biophys Res Commun. 1987;149(3):960–968. doi:10.1016/0006-291x(87)90502-x
11. Gross G, Waks T, Eshhar Z. Expression of immunoglobulin-T-cell receptor chimeric molecules as functional receptors with antibody-type specificity. Proc Natl Acad Sci U S A. 1989;86(24):10024–10028. doi:10.1073/pnas.86.24.10024
12. Eshhar Z, Waks T, Gross G, et al. Specific activation and targeting of cytotoxic lymphocytes through chimeric single chains consisting of antibody-binding domains and the gamma or zeta subunits of the immunoglobulin and T-cell receptors. Proc Natl Acad Sci U S A. 1993;90(2):720–724. doi:10.1073/pnas.90.2.720
13. Moritz D, Wels W, Mattern J, et al. Cytotoxic T lymphocytes with a grafted recognition specificity for ERBB2-expressing tumor cells. Proc Natl Acad Sci U S A. 1994;91(10):4318–4322. doi:10.1073/pnas.91.10.4318
14. Hwu P, Shafer GE, Treisman J, et al. Lysis of ovarian cancer cells by human lymphocytes redirected with a chimeric gene composed of an antibody variable region and the Fc receptor gamma chain. J Exp Med. 1993;178(1):361–366. doi:10.1084/jem.178.1.361
15. Hwu P, Yang JC, Cowherd R, et al. In vivo antitumor activity of T cells redirected with chimeric antibody/T-cell receptor genes. Cancer Res. 1995;55(15):3369–3373.
16. Kershaw MH, Westwood JA, Parker LL, et al. A phase I study on adoptive immunotherapy using gene-modified T cells for ovarian cancer. Clin Cancer Res. 2006;12(20 Pt 1):6106–6115. doi:10.1158/1078-0432.Ccr-06-1183
17. Lamers CH, Sleijfer S, Vulto AG, et al. Treatment of metastatic renal cell carcinoma with autologous T-lymphocytes genetically retargeted against carbonic anhydrase IX: first clinical experience. J Clin Oncol. 2006;24(13):e20–2. doi:10.1200/jco.2006.05.9964
18. Zhao Z, Condomines M, van der Stegen SC, et al. Structural design of engineered costimulation determines tumor rejection kinetics and persistence of CAR T cells. Cancer Cell. 2015;28(4):415–428. doi:10.1016/j.ccell.2015.09.004
19. Song DG, Ye Q, Poussin M, et al. CD27 costimulation augments the survival and antitumor activity of redirected human T cells in vivo. Blood. 2012;119(3):696–706. doi:10.1182/blood-2011-03-344275
20. Guedan S, Chen X, Madar A, et al. ICOS-based chimeric antigen receptors program bipolar TH17/TH1 cells. Blood. 2014;124(7):1070–1080. doi:10.1182/blood-2013-10-535245
21. Fujiwara K, Kitaura M, Tsunei A, et al. Structure of the signal transduction domain in second-generation CAR regulates the input efficiency of CAR signals. Int J Mol Sci. 2021;22(5):2476. doi:10.3390/ijms22052476
22. Gu A, Bai Y, Zhang C, et al. IL13Rα2-targeted third-generation CAR-T cells with CD28 transmembrane domain mediate the best anti-glioblastoma efficacy. Cancer Immunol Immunother. 2023;72(7):2393–2403. doi:10.1007/s00262-023-03423-5
23. Zhang S, Gu C, Huang L, et al. The third-generation anti-CD30 CAR T-cells specifically homing to the tumor and mediating powerful antitumor activity. Sci Rep. 2022;12(1):10488. doi:10.1038/s41598-022-14523-0
24. Chmielewski M, Abken H. TRUCKs: the fourth generation of CARs. Expert Opin Biol Ther. 2015;15(8):1145–1154. doi:10.1517/14712598.2015.1046430
25. Zhao L, Li S, Wei X, et al. A novel loop-structure-based bispecific CAR that targets CD19 and CD22 with enhanced therapeutic efficacy against B-cell malignancies. Protein and Cell. 2024;16(3):227–231. doi:10.1093/procel/pwae034
26. Huang R, Li X, He Y, et al. Recent advances in CAR-T cell engineering. J Hematol Oncol. 2020;13(1):86. doi:10.1186/s13045-020-00910-5
27. Sterner RC, Sterner RM. CAR-T cell therapy: current limitations and potential strategies. Blood Cancer J. 2021;11(4):69. doi:10.1038/s41408-021-00459-7
28. Bailey SR, Maus MV. Gene editing for immune cell therapies. Nat Biotechnol. 2019;37(12):1425–1434. doi:10.1038/s41587-019-0137-8
29. Martinez LR, Moussai D, Casadevall A. Antibody to Cryptococcus neoformans glucuronoxylomannan inhibits the release of capsular antigen. Infect Immun. 2004;72(6):3674–3679. doi:10.1128/iai.72.6.3674-3679.2004
30. Sarén T, Saronio G, Marti Torrell P, et al. Complementarity-determining region clustering may cause CAR-T cell dysfunction. Nat Commun. 2023;14(1):4732. doi:10.1038/s41467-023-40303-z
31. Rafiq S, Hackett CS, Brentjens RJ. Engineering strategies to overcome the current roadblocks in CAR T cell therapy. Nat Rev Clin Oncol. 2020;17(3):147–167. doi:10.1038/s41571-019-0297-y
32. Folimonova V, Chen X, Negi H, et al. CD28 hinge used in chimeric antigen receptor (CAR) T-cells exhibits local structure and conformational exchange amidst global disorder. Commun Biol. 2024;7(1):1072. doi:10.1038/s42003-024-06770-w
33. Leick MB, Silva H, Scarfò I, et al. Non-cleavable hinge enhances avidity and expansion of CAR-T cells for acute myeloid leukemia. Cancer Cell. 2022;40(5):494–508.e5. doi:10.1016/j.ccell.2022.04.001
34. Guedan S, Posey AD, Shaw C, et al. Enhancing CAR T cell persistence through ICOS and 4-1BB costimulation. JCI Insight. 2018;3(1). doi:10.1172/jci.insight.96976
35. Chabannon C, Bonini C. Structure of and Signalling Through Chimeric Antigen Receptor. In: Kröger N, editor. The EBMT/EHA CAR-T Cell Handbook. Springer Copyright 2022, The Author(s). Cham (CH); 2022:3–5.
36. Starr R, Aguilar B, Gumber D, et al. Inclusion of 4-1BB costimulation enhances selectivity and functionality of IL13Rα2-targeted chimeric antigen receptor T cells. Cancer Res Commun. 2023;3(1):66–79. doi:10.1158/2767-9764.Crc-22-0185
37. Cappell KM, Kochenderfer JN. A comparison of chimeric antigen receptors containing CD28 versus 4-1BB costimulatory domains. Nat Rev Clin Oncol. 2021;18(11):715–727. doi:10.1038/s41571-021-00530-z
38. Benmebarek MR, Karches CH, Cadilha BL, et al. Killing mechanisms of chimeric antigen receptor (CAR) T cells. Int J Mol Sci. 2019;20(6):1283. doi:10.3390/ijms20061283
39. de Saint Basile G, Ménasché G, Fischer A. Molecular mechanisms of biogenesis and exocytosis of cytotoxic granules. Nat Rev Immunol. 2010;10(8):568–579. doi:10.1038/nri2803
40. Textor A, Listopad JJ, Wührmann LL, et al. Efficacy of CAR T-cell therapy in large tumors relies upon stromal targeting by IFNγ. Cancer Res. 2014;74(23):6796–6805. doi:10.1158/0008-5472.Can-14-0079
41. Kägi D, Vignaux F, Ledermann B, et al. Fas and perforin pathways as major mechanisms of T cell-mediated cytotoxicity. Science. 1994;265(5171):528–530. doi:10.1126/science.7518614
42. Lowin B, Hahne M, Mattmann C, et al. Cytolytic T-cell cytotoxicity is mediated through perforin and Fas lytic pathways. Nature. 1994;370(6491):650–652. doi:10.1038/370650a0
43. Liu X, Jiang S, Fang C, et al. Affinity-tuned ErbB2 or EGFR chimeric antigen receptor T cells exhibit an increased therapeutic index against tumors in mice. Cancer Res. 2015;75(17):3596–3607. doi:10.1158/0008-5472.Can-15-0159
44. Hudecek M, Lupo-Stanghellini M-T, Kosasih PL, et al. Receptor affinity and extracellular domain modifications affect tumor recognition by ROR1-specific chimeric antigen receptor T cells. Clin Cancer Res. 2013;19(12):3153–3164. doi:10.1158/1078-0432.Ccr-13-0330
45. Lynn RC, Feng Y, Schutsky K, et al. High-affinity FRβ-specific CAR T cells eradicate AML and normal myeloid lineage without HSC toxicity. Leukemia. 2016;30(6):1355–1364. doi:10.1038/leu.2016.35
46. Caruso HG, Hurton LV, Najjar A, et al. Tuning sensitivity of CAR to EGFR density limits recognition of normal tissue while maintaining potent antitumor activity. Cancer Res. 2015;75(17):3505–3518. doi:10.1158/0008-5472.Can-15-0139
47. He C, Mansilla-Soto J, Khanra N, et al. CD19 CAR antigen engagement mechanisms and affinity tuning. Sci Immunol. 2023;8(81):eadf1426. doi:10.1126/sciimmunol.adf1426
48. Chen J, Qiu S, Li W, et al. Tuning charge density of chimeric antigen receptor optimizes tonic signaling and CAR-T cell fitness. Cell Res. 2023;33(5):341–354. doi:10.1038/s41422-023-00789-0
49. Heads JT, Lamb R, Kelm S, et al. Electrostatic interactions modulate the differential aggregation propensities of IgG1 and IgG4P antibodies and inform charged residue substitutions for improved developability. Protein Eng Des Sel. 2019;32(6):277–288. doi:10.1093/protein/gzz046
50. Machado MP, Dos Santos MH, Guimarães JG, et al. GXMR-CAR containing distinct GXM-specific single-chain variable fragment (scFv) mediated the cell activation against Cryptococcus spp. And had difference in the strength of tonic signaling. Bioengineered. 2023;14(1):2281059. doi:10.1080/21655979.2023.2281059
51. Dos Santos MH, Machado MP, Kumaresan PR, et al. Modification of hinge/transmembrane and signal transduction domains improves the expression and signaling threshold of GXMR-CAR specific to Cryptococcus spp. Cells. 2022;11(21):3386. doi:10.3390/cells11213386
52. Alabanza L, Pegues M, Geldres C, et al. Function of novel anti-CD19 chimeric antigen receptors with human variable regions is affected by hinge and transmembrane domains. Mol Ther. 2017;25(11):2452–2465. doi:10.1016/j.ymthe.2017.07.013
53. Shargian L, Raanani P, Yeshurun M, et al. Chimeric antigen receptor T-cell therapy is superior to standard of care as second-line therapy for large B-cell lymphoma: a systematic review and meta-analysis. Br J Haematol. 2022;198(5):838–846. doi:10.1111/bjh.18335
54. Mellman I, Coukos G, Dranoff G. Cancer immunotherapy comes of age. Nature. 2011;480(7378):480–489. doi:10.1038/nature10673
55. Jin X, Liu Z, Yang D, et al. Recent progress and future perspectives of immunotherapy in advanced gastric cancer. Front Immunol. 2022;13:948647. doi:10.3389/fimmu.2022.948647
56. Budde LE, Berger C, Lin Y, et al. Combining a CD20 chimeric antigen receptor and an inducible caspase 9 suicide switch to improve the efficacy and safety of T cell adoptive immunotherapy for lymphoma. PLoS One. 2013;8(12):e82742. doi:10.1371/journal.pone.0082742
57. Atar D, Ruoff L, Mast A-S, et al. Rational combinatorial targeting by adapter CAR-T-cells (AdCAR-T) prevents antigen escape in acute myeloid leukemia. Leukemia. 2024;38(10):2183–2195. doi:10.1038/s41375-024-02351-2
58. Chekol Abebe E, Yibeltal Shiferaw M, Tadele Admasu F, et al. Ciltacabtagene autoleucel: the second anti-BCMA CAR T-cell therapeutic armamentarium of relapsed or refractory multiple myeloma. Front Immunol. 2022;13:991092. doi:10.3389/fimmu.2022.991092
59. Manni S, Del Bufalo F, Merli P, et al. Neutralizing IFNγ improves safety without compromising efficacy of CAR-T cell therapy in B-cell malignancies. Nat Commun. 2023;14(1):3423. doi:10.1038/s41467-023-38723-y
60. Arun Kumar S, Gao J, Patel SA. The shifting therapeutic paradigm for relapsed/refractory mantle cell lymphoma. Leuk Res. 2023;134:107385. doi:10.1016/j.leukres.2023.107385
61. Roddie C, O’Reilly M, Dias Alves Pinto J, et al. Manufacturing chimeric antigen receptor T cells: issues and challenges. Cytotherapy. 2019;21(3):327–340. doi:10.1016/j.jcyt.2018.11.009
62. Shah NN, Lee DW, Yates B, et al. Long-term follow-up of CD19-CAR T-cell therapy in children and young adults with B-ALL. J Clin Oncol. 2021;39(15):1650–1659. doi:10.1200/jco.20.02262
63. Denlinger N, Bond D, Jaglowski S. CAR T-cell therapy for B-cell lymphoma. Curr Probl Cancer. 2022;46(1):100826. doi:10.1016/j.currproblcancer.2021.100826
64. Wang Y, Zhang W-Y, Han Q-W, et al. Effective response and delayed toxicities of refractory advanced diffuse large B-cell lymphoma treated by CD20-directed chimeric antigen receptor-modified T cells. Clin Immunol. 2014;155(2):160–175. doi:10.1016/j.clim.2014.10.002
65. Yin Z, Zhang Y, Wang X. Advances in chimeric antigen receptor T-cell therapy for B-cell non-Hodgkin lymphoma. Biomark Res. 2021;9(1):58. doi:10.1186/s40364-021-00309-5
66. Fry TJ, Shah NN, Orentas RJ, et al. CD22-targeted CAR T cells induce remission in B-ALL that is naive or resistant to CD19-targeted CAR immunotherapy. Nat Med. 2018;24(1):20–28. doi:10.1038/nm.4441
67. Singh N, Frey NV, Engels B, et al. Antigen-independent activation enhances the efficacy of 4-1BB-costimulated CD22 CAR T cells. Nat Med. 2021;27(5):842–850. doi:10.1038/s41591-021-01326-5
68. Shah NN, Highfill SL, Shalabi H, et al. CD4/CD8 T-cell selection affects chimeric antigen receptor (CAR) T-cell potency and toxicity: updated results from a phase I Anti-CD22 CAR T-cell trial. J Clin Oncol. 2020;38(17):1938–1950. doi:10.1200/jco.19.03279
69. Yang X, Wang GX, Zhou JF. CAR T Cell Therapy for Hematological Malignancies. Curr Med Sci. 2019;39(6):874–882. doi:10.1007/s11596-019-2118-z
70. Yan W, Liu Z, Liu J, et al. Application of chimeric antigen receptor T cells in the treatment of hematological malignancies. Biomed Res Int. 2020;2020(1):4241864. doi:10.1155/2020/4241864
71. Rodríguez-Zúñiga MJM, Cortez-Franco F, Qujiano-Gomero E. Adult T-cell leukemia/lymphoma. review of the literature. Actas Dermosifiliogr. 2018;109(5):399–407. doi:10.1016/j.ad.2017.08.014
72. Whitlock JA, Malvar J, Dalla‐Pozza L, et al. Nelarabine, etoposide, and cyclophosphamide in relapsed pediatric T-acute lymphoblastic leukemia and T-lymphoblastic lymphoma (study T2008-002 NECTAR). Pediatr Blood Cancer. 2022;69(11):e29901. doi:10.1002/pbc.29901
73. Lato MW, Przysucha A, Grosman S, et al. The new therapeutic strategies in pediatric T-cell acute lymphoblastic leukemia. Int J Mol Sci. 2021;22(9):4502. doi:10.3390/ijms22094502
74. Shimony S, DeAngelo DJ, Luskin MR. Nelarabine: when and how to use in the treatment of T-cell acute lymphoblastic leukemia. Blood Adv. 2024;8(1):23–36. doi:10.1182/bloodadvances.2023010303
75. Hu Y, Zhou Y, Zhang M, et al. Genetically modified CD7-targeting allogeneic CAR-T cell therapy with enhanced efficacy for relapsed/refractory CD7-positive hematological malignancies: a phase I clinical study. Cell Res. 2022;32(11):995–1007. doi:10.1038/s41422-022-00721-y
76. Watanabe N, Mo F, Zheng R, et al. Feasibility and preclinical efficacy of CD7-unedited CD7 CAR T cells for T cell malignancies. Mol Ther. 2023;31(1):24–34. doi:10.1016/j.ymthe.2022.09.003
77. Gomes-Silva D, Srinivasan M, Sharma S, et al. CD7-edited T cells expressing a CD7-specific CAR for the therapy of T-cell malignancies. Blood. 2017;130(3):285–296. doi:10.1182/blood-2017-01-761320
78. Lu P, Liu Y, Yang J, et al. Naturally selected CD7 CAR-T therapy without genetic manipulations for T-ALL/LBL: first-in-human phase 1 clinical trial. Blood. 2022;140(4):321–334. doi:10.1182/blood.2021014498
79. Zhang Y, Li C, Du M, et al. Allogenic and autologous anti-CD7 CAR-T cell therapies in relapsed or refractory T-cell malignancies. Blood Cancer J. 2023;13(1):61. doi:10.1038/s41408-023-00822-w
80. Ghobadi A, Aldoss I, Maude S, et al. P356: Phase 1/2 dose-escalation study of anti-cd7 allogenic CAR-T cell in relapsed or refractory(R/R) T-cell acute lymphoblastic leukemia/lymphoblastic lymphoma(T-ALL/LBL). HemaSphere. 2023;7(S3):e1789302. doi:10.1097/01.Hs9.0000968336.17893.02
81. Feng J, Xu H, Cinquina A, et al. Treatment of aggressive T cell lymphoblastic lymphoma/leukemia using anti-CD5 CAR T cells. Stem Cell Rev Rep. 2021;17(2):652–661. doi:10.1007/s12015-020-10092-9
82. Frigault MJ, Chen Y-B, Gallagher KME, et al. Phase 1 study of CD37-directed CAR T cells in patients with relapsed or refractory CD37+ hematologic malignancies. Blood. 2021;138(Supplement 1):653. doi:10.1182/blood-2021-146236
83. Cwynarski K, Iacoboni G, Tholouli E, et al. First in human study of AUTO4, a TRBC1-targeting CAR T-cell therapy in relapsed/refractory TRBC1-positive peripheral T-cell lymphoma. Blood. 2022;140(Supplement 1):10316–10317. doi:10.1182/blood-2022-165971
84. Testa U, Chiusolo P, Pelosi E, et al. CAR-T cell therapy for T-cell malignancies. Mediterr J Hematol Infect Dis. 2024;16(1):e2024031. doi:10.4084/mjhid.2024.031
85. Callander NS, Baljevic M, Adekola K, et al. NCCN guidelines® insights: multiple myeloma, version 3.2022. J Natl Compr Canc Netw. 2022;20(1):8–19. doi:10.6004/jnccn.2022.0002
86. Imai Y. Latest development in multiple myeloma. Cancers. 2020;12(9):2544. doi:10.3390/cancers12092544
87. Zhang X, Zhang H, Lan H, et al. CAR-T cell therapy in multiple myeloma: current limitations and potential strategies. Front Immunol. 2023;14:1101495. doi:10.3389/fimmu.2023.1101495
88. Montefusco V, Mussetti A, Salas MQ, et al. Old and new generation proteasome inhibitors in multiple myeloma. Panminerva Med. 2020;62(4):193–206. doi:10.23736/s0031-0808.20.04148-8
89. Ito S. Proteasome inhibitors for the treatment of multiple myeloma. Cancers. 2020;12(2):265. doi:10.3390/cancers12020265
90. Heider M, Nickel K, Högner M, et al. Multiple myeloma: molecular pathogenesis and disease evolution. Oncol Res Treat. 2021;44(12):672–681. doi:10.1159/000520312
91. Shah N, Chari A, Scott E, et al. B-cell maturation antigen (BCMA) in multiple myeloma: rationale for targeting and current therapeutic approaches. Leukemia. 2020;34(4):985–1005. doi:10.1038/s41375-020-0734-z
92. Sanchez E, Li M, Kitto A, et al. Serum B -cell maturation antigen is elevated in multiple myeloma and correlates with disease status and survival. Br J Haematol. 2012;158(6):727–738. doi:10.1111/j.1365-2141.2012.09241.x
93. Manier S, Ingegnere T, Escure G, et al. Current state and next-generation CAR-T cells in multiple myeloma. Blood Rev. 2022;54:100929. doi:10.1016/j.blre.2022.100929
94. Anderson Jr LD Jr. Idecabtagene vicleucel (ide-cel) CAR T-cell therapy for relapsed and refractory multiple myeloma. Future Oncol. 2022;18(3):277–289. doi:10.2217/fon-2021-1090
95. Munshi NC, Anderson LD, Shah N, et al. Idecabtagene Vicleucel in Relapsed and Refractory Multiple Myeloma. N Engl J Med. 2021;384(8):705–716. doi:10.1056/NEJMoa2024850
96. Rodriguez-Otero P, Ailawadhi S, Arnulf B, et al. Ide-cel or Standard Regimens in Relapsed and Refractory Multiple Myeloma. N Engl J Med. 2023;388(11):1002–1014. doi:10.1056/NEJMoa2213614
97. Alsina M, Shah N, Raje NS, et al. Updated Results from the Phase I CRB-402 Study of Anti-Bcma CAR-T Cell Therapy bb21217 in Patients with Relapsed and Refractory Multiple Myeloma: correlation of Expansion and Duration of Response with T Cell Phenotypes. Blood. 2020;136(Supplement 1):25–26. doi:10.1182/blood-2020-140410
98. Berdeja JG, Madduri D, Usmani SZ, et al. Ciltacabtagene autoleucel, a B-cell maturation antigen-directed chimeric antigen receptor T-cell therapy in patients with relapsed or refractory multiple myeloma (CARTITUDE-1): a phase 1b/2 open-label study. Lancet. 2021;398(10297):314–324. doi:10.1016/s0140-6736(21)00933-8
99. Cohen AD, Mateos M-V, Cohen YC, et al. Efficacy and safety of cilta-cel in patients with progressive multiple myeloma after exposure to other BCMA-targeting agents. Blood. 2023;141(3):219–230. doi:10.1182/blood.2022015526
100. Gregory T, Cohen AD, Costello CL, et al. Efficacy and Safety of P-Bcma-101 CAR-T Cells in Patients with Relapsed/Refractory (r/r) Multiple Myeloma (MM). Blood. 2018;132(Supplement 1):1012. doi:10.1182/blood-2018-99-111419
101. Rodriguez-Otero P, van de Donk NWCJ, Pillarisetti K, et al. GPRC5D as a novel target for the treatment of multiple myeloma: a narrative review. Blood Cancer J. 2024;14(1):24. doi:10.1038/s41408-023-00966-9
102. Smith EL, Harrington K, Staehr M, et al. GPRC5D is a target for the immunotherapy of multiple myeloma with rationally designed CAR T cells. Sci Transl Med. 2019;11(485). doi:10.1126/scitranslmed.aau7746
103. Zhang M, Wei G, Zhou L, et al. GPRC5D CAR T cells (OriCAR-017) in patients with relapsed or refractory multiple myeloma (POLARIS): a first-in-human, single-centre, single-arm, phase 1 trial. Lancet Haematol. 2023;10(2):e107–e116. doi:10.1016/s2352-3026(22)00372-6
104. Marofi F, Motavalli R, Safonov VA, et al. CAR T cells in solid tumors: challenges and opportunities. Stem Cell Res Ther. 2021;12(1):81. doi:10.1186/s13287-020-02128-1
105. Kaczanowska S, Murty T, Alimadadi A, et al. Immune determinants of CAR-T cell expansion in solid tumor patients receiving GD2 CAR-T cell therapy. Cancer Cell. 2024;42(1):35–51.e8. doi:10.1016/j.ccell.2023.11.011
106. Zhao Z, Li Y, Liu W, et al. Engineered IL-7 receptor enhances the therapeutic effect of AXL-CAR-T cells on triple-negative breast cancer. Biomed Res Int. 2020;2020(1):4795171. doi:10.1155/2020/4795171
107. Del Bufalo F, De Angelis B, Caruana I, et al. GD2-CART01 for relapsed or refractory high-risk neuroblastoma. N Engl J Med. 2023;388(14):1284–1295. doi:10.1056/NEJMoa2210859
108. Liu Z, Zhou J, Yang X, et al. Safety and antitumor activity of GD2-Specific 4SCAR-T cells in patients with glioblastoma. Mol Cancer. 2023;22(1):3. doi:10.1186/s12943-022-01711-9
109. O’Rourke DM, Nasrallah MP, Desai A, et al. A single dose of peripherally infused EGFRvIII-directed CAR T cells mediates antigen loss and induces adaptive resistance in patients with recurrent glioblastoma. Sci Transl Med. 2017;9(399). doi:10.1126/scitranslmed.aaa0984
110. Zhou R, Yazdanifar M, Roy LD, et al. CAR T cells targeting the tumor MUC1 glycoprotein reduce triple-negative breast cancer growth. Front Immunol. 2019;10:1149. doi:10.3389/fimmu.2019.01149
111. Qi C, Gong J, Li J, et al. Claudin18.2-specific CAR T cells in gastrointestinal cancers: phase 1 trial interim results. Nat Med. 2022;28(6):1189–1198. doi:10.1038/s41591-022-01800-8
112. Zhao W, Jia L, Zhang M, et al. The killing effect of novel bi-specific Trop2/PD-L1 CAR-T cell targeted gastric cancer. Am J Cancer Res. 2019;9(8):1846–1856.
113. Zhang Y, Liang K, Zhou X, et al. Combination therapy of DKK1 inhibition and NKG2D chimeric antigen receptor T cells for the treatment of gastric cancer. Cancer Sci. 2023;114(7):2798–2809. doi:10.1111/cas.15828
114. Kim M, Pyo S, Kang CH, et al. Folate receptor 1 (FOLR1) targeted chimeric antigen receptor (CAR) T cells for the treatment of gastric cancer. PLoS One. 2018;13(6):e0198347. doi:10.1371/journal.pone.0198347
115. Ye J, Liu Q, He Y, et al. Combined therapy of CAR-IL-15/IL-15Rα-T cells and GLIPR1 knockdown in cancer cells enhanced anti-tumor effect against gastric cancer. J Transl Med. 2024;22(1):171. doi:10.1186/s12967-024-04982-6
116. Wang S, Wei W, Yuan Y, et al. Cell-surface GRP78-targeted chimeric antigen receptor T cells eliminate lung cancer tumor xenografts. Int J Mol Sci. 2024;25(1). doi:10.3390/ijms25010564
117. Yaga M, Hasegawa K, Ikeda S, et al. CD98 heavy chain protein is overexpressed in non-small cell lung cancer and is a potential target for CAR T-cell therapy. Sci Rep. 2024;14(1):17917. doi:10.1038/s41598-024-68779-9
118. Wallstabe L, Göttlich C, Nelke LC, et al. ROR1-CAR T cells are effective against lung and breast cancer in advanced microphysiologic 3D tumor models. JCI Insight. 2019;4(18). doi:10.1172/jci.insight.126345
119. Chinsuwan T, Hirabayashi K, Mishima S, et al. Ligand-based, piggyBac-engineered CAR-T cells targeting EGFR are safe and effective against non-small cell lung cancers. Mol Ther Oncolytics. 2023;31:100728. doi:10.1016/j.omto.2023.100728
120. Zhou D, Byers LA, Sable B, et al. Clinical pharmacology profile of AMG 119, the first chimeric antigen receptor T (CAR-T) cell therapy targeting delta-like ligand 3 (DLL3), in patients with relapsed/refractory small cell lung cancer (SCLC). J Clin Pharmacol. 2024;64(3):362–370. doi:10.1002/jcph.2346
121. Nie F, Chen Y, Hu Y, et al. TREM1 / DAP12 based novel multiple chain CAR-T cells targeting DLL3 show robust anti-tumour efficacy for small cell lung cancer. Immunology. 2024;172(3):362–374. doi:10.1111/imm.13776
122. Katz SC, Hardaway J, Prince E, et al. HITM-SIR: phase Ib trial of intraarterial chimeric antigen receptor T-cell therapy and selective internal radiation therapy for CEA(+) liver metastases. Cancer Gene Ther. 2020;27(5):341–355. doi:10.1038/s41417-019-0104-z
123. Liu X, Gao F, Jiang L, et al. 32A9, a novel human antibody for designing an immunotoxin and CAR-T cells against glypican-3 in hepatocellular carcinoma. J Transl Med. 2020;18(1):295. doi:10.1186/s12967-020-02462-1
124. Baybutt TR, Entezari AA, Caspi A, et al. CD8α structural domains enhance GUCY2C CAR-T cell efficacy. Cancer Biol Ther. 2024;25(1):2398801. doi:10.1080/15384047.2024.2398801
125. Cao Y, Efetov SK, He M, et al. Updated clinical perspectives and challenges of chimeric antigen receptor-T cell therapy in colorectal cancer and invasive breast cancer. Arch Immunol Ther Exp. 2023;71(1):19. doi:10.1007/s00005-023-00684-x
126. Yue J, Shao S, Zhou J, et al. A bispecific antibody targeting HER2 and CLDN18.2 eliminates gastric cancer cells expressing dual antigens by enhancing the immune effector function. Invest New Drugs. 2024;42(1):106–115. doi:10.1007/s10637-024-01417-3
127. Zhang Y, Tacheva-Grigorova SK, Sutton J, et al. Allogeneic CAR T cells targeting DLL3 are efficacious and safe in preclinical models of small cell lung cancer. Clin Cancer Res. 2023;29(5):971–985. doi:10.1158/1078-0432.Ccr-22-2293
128. Dagar G, Gupta A, Masoodi T, et al. Harnessing the potential of CAR-T cell therapy: progress, challenges, and future directions in hematological and solid tumor treatments. J Transl Med. 2023;21(1):449. doi:10.1186/s12967-023-04292-3
129. Krenciute G, Prinzing BL, Yi Z, et al. Transgenic expression of IL15 improves antiglioma activity of IL13Rα2-CAR T cells but results in antigen loss variants. Cancer Immunol Res. 2017;5(7):571–581. doi:10.1158/2326-6066.Cir-16-0376
130. Hu Y, Zhou Y, Zhang M, et al. CRISPR/Cas9-engineered universal CD19/CD22 dual-targeted CAR-T cell therapy for relapsed/refractory B-cell acute lymphoblastic leukemia. Clin Cancer Res. 2021;27(10):2764–2772. doi:10.1158/1078-0432.Ccr-20-3863
131. Choi BD, Yu X, Castano AP, et al. CAR-T cells secreting BiTEs circumvent antigen escape without detectable toxicity. Nat Biotechnol. 2019;37(9):1049–1058. doi:10.1038/s41587-019-0192-1
132. Correia MP, Stojanovic A, Wels WS, et al. Innate-like NKp30 + CD8 + T cells armed with TCR/CAR target tumor heterogeneity. Oncoimmunology. 2021;10(1):1973783. doi:10.1080/2162402x.2021.1973783
133. Adusumilli PS, Zauderer MG, Rivière I, et al. A phase I trial of regional mesothelin-targeted CAR T-cell therapy in patients with malignant pleural disease, in combination with the Anti-PD-1 agent pembrolizumab. Cancer Discov. 2021;11(11):2748–2763. doi:10.1158/2159-8290.Cd-21-0407
134. Zwart ES, van Ee T, Affandi AJ, et al. Spatial immune composition of tumor microenvironment in patients with pancreatic cancer. Cancer Immunol Immunother. 2023;72(12):4385–4397. doi:10.1007/s00262-023-03573-6
135. Zuo C, Xia J, Chen L. Dissecting tumor microenvironment from spatially resolved transcriptomics data by heterogeneous graph learning. Nat Commun. 2024;15(1):5057. doi:10.1038/s41467-024-49171-7
136. Zullo L, Rossi G, Dellepiane C, et al. Safety and efficacy of immune checkpoint inhibitors in non-small-cell lung cancer: focus on challenging populations. Immunotherapy. 2021;13(6):509–525. doi:10.2217/imt-2020-0226
137. Chen J, Zhu T, Jiang G, et al. Target delivery of a PD-1-TREM2 scFv by CAR-T cells enhances anti-tumor efficacy in colorectal cancer. Mol Cancer. 2023;22(1):131. doi:10.1186/s12943-023-01830-x
138. Agarwal S, Aznar MA, Rech AJ, et al. Deletion of the inhibitory co-receptor CTLA-4 enhances and invigorates chimeric antigen receptor T cells. Immunity. 2023;56(10):2388–2407.e9. doi:10.1016/j.immuni.2023.09.001
139. Tang L, Pan S, Wei X, et al. Arming CAR-T cells with cytokines and more: innovations in the fourth-generation CAR-T development. Mol Ther. 2023;31(11):3146–3162. doi:10.1016/j.ymthe.2023.09.021
140. Chen Y, Sun C, Landoni E, et al. Eradication of neuroblastoma by T cells redirected with an optimized GD2-specific chimeric antigen receptor and interleukin-15. Clin Cancer Res. 2019;25(9):2915–2924. doi:10.1158/1078-0432.Ccr-18-1811
141. Chmielewski M, Abken H. CAR T Cells Releasing IL-18 Convert to T-Bet(high) FoxO1(low) Effectors that Exhibit Augmented Activity against Advanced Solid Tumors. Cell Rep. 2017;21(11):3205–3219. doi:10.1016/j.celrep.2017.11.063
142. Zhang N, Liu X, Qin J, et al. LIGHT/TNFSF14 promotes CAR-T cell trafficking and cytotoxicity through reversing immunosuppressive tumor microenvironment. Mol Ther. 2023;31(9):2575–2590. doi:10.1016/j.ymthe.2023.06.015
143. Lu LL, Xiao S-X, Lin Z-Y, et al. GPC3-IL7-CCL19-CAR-T primes immune microenvironment reconstitution for hepatocellular carcinoma therapy. Cell Biol Toxicol. 2023;39(6):3101–3119. doi:10.1007/s10565-023-09821-w
144. Kawalekar OU, O’ Connor RS, Fraietta JA, et al. Distinct signaling of coreceptors regulates specific metabolism pathways and impacts memory development in CAR T cells. Immunity. 2016;44(3):712. doi:10.1016/j.immuni.2016.02.023
145. Moreno-Cortes E, Franco-Fuquen P, Garcia-Robledo JE, et al. ICOS and OX40 tandem co-stimulation enhances CAR T-cell cytotoxicity and promotes T-cell persistence phenotype. Front Oncol. 2023;13:1200914. doi:10.3389/fonc.2023.1200914
146. Alvanou M, Lysandrou M, Christophi P, et al. Empowering the potential of CAR-T cell immunotherapies by epigenetic reprogramming. Cancers. 2023;15(7):1935. doi:10.3390/cancers15071935
147. Zebley CC, Gottschalk S, Youngblood B. Rewriting history: epigenetic reprogramming of CD8(+) T cell differentiation to enhance immunotherapy. Trends Immunol. 2020;41(8):665–675. doi:10.1016/j.it.2020.06.008
148. Scharping NE, Menk AV, Moreci RS, et al. The tumor microenvironment represses T cell mitochondrial biogenesis to drive intratumoral T cell metabolic insufficiency and dysfunction. Immunity. 2016;45(2):374–388. doi:10.1016/j.immuni.2016.07.009
149. Pauken KE, Sammons MA, Odorizzi PM, et al. Epigenetic stability of exhausted T cells limits durability of reinvigoration by PD-1 blockade. Science. 2016;354(6316):1160–1165. doi:10.1126/science.aaf2807
150. Zebley CC, Brown C, Mi T, et al. CD19-CAR T cells undergo exhaustion DNA methylation programming in patients with acute lymphoblastic leukemia. Cell Rep. 2021;37(9):110079. doi:10.1016/j.celrep.2021.110079
151. Chen GM, Chen C, Das RK, et al. Integrative Bulk and Single-Cell Profiling of Premanufacture T-cell Populations Reveals Factors Mediating Long-Term Persistence of CAR T-cell Therapy. Cancer Discov. 2021;11(9):2186–2199. doi:10.1158/2159-8290.Cd-20-1677
152. Ghoneim HE, Fan Y, Moustaki A, et al. De novo epigenetic programs inhibit PD-1 blockade-mediated T Cell rejuvenation. Cell. 2017;170(1):142–157.e19. doi:10.1016/j.cell.2017.06.007
153. Wang Y, Tong C, Dai H, et al. Low-dose decitabine priming endows CAR T cells with enhanced and persistent antitumour potential via epigenetic reprogramming. Nat Commun. 2021;12(1):409. doi:10.1038/s41467-020-20696-x
154. Liu Z, Li X, Gao Y, et al. Epigenetic reprogramming of Runx3 reinforces CD8 + T-cell function and improves the clinical response to immunotherapy. Mol Cancer. 2023;22(1):84. doi:10.1186/s12943-023-01768-0
155. Weber EW, Parker KR, Sotillo E, et al. Transient rest restores functionality in exhausted CAR-T cells through epigenetic remodeling. Science. 2021;372(6537). doi:10.1126/science.aba1786
156. Pace L, Goudot C, Zueva E, et al. The epigenetic control of stemness in CD8 + T cell fate commitment. Science. 2018;359(6372):177–186. doi:10.1126/science.aah6499
157. Saitakis M. Epigenetic reprogramming of CAR T cells for in vivo functional persistence against solid tumors. Genes Immun. 2024;25(5):434–436. doi:10.1038/s41435-024-00262-x
158. Wang Y, Zhao G, Wang S, et al. Deleting SUV39H1 in CAR-T cells epigenetically enhances the antitumor function. MedComm. 2024;5(5):e552. doi:10.1002/mco2.552
159. Niborski LL, Gueguen P, Ye M, et al. CD8+T cell responsiveness to anti-PD-1 is epigenetically regulated by Suv39h1 in melanomas. Nat Commun. 2022;13(1):3739. doi:10.1038/s41467-022-31504-z
160. Vo LT, Kinney MA, Liu X, et al. Regulation of embryonic haematopoietic multipotency by EZH1. Nature. 2018;553(7689):506–510. doi:10.1038/nature25435
161. Shen X, Liu Y, Hsu Y-J, et al. EZH1 mediates methylation on histone H3 lysine 27 and complements EZH2 in maintaining stem cell identity and executing pluripotency. Mol Cell. 2008;32(4):491–502. doi:10.1016/j.molcel.2008.10.016
162. Jing R, Scarfo I, Najia MA, et al. EZH1 repression generates mature iPSC-derived CAR T cells with enhanced antitumor activity. Cell Stem Cell. 2022;29(8):1181–1196.e6. doi:10.1016/j.stem.2022.06.014
163. Verma V, Jafarzadeh N, Boi S, et al. MEK inhibition reprograms CD8(+) T lymphocytes into memory stem cells with potent antitumor effects. Nat Immunol. 2021;22(1):53–66. doi:10.1038/s41590-020-00818-9
164. Scott AC, Dündar F, Zumbo P, et al. TOX is a critical regulator of tumour-specific T cell differentiation. Nature. 2019;571(7764):270–274. doi:10.1038/s41586-019-1324-y
165. Sekine T, Perez-Potti A, Nguyen S, et al. TOX is expressed by exhausted and polyfunctional human effector memory CD8 + T cells. Sci Immunol. 2020;5(49). doi:10.1126/sciimmunol.aba7918
166. Beltra JC, Abdel-Hakeem MS, Manne S, et al. Stat5 opposes the transcription factor Tox and rewires exhausted CD8(+) T cells toward durable effector-like states during chronic antigen exposure. Immunity. 2023;56(12):2699–2718.e11. doi:10.1016/j.immuni.2023.11.005
167. Chapman NM, Boothby MR, Chi H. Metabolic coordination of T cell quiescence and activation. Nat Rev Immunol. 2020;20(1):55–70. doi:10.1038/s41577-019-0203-y
168. Franco F, Jaccard A, Romero P, et al. Metabolic and epigenetic regulation of T-cell exhaustion. Nat Metab. 2020;2(10):1001–1012. doi:10.1038/s42255-020-00280-9
169. Wang G, Wang -J-J, Guan R, et al. Targeting strategies for glucose metabolic pathways and T cells in colorectal cancer. Curr Cancer Drug Targets. 2019;19(7):534–550. doi:10.2174/1568009618666181015150138
170. Davern M, Donlon NE, O’Connell F, et al. Nutrient deprivation and hypoxia alter T cell immune checkpoint expression: potential impact for immunotherapy. J Cancer Res Clin Oncol. 2023;149(8):5377–5395. doi:10.1007/s00432-022-04440-0
171. Yan Y, Zhang G-X, Gran B, et al. IDO upregulates regulatory T cells via tryptophan catabolite and suppresses encephalitogenic T cell responses in experimental autoimmune encephalomyelitis. J Immunol. 2010;185(10):5953–5961. doi:10.4049/jimmunol.1001628
172. Wang W, Guo M-N, Li N, et al. Glutamine deprivation impairs function of infiltrating CD8 + T cells in hepatocellular carcinoma by inducing mitochondrial damage and apoptosis. World J Gastrointest Oncol. 2022;14(6):1124–1140. doi:10.4251/wjgo.v14.i6.1124
173. Facciabene A, Peng X, Hagemann IS, et al. Tumour hypoxia promotes tolerance and angiogenesis via CCL28 and T(reg) cells. Nature. 2011;475(7355):226–230. doi:10.1038/nature10169
174. Lopez Krol A, Nehring HP, Krause FF, et al. Lactate induces metabolic and epigenetic reprogramming of pro-inflammatory Th17 cells. EMBO Rep. 2022;23(12):e54685. doi:10.15252/embr.202254685
175. Ma X, Bi E, Lu Y, et al. Cholesterol induces CD8(+) T cell exhaustion in the tumor microenvironment. Cell Metab. 2019;30(1):143–156.e5. doi:10.1016/j.cmet.2019.04.002
176. Kleinfeld AM, Okada C. Free fatty acid release from human breast cancer tissue inhibits cytotoxic T-lymphocyte-mediated killing. J Lipid Res. 2005;46(9):1983–1990. doi:10.1194/jlr.M500151-JLR200
177. Corn KC, Windham MA, Rafat M. Lipids in the tumor microenvironment: from cancer progression to treatment. Prog Lipid Res. 2020;80:101055. doi:10.1016/j.plipres.2020.101055
178. Guerrero JA, Klysz DD, Chen Y, et al. GLUT1 overexpression in CAR-T cells induces metabolic reprogramming and enhances potency. Nat Commun. 2024;15(1):8658. doi:10.1038/s41467-024-52666-y
179. Liu Y, Liang X, Dong W, et al. Tumor-repopulating cells induce PD-1 expression in CD8(+) T cells by transferring kynurenine and AhR activation. Cancer Cell. 2018;33(3):480–494.e7. doi:10.1016/j.ccell.2018.02.005
180. Jeng MY, Hull PA, Fei M, et al. Metabolic reprogramming of human CD8(+) memory T cells through loss of SIRT1. J Exp Med. 2018;215(1):51–62. doi:10.1084/jem.20161066
181. Peng JJ, Wang L, Li Z, et al. Metabolic challenges and interventions in CAR T cell therapy. Sci Immunol. 2023;8(82):eabq3016. doi:10.1126/sciimmunol.abq3016
182. Zhang M, Jin X, Sun R, et al. Optimization of metabolism to improve efficacy during CAR-T cell manufacturing. J Transl Med. 2021;19(1):499. doi:10.1186/s12967-021-03165-x
183. Zhang T, Zhang Z, Li F, et al. miR-143 regulates memory T cell differentiation by reprogramming T cell metabolism. J Immunol. 2018;201(7):2165–2175. doi:10.4049/jimmunol.1800230
184. Zang J, Yang Y, Zheng X, et al. Dynamic tagging to drive arginine nano-assembly to metabolically potentiate immune checkpoint blockade therapy. Biomaterials. 2023;292:121938. doi:10.1016/j.biomaterials.2022.121938
185. Zhu L, Shi Y, Feng Z, et al. Fatostatin promotes anti-tumor immunity by reducing SREBP2 mediated cholesterol metabolism in tumor-infiltrating T lymphocytes. Eur J Pharmacol. 2024;971:176519. doi:10.1016/j.ejphar.2024.176519
186. Zhao L, Li J, Liu Y, et al. Cholesterol esterification enzyme inhibition enhances antitumor effects of human chimeric antigen receptors modified T cells. J Immunother. 2018;41(2):45–52. doi:10.1097/cji.0000000000000207
187. Zhang Y, Kurupati R, Liu L, et al. Enhancing CD8(+) T cell fatty acid catabolism within a metabolically challenging tumor microenvironment increases the efficacy of melanoma immunotherapy. Cancer Cell. 2017;32(3):377–391.e9. doi:10.1016/j.ccell.2017.08.004
188. Vodnala SK, Eil R, Kishton RJ, et al. T cell stemness and dysfunction in tumors are triggered by a common mechanism. Science. 2019;363(6434). doi:10.1126/science.aau0135
189. Shao M, Teng X, Guo X, et al. Inhibition of calcium signaling prevents exhaustion and enhances anti-leukemia efficacy of CAR-T Cells via SOCE-calcineurin-NFAT and glycolysis pathways. Adv Sci. 2022;9(9):e2103508. doi:10.1002/advs.202103508
190. Carnevale J, Shifrut E, Kale N, et al. RASA2 ablation in T cells boosts antigen sensitivity and long-term function. Nature. 2022;609(7925):174–182. doi:10.1038/s41586-022-05126-w
191. Ajmal I, Farooq MA, Duan Y, et al. Intrinsic ADRB2 inhibition improves CAR-T cell therapy efficacy against prostate cancer. Mol Ther. 2024;32(10):3539–3557. doi:10.1016/j.ymthe.2024.08.028
192. Zhao Y, Chen J, Andreatta M, et al. IL-10-expressing CAR T cells resist dysfunction and mediate durable clearance of solid tumors and metastases. Nat Biotechnol. 2024;42(11):1693–1704. doi:10.1038/s41587-023-02060-8
193. Wei J, Long L, Zheng W, et al. Targeting REGNASE-1 programs long-lived effector T cells for cancer therapy. Nature. 2019;576(7787):471–476. doi:10.1038/s41586-019-1821-z
194. Wenes M, Jaccard A, Wyss T, et al. The mitochondrial pyruvate carrier regulates memory T cell differentiation and antitumor function. Cell Metab. 2022;34(5):731–746.e9. doi:10.1016/j.cmet.2022.03.013
195. Mai D, Johnson O, Reff J, et al. Combined disruption of T cell inflammatory regulators Regnase-1 and Roquin-1 enhances antitumor activity of engineered human T cells. Proc Natl Acad Sci U S A. 2023;120(12):e2218632120. doi:10.1073/pnas.2218632120
196. Zahid A, Siegler EL, Kenderian SS. CART cell toxicities: new insight into mechanisms and management. Clin Hematol Int. 2020;2(4):149–155. doi:10.2991/chi.k.201108.001
197. Zhou X, Rasche L, Kortüm KM, et al. Toxicities of chimeric antigen receptor T cell therapy in multiple myeloma: an overview of experience from clinical trials. Pathophysiology, and Management Strategies Front Immunol. 2020;11:620312. doi:10.3389/fimmu.2020.620312
198. Santomasso BD, Park JH, Salloum D, et al. Clinical and biological correlates of neurotoxicity associated with CAR T-cell therapy in patients with B-cell acute lymphoblastic leukemia. Cancer Discov. 2018;8(8):958–971. doi:10.1158/2159-8290.Cd-17-1319
199. Santomasso B, Bachier C, Westin J, et al. The other side of CAR T-cell therapy: cytokine release syndrome, neurologic toxicity, and financial burden. Am Soc Clin Oncol Educ Book. 2019;39:433–444. doi:10.1200/edbk_238691
200. Zia K, Nur-e-Alam M, Ahmad A, et al. Taming the cytokine storm: small molecule inhibitors targeting IL-6/IL-6α receptor. Mol Divers. 2024;28(6):4151–4165. doi:10.1007/s11030-023-10805-5
201. Zhong G, Zhang X, Guo Z, et al. Complete remission of advanced pancreatic cancer induced by claudin18.2-targeted CAR-T cell therapy: a case report. Front Immunol. 2024;15:1325860. doi:10.3389/fimmu.2024.1325860
202. Nellan A, McCully CML, Cruz Garcia R, et al. Improved CNS exposure to tocilizumab after cerebrospinal fluid compared to intravenous administration in rhesus macaques. Blood. 2018;132(6):662–666. doi:10.1182/blood-2018-05-846428
203. Wehrli M, Gallagher K, Chen Y-B, et al. Single-center experience using anakinra for steroid-refractory immune effector cell-associated neurotoxicity syndrome (ICANS). J Immunother Cancer. 2022;10(1):e003847. doi:10.1136/jitc-2021-003847
204. Zhang L, Wang S, Xu J, et al. Etanercept as a new therapeutic option for cytokine release syndrome following chimeric antigen receptor T cell therapy. Exp Hematol Oncol. 2021;10(1):16. doi:10.1186/s40164-021-00209-2
205. Teachey DT, Lacey SF, Shaw PA, et al. Identification of Predictive Biomarkers for Cytokine Release Syndrome after Chimeric Antigen Receptor T-cell Therapy for Acute Lymphoblastic Leukemia. Cancer Discov. 2016;6(6):664–679. doi:10.1158/2159-8290.Cd-16-0040
206. Sterner RM, Sakemura R, Cox MJ, et al. GM-CSF inhibition reduces cytokine release syndrome and neuroinflammation but enhances CAR-T cell function in xenografts. Blood. 2019;133(7):697–709. doi:10.1182/blood-2018-10-881722
207. Yakoub-Agha I, Chabannon C, Bader P, et al. Management of adults and children undergoing chimeric antigen receptor T-cell therapy: best practice recommendations of the European Society for Blood and Marrow Transplantation (EBMT) and the Joint Accreditation Committee of ISCT and EBMT (JACIE). Haematologica. 2020;105(2):297–316. doi:10.3324/haematol.2019.229781
208. Davila ML, Riviere I, Wang X, et al. Efficacy and toxicity management of 19-28z CAR T cell therapy in B cell acute lymphoblastic leukemia. Sci Transl Med. 2014;6(224):224ra25. doi:10.1126/scitranslmed.3008226
209. Zhang X, Wang X, Sun L, et al. Tofacitinib reduces acute lung injury and improves survival in a rat model of sepsis by inhibiting the JAK-STAT/NF-κB pathway. J Inflamm (Lond). 2023;20(1):5. doi:10.1186/s12950-023-00332-3
210. Zi FM, Ye LL, Zheng JF, et al. Using JAK inhibitor to treat cytokine release syndrome developed after chimeric antigen receptor T cell therapy for patients with refractory acute lymphoblastic leukemia: a case report. Medicine. 2021;100(19):e25786. doi:10.1097/md.0000000000025786
211. Mascarenhas J, Hoffman R. Ruxolitinib: the first FDA approved therapy for the treatment of myelofibrosis. Clin Cancer Res. 2012;18(11):3008–3014. doi:10.1158/1078-0432.Ccr-11-3145
212. Ruella M, Kenderian SS, Shestova O, et al. Kinase inhibitor ibrutinib to prevent cytokine-release syndrome after anti-CD19 chimeric antigen receptor T cells for B-cell neoplasms. Leukemia. 2017;31(1):246–248. doi:10.1038/leu.2016.262
213. Liu M, Deng H, Mu J, et al. Ibrutinib improves the efficacy of anti-CD19-CAR T-cell therapy in patients with refractory non-Hodgkin lymphoma. Cancer Sci. 2021;112(7):2642–2651. doi:10.1111/cas.14915
214. Zhang A, Sun Y, Du J, et al. Reducing Hinge Flexibility of CAR-T Cells Prolongs Survival In Vivo With Low Cytokines Release. Front Immunol. 2021;12:724211. doi:10.3389/fimmu.2021.724211
215. Zhang Y, Qin D, Shou AC, et al. Exploring CAR-T Cell Therapy Side Effects: mechanisms and Management Strategies. J Clin Med. 2023;12(19):6124. doi:10.3390/jcm12196124
216. Parker KR, Migliorini D, Perkey E, et al. Single-Cell Analyses Identify Brain Mural Cells Expressing CD19 as Potential Off-Tumor Targets for CAR-T Immunotherapies. Cell. 2020;183(1):126–142.e17. doi:10.1016/j.cell.2020.08.022
217. Uy NF, Pequignot E, Frey NV, et al. Hypogammaglobulinemia and Infection Risk in Chronic Lymphocytic Leukemia (CLL) Patients Treated with CD19-Directed Chimeric Antigen Receptor T (CAR-T) Cells. Blood. 2020;136(Supplement 1):30–32. doi:10.1182/blood-2020-141224
218. Feng K, Liu Y, Guo Y, et al. Phase I study of chimeric antigen receptor modified T cells in treating HER2-positive advanced biliary tract cancers and pancreatic cancers. Protein Cell. 2018;9(10):838–847. doi:10.1007/s13238-017-0440-4
219. Lamers CH, Sleijfer S, van Steenbergen S, et al. Treatment of metastatic renal cell carcinoma with CAIX CAR-engineered T cells: clinical evaluation and management of on-target toxicity. Mol Ther. 2013;21(4):904–912. doi:10.1038/mt.2013.17
220. Sasaki T, Sakoda Y, Adachi K, et al. Therapeutic effects of anti-GM2 CAR-T cells expressing IL-7 and CCL19 for GM2-positive solid cancer in xenograft model. Cancer Med. 2023;12(11):12569–12580. doi:10.1002/cam4.5907
221. Wu CY, Roybal KT, Puchner EM, et al. Remote control of therapeutic T cells through a small molecule-gated chimeric receptor. Science. 2015;350(6258):aab4077. doi:10.1126/science.aab4077
222. Halliwell E, Vitali A, Muller H, et al. Targeting of low ALK antigen density neuroblastoma using AND logic-gate engineered CAR-T cells. Cytotherapy. 2023;25(1):46–58. doi:10.1016/j.jcyt.2022.10.007
223. Zhu L, Man C-W, Harrison RES, et al. Engineering a Programmed Death-Ligand 1-Targeting Monobody Via Directed Evolution for SynNotch-Gated Cell Therapy. ACS Nano. 2024;18(11):8531–8545. doi:10.1021/acsnano.4c01597
224. Qin C, Dong M-H, Zhou L-Q, et al. Single-cell analysis of refractory anti-SRP necrotizing myopathy treated with anti-BCMA CAR-T cell therapy. Proc Natl Acad Sci U S A. 2024;121(6):e2315990121. doi:10.1073/pnas.2315990121
225. Taubmann J, Müller F, Yalcin Mutlu M, et al. CD19 chimeric antigen receptor T cell treatment: unraveling the role of B cells in systemic lupus erythematosus. Arthritis Rheumatol. 2024;76(4):497–504. doi:10.1002/art.42784
226. Wang W, He S, Zhang W, et al. BCMA-CD19 compound CAR T cells for systemic lupus erythematosus: a phase 1 open-label clinical trial. Ann Rheum Dis. 2024;83(10):1304–1314. doi:10.1136/ard-2024-225785
 © 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.