Back to Journals » OncoTargets and Therapy » Volume 13
Crosstalk Between AR and Wnt Signaling Promotes Castration-Resistant Prostate Cancer Growth
Authors Luo J, Wang D, Wan X, Xu Y, Lu Y, Kong Z, Li D, Gu W, Wang C, Li Y, Ji C, Gu S, Xu Y
Received 13 January 2020
Accepted for publication 24 July 2020
Published 18 September 2020 Volume 2020:13 Pages 9257—9267
DOI https://doi.org/10.2147/OTT.S245861
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr XuYu Yang
Jun Luo1 ,* Dan Wang2 ,* Xuechao Wan,2 Yangguang Xu,2 Yali Lu,2 Zhe Kong,2 Dujian Li,1 Wei Gu,1 Chenji Wang,2 Yao Li,2 Chaoneng Ji,2 Shaohua Gu,2 Yaoting Xu1
1Department of Urology, Shanghai Fourth People’s Hospital Affiliated to Tongji University School of Medicine, Shanghai, People’s Republic of China; 2Shanghai Engineering Research Center of Industrial Microorganisms, School of Life Science, Fudan University, Shanghai 200433, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Shaohua Gu; Yaoting Xu Email [email protected]; [email protected]
Introduction: Prostate cancer (PCa) is the most commonly diagnosed cancer and the third leading cause of cancer-related death in males in the United States. Despite the initial efficacy of androgen deprivation therapy in prostate cancer (PCa) patients, most patients progress to castration-resistant prostate cancer. However, the mechanisms underlying the androgen-independent progression of PCa remain largely unknown.
Methods: In this study, we established a PCa cell line (LNCaP-AI) by maintaining LNCaP cells under androgen-depleted conditions. To explore the cellular and molecular mechanisms of androgen-independent growth of PCa, we analyzed the gene expression patterns in androgen-independent prostate cancer (AIPC) compared with that in androgen-dependent prostate cancer (ADPC). KEGG pathway analysis revealed that Wnt signaling pathways were activated after androgen deprivation therapy (ADT). In vitro experiments showed that the inhibition of Wnt pathway reduced AIPC cell growth by inhibiting cell cycle progression and promoting apoptosis. Furthermore, WNT5A, LEF1 were identified as direct targets of AR by chromatin immunoprecipitation (ChIP) assay and public ChIP-seq datasets analysis.
Results: In the present study, we found a regulatory mechanism through which crosstalk between androgen receptor (AR) and Wnt signals promoted androgen-independent conversion of PCa. The Wnt pathway was inhibited by androgen in androgen-dependent prostate cancer cells, but this blocking effect was not elicited in androgen-independent prostate cancer (AIPC) cells. Moreover, Wnt pathway genes WNT5A and LEF1 were directly downregulated by AR. In vitro experiments showed that inhibition of the Wnt pathways repressed AIPC cell growth by inhibiting cell cycle progression and promoting apoptosis. We found that WNT5A and LEF1 were downregulated in low-grade PCa but upregulated in metastatic PCa.
Conclusion: In summary, we revealed that crosstalk between AR and Wnt signaling pathways promotes androgen-independent growth of PCa, which may provide novel therapeutic opportunities for castration-resistant prostate cancer.
Keywords: Wnt pathway, prostate cancer, androgen receptor, androgen deprivation therapy
Introduction
Prostate cancer (PCa) is a frequently diagnosed cancer and the third leading cause of cancer-related death among males in the US.1 Androgen deprivation therapy (ADT), a standard therapy for PCa, restrains the progression of androgen-dependent prostate cancer (ADPC) by disrupting the androgen receptor (AR) signaling pathway.2,3 Unfortunately, PCa can develop resistance to ADT after 18–24 months of treatment, resulting in a poor prognosis.4,5 Thus, identifying biological mechanisms contributing to hormone-independent growth of PCa is crucial.
The exact molecular mechanisms of PCa remain unclear. AR plays a critical role in prostate carcinogenesis by regulating transcriptional targets, gene fusions, and genomic stability.6,7 Therapies targeting the AR pathway have recently been proven to be effective against castration-resistant prostate cancer (CRPC). However, current clinical inhibitors targeting AR alone only provide a 3–4 month survival benefit for CRPC patients,8 suggesting that other pathways may be activated during ADT.
Emerging evidence has demonstrated that several pathways, including mTOR and Wnt pathways, are related to CRPC. Several reports have shown that upregulation of PI3K/AKT/mTOR pathways is associated with the progression of prostate cancer owing to PTEN loss.9–11 These kinase pathways also enhance the activity of AR by mediating the phosphorylation of coactivators.12,13 The roles of the Wnt/β-catenin pathway in various types of cancers have been widely reported.14–16 Recent microarray and RNA-sequencing studies have revealed activation of Wnt signaling in CRPC.17 Thus, elucidating the molecular crosstalk between the androgen signaling pathway and these pathways may facilitate identification of novel therapeutic targets for PCa.
In the present study, we established a PCa cell line (LNCaP-AI) by maintaining LNCaP cells under androgen-depleted conditions. GSE8702 and GSE33316 are used to identify important functional pathways in androgen-independent transformation of PCa. Using bioinformatics analysis, we identify two main hub genes (LEF1 and WNT5A) that regulated the crosstalk between AR and Wnt signaling to promote CRPC, thereby providing novel therapeutic targets for clinical treatment of CRPC.
Materials and Methods
Microarray Data and Data Preprocessing
Microarray data of GSE33316, GSE8702, GSE6919 were download from the National Center for Biotechnology Information Gene Expression Omnibus (GEO) database (http://www.ncbi.nlm.nih.gov/geo/) and a log2 transformation was performed. All sample data were normalized using the limma package in R software version 3.5.1. The differentially expressed genes (DEGs) were obtained with thresholds of |logFC|>1.5 and P<0.05, using linear models and empirical Bayes methods. Clustering analysis was performed using the DEGs and a heatmap of different groups was constructed.
KEGG Pathway Analysis
KEGG pathway enrichment analysis was also performed to identify pathways enriched in PCa using the MAS3.0 system (http://bioinfo.capitalbio.com/mas3/). The P-value was calculated by hypergeometric distribution and a pathway with P < 0.05 was considered as significant.
Cell Culture
LNCaP cells were purchased from the American Type Culture Collection. The WPMY-1 were obtained from Cell Bank of Chinese Academy of Sciences (Shanghai, China). The two cell lines (LNCaP and WPMY-1 cells) were maintained in RPMI medium with 10% FBS, 1% penicillin/streptomycin, 1% nonessential amino acids and 1% sodium pyruvate, and were cultured at 37°C in 5% CO2. LNCaP-AI cells were established by LNCaP cells in RPMI medium supplemented with 10% charcoal-dextran-stripped FBS for more than 12 months. Although more than 99% of cells undergoing apoptosis during 3 months of cell culture in DCC-FBS, the remaining new cell line, LNCaP-AI, begun to grow after these 3 months.
RNA Interference and Transient Transfection
Cells were seeded in plates and transfected with siRNAs using Lipofectamine 3000, following the instructions. All sequences of synthetic oligonucleotides are listed inSupplementary Table 1.
Real-Time Reverse Transcription PCR (qRT-PCR) Analysis
QRT-PCR for mRNAs was performed as described previously.18 Total RNA was extracted from PCa cells with Trizol (Invitrogen, Carlsbad, CA, USA), and reverse transcription was performed according to the manual of the PrimeScript RT reagent kit (TaKaRa, Tokyo, Japan). Real-time quantitative PCR was tried out with Ultra SYBR Mixture (with ROX) kit (CWBIO, Beijing, China) on the LightCycler 480II (Roche Applied Science, Basel, Switzerland) instrument. The Ct values were normalized using β-actin or RNU6 as an internal control to estimate the different expression of genes. Relative mRNA expression was calculated using the 2−ΔΔCt method. Each sample was run in triplicate to ensure quantitative accuracy. All sequences of synthetic oligonucleotides are listed in Supplementary Table 2.
Chromatin Immunoprecipitation (ChIP) Assay
ChIP was performed as described previously.19 Ethanol and dimethyl sulphoxide (DMSO) were used as vehicles of dihydrotestosterone (DHT) and Casodex (CDX), respectively. LNCaP cells were treated with DHT (100 nM), CDX (10 μM), DHT-CDX combination, or vehicles for 4 h before harvesting. DNA fragments were purified and analyzed by real-time PCR.
Western Blotting Analysis
The Western blotting analysis was performed as described previously.19 Cells were lysed in RIPA buffer (CWBIO, Beijing, China) supplemented with protease inhibitors (Complete, EDTA-free; Roche Diagnostics) and PMSF (Calbiochem). Lysates were separated on a 12% acrylamide gel and subjected to Western blot analysis. Immunoblots were incubated overnight at 4°C with the primary antibodies and anti-Actin antibodies (goat polyclonal; Santa Cruz Biotechnology; 1:2, 000). Goat anti-mouse IgG-HRP and goat anti-rabbit IgG-HRP (Sigma–Aldrich, USA) secondary antibodies were used to visualize bands using Amersham ECL Prime (GE Healthcare, UK). The signal intensity of Western blots was quantified by Quantity One Software (Bio-Rad, USA).
Cell Proliferation Assay
Cell proliferation assay was performed as described previously using Cell Counting Kit-8 according to the manufacturer’s instructions. Absorbance was measured at 450 nm with Microplate Reader ELx808. The absorbance at 630 nm was used as a reference.
Cell Cycle and Apoptosis Assay
Cells were harvested after 48-hr transfection. For cycle assay, cells were incubated with 0.03% Triton X-100 and propidium iodide (PI) (50 ng/mL) for 15 min; the percentages of cells in different phases of the cell cycle were measured with a FACScalibur flow cytometer and analyzed with ModFit software. For apoptosis assay, cells were assayed with FITC Annexin V Apoptosis Detection Kit and analyzed by flow cytometry.
Statistical Analysis
The numerical data were presented as mean ± standard deviation (SD) of at least three determinations. Statistical comparisons between groups of normalized data were performed using T-test or Mann–Whitney U-test according to the test condition. A p < 0.05 was considered statistical significance with a 95% confidence level.
Results
AR Transcriptional Activity Downregulates Significantly During Androgen Deprivation in LNCaP-AI Cells Compared with LNCaP Cells
Previous studies have shown that PCa cells under long-term androgen-deprived (LTAD) conditions survive hormone deprivation therapy and gain insensitivity to bicalutamide (CDX), an AR antagonist.20–22 To explore the molecular mechanisms underlying LTAD, we established an androgen-independent LNCaP-AI cell line derived from LNCaP cells. LNCaP cells were cultivated for 12 months in medium supplemented with 10% dextran-coated charcoal-treated fetal bovine serum. We found that >99% of the cells underwent apoptosis during 12 months of LTAD treatment. The remaining cells, which were named as LNCaP-AI, grew in an androgen-independent environment.
The ability of LNCaP and LNCaP-AI cells to proliferate under dihydrotestosterone (DHT) stimulation was examined by CCK-8 assays (Figure 1A). As expected, the proliferation rate of LNCaP cells was significantly lower than that of LNCaP-AI cells in the androgen-deprivation medium. Thus, LNCaP-AI cells were less sensitive to androgen stimulation than LNCaP cells. We performed CCK-8 assays to assess the effect of CDX on the proliferation of LNCaP and LNCaP-AI cells, and observed that LNCaP-AI cells were less sensitive to CDX treatment compared with LNCaP cells (Figure 1B). Next, we examined the expression of AR during androgen deprivation of LNCaP cells. However, AR protein showed no significant change in LNCaP-AI cells compared with LNCaP cells (Figure 1C).
 |
Figure 1 AR transcriptional activity was significantly downregulated during androgen deprivation in LNCaP-AI cells comparing to LNCaP cells. (A) Cell proliferation analysis was performed to detect the growth rate of LNCaP and LNCaP-AI cells under DHT stimulation. (B) Cell proliferation analysis was performed to detect the growth rate of LNCaP and LNCaP-AI cells at different concentration gradient after CDX treatment. (C) AR protein expression levels in LNCaP, LNCaP-AI, and WPMY-1 cell lines. Actin was used as the internal control. (D–F) qRT-PCR analysis of KLK2 (F), TMPPSS2 (G), KLK3 (H) expression levels in LNCaP and LNCaP-AI cell lines at different time points after DHT treatment. (G–I) qRT-PCR analysis of KLK2 (G), TMPPSS2 (H), KLK3 (I) expression levels in LNCaP and LNCaP-AI cell lines at different concentration gradient of DHT treatment. Data are presented as the mean ± SD (n = 3). Significance was defined as p<0.05 (*p < 0.05; ***p < 0.001); ns means not significant. |
KLK2, TMPRSS2, and KLK3 are direct targets of AR. We conducted qRT-PCR experiments to detect the expression of KLK2, TMPRSS2, and KLK3 in LNCaP and LNCaP-AI cells stimulated by DHT and assessed changes in AR transcriptional activity during androgen deprivation. We observed increased expression of KLK2, TMPRSS2, and KLK3 in both LNCaP and LNCaP-AI cells in a time- and dose-dependent manner. However, the expression of KLK2, TMPRSS2, and KLK3 was significantly higher in LNCaP cells than in LNCaP-AI cells (Figure 1D–I). Taken together, these findings showed that the sensitivity of AR transcriptional activity to DHT stimulation was significantly reduced during long-term androgen deprivation. To understand the potential mechanism suppressing AR transcriptional activity, we determined the expression pattern of AR coregulators under long-term androgen-deprived (LTAD) conditions. We conducted bioinformatics analysis of GSE8702 and found that the expression of AR corepressors, such as NR2C1,23 REL,24 NR2C2,25 and JUN,26,27 was significantly increased in LNCaP-AI cells (Supplementary Figure 1).
Androgen Deprivation Activates the Wnt Pathway
To explore the mechanisms of the androgen-independent behavior of PCa, we identified genes expressed differentially during androgen deprivation by searching two public datasets, GSE870228 and GSE33316.29 A total of 1282 genes were commonly upregulated (Figure 2A), and 854 genes were downregulated during androgen deprivation in these two datasets (Figure 2B). Hierarchical clustering of systematic variations in the expression of genes during androgen deprivation is shown in Figure 2C and D. To identify biological pathways disrupted by ADT, we performed enrichment analysis of up- and down-regulated genes (false discovery rate <0.05) on the basis of KEGG pathway analysis using GSE8702 and GSE33316. MAPK, PI3K/AKT/mTOR, Wnt/β-catenin, and TGF-β pathways contained the highest number of significantly upregulated genes among the cluster of upregulated cancer pathways (Figure 2E, G, H). Previous studies had indicated that these red circles labeled pathways are related to the androgen independent PCa progression. In contrast, downregulated genes were mainly related to p53, insulin, and TGF-β signaling pathways (Figure 2F). To explore the potential direct target genes of AR, we analyzed two public ChIP-seq datasets from LNCaP cells. A total of 11 androgen-responsive genes were identified with androgen response elements (AREs) by intersecting two sets of expression data and two sets of ChIP-seq data, suggesting that these genes were directly regulated by AR (Figure 3A). The resulting heatmap showed that mRNA expression of the 11 candidate genes had changed dynamically after androgen deprivation (Figure 3B and C).
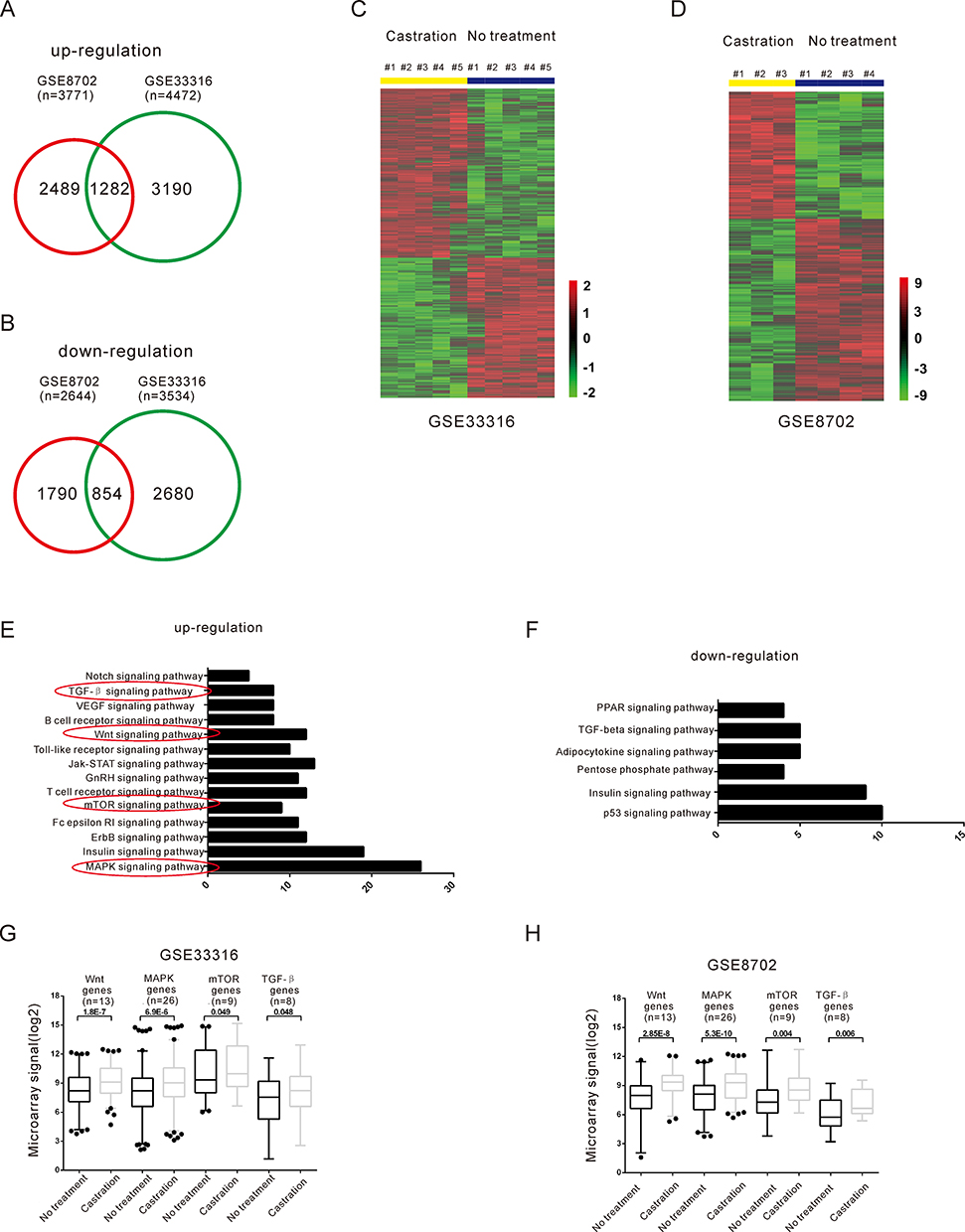 |
Figure 2 Identification of the candidate crosstalk pathways with the AR pathway by analysis of differentially expressed genes during androgen deprivation. (A–B) Venn diagrams display the overlap of up- (A) and down-regulated (B) genes in GSE33316 and GSE8702. (C–D) Hierarchical clustering of differentially expressed genes from GSE33316 (C) and GSE8702 (D). Red color represents the up-regulated genes, and green color represents the down-regulated genes. (E–F) KEGG pathway analysis of up- (E) and down-expressed regulation (F) genes after androgen deprivation. The red circles labeled pathways represent that these pathways are related to the androgen independent PCa progression. The number of genes and P value of each GO category are indicated at the x-axis and next to the bar, respectively. (G–H) The relative expression level of Wnt and MAPK pathways related genes after DHT deprivation compared with control in public gene expression data GSE33316 and GSE8702. |
 |
Figure 3 LEF1 and WNT5A were direct targets of AR. (A) Venn diagrams for androgen-responsive genes, pathways related genes and AR ChIP-seq dataset used to identify candidates mediated the crosstalk between AR and other pathways. (B–C) Heatmap shows 11 candidate genes expression changes after androgen deprivation in GSE33316 (B) and GSE8702 (C). (D–E) qRT-PCR analysis the expression of LEF1 (D), WNT5A (F) after androgen deprivation therapy at indicated times in LNCaP cells. (F–G) qRT-PCR analysis of LEF1 (E), WNT5A (G) expression after DHT stimulation at different time points in LNCaP cells. (H) qRT-PCR analysis the expression of LEF1, WNT5A in LNCaP cells following knockdown AR. (I) CHIP assay of AR-binding on candidate AREs of LEF1, WNT5A. (J) Schematic locations of potential androgen response elements (AREs) in the upstream within 10kb of LEF1, WNT5A transcription start site (TSS). Data are presented as the mean ± SD (n = 3). Significance was defined as p<0.05 (*p < 0.05; **p < 0.01; ***p < 0.001); ns means not significant. |
In this study, we focused on potential regulation of Wnt-pathway-related genes LEF1 and WNT5A via androgen deprivation. Our qRT-PCR analysis confirmed that the mRNA expression levels of LEF1 and WNT5A increased after DHT deprivation (Figure 3D and E). Moreover, mRNA expression of LEF1 and WNT5A was reduced by DHT in a time- and dose-dependent manner in LNCaP cells (Figure 3F and G). The mRNA expression of LEF1 and WNT5A was markedly elevated by AR knockdown in LNCaP cells (Figure 3H). To investigate whether AR regulates the mRNA expression of LEF1 and WNT5A by directly binding to their promoters, we predicted the locations of several potential AREs within 10 kb upstream of the LEF1 and WNT5A transcription start sites using the Genomatix database (Figure 3I). CHIP qRT-PCR analysis indicated that androgen-activated AR was significantly recruited to the predicted AREs of LEF1 and WNT5A under DHT stimulation for 4 h (Figure 3J). Taken together, these results suggested that Wnt-pathway-related genes LEF1 and WNT5A were upregulated by androgen deprivation.
The Wnt Pathway is Critical for AIPC Cells
To examine the functional effects of the Wnt pathway in CRPC, we performed CCK-8 proliferation assays of LNCaP (androgen-dependent) and LNCaP-AI (androgen-independent) cells in the presence of Wnt pathway inhibitors (KY02111 and XAV-939). The CCK-8 assays demonstrated that the proliferation rates of ADPC cells (LNCaP) were not affected by Wnt pathway inhibitors KY02111 and XAV-939 (Figure 4A and B). In contrast, these inhibitors significantly inhibited the proliferation of AIPC cells (LNCaP-AI) (Figure 4C and D).
 |
Figure 4 Wnt pathways inhibitor attenuate LNCaP-AI cells proliferation and cell cycle but have no effect on LNCaP cells. (A–B) Cell proliferation analysis was performed to detect the effect of KY02111 (A), XAV-939 (B) on LNCaP cells. (C–D) Cell proliferation analysis was performed to detect the effect of KY02111 (C) and XAV-939 (D) on LNCaP-AI cells. (E–F) Cell cycle analysis was performed to detect the effect of KY02111 (E), XAV-939 (F) on LNCaP cells. (G–H) Cell cycle analysis was performed to detect the effect of KY02111 (G) and XAV-939 (H) on LNCaP-AI cells. (I–J) Cell apoptosis assay was performed in LNCaP and LNCaP-AI cells treated with KY02111 (I) and XAV-939 (J). Data are presented as the mean ± SD (n = 3). Significance was defined as p<0.05 (*p < 0.05; **p < 0.01; ***p < 0.001); ns means not significant. |
To determine whether the pathway effects on cell proliferation reflected changes in cell cycle progression, we assessed the effects of the pathway-specific inhibitors on the cell cycle profiles of LNCaP and LNCaP-AI cells by flow cytometry. We found that differences in the proportions of LNCaP cells in G0/G1, S, and G2/M (Figure 4E and F) phases following treatment with KY02111and XAV-939 were not significantly different (p > 0.05). However, KY02111 and XAV-939 significantly increased AIPC cells, including LNCaP-AI cells in G1 phase and decreased cells in S phase (Figure 4G and H). The results showed that treatment with KY02111 and XAV-939 increased apoptosis of LNCaP-AI cells compared with that of the DMSO-treated control, but did not affect LNCaP cells (p < 0.05, Figure 4I and J). Taken together, these results revealed that Wnt/β-catenin pathway played a key role in the androgen-dependent to non-dependent transition process.
We next measured the expression of WNT5A and LEF1 in LNCaP and LNCaP-AI cells. The results showed that the basal expression levels of LEF1 and Wnt5A in LNCaP-AI cells were significantly higher than those in LNCaP cells (Figure 5A).
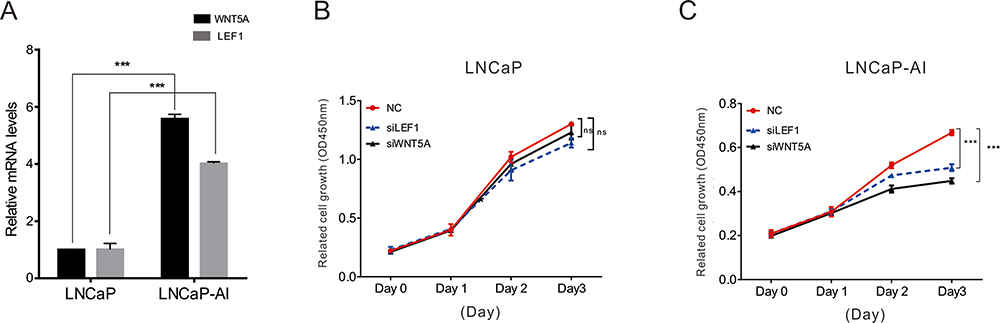 |
Figure 5 The function of Wnt5A and LEF1 in LNCaP and LNCaP-AI cells. (A) qRT-PCR analysis the expression of WNT5A and LEF1 in LNCaP and LNCaP-AI cells. (B–C) Cell proliferation analysis was performed to detect the effect of knockdown WNT5A and LEF1 in LNCaP and LNCaP-AI cells. Data are presented as the mean ± SD (n = 3). Significance was defined as p<0.05 (***p < 0.001); ns means not significant. |
We determined the functions of LEF1 and Wnt5A in LNCaP and LNCaP-AI cells by siRNA-mediated knockdown. As shown in Figure 5B and C, knockdown of LEF1 or Wnt5A had little effect on LNCaP cell proliferation, but the proliferation of LNCaP-AI cells was significantly different, indicating that LEF1 and Wnt5A play an important role in AIPC cell proliferation.
Integrative Analysis Reveals Dysregulation of LEF1 and WNT5A in PCa Progression
To determine the potential roles of LEF1 and WNT5A in the pathogenesis of PCa, we further investigated their expression levels at different stages of PCa using two publicly available gene expression datasets: GSE6919 and The Cancer Genome Atlas (TCGA). The GSE6919 dataset showed that LEF1 and WNT5A were significantly upregulated in metastatic PCa samples compared with normal tissues (Figure 6A and B). The TCGA database showed that LEF1 and WNT5A were both overexpressed in high Gleason score tumors (≥8) compared with normal samples (Figure 6C and 6D). These data suggest that LEF1 and WNT5A were dysregulated during PCa progression.
 |
Figure 6 Expression pattern of LEF1 and WNT5A in PCa samples and the model that summarizes the crosstalk between AR and Wnt signaling promotes androgen-independent growth of Prostate Cancer. (A-B) Expression levels of LEF1 (A), WNT5A (B), in metastatic PCa samples compared to normal prostate tissues by analyzing GSE6919 dataset. (C–D) Expression levels of LEF1 (C), WNT5A (D) in normal prostate tissues, low grade, and high-grade PCa samples by analyzing TCGA dataset. Data are presented as the mean ± SD (n = 3). Significance was defined as p<0.05 (*p < 0.05; **p < 0.01; ***p < 0.001); ns means not significant. (E) In androgen-dependent PCa cells, AR inhibits Wnt signaling by repressing WNT5A and LEF1 expression. However, the inhibitory effect of AR on the WNT5A and LEF1 is blocked during androgen deprivation therapy. The up-regulation of these genes could activate Wnt signaling promotes androgen-independent growth of prostate cancer. |
Discussion
The main challenge in PCa treatment is progression to resistance against ADT.30,31 However, the underlying molecular mechanisms of CRPC progression remain unclear. In this study, we established a prostate cancer cell line (LNCaP-AI) by maintaining LNCaP cells under androgen-depleted conditions. By analyzing two previously published datasets, we found several pathways, including mTOR, TGF-β, MAPK, and Wnt/β-catenin pathways, which were perturbed by ADT. In this study, we found a regulatory mechanism of crosstalk between AR and Wnt signaling, promoting androgen-independent growth of PCa (Figure 6E).
We explored functionally important pathways contributing to PCa hormone-independent growth. KEGG pathway analysis showed that the components of the Wnt signaling pathways were remarkably upregulated after ADT, suggesting that the Wnt pathway was activated in CRPC. Upregulation of the Wnt pathway in advanced CRPC has been shown in previous studies.32–34 Similarly, single cell analysis of prostate circulating tumor cells (CTCs) revealed that non-canonical Wnt signaling was activated in CRPC.35 Lee et al showed that the Wnt/β-catenin pathway was activated in prostate cancer cells after androgen deprivation to promote androgen-independent growth, which was mediated partly via an enhanced interaction of β-catenin with TCF4.36 Seo et al found that Wnt signaling promoted androgen-independent prostate cancer cell proliferation via upregulation of the Hippo pathway effector YAP.37 However, the molecular functions of these pathways in clinical CRPC remained unclear. We examined the functional effects of Wnt pathways in CRPC and observed a potent inhibitory effect of Wnt pathway inhibitor on AIPC cells (LNCaP-AI). We also found that two ADT-induced genes, LEF1 and WNT5A, were direct targets of AR. Interestingly, previous studies had indicated that both genes were affected by AR. For example, Miyamoto et al found that non-canonical Wnt signaling was activated by anti-androgen resistance by analyzing RNA-Seq data of single prostate CTCs.38 Ectopic expression of Wnt5A in prostate cancer cells attenuated the anti-proliferative effect of AR inhibition. These genes likely mediate the crosstalk between androgen signaling and the Wnt pathway. We further determined that the crosstalk between AR and Wnt signaling promoted the androgen-independent growth of prostate cancer. We also revealed that knockdown LEF1 and WNT5A suppressed androgen-independent, but not androgen-dependent, cell proliferation. Our results may provide novel therapeutic opportunities for CRPC by inhibiting the Wnt pathway.
Author Contributions
Jun Luo, Dan Wang, Shaohua Gu, Yaoting Xu, Yali Lu, Zhe Kong, Dujian Li, Wei Gu, Xuechao Wan conceived and designed the study. Jun Luo, Dan Wang, Xuechao Wan, Yangguang Xu, Yali Lu, Zhe Kong, Dujian Li, Chenji Wang, Yao Li performed the experiments. Shaohua Gu, Yaoting Xu, Chaoneng Ji provided experimental technical guidance. Chenji Wang, Chaoneng Ji, Shaohua Gu performed software analysis and visualization. All authors made substantial contributions to conception and design, acquisition of data, or analysis and interpretation of data; took part in drafting the article or revising it critically for important intellectual content; gave final approval of the version to be published; and agree to be accountable for all aspects of the work.
Disclosure
The authors declare no financial or non-financial conflicts of interest for this work.
References
1. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2018. CA Cancer J Clin. 2018;68(1):7–30. doi:10.3322/caac.21442
2. Prasanna Sooriakumaran TN, Akre O, Widmark A, et al. Survival among men at high risk of disseminated prostate cancer receiving initial locally directed radical treatment or initial androgen deprivation therapy. Eur Urol. 2017;72(3):345–351. doi:10.1016/j.eururo.2017.04.002
3. Jason WD, Hearn GA, Reichard CA, et al. HSD3B1 and resistance to androgen deprivation therapy in prostate cancer: a multi-cohort study. Lancet Oncol. 2016;17(10):1435–1444. doi:10.1016/S1470-2045(16)30227-3
4. COC LK, Ding K, Toren P, et al. Nadir testosterone within first year of androgen-deprivation therapy (ADT) predicts for time to castration-resistant progression: a secondary analysis of the PR-7 trial of intermittent versus continuous ADT. J Clin Oncol. 2015;33(10):1151–1156. doi:10.1200/JCO.2014.58.2973
5. Franklin M, Chu OS, Gomella L, et al. A randomised, double-blind study comparing the addition of bicalutamide with or without dutasteride to GnRH analogue therapy in men with non-metastatic castrate-resistant prostate cancer. Eur J Cancer. 2015;51(12):1555–1569.
6. Jelani C, Zarif CKM. The importance of non-nuclear AR signaling in prostate cancer progression and therapeutic resistance. Cell Signal. 2016;28(5):348–356. doi:10.1016/j.cellsig.2016.01.013
7. Michael A, Augello RBD, Knudsen KE. AR function in promoting metastatic prostate cancer. Cancer Metastasis Rev. 2014;33(2–3):399–411.
8. de Bono JS, Sonpavde G, Maroto JP, et al. Phase II study of eribulin mesylate (E7389) in patients with metastatic castration-resistant prostate cancer stratified by prior taxane therapy. Ann Oncol. 2012;23(5):1241–1249. doi:10.1093/annonc/mdr380
9. CAB MK, Yusuke Imamura JK, Leung DP, Caley JW, Mawji NR, Sadar MD. Cotargeting androgen receptor splice variants and mTOR signaling pathway for the treatment of castration-resistant prostate cancer. Clin Cancer Res. 2015;22(11):2744–2754.
10. Hongzoo Park YK, Sul J-W, Jeong IG, et al. Synergistic anticancer efficacy of MEK inhibition and dual PI3K/mTOR inhibition in castration-resistant prostate cancer. Prostate. 2015;75(15):1747–1759. doi:10.1002/pros.23057
11. Chiara Ciccarese FM, Iacovelli R, Fiorentino M, et al. Prostate cancer heterogeneity: discovering novel molecular targets for therapy. Cancer Treat Rev. 2017;54:68–73. doi:10.1016/j.ctrv.2017.02.001
12. Stone L. Prostate cancer: co-targeting mTOR and AR-Vs is efficacious for CRPC. Nat Rev Urol. 2016;13(3):122. doi:10.1038/nrurol.2016.12
13. Mirjam Blattner DL, Robinson BD, Huang D, et al. Mutation drives prostate tumorigenesis in vivo through coordinate regulation of PI3K/mTOR and AR signaling. Cancer Cell. 2017;31(3):436–451. doi:10.1016/j.ccell.2017.02.004
14. Zhan T, M Boutros. Wnt signaling in cancer. Oncogene. 2017;36(11):1461–1473.
15. Jamie N,Anastas RTM. WNT signalling pathways as therapeutic targets in cancer. Nat Rev Cancer. 2013;13(1):11–26. doi:10.1038/nrc3419
16. Stewart J, Wnt Signaling D. Pathway in non-small cell lung cancer. J Natl Cancer Inst. 2014;106(1):t356. doi:10.1093/jnci/djt356
17. Zi X, Anne YG, Simoneau R, et al. Expression of Frzb/Secreted Frizzled-Related Protein 3, a secreted Wnt antagonist, in human androgen-independent prostate cancer PC-3 cells suppresses tumor growth and cellular invasiveness. Cancer Res. 2005;65(21):9762–9770. doi:10.1158/0008-5472.CAN-05-0103
18. Xuechao Wan WH, Yang S, Zhang Y, et al. Androgen-induced miR-27A acted as a tumor suppressor by targeting MAP2K4 and mediated prostate cancer progression. Int J Biochem Cell Biol. 2016;79:249–260. doi:10.1016/j.biocel.2016.08.043
19. Shu Yang JZ, Zhang Y, Wan X, et al. KDM1A triggers androgen-induced miRNA transcription via H3K4me2 demethylation and DNA oxidation. PROSTATE. 2015;75(9):936–946. doi:10.1002/pros.22977
20. Naoki Harada KI, Yamaji R, Nakano Y, Inui H. Androgen deprivation causes truncation of the C-terminal region of androgen receptor in human prostate cancer LNCaP cells. Cancer Sci. 2012;103(6):1022–1027. doi:10.1111/j.1349-7006.2012.02250.x
21. Li M, Di L, Na YQ, GaoGF, Zhijun X. Autophagy protects LNCaP cells under androgen deprivation conditions. Autophagy. 2008;4(1):54–60. doi:10.4161/auto.5209
22. Daniel Seiler JZ, Liu G, Wang S, Joyce Yamashiro RE, Reiter JH, Zeng G. Enrichment of putative prostate cancer stem cells after androgen deprivation: upregulation of pluripotency transactivators concurs with resistance to androgen deprivation in LNCaP cell lines. Prostate. 2013;73(13):1378–1390. doi:10.1002/pros.22685
23. Mu X, Chang C. TR2 orphan receptor functions as negative modulator for androgen receptor in prostate cancer cells PC-3. Prostate. 2003;57(2):129–133. doi:10.1002/pros.10282
24. Mukhopadhyay NK, Ferdinand AS, Mukhopadhyay L, et al. Unraveling androgen receptor interactomes by an array-based method: discovery of proto-oncoprotein c-Rel as a negative regulator of androgen receptor. Exp Cell Res. 2006;312(19):3782–3795. doi:10.1016/j.yexcr.2006.07.017
25. Lee YF, Shyr CR, Thin TH, Lin WJ, Chang C. Convergence of two repressors through heterodimer formation of androgen receptor and testicular orphan receptor-4: a unique signaling pathway in the steroid receptor superfamily. Proc Natl Acad Sci U S A. 1999;96(26):14724–14729. doi:10.1073/pnas.96.26.14724
26. Bubulya A, Wise SC, Shen XQ, Burmeister LA, Shemshedini L. c-Jun can mediate androgen receptor-induced transactivation. J Biol Chem. 1996;271(40):24583–24589. doi:10.1074/jbc.271.40.24583
27. Chen SY, Cai C, Fisher CJ, et al. c-Jun enhancement of androgen receptor transactivation is associated with prostate cancer cell proliferation. Oncogene. 2006;25(54):7212–7223. doi:10.1038/sj.onc.1209705
28. Jason M, D’Antonio CM, Monzon FA, Pflug BR. Longitudinal analysis of androgen deprivation of prostate cancer cells identifies pathways to androgen independence. Prostate. 2008;68(7):698–714. doi:10.1002/pros.20677
29. B-EW YS, Leong KG, Yue P, et al. Androgen deprivation causes epithelial–mesenchymal transition in the prostate: implications for androgen-deprivation therapy. Cancer Res. 2012;72(2):527–536. doi:10.1158/0008-5472.CAN-11-3004
30. Karantanos T, Thompson TC. Prostate cancer progression after androgen deprivation therapy: mechanisms of castrate resistance and novel therapeutic approaches. Oncogene. 2013;32(49):5501–5511. doi:10.1038/onc.2013.206
31. Wadosky KM. SK. Molecular mechanisms underlying resistance to androgen deprivation therapy in prostate cancer. Oncotarget. 2016;7(39):64447–64470. doi:10.18632/oncotarget.10901
32. Li DK Y, Wang Z, Ahmad A, Bao B, Padhye S, Sarkar FH. Inactivation of AR/TMPRSS2-ERG/Wnt signaling networks attenuates the aggressive behavior of prostate cancer cells. Cancer Prev Res (Phila). 2011;4(9):1495–1506. doi:10.1158/1940-6207.CAPR-11-0077
33. Li AK J, Hodges KB, Ahmad N, et al. Co-targeting Polo-like kinase 1(Plk1) and the Wnt/β-catenin signaling pathway in castration-resistant prostate cancer. Mol Cell Biol. 2015;35(24):4185–4198. doi:10.1128/MCB.00825-15
34. Hubert Pakula DX, Zhe L. A tale of two signals: AR and WNT in development and tumorigenesis of prostate and mammary gland. Cancers (Basel). 2017;9:2.
35. IMS PR, Eugenia M, Villasevil M, et al. Next-generation sequencing of advanced prostate cancer treated with androgen-deprivation therapy. Eur Urol. 2014;66(1):32–39.
36. Lee E, Ha S, Logan SK. Divergent androgen receptor and beta-catenin signaling in prostate cancer cells. PLoS One. 2015;10(10):e0141589. doi:10.1371/journal.pone.0141589
37. Seo WI, Park S, Gwak J, et al. Wnt signaling promotes androgen-independent prostate cancer cell proliferation through up-regulation of the hippo pathway effector YAP. Biochem Biophys Res Commun. 2017;486(4):1034–1039. doi:10.1016/j.bbrc.2017.03.158
38. Miyamoto DT, Zheng Y, Wittner BS, et al. RNA-Seq of single prostate CTCs implicates noncanonical Wnt signaling in antiandrogen resistance. Science. 2015;349(6254):1351–1356. doi:10.1126/science.aab0917
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.