Back to Journals » Breast Cancer: Targets and Therapy » Volume 14
Cross-Platform in-silico Analyses Exploring Tumor Immune Microenvironment with Prognostic Value in Triple-Negative Breast Cancer
Authors Kok VC ![]() , Wang CCN, Liao SH, Chen DL
, Wang CCN, Liao SH, Chen DL ![]()
Received 21 January 2022
Accepted for publication 5 April 2022
Published 12 April 2022 Volume 2022:14 Pages 85—99
DOI https://doi.org/10.2147/BCTT.S359346
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Pranela Rameshwar
Victor C Kok,1,2 Charles CN Wang,2,3 Szu-Han Liao,2 De-Lun Chen2
1Division of Medical Oncology, Kuang Tien General Hospital Cancer Center, Taichung, 43303, Taiwan; 2Department of Bioinformatics and Medical Engineering, Asia University, Taichung, 41354, Taiwan; 3Center for Artificial Intelligence and Precision Medicine Research, Asia University, Taichung, 41354, Taiwan
Correspondence: Victor C Kok; Charles CN Wang, Email [email protected]; [email protected]
Introduction: Only a proportion of triple-negative breast cancer (TNBC) is immunotherapy-responsive. We hypothesized that the tumor microenvironment (TME) influences the outcomes of TNBC and investigated the relevant signaling pathways.
Materials and Methods: Immune score (IS) and stromal score (SS) were calculated using the ESTIMATE and correlated with the overall survival (OS) in TNBC. RNA-seq data from 115 TNBC samples and 112 normal adjacent tissues were retrieved. Validations in the methylation levels in 10 TNBC and five non-TNBC cell lines were obtained. Cox model overall survival (OS) validated the derived transcription factor (TF) genes in cBioPortal breast cancer patients.
Results: SS-low predicts a higher OS compared with SS-high patients (P = 0.0081 IS-high/SS-low patients had better OS (P = 0.045) than IS-low/SS-high patients. More macrophages were polarized to the M2 state in patients with IS-low/SS-high patients (P < 0.001). Moreover, CIBERSORTx showed more CD8+ cytotoxic T-cells in IS-high/SS-low patients (p = 0.0286) and more resting NK cells in the IS-low/SS-high TME (P = 0.0108). KEGG pathway analysis revealed that overexpressed genes were enriched in the IL-17 and cytokine-cytokine receptor interaction pathways. The lncRNA DRAIC, a tumor suppressor, was consistently deactivated in the 10 TNBC cell lines. On the cBioPortal platform, we validated that 13% of ER-negative, HER2-unamplified BC harbored IL17RA deep deletion and 25% harbored TRAF3IP2 amplification. On cBioPortal datasets, the nine altered TF genes derived from the X2K analysis showed significantly worse relapse-free survival in 2377 patients and OS in 4819 invasive BC patients than in the unaltered cohort.
Conclusion: Of note, the results of this integrated in silico study can only be generalized to approximately 17% of patients with TNBC, in which infiltrating stromal cells and immune cells play a determinant prognostic role.
Keywords: triple-negative breast cancer, immune cells, tumor microenvironment, IL-17 signaling pathway, immune evasion, in silico study
Introduction
In 2020, the global incidence of breast cancer was 2,261,419 diagnosed across 185 countries, accounting for 11.7% of all cancer types.1 Triple-negative breast cancer (TNBC) is characterized as estrogen receptor-negative, progesterone receptor-negative, and a lack of HER2 expression or amplification and accounts for approximately 15–20% of all breast cancers.2,3 TNBCs are heterogeneous (in terms of genomics, transcriptomics, and histopathology) and demonstrate the heterogeneity of response to anti-programmed death-1/ligand-1 (anti-PD-1/PD-L1) checkpoint inhibition immunotherapy.3–5
In fact, recent studies by Schmid et al revealed that PD-1 or PD-L1 checkpoint blockade immunotherapy combined with neoadjuvant chemotherapy (NAC) improved the pathological complete response (pCR) rate in the neoadjuvant setting; therefore, they proposed such an approach would probably improve the progression-free survival (PFS) of patients with advanced or metastatic TNBC staining positive for PD-L1 in tumor-infiltrating immune cells, if used as the first-line therapy.6–8 In contrast, other tumors are weakly immunogenic depending on the composition of the infiltrating immune cell populations and extrinsic factors (eg different metabolites or specific cytokines) enriched in the immune tumor microenvironment (TME). Of note, recent bioinformatics research, based on bulk tumor gene expression data demonstrated that a low abundance of regulatory CD4+ T cells (Treg) was significantly associated with an increased pCR rate in TNBC patients after NAC.9
In a TNBC surgical specimen, untreated tumor cells typically represent approximately 60% of the cellular component, lymphoid and immune cells account for 20%, and stromal cells such as fibroblasts, histiocytes, endothelial cells, myofibroblasts, and adipocytes represent the remaining 20%.10 The primary function of stromal cells is to establish an immune response. The TME creates a chemokine-rich milieu inside, promoting the encounter between the boundary tumor cells and a variety of surrounding infiltrating immune cells in addition to cancer-associated fibroblasts, neo-vessels, neo-neurites, and other supportive tissues.11 Prior research has demonstrated that the prognosis of TNBC in terms of disease-free survival and disease-specific survival is worse in the basal-like immunosuppressed subtype and fare better in the basal-like immune-activated subtype, indicating that the immune TME plays a crucial role in the formation of either a tumor-permissive or tumor-expulsive milieu4 A clinicopathological study also demonstrated that TNBC with a high number of tumor-infiltrating CD56-positive natural killer (NK) cells was associated with a more favorable disease-free survival.12 Additionally, research focusing on cancer-associated chemokines revealed that NK cells expressing abundant CXCR3 (also known as GPR9 and CD183) molecules on the cell surface are recruited by the chemokines CXCL9, CXCL10, or CXCL11 secreted by immune cells or stromal cells in the TME.13 However, it is still unclear what driving force explains the immunogenic or immunosuppressive phenotype in the TME in patients with TNBC.
Recently, there have been marked improvements in the research efficiency using cross-platform in-silico bioinformatics analyses of multi-omics, multi-layer cancer data.4,10,14–17 Hence, here, we leveraged multi-platform in-silico analyses of genomic data to investigate which infiltrating immune cell populations would be associated with the immune phenotype, and which signaling pathways could determine a subgroup of TNBC that is strongly immunogenic, thus, having a better prognosis. Lastly, we also aimed to determine whether transcription factor signatures derived from the RNA-seq data have prognostic value.
Materials and Methods
Ethics Statement and Study Design
This human data-based research study leveraged multiple publicly accessible RNA-seq datasets containing only mature, anonymous, and de-identified genetic and demographic data. The Institute Review Board of Kuang Tien General Hospital approved the study with a Certificate of Approval numbered KTGH-10458. This study was performed in accordance with the Declaration of Helsinki.
The schematic diagram (Figure 1) shows the study design and methodology adopted in this study. First, we downloaded from TCGA, a breast cancer gene expression RNA-seq dataset (TCGA-BRCA). Using this dataset, we sorted 115 patients with non-metaplastic TNBC, characterized by the lack of estrogen receptors, progesterone receptors, and HER2 non-overexpression or HER2-FISH un-amplification.
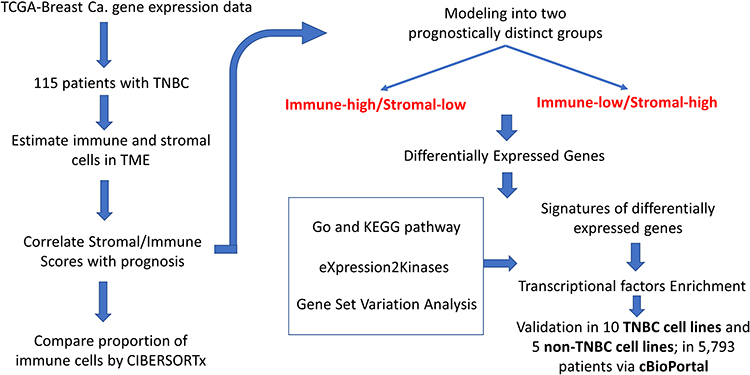 |
Figure 1 Study flow diagram. The schematic diagram represents the study design and methodology adopted. |
Estimation of the Immune/Stromal Scores
As mentioned above, the gene expression data and corresponding clinical information from a total of 1222 cases were retrieved from a publicly available dataset (TCGA-BRCA).18 After extracting the ER, PgR, and HER2 information of each sample, a total of 115 TNBC cases, 112 normal cases, and 994 non-TNBC cases were identified. To predict TNBC purity, we applied the Estimation of STromal and Immune cells in MAlignant Tumor tissues using Expression data (ESTIMATE) algorithm to the normalized expression matrix to determine the immune/stromal scores of each TNBC patients. The immune score (IS; derived from the immune signature of 141 genes) and stromal score (SS; derived from the stromal signature of 141 genes) were calculated using the r-ESTIMATE package in R.19 Supplementary Table S1 presents a gene list of these immune-signature and stromal-signature genes. This algorithm rank-normalizes and rank-orders a set of gene expression values in a given sample and calculates the empirical cumulative distribution function from the signature gene set of the remaining genes; therefore, ESTIMATE can calculate the SS and IS from the RNA-seq data. The algorithm also allows the combination of these scores into an estimate score, used to infer DNA copy number-based tumor purity, as per the following formula: Fraction of tumor cells in a clinical sample (namely, transcriptomic-based tumor purity) = cos (0.6049872018 + 0.0001467884 × [combined stromal-immune score]).
Correlation Analyses
The overall survival (primary prognosis endpoint-based) was estimated using Kaplan-Meier survival analysis. Log rank test was used to compare the survivals stratified by stage. Based on the calculated SS and IS, the corresponding patients were classified into two groups, and their prognoses were individually examined. A previously published method (maximally selected rank statistics), was employed for optimal cutoff identification using the survival in R to explore the relationship between the overall survival and the two groups of TNBC cases.
CIBERSORT Analysis
We used CIBERSORT X (https://cibersort.stanford.edu) to compare the proportion of infiltrating immune cells in the TME of IS-low/SS-high and IS-high/SS-low TNBC patients.20,21 The CIBERSORT accurately allows the evaluation of the relative levels of 22 immune cell phenotypes; such and analysis was performed using the immunedeconv package in R.22 The immune cell fraction level divided by the cutoff value was 0 or 1 in the subsequent scoring formula.
Differential Expression Analysis
According to the ESTIMATE results, the intersection genes were selected based on the stromal/immune scores. The limma package in R was used to screen DEGs between normal and TNBC samples.23 Genes with a p-value ≤ 0.0001 and absolute log2 fold-change ≥ 4 were considered to be differentially expressed and extracted further for network construction. Heatmaps were generated using the pheatmap package in R.24 The generated heatmaps and volcano plots show the differentially expressed genes in either the IS-high/SS-low or IS-low/SS-high TNBC groups.
Gene Ontology and KEGG Pathway Enrichment Analyses
Using these DEGs defined as above for the two TNBC groups, we investigated the enriched signaling pathways based on GO terms and KEGG pathways. Functional enrichment analyses of GO terms, including cellular components, molecular functions, and biological processes, and KEGG pathways were performed using the clusterProfiler package in R.25 The functions among genes of interest were adjusted with a cutoff criterion of p < 0.05.
eXpression2Kinases Analysis
We utilized eXpression2Kinases (X2K), as reported elsewhere, to disclose the potential enrichment of transcription factors.26,27
Gene Set Variation Analysis
Gene set variation analysis (GSVA) is a bioinformatics framework that organizes gene expression data in the form of a pathway or signature summary.28 GSVA is a popular pathway-related immune infiltration and tumor mutational burden immune-related analysis. Here, we leveraged GSVA to provide an accurate definition of pathway enrichment between samples from different groups. We used the GSVA package in R to evaluate the t score and assigned the pathway activity conditions. We then used the pheatmap package in R to display the distinctions in pathway activation between normal tissues and those of IS-high/SS-low and IS-low/SS-high patients.
Validation in TNBC Cancer Cell Lines and Non-TNBC Cancer Cell Lines
In the Depmap platform (https://depmap.org/portal/), we used the methylation dataset (1 kb upstream transcription start sites) to compare the lncRNA methylation between the TNBC cell lines and non-TNBC cell lines (ER+/HER2+ or HER2-negative). We defined fractional methylation > 0.6 indicates “methylated” and < 0.6 “unmethylated”. Ten TNBC cell lines used were HCC2157, BT20, MDAMB231, HCC1395, HCC1937, HCC1599, MDAMB436, MDAMB468, HCC1806, and HCC1143. Five non-TNBC cell lines were BT474, UACC812, EFM192A, EFM19, and ZR751.
Validation in cBioPortal for Cancer Genomics
Finally, we validated our findings in a different, more extensive dataset of breast cancer via cBioPortal analysis.29,30
Statistical Analyses
We used the R package statistical software to implement the survival analysis with relevant R functions as survfit(), survdiff(), coxfit1. In addition, we used the StatsDirect version 3.3 to compute the Mann–Whitney U-test and the grouped linear regression. Finally, the built-in default statistical software performed all other statistical analyses on each platform.
Results
From the TCGA Breast Cancer dataset, gene expression data for 115 patients with triple-negative breast cancer and 112 normal adjacent tissues were retrieved. Figure 1 demonstrates the study design, flow, and methodology used in the current study. In this cohort of 115 TNBC patients, favorable overall survival as per the Kaplan-Meier curves was significantly correlated with an earlier disease stage (P = 0.0012; Figure 2).
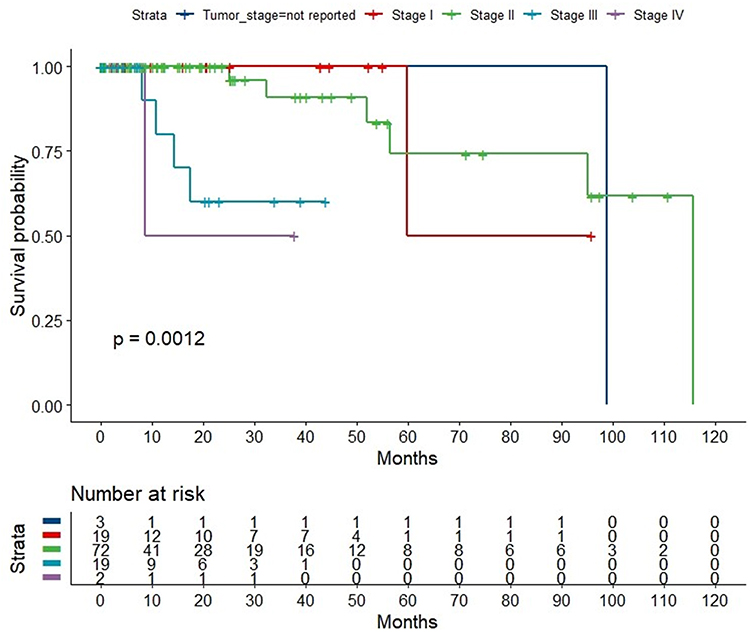 |
Figure 2 Overall survival of 115 patients with triple-negative breast cancer stratified by cancer stage at diagnosis. Kaplan-Meier curves referring to different overall TNBC stages. Log rank test was used to compare the survivals stratified by stage. The higher stage correlates significantly with poorer survival (P = 0.0012). |
The ESTIMATE algorithm computationally estimates the fraction of stromal and immune cells in the TME of all the 115 TNBC patients. The SS was high (SS-high) in 10 patients, and the IS was high (IS-high) in the ten other patients. There were no significant correlations between the SS or IS and the cancer stage in all the 115 patients (Supplementary Figure S1). However, as per the Kaplan-Meier analyses, SS-low patients showed a higher overall survival (OS) than that of SS-high patients (P = 0.0081; Supplementary Figure S2), while IS-high patients showed a higher OS than that of IS-low patients (P = 0.2; too few cases in the IS-high; Supplementary Figure S3). Of note, a strong correlation between both the SS and IS and the patients’ overall prognosis was observed. Expectedly, when compared with IS-low/SS-high patients, IS-high/SS-low patients showed a better OS (P = 0.045) (Figure 3). Cytoscape revealed the immune cell infiltration levels between samples grouped by IS-high/SS-low or IS-low/SS-high (Supplementary Figure S4). Interestingly, grouped linear regression showed a statistically significant increase in M2 macrophages in TNBC patients with the IS-high/SS-low phenotype. In line with these results, in the tumor microenvironment (TME), a higher proportion of M2 was also found in IS-low/SS-high patients, as estimated by CYBERSORTx (P < 0.001) (Supplementary Figure S5). Additionally, significantly more CD8+ cytotoxic T-cells, memory B cells (P = 0.0304), activated CD4+ memory T cells (P = 0.0056), follicular helper T cells (P = 0.0044), and activated NK cells (P = 0.0511) were also observed in IS-high/SS-low patients (14% vs 5.3%, p = 0.0143) (Table 1). On the other hand, more resting NK cells were observed in IS-low/SS-high patients (P = 0.0108).
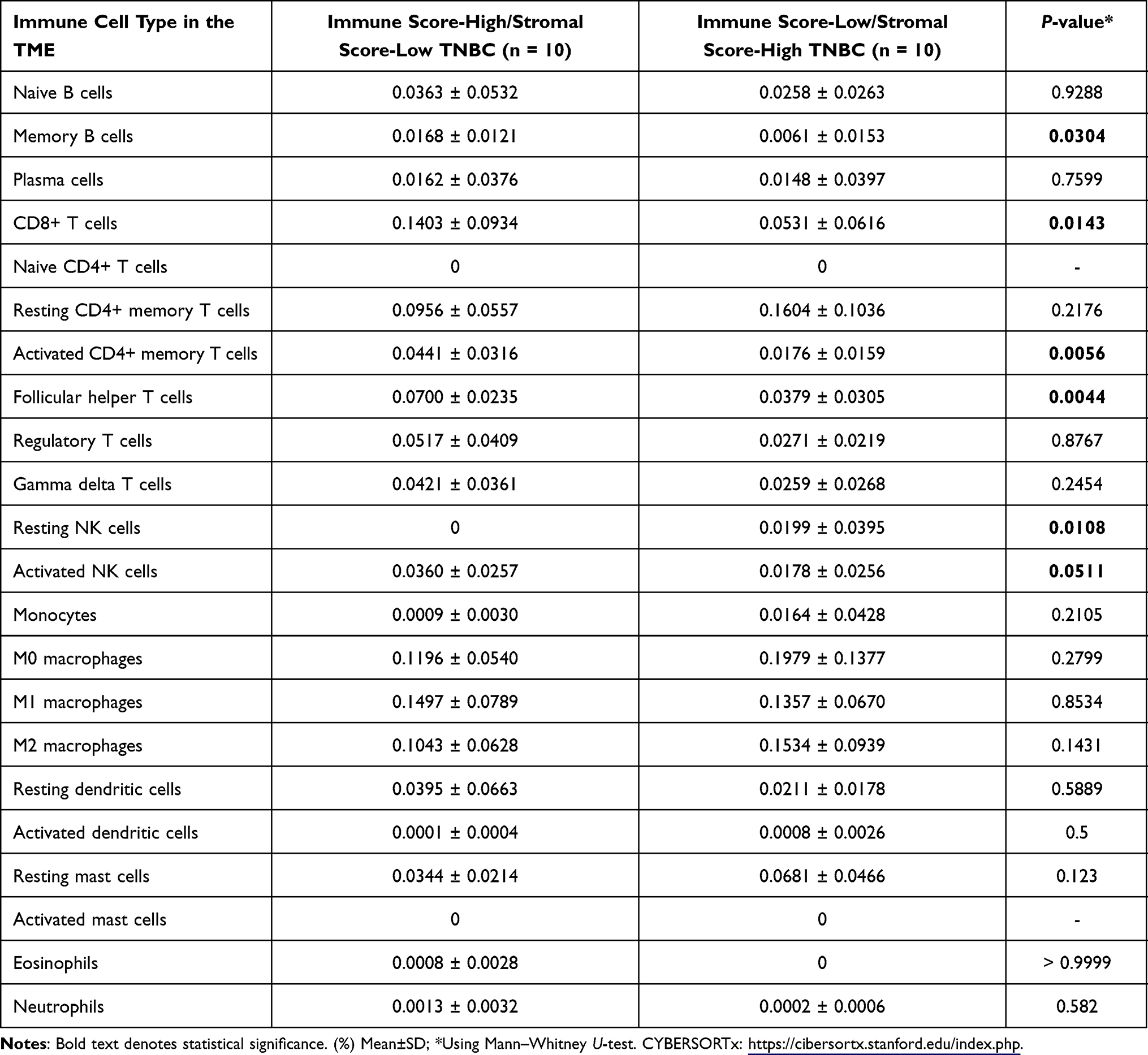 |
Table 1 Comparison of the Immune Cell Profiles in the Tumor Microenvironment (TME) of IS-High/SS-Low and IS-Low/SS-High Triple-Negative Breast Cancer Patients, Estimated Using CYBERSORTx |
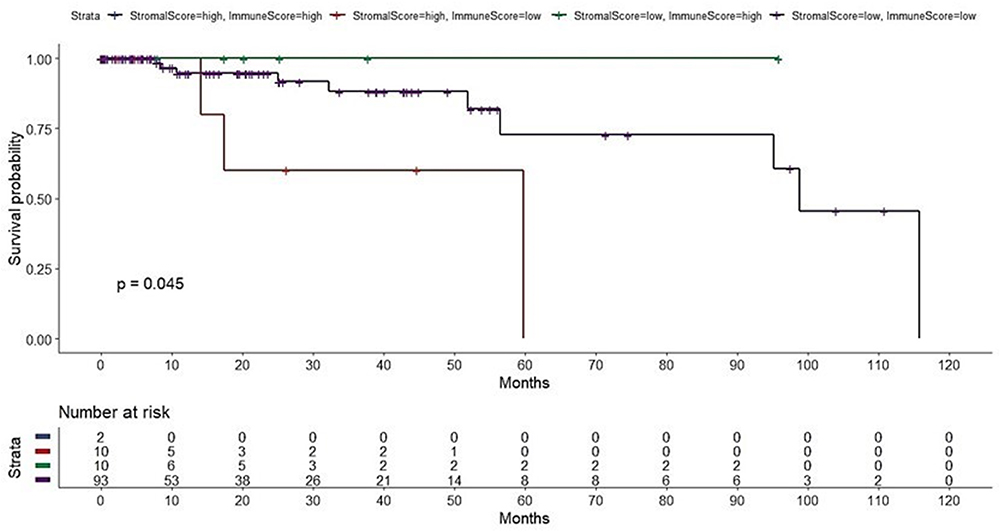 |
Figure 3 Overall survival of 115 patients with triple-negative breast cancer stratified by the combination of the Stromal Score and Immune Score. Both scores were inferred by the ESTIMATE gene expression signatures. SS-low/IS-high patients were associated with excellent overall survival, whereas SS-high/IS-low patients showed the worst overall survival. |
Moreover, DEG analysis showed 651 DEGs (284 upregulated, and 367 downregulated) in IS-high/SS-low patients, and 370 DEGs (187 upregulated, and 183 downregulated) in IS-low/SS-high patients (Supplementary Figure S6). Heatmaps and volcano plots of the DEGs in the context of these two groups are shown in Figure 4A–D. DESeq2 uses the Wald test to identify differentially expressed genes by comparing the immunogenic or immunotolerant classes and the normal adjacent tissue. To put things in context, we displayed the top 30 DEGs ranked by the Wald statistic for the four categories by the combined immune and stromal scores. In IS-high/SS-low TNBC tumors, the top 30 upregulated DEGs were LAG3, CCNE1, GBP5, CDCA8, ETV7, CDCA3, CXCL10, KCNJ10, IDO1, PTTG1, SKA1, CXCL11, IFNG, LOC105373098, TPX2, PLK1, WFDC21P, A2ML1, KIF2C, MMP11, IL21R, NDC80, IL4I1, AIM2, CXCL9, CDC20, CCL25, RTP3, NUF2, and CENPA; in contrast, the downregulated DEGs were MAMDC2, FAXDC2, VEGFD, TGFBR3, NOVA1, ADAMTS5, ECRG4, LMOD1, CAVIN2, ADAM33, TMTC1, DACH1, RERGL, MAB21L1, SLC7A2, AGTR1, PGR, SCN2B, PAMR1, MYH11, ATP1A2, ADAMTS9-AS2, CD300LG, CA4, CHL1, GPC3, ARHGAP20, CCL14, ABCA8, and ADGRL3. In IS-low/SS-high TNBC tumors, the top 30 upregulated DEGs were MMP11, CEMIP, COL11A1, COL10A1, CA9, LOC101929128, LOC157273, INHBA, CCL11, OLAH, CILP2, LYPD1, MMP1, HAPLN1, HOXB9, LINC02487, EPYC, GBP5, TDO2, CCKBR, CCN4, CXCL11, LIPG, LINC01615, ANLN, GJB2, LINC00511, LINC00673, CXCL10, and SHISAL1; whereas, the downregulated DEGs were ABCA10, CAVIN2, ECRG4, SCN2B, ANGPTL7, VEGFD, MYH11, CD300LG, SCARA5, HPSE2, MYOC, HEPACAM2, LINC00993, HIF3A, SLC16A12, ATP1A2, PGM5-AS1, BTNL9, TNXB, ANKRD30A, GDF10, PGR, PTPRQ, B3GALT1, NPY2R, PLPPR1, HSD17B13, DEFB132, LEP, and GLYAT.
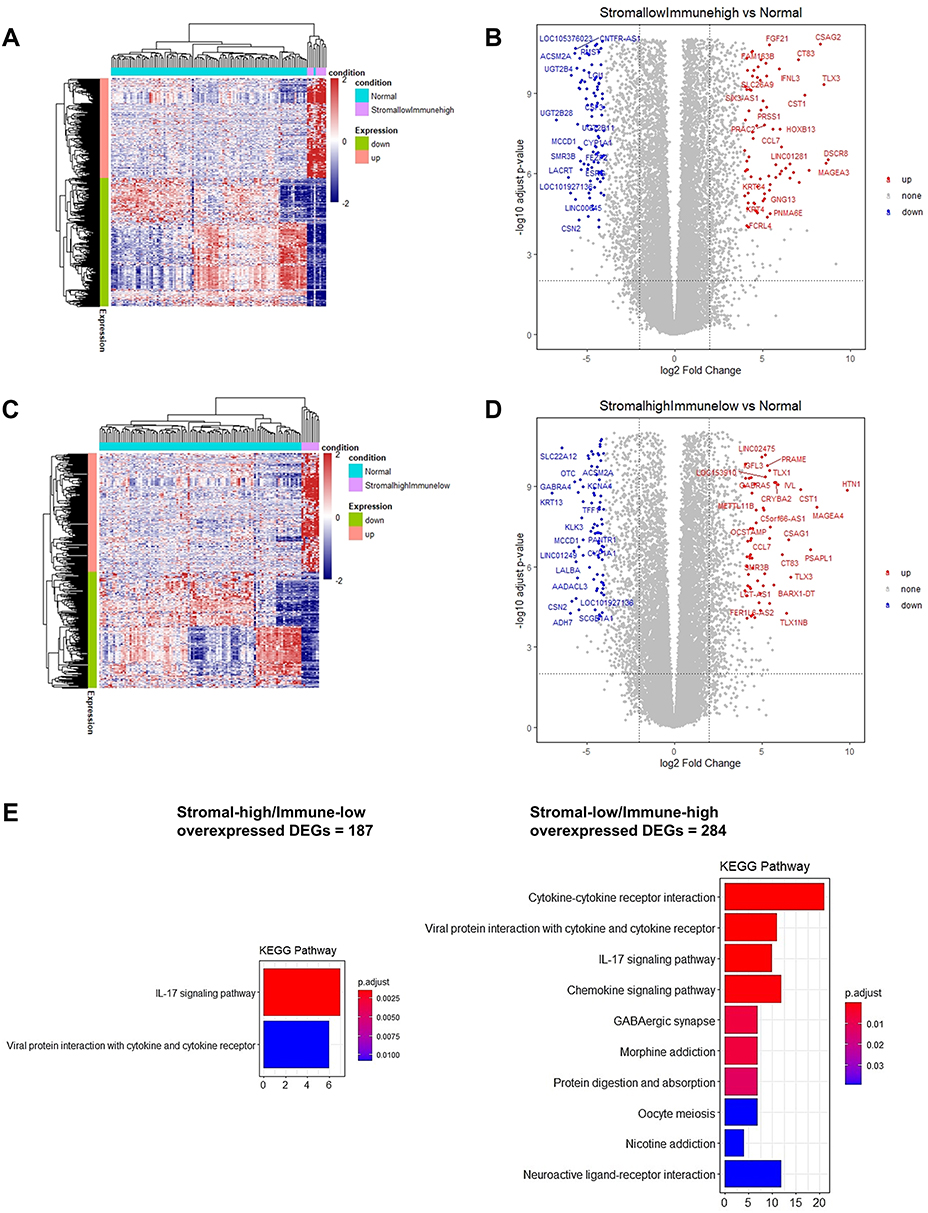 |
Figure 4 Heatmaps and volcano plots of the differentially expressed genes in SS-low/IS-high (A and B) and SS-high/IS-low (C and D) TNBC patients, and the corresponding KEGG pathway analysis (E). We labeled the DEGs satisfying the conditions of having an adjusted P-value < 0.0001 and |log2FoldChange| ≥ 5 in panels (B and D). The overexpressed DEGs in the context of both phenotypes are enriched in the IL-17 and cytokine-cytokine receptor interactions’ signaling pathways. |
KEGG pathway enrichment analysis showed that the overexpressed DEGs from both phenotypes were enriched in the IL-17 signaling pathway and viral protein interaction cytokine and cytokine receptor genes. Notably, the overexpressed DEGs in the context of the SS-low/IS-high phenotype were also enriched in other two cytokine-related pathways: cytokine-cytokine receptor interactions pathway and chemokine signaling pathway (Figure 4E).
Using X2K, we also inferred the transcription factors associated with the two immune phenotypes. In SS-high/IS-low TNBC patients, the inferred TFs were PPARG, HNF4A (also known as farnesoid X receptor, FXR), NR1H4, NR0B2, and MLXIPL, whereas the TF signature of SS-low/IS-high TNBC patients was composed of the PPARG, CEBPA, and MLXIPL TFs (Figure 5). Additionally, we performed a literature search on PubMed and discovered nine TF genes linked to IL-17-mediated signaling, including PPARG, CEBPA, MEOX1, KLF15, CD36, ZNF750, EZH2, HNF4A, and NR0B2 (Table 2).
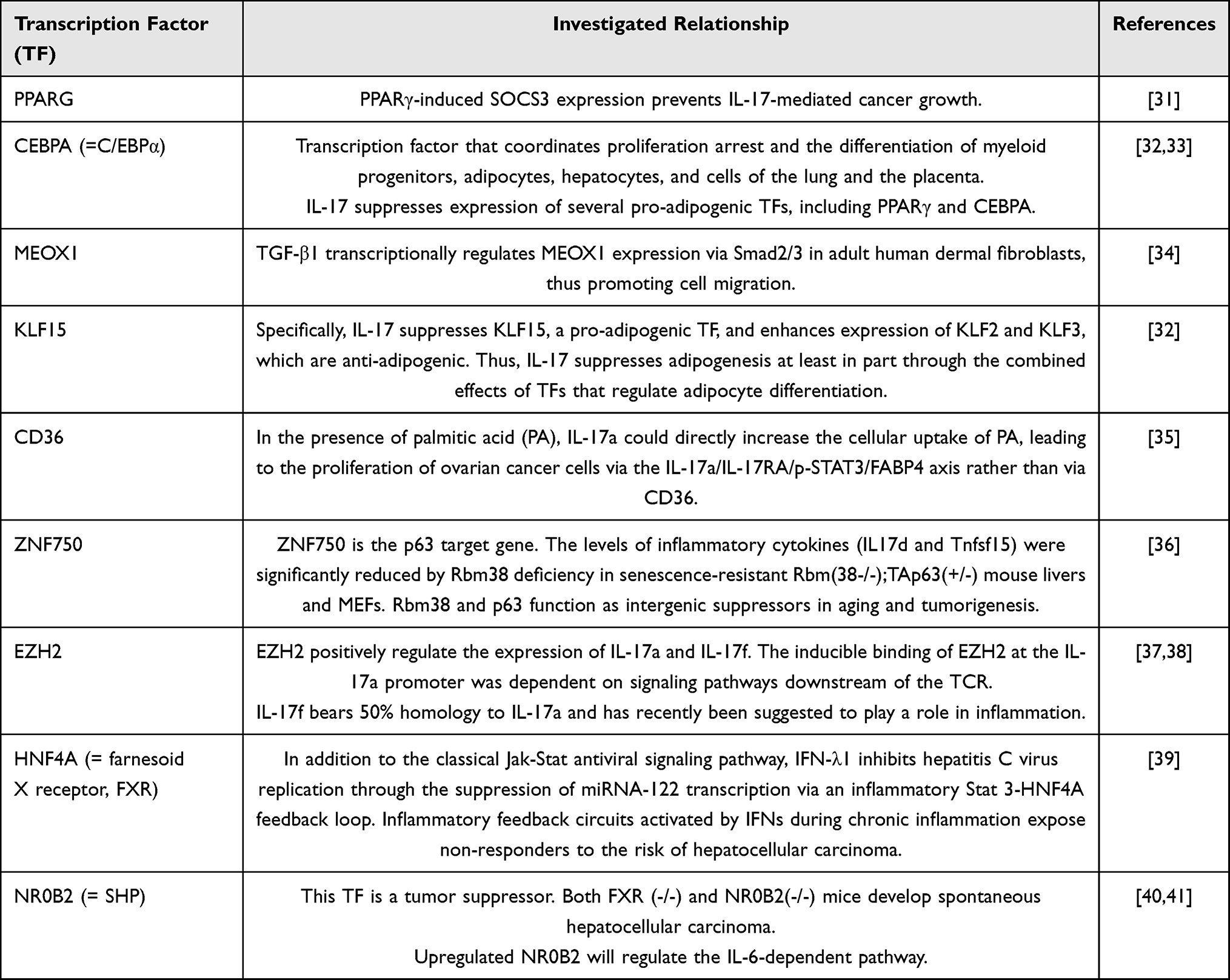 |
Table 2 Selection of the Transcription Factors Linked to Interleukin-17-Mediated Signaling |
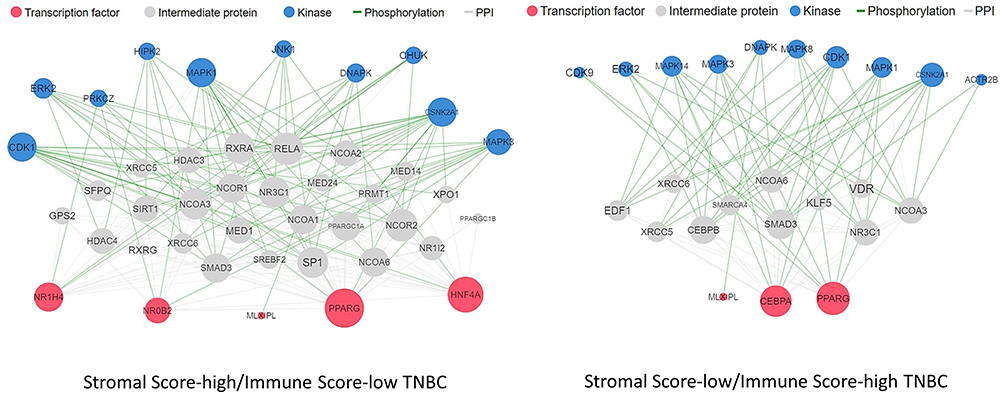 |
Figure 5 Transcription factors inferred from the X2K in the context of SS-high/IS-low or SS-low/IS-high TNBC patients. |
In addition, GSVA also suggested the involvement of additional pathways, including the JAK-STAT signaling, T cell receptor signaling, B cell receptor signaling, cytokine-cytokine receptor interaction, TGF-β signaling, and PPAR signaling pathways (Supplementary Figure S7; Figure S7A shows the GSVA of the 370 DEGs of the SS-high/IS-low subgroup and Figure S7B shows the GSVA of the 651 DEGs of the SS-low/IS-high subgroup).
Validation of the Results in the TNBC Cell Lines
IL-17 signaling will kick off the NFκB signaling and induce M1 to M2-like transformation in TME macrophages.42 Long non-coding RNA, DRAIC, a tumor suppressor, would interact with the IκB kinase (IKK) to inhibit NFκB activation.43 In the Depmap platform, we used the methylation dataset (1 kb upstream transcription start sites) to compare the lncRNA DRAIC methylation between the ten TNBC cell lines and five non-TNBC cell lines (ER+/HER2+ or HER2-negative). We defined fractional methylation > 0.6 indicates “methylated” and < 0.6 “unmethylated”. Our study results showed that 10 TNBC cell lines had methylated lncRNA DRAIC, whereas all five non-TNBC cell lines were unmethylated (Figure 6). Therefore, the lncRNA DRAIC tumor suppressor was consistently deactivated in the 10 TNBC cell lines.
 |
Figure 6 Methylation fraction of the lncRNA DRAIC was compared between 10 TNBC cancer cell lines and 5 non-TNBC cancer cell lines. Long non-coding RNA, DRAIC, a tumor suppressor, would interact with IKK to inhibit NFκB activation. In the Depmap platform, we used the methylation dataset (1 kb upstream transcription start sites) to compare the PPARG methylation between the TNBC cell lines and non-TNBC cell lines (ER+/HER2+ or HER2-negative). We defined fractional methylation > 0.6 indicates “methylated” and < 0.6 “unmethylated”. Our study results showed that 10 TNBC cell lines (HCC2157, BT20, MDAMB231, HCC1395, HCC1937, HCC1599, MDAMB436, MDAMB468, HCC1806, and HCC1143) had methylated lncRNA DRAIC, whereas all five non-TNBC cell lines (BT474, UACC812, EFM192A, EFM19, and ZR751) were unmethylated. |
Validation of the Results in the cBioPortal
IL-17 is a proinflammatory cytokine that signals mainly via the TRAF3 Interacting protein 2 (TRAF3IP2), as reported previously.44,45 TRAF3IP2 is an inflammatory mediator and upstream regulator of several crucial transcription factors, such as AP-1 and NF-κB.45 Act1, an essential component in IL-17 signaling complex, is encoded by the gene TRAF3IP2. Finally, we used the Oncoprint from cBioPortal to validate the frequency of IL-17 genes in TNBC. We discovered that 13% of ER-negative and HER2-FISH unamplified breast cancers harbored IL17RA deep deletions and 25% harbored TRAF3IP2 amplifications (Supplementary Figure S8).
Interestingly, we also discovered using the cBioPortal platform that aberrations in the nine TF genes mentioned above are associated with a worse prognosis, as per the relapse-free survival of 2377 patients (log-rank, P = 0.00007) and the overall survival of a larger group of 4819 patients (log-rank, P = 0.001697) (Figure 7). Finally, we interrogated the differential expression of all the related genes of the IL-17 signaling pathway in the two polarized immune-stroma-scored TNBCs. The hierarchical clustering heatmaps show evidence in supporting that most IL-17 pathway genes had differentially expressed in the tumor specimens than the normal adjacent tissue counterparts (Supplementary Figure S9). In addition, it is notable that IL-17D and IL-17F were more differentially expressed than the normal adjacent tissues in the stromal-low immune-high tumors.
 |
Figure 7 Validation using the cBioPortal demonstrates the relapse-free survival in 2377 patients and the overall survival in 4819 patients with invasive breast carcinoma stratified based on the alteration of the transcription factor genes PPARG, CEBPA, MEOX1, CD36, ZNF750, KLF15, EZH2, HNF4A, and NR0B2. The altered group refers to any alteration in one of these transcription factors. |
Discussion
Several recent studies have shed light on the pivotal role of the TME in the shaping of the tumor behavior in TNBC.46–53 Notably, Karn et al reported that lymphocyte-rich TNBC with high immune metagene expression had lower clonal heterogeneity, fewer somatic mutations, decreased somatic copy number alteration levels, and lower neoantigen counts.52 Given the paucity of studies investigating the mechanism behind the strikingly different prognoses of immunosuppressive or immunogenic TNBCs, our study aimed to explore the differences in stromal cells (particularly immune cells) within the TME, as well as in the enriched DEGs in the search for the driving force behind the formation of these two different phenotypes. Interestingly, we discovered that the composition of immune cells reacting to TNBC tumor cells was strikingly different in the highly immuno-weak stromal TME and the strong stromal-weak immunogenic TME. Additionally, we discovered that the immunosuppressive type (SS-high/IS-low) showed gene signatures enriched in the IL-17, and viral-cytokine and cytokine receptor interactions’ pathways. Of note, this study is unique considering the demonstration that altered TF genes derived from IL-17-mediated signaling showed strong significance with respect to both the relapse-free and overall survival, as per the analysis of another extensive dataset.
More than 1600 known (or likely) human TF genes represent approximately 8% of the human genes.54 Mutations in TF genes are often highly deleterious in humans.54 Nine TF genes, PPARG, CEBPA, MEOX1, CD36, ZNF750, KLF15, EZH2, HNF4A, and NR0B2, were enriched in our bioinformatics analysis. Importantly, this is the first time that the alteration in one or more of the nine TF in the context of TNBC was linked to the IL-17 signaling pathway and associated with significantly poorer prognosis in terms of relapse-free survival and overall survival in a large number of breast cancer patients, excluding ER-positive and HER2-amplified cases.
In cancer tissues, activated T cells secrete the proinflammatory cytokine IL-17, which through the regulation of the MAPKs and NF-κB activities, gives rise to the increased expression of IL-6 and cyclooxygenase-2.55 A score of research recapitulated these observations and revealed a more detailed mechanistic understanding. A plethora of studies have revealed increased IL-17A levels in ER-negative or triple-negative breast cancer.56 In fact, the upregulation of IL-17A signaling is associated with increased expression of programmed cell death protein 1 (PD-1) and programmed death-ligand 1 (PD-L1) in breast cancer with low ER expression, which may elevate the infiltration of CD8+ T cells in the tumor tissues.56 Of note, CD4+ T cells among tumor-infiltrating lymphocytes in the TME are the main source of IL-17.57,58 IL-17-mediated downstream signaling plays a critical role in the TME, inducing the expression of different genes either to switch on pro-tumor effector cytokines or to inhibit tumor growth in a context- and system-dependent manner.57 For example, IL-17 induces the production of IL-6, which in turn induces STAT3 (signal transducer and activator of transcription 3);57 in fact, IL-6 orchestrates both the increase in the recruitment of suppressive tumor-associated myeloid cells (MDSCs) and impacts their ability to inhibit anti-tumor T-cell responses.59 Early on, a recent study uncovered how an IL-17–mediated paracrine network could recruit immature myeloid cells into the TME, causing tumor progression; this study demonstrated that through NF-κB and ERK signaling, tumor-infiltrating T helper type 17 (Th17) cells and IL-17 would induce granulocyte colony-stimulating factor expression, which is crucial for the mobilization of immature myeloid cells into the TME.47 A recent study of the role IL-17A plays in the TNBC’s TME identified that IL-17A stimulates the angiogenic and migratory activity of TNBC cells and modulates the TME’s immune landscape towards a pro-metastatic phenotype.55 In contrast, Bianchini et al demonstrated that a subset of highly proliferative, ER-negative breast cancers with high expression of a B-cell/plasma cell stromal metagene corresponding to immune functions and extracellular matrix components were associated with a favorable prognosis.10 These studies lend support to the composition of cellular constituents in the TME, and their association with the patient’s prognosis.
Using the methylation dataset, our in-silico cell line study in the DepMap platform demonstrates that 10 TNBC cell lines had methylated lncRNA DRAIC. In contrast, all five non-TNBC cell lines were unmethylated. This tumor suppressor, consistently deactivated in the 10 TNBC cell lines, shall require further research and investigation into its clinical significance.
Still, regarding the TF genes associated with TNBC, a previous study suggested that EZH2 plays decisive roles in immune cells (eg, T cells, NK cells, dendritic cells, and macrophages) in the tumor microenvironment.60 Liu et al demonstrated a novel function of PPARγ in lymphocyte trafficking and the cross-talk between Th17 and B cells.61 Additionally, investigators from the Institut Curie and INSERM found that a high Th17 metagene was associated with a good prognosis in T cell non-inflamed type TNBC, suggesting Th17 is a novel prognostic composite biomarker. Altogether, these studies clearly support the notion that integrating immune cells and tumor molecular diversity is an efficient strategy for the prognostic stratification of cancer patients.62
Several recently published studies provide some functional data to show how the IL-17-mediated signaling pathway drives immune response and modulates the therapeutic response to chemotherapy or immunotherapy in solid tumors, including breast cancers.63–70 These data will inform future therapeutic strategy design both pre-clinically and clinically. Some exciting and relevant research findings are discussed. In the TME, adenosine is a well-known immunosuppressive molecule. Adenosine and adenosine triphosphate (ATP) are the most abundant metabolites within a cell and extracellular space, acting as an autocrine and paracrine messenger. While ATP acts as an accelerator to promote proinflammatory activities, adenosine, via the Gs-coupled A2a and A2b receptors, suppresses various immune cells.11 Thibaudin et al identified that in breast cancer tumors, Th17 cells would express the ectonucleotidases, CD73 and CD39, which are the enzymes that degrade extracellular ATP into adenosine. The investigators further observed that the adenosine molecule would suppress T cell immune responses.69 Wang et al investigated how IL-17A regulated natural killer (NK) cell activity and found that by restraining IL-15-driven NK cell terminal maturation, IL-17A constrained the NK cell’s anti-cancer activity.70 Th17 cells also contribute to the acquired resistance to PD-L1 blockade immunotherapy and MEK inhibitor in KRAS/p53-mutant lung cancers.66,68 Th17 cells create resistance by secreting IL-17 and IL-22 cytokines. The investigators established an in vivo model to prove the principle that through antibody depletion of IL-17A, combining the MEK inhibition and PD-L1 blockade markedly reduce the therapy-induced acquired resistance.68
Finally, the purpose of the current research was neither to develop a predictive molecular panel biomarker, nor to identify innate resistance versus sensitive phenotypes to checkpoint inhibitor immunotherapy; we aimed to investigate the factors behind the TME’s immune-stromal state and to look for the systemic functions of the involved molecular signaling pathways. However, this study is not without limitations. One of them is the available low number of cases used to separate tumors into two extreme immune-stromal phenotypes.
Conclusions
Overall, our data suggest that the disclosure of the distinct molecular anatomy of the TME in patients with TNBC (not of the cancer cells per se) will assist in the determination of the ultimate tumor behavior. Of note, the results of this integrated in silico study can only be generalized to approximately 17% of patients with TNBC, in which infiltrating stromal cells and immune cells play a determinant prognostic role. Remarkably, our data suggest that the determining factor for the differences in the tumor immune microenvironment (immunogenic versus immunosuppressive) can be explained by IL-17 signaling and its paracrine network.
Data Sharing Statement
The datasets used and/or analyzed during the current study are available from the corresponding author upon reasonable request.
Acknowledgments
The results published here are based on data generated by the TCGA Research Network: https://www.cancer.gov/tcga. Part of the results in this study were presented as an e-poster at the American Association for Cancer Research 2021 Annual Meeting, held during April 10–15, 2021. The prior version without peer review has been posted as a preprint with DOI: 10.21203/rs.3.rs-495771/v1.
Funding
This study was partly supported by a research fund from the Ministry of Science and Technology, Taiwan, with the grant number as MOST 109-2321-B-468-001. The funder had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Disclosure
The authors declare that they have no competing interests in this work.
References
1. Sung H, Ferlay J, Siegel RL, et al. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2021;71(3):209–249. doi:10.3322/caac.21660
2. Glodzik D, Bosch A, Hartman J, et al. Comprehensive molecular comparison of BRCA1 hypermethylated and BRCA1 mutated triple negative breast cancers. Nat Commun. 2020;11(1):3747. doi:10.1038/s41467-020-17537-2
3. Marra A, Trapani D, Viale G, Criscitiello C, Curigliano G. Practical classification of triple-negative breast cancer: intratumoral heterogeneity, mechanisms of drug resistance, and novel therapies. NPJ Breast Cancer. 2020;6:54. doi:10.1038/s41523-020-00197-2
4. Burstein MD, Tsimelzon A, Poage GM, et al. Comprehensive genomic analysis identifies novel subtypes and targets of triple-negative breast cancer. Clin Cancer Res. 2015;21(7):1688–1698. doi:10.1158/1078-0432.Ccr-14-0432
5. Kok VC. Hepatic metastasectomy and paclitaxel provide long-term survival for a young woman with recurrent triple-negative metastatic breast cancer: 16 years follow-up. J Cancer Res Ther. 2018;14(3):722–723. doi:10.4103/0973-1482.179087
6. Schmid P, Adams S, Rugo HS, et al. Atezolizumab and Nab-paclitaxel in advanced triple-negative breast cancer. N Engl J Med. 2018;379(22):2108–2121. doi:10.1056/NEJMoa1809615
7. Schmid P, Cortes J, Pusztai L, et al. Pembrolizumab for early triple-negative breast cancer. N Engl J Med. 2020;382(9):810–821. doi:10.1056/NEJMoa1910549
8. Schmid P, Rugo HS, Adams S, et al. Atezolizumab plus nab-paclitaxel as first-line treatment for unresectable, locally advanced or metastatic triple-negative breast cancer (IMpassion130): updated efficacy results from a randomised, double-blind, placebo-controlled, Phase 3 trial. Lancet Oncol. 2020;21(1):44–59. doi:10.1016/s1470-2045(19)30689-8
9. Oshi M, Asaoka M, Tokumaru Y, et al. Abundance of regulatory T cell (Treg) as a predictive biomarker for neoadjuvant chemotherapy in triple-negative breast cancer. Cancers (Basel). 2020;12(10). doi:10.3390/cancers12103038
10. Bianchini G, Qi Y, Alvarez RH, et al. Molecular anatomy of breast cancer stroma and its prognostic value in estrogen receptor-positive and -negative cancers. J Clin Oncol. 2010;28(28):4316–4323. doi:10.1200/jco.2009.27.2419
11. Kok VC. Current understanding of the mechanisms underlying immune evasion from PD-1/PD-L1 immune checkpoint blockade in head and neck cancer. Front Oncol. 2020;10:268. doi:10.3389/fonc.2020.00268
12. Bouzidi L, Triki H, Charfi S, et al. Prognostic value of natural killer cells besides tumor-infiltrating lymphocytes in breast cancer tissues. Clin Breast Cancer. 2021;21(6):e738–e747. doi:10.1016/j.clbc.2021.02.003
13. Nagarsheth N, Wicha MS, Zou W. Chemokines in the cancer microenvironment and their relevance in cancer immunotherapy. Nat Rev Immunol. 2017;17(9):559–572. doi:10.1038/nri.2017.49
14. Sivadas A, Kok VC, Ng KL. Multi-omics analyses provide novel biological insights to distinguish lobular ductal types of invasive breast cancers. Breast Cancer Res Treat. 2022. doi:10.1007/s10549-022-06567-7
15. Mapiye DS, Christoffels AG, Gamieldien J. Identification of phenotype-relevant differentially expressed genes in breast cancer demonstrates enhanced quantile discretization protocol’s utility in multi-platform microarray data integration. J Bioinform Comput Biol. 2016;14(5):1650022. doi:10.1142/s0219720016500220
16. Lehmann BD, Colaprico A, Silva TC, et al. Multi-omics analysis identifies therapeutic vulnerabilities in triple-negative breast cancer subtypes. Nat Commun. 2021;12(1):6276. doi:10.1038/s41467-021-26502-6
17. De Mattos-arruda L, Sammut SJ, Ross EM, et al. The genomic and immune landscapes of lethal metastatic breast cancer. Cell Rep. 2019;27(9):2690–2708.e10. doi:10.1016/j.celrep.2019.04.098
18. Wang CCN, Li CY, Cai JH, et al. Identification of prognostic candidate genes in breast cancer by integrated bioinformatic analysis. J Clin Med. 2019;8(8). doi:10.3390/jcm8081160
19. Yoshihara K, Shahmoradgoli M, Martinez E, et al. Inferring tumour purity and stromal and immune cell admixture from expression data. Nat Commun. 2013;4:2612. doi:10.1038/ncomms3612
20. Newman AM, Liu CL, Green MR, et al. Robust enumeration of cell subsets from tissue expression profiles. Nat Methods. 2015;12(5):453–457. doi:10.1038/nmeth.3337
21. Newman AM, Steen CB, Liu CL, et al. Determining cell type abundance and expression from bulk tissues with digital cytometry. Nat Biotechnol. 2019;37(7):773–782. doi:10.1038/s41587-019-0114-2
22. Sturm G, Finotello F, List M. Immunedeconv: an R package for unified access to computational methods for estimating immune cell fractions from bulk RNA-sequencing data. Methods Mol Biol. 2020;2120:223–232. doi:10.1007/978-1-0716-0327-7_16
23. Ritchie ME, Phipson B, Wu D, et al. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015;43(7):e47. doi:10.1093/nar/gkv007
24. Galili T, O’Callaghan A, Sidi J, Sievert C. heatmaply: an R package for creating interactive cluster heatmaps for online publishing. Bioinformatics. 2018;34(9):1600–1602. doi:10.1093/bioinformatics/btx657
25. Yu G, Wang LG, Han Y, He QY. clusterProfiler: an R package for comparing biological themes among gene clusters. Omics. 2012;16(5):284–287. doi:10.1089/omi.2011.0118
26. Chen EY, Xu H, Gordonov S, Lim MP, Perkins MH, Ma’ayan A. Expression2Kinases: mRNA profiling linked to multiple upstream regulatory layers. Bioinformatics. 2012;28(1):105–111. doi:10.1093/bioinformatics/btr625
27. Clarke DJB, Kuleshov MV, Schilder BM, et al. eXpression2Kinases (X2K) Web: linking expression signatures to upstream cell signaling networks. Nucleic Acids Res. 2018;46(W1):W171–w179. doi:10.1093/nar/gky458
28. Hänzelmann S, Castelo R, Guinney J. GSVA: gene set variation analysis for microarray and RNA-seq data. BMC Bioinform. 2013;14:7. doi:10.1186/1471-2105-14-7
29. Cerami E, Gao J, Dogrusoz U, et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012;2(5):401–404. doi:10.1158/2159-8290.Cd-12-0095
30. Gao J, Aksoy BA, Dogrusoz U, et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal. 2013;6(269):pl1. doi:10.1126/scisignal.2004088
31. Berger H, Vegran F, Chikh M, et al. SOCS3 transactivation by PPARgamma prevents IL-17-driven cancer growth. Cancer Res. 2013;73(12):3578–3590. doi:10.1158/0008-5472.CAN-12-4018
32. Ahmed M, Gaffen SL. IL-17 inhibits adipogenesis in part via C/EBPalpha, PPARgamma and Kruppel-like factors. Cytokine. 2013;61(3):898–905. doi:10.1016/j.cyto.2012.12.007
33. Pabst T, Mueller BU, Zhang P, et al. Dominant-negative mutations of CEBPA, encoding CCAAT/enhancer binding protein-alpha (C/EBPalpha), in acute myeloid leukemia. Nat Genet. 2001;27(3):263–270. doi:10.1038/85820
34. Wei ZY, Li HS, Zhou JY, et al. [Mechanism of transcriptional regulation of Meox1 by transforming growth factor beta (1) and its effect on cell migration of adult human dermal fibroblasts]. Zhonghua Shao Shang Za Zhi. 2020;36(3):224–233, Chinese. doi:10.3760/cma.j.cn501120-20200109-00014
35. Yu C, Niu X, Du Y, et al. IL-17A promotes fatty acid uptake through the IL-17A/IL-17RA/p-STAT3/FABP4 axis to fuel ovarian cancer growth in an adipocyte-rich microenvironment. Cancer Immunol Immunother. 2020;69(1):115–126. doi:10.1007/s00262-019-02445-2
36. Jiang Y, Xu E, Zhang J, Chen M, Flores E, Chen X. The Rbm38-p63 feedback loop is critical for tumor suppression and longevity. Oncogene. 2018;37(21):2863–2872. doi:10.1038/s41388-018-0176-5
37. Burns LA, Maroof A, Marshall D, et al. Presence, function, and regulation of IL-17F-expressing human CD4(+) T cells. Eur J Immunol. 2020;50(4):568–580. doi:10.1002/eji.201948138
38. Hod-Dvorai R, Jacob E, Boyko Y, Avni O. The binding activity of Mel-18 at the Il17a promoter is regulated by the integrated signals of the TCR and polarizing cytokines. Eur J Immunol. 2011;41(8):2424–2435. doi:10.1002/eji.201141620
39. Aboulnasr F, Hazari S, Nayak S, et al. IFN-lambda inhibits MiR-122 transcription through a Stat3-HNF4alpha inflammatory feedback loop in an IFN-alpha resistant HCV cell culture system. PLoS One. 2015;10(12):e0141655. doi:10.1371/journal.pone.0141655
40. Kim YD, Kim YH, Cho YM, et al. Metformin ameliorates IL-6-induced hepatic insulin resistance via induction of orphan nuclear receptor small heterodimer partner (SHP) in mouse models. Diabetologia. 2012;55(5):1482–1494. doi:10.1007/s00125-012-2494-4
41. Li G, Kong B, Zhu Y, et al. Small heterodimer partner overexpression partially protects against liver tumor development in farnesoid X receptor knockout mice. Toxicol Appl Pharmacol. 2013;272(2):299–305. doi:10.1016/j.taap.2013.06.016
42. Shen J, Sun X, Pan B, et al. IL-17 induces macrophages to M2-like phenotype via NF-κB. Cancer Manag Res. 2018;10:4217–4228. doi:10.2147/cmar.S174899
43. Saha S, Zhang Y, Wilson B, Abounader R, Dutta A. The tumor-suppressive long noncoding RNA DRAIC inhibits protein translation and induces autophagy by activating AMPK. J Cell Sci. 2021;134(24). doi:10.1242/jcs.259306
44. Alt EU, Wörner PM, Pfnür A, et al. Targeting TRAF3IP2, compared to Rab27, is more effective in suppressing the development and metastasis of breast cancer. Sci Rep. 2020;10(1):8834. doi:10.1038/s41598-020-64781-z
45. Herjan T, Hong L, Bubenik J, et al. IL-17-receptor-associated adaptor Act1 directly stabilizes mRNAs to mediate IL-17 inflammatory signaling. Nat Immunol. 2018;19(4):354–365. doi:10.1038/s41590-018-0071-9
46. Bareche Y, Buisseret L, Gruosso T, et al. Unraveling triple-negative breast cancer tumor microenvironment heterogeneity: towards an optimized treatment approach. J Natl Cancer Inst. 2020;112(7):708–719. doi:10.1093/jnci/djz208
47. Chung AS, Wu X, Zhuang G, et al. An interleukin-17-mediated paracrine network promotes tumor resistance to anti-angiogenic therapy. Nat Med. 2013;19(9):1114–1123. doi:10.1038/nm.3291
48. Deng J, Thennavan A, Shah S, et al. Serial single-cell profiling analysis of metastatic TNBC during Nab-paclitaxel and pembrolizumab treatment. Breast Cancer Res Treat. 2020;185:85–94. doi:10.1007/s10549-020-05936-4
49. Deng L, Lu D, Bai Y, Wang Y, Bu H, Zheng H. Immune profiles of tumor microenvironment and clinical prognosis among women with triple-negative breast cancer. Cancer Epidemiol Biomarkers Prev. 2019;28(12):1977–1985. doi:10.1158/1055-9965.Epi-19-0469
50. Vangangelt KMH, van Pelt GW, Engels CC, et al. Prognostic value of tumor-stroma ratio combined with the immune status of tumors in invasive breast carcinoma. Breast Cancer Res Treat. 2018;168(3):601–612. doi:10.1007/s10549-017-4617-6
51. Zheng S, Zou Y, Xie X, et al. Development and validation of a stromal immune phenotype classifier for predicting immune activity and prognosis in triple-negative breast cancer. Int J Cancer. 2020;147(2):542–553. doi:10.1002/ijc.33009
52. Karn T, Jiang T, Hatzis C, et al. Association between genomic metrics and immune infiltration in triple-negative breast cancer. JAMA Oncol. 2017;3(12):1707–1711. doi:10.1001/jamaoncol.2017.2140
53. Deng J, Thennavan A, Shah S, et al. Serial single-cell profiling analysis of metastatic TNBC during Nab-paclitaxel and pembrolizumab treatment. Breast Cancer Res Treat. 2021;185(1):85–94. doi:10.1007/s10549-020-05936-4
54. Lambert SA, Jolma A, Campitelli LF, et al. The human transcription factors. Cell. 2018;172(4):650–665. doi:10.1016/j.cell.2018.01.029
55. Tsai YF, Huang CC, Lin YS, et al. Interleukin 17A promotes cell migration, enhances anoikis resistance, and creates a microenvironment suitable for triple negative breast cancer tumor metastasis. Cancer Immunol Immunother. 2021;70(8):2339–2351. doi:10.1007/s00262-021-02867-x
56. Shuai C, Yang X, Pan H, Han W. Estrogen receptor downregulates expression of PD-1/PD-L1 and infiltration of CD8(+) T cells by inhibiting IL-17 signaling transduction in breast cancer. Front Oncol. 2020;10:582863. doi:10.3389/fonc.2020.582863
57. Wang L, Yi T, Kortylewski M, Pardoll DM, Zeng D, Yu H. IL-17 can promote tumor growth through an IL-6-Stat3 signaling pathway. J Exp Med. 2009;206(7):1457–1464. doi:10.1084/jem.20090207
58. Su X, Ye J, Hsueh EC, Zhang Y, Hoft DF, Peng G. Tumor microenvironments direct the recruitment and expansion of human Th17 cells. J Immunol. 2010;184(3):1630–1641. doi:10.4049/jimmunol.0902813
59. Ruhland MK, Loza AJ, Capietto AH, et al. Stromal senescence establishes an immunosuppressive microenvironment that drives tumorigenesis. Nat Commun. 2016;7:11762. doi:10.1038/ncomms11762
60. Gan L, Yang Y, Li Q, Feng Y, Liu T, Guo W. Epigenetic regulation of cancer progression by EZH2: from biological insights to therapeutic potential. Biomark Res. 2018;6:10. doi:10.1186/s40364-018-0122-2
61. Liu YH, Tsai YS, Lin SC, et al. Quantitative PPARγ expression affects the balance between tolerance and immunity. Sci Rep. 2016;6:26646. doi:10.1038/srep26646
62. Faucheux L, Grandclaudon M, Perrot-Dockès M, et al. A multivariate Th17 metagene for prognostic stratification in T cell non-inflamed triple negative breast cancer. Oncoimmunology. 2019;8(9):e1624130. doi:10.1080/2162402x.2019.1624130
63. Chen J, Ye X, Pitmon E, et al. IL-17 inhibits CXCL9/10-mediated recruitment of CD8(+) cytotoxic T cells and regulatory T cells to colorectal tumors. J Immunother Cancer. 2019;7(1):324. doi:10.1186/s40425-019-0757-z
64. Dadaglio G, Fayolle C, Oberkampf M, et al. IL-17 suppresses the therapeutic activity of cancer vaccines through the inhibition of CD8(+) T-cell responses. Oncoimmunology. 2020;9(1):1758606. doi:10.1080/2162402x.2020.1758606
65. Kaewkangsadan V, Verma C, Eremin JM, Cowley G, Ilyas M, Eremin O. Crucial contributions by T lymphocytes (effector, regulatory, and checkpoint inhibitor) and cytokines (TH1, TH2, and TH17) to a pathological complete response induced by neoadjuvant chemotherapy in women with breast cancer. J Immunol Res. 2016;2016:4757405. doi:10.1155/2016/4757405
66. Li Q, Ngo PT, Egilmez NK. Anti-PD-1 antibody-mediated activation of type 17 T-cells undermines checkpoint blockade therapy. Cancer Immunol Immunother. 2021;70(6):1789–1796. doi:10.1007/s00262-020-02795-2
67. Moaaz M, Lotfy H, Motawea MA, Fadali G. The interplay of interleukin-17A and breast cancer tumor microenvironment as a novel immunotherapeutic approach to increase tumor immunogenicity. Immunobiology. 2021;226(2):152068. doi:10.1016/j.imbio.2021.152068
68. Peng DH, Rodriguez BL, Diao L, et al. Th17 cells contribute to combination MEK inhibitor and anti-PD-L1 therapy resistance in KRAS/p53 mutant lung cancers. Nat Commun. 2021;12(1):2606. doi:10.1038/s41467-021-22875-w
69. Thibaudin M, Chaix M, Boidot R, et al. Human ectonucleotidase-expressing CD25(high) Th17 cells accumulate in breast cancer tumors and exert immunosuppressive functions. Oncoimmunology. 2016;5(1):e1055444. doi:10.1080/2162402x.2015.1055444
70. Wang X, Sun R, Hao X, Lian ZX, Wei H, Tian Z. IL-17 constrains natural killer cell activity by restraining IL-15-driven cell maturation via SOCS3. Proc Natl Acad Sci U S A. 2019;116(35):17409–17418. doi:10.1073/pnas.1904125116
 © 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.