Back to Journals » Neuropsychiatric Disease and Treatment » Volume 16
CPCGI Reduces Gray and White Matter Injury by Upregulating Nrf2 Signaling and Suppressing Calpain Overactivation in a Rat Model of Controlled Cortical Impact
Authors Niu F, Qian K ![]() , Qi H, Zhao Y, Jiang Y
, Qi H, Zhao Y, Jiang Y ![]() , Jia W, Sun M
, Jia W, Sun M
Received 4 June 2020
Accepted for publication 28 July 2020
Published 12 August 2020 Volume 2020:16 Pages 1929—1941
DOI https://doi.org/10.2147/NDT.S266136
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Yu-Ping Ning
Fei Niu,1,* Ke Qian,2,* Hongyan Qi,3 Yumei Zhao,4 Yingying Jiang,4 Wang Jia,2 Ming Sun4
1Department of Neurotrauma, Beijing Key Laboratory of Central Nervous System Injury, Beijing Neurosurgical Institute, Capital Medical University, Beijing 100070, People’s Republic of China; 2Department of Neurosurgery, Beijing Tiantan Hospital, Capital Medical University, Beijing 100070, People’s Republic of China; 3Department of Acupuncture, Lianyungang TCM Hospital Affiliated to Nanjing University of Chinese Medicine, Lianyungang City 222000, Jiangsu Province, People’s Republic of China; 4Department of Neuropharmacology, Beijing Key Laboratory of Central Nervous System Injury, Beijing Neurosurgical Institute, Capital Medical University, Beijing 100070, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Ming Sun
Department of Neuropharmacology, Beijing Key Laboratory of Central Nervous System Injury, Beijing Neurosurgical Institute, Capital Medical University, 119 South Fourth Ring West Road, Fengtai District, Beijing 100070, People’s Republic of China
Tel/ Fax +86-10-59975497
Email [email protected]
Background: Compound porcine cerebroside and ganglioside injection (CPCGI), which involves injection of a neurotrophic drug, has been widely used to treat certain brain disorders in the clinic; however, the detailed mechanism is unknown. This study investigated whether CPCGI protects the brain from trauma by stimulating antioxidative nuclear factor erythroid-2-related factor 2 (Nrf2) signaling and suppressing calpain overactivation in a rat model of controlled cortical impact (CCI).
Materials and Methods: The rat model of CCI was used. Neurological deficits, contusion, and white matter damage were evaluated 3 days after CCI. Calpain activation, Nrf2 signaling and oxidative stress were determined 24 h after CCI.
Results: CPCGI dose-dependently reduced neurological deficits, attenuated axonal and myelin sheath injury, and decreased contusion volume 3 days post-CCI. Moreover, CPCGI reduced calpain activity, and enhanced the cytosolic levels of calpastatin, αII-spectrin, microtubule-associated protein 2 (MAP2), neurofilament heavy chain (NF-H) and myelin basic protein (MBP) in traumatic tissues 24 h post-CCI. Furthermore, CPCGI reduced the levels of nuclear Kelch-like ECH-associated protein 1 (Keap1) and thioredoxin interacting protein (TXNIP); increased the levels of cytosolic Nrf2 and thioredoxin 1 (Trx 1) and nuclear Nrf2; increased the cytosolic and nuclear Nrf2/Keap1 and Trx 1/TXNIP ratios; enhanced the levels of heme oxygenase 1 (HO-1), glutathione (GSH), superoxide dismutase activity, and total antioxidative capacity; and reduced the levels of malondialdehyde in TBI tissues.
Conclusion: These data confirm the neuroprotective effect of CPCGI against gray and white matter damage due to CCI and suggest that activating Nrf2 signaling and alleviating oxidative stress-mediated calpain activation could be one mechanism by which CPCGI protects against brain trauma.
Keywords: compound porcine cerebroside and ganglioside injection, traumatic brain injury, Nrf2 signaling, oxidative stress, calpain
Introduction
Traumatic brain injury (TBI) is a major global health problem with high morbidity and mortality, and the incidence is rising among both young and elderly individuals.1 Secondary injury, triggered by primary injury, causes neurological dysfunction and neuronal damage. The mechanism of secondary injury due to TBI is very complex, involving excitotoxicity, intracellular calcium overload, oxidative stress, inflammation, apoptosis, and so on. In particular, oxidative stress is reported to play a vital role in the acute, subacute and chronic phases of TBI.2,3
The nuclear factor erythroid-2-related factor 2 (Nrf2) pathway controls an array of endogenous cellular defense mechanisms against oxidative stress.4 Under physiological conditions, Nrf2 is conjugated by Kelch-like ECH-associated protein 1 (Keap1) and isolated in the cytoplasm of cells; under external stimulation, Nrf2 is released from the Nrf2/Keap1 dimer and transfers from the cytosol into the nucleus, where it binds with the antioxidant response element (ARE) in DNA, upregulating the expression of antioxidant enzymes to resist the oxidation of free radicals. The downstream Phase II detoxification enzyme and antioxidation enzymes include superoxide dismutase (SOD), catalase, glutathione (GSH) peroxidase, heme oxygenase 1 (HO-1), thioredoxin reductase, and thioredoxin (Trx).5 Studies have confirmed the involvement of Nrf2 signaling in the antioxidant protective effects after TBI,6,7 suggesting a potential role of Nrf2 as a therapeutic target of TBI.
In recent decades, the role of calpain in brain injury has been widely recognized. Calpain is a calcium-dependent neutral protease that is strictly regulated by calcium and calpastatin, an endogenous calpain inhibitor.8 For example, the overactivation of calpain induced by TBI degrades a number of target proteins, and results in the neuronal death. Various calpain inhibitors have been reported to improve the neurological function and reduce neuronal death due to TBI. Therefore, calpain inhibition has become a target in the research for new drugs to protect the brain against TBI.9
In 2010, the China Food and Drug Administration approved compound porcine cerebroside and ganglioside injection (CPCGI), a neurotrophic drug, for use in the treatment of stroke, Alzheimer’s disease, and central and peripheral nerve injuries.10–13 The main components of CPCGI include polypeptides, various gangliosides, and hypoxanthine, etc. CPCGI has been reported to improve cerebral blood circulation and neurological function, reduce apoptotic cell death, and prompt the recovery of synaptic and mitochondrial function in a rat model of focal cerebral ischemia.10,13 However, there is no report about the protective effects of CPCGI against TBI. In the present study, we primarily investigated whether CPCGI protected the brain against TBI through upregulating Nrf2 signaling and suppressing calpain overactivation due to oxidative stress in a rat model of controlled cortical impact (CCI).
Materials and Methods
Controlled Cortical Impact Models
All experiments were performed in accordance with the guidelines established by the National Institutes of Health for the Care and Use of Laboratory Animals and were approved by the Animal Care Committee of the Beijing Neurosurgical Institute. Male Sprague-Dawley rats (weighing 280–320 g, Vital River Laboratories, Beijing, China) were housed under a 12 h light/12 h dark cycle at a constant temperature (22 °C) with free access to food and water. The animals were anesthetized by isoflurane inhalation and fixed in a stereotaxic frame (RWD Life Sciences Co., Shenzhen, China). The body temperature was maintained at 37 ± 0.5 °C by a thermal pad throughout the surgical procedure. CCI was induced by a PCI3000 PinPoint Precision Cortical Impactor system (Hatteras Instruments, Cary, NC, USA). A circular craniotomy of approximately 5 mm in diameter was made in the middle of the right parietal bone between the coronal and lambdoid sutures, and approximately 0.5 mm from the sagittal suture, leaving the dura intact. The CCI was produced with a 4.0 mm-diameter impactor, and the parameters were as follows: speed = 2.0 m/sec; impact duration = 85 ms; and vertical deformation depth = 2.5 mm, which is consistent with the moderate brain injury model.14 After each impact, the impact tip was wiped clean with sterile alcohol. The sham-operated rats only underwent a craniotomy. After surgery, the incision was closed with 4–0 silk sutures.
Experimental Protocols
CPCGI (provided by Buchang Pharmaceutical Co. Ltd, Xian, Shanxi, China) was dissolved in normal saline (NS). In the experiment performed to calculate the neurological severity score (NSS), contusion volume and corpus callosum damage, the rats were randomly divided into five groups treated with different doses of CPCGI or vehicle: (1) Low-dose CPCGI (CPCGI-L): 0.2 mL/kg; (2) Medium-dose CPCGI (CPCGI-M): 0.6 mL/kg; (3) High-dose CPCGI (CPCGI-H): 2 mL/kg; (4) vehicle: a similar volume of NS (2 mL/kg); and (5) sham: NS (2 mL/kg). NS or CPCGI was administered intraperitoneally (i.p.) in a volume of 2 mL/kg 30 min after induction of CCI. On the second and third days after induction of trauma, NS or CPCGI was administered at 9 am again. The NSS was evaluated 6 h, 24 h, and 3 days after CCI (n = 14 per group). The brains were collected 72 h post-CCI, and the contusion volumes and corpus callosum (CC) damage were assayed (n = 13 per group).
For calpain activity assay, oxidative injury, and Western blotting, rats were randomly divided into three groups and treated with CPCGI or vehicle: (1) CPCGI: 0.6 mL/kg; (2) vehicle, NS (2 mL/kg); and (3) sham: NS (2 mL/kg). The traumatic brain tissues were collected 24 h post-CCI (n = 6 per group).
Evaluation of Neurological Deficits
In all animals, neurobehavioral tests were performed before induction of CCI and 6 h, 24 h, 72 h post-CCI. Neurological function was assessed using the modified neurological severity score (NSS), a scale of 0–18 (normal score = 0; maximal deficit score = 18) that includes motor, sensory, balance and reflex tests.15 All behavioral scoring was performed by two trained observers who were blinded to the experimental groups.
Determination of Contusion Volume and White Matter Damage
Sham-operated or traumatic rats were anesthetized and transcardially perfused with heparinized normal saline and 4% buffered paraformaldehyde 3 days after induction of CCI. The intact brains were dissected and fixed in 4% buffered paraformaldehyde for 24 h and embedded in paraffin, and 5 μm-thick sections at 0.6 mm intervals from the frontal pole were collected. The sections were stained with hematoxylin and eosin (H&E) and examined under a light microscope, and pictures were taken by a CCD camera. The contusion brain regions were distinguished from the normal brain by pyknotic neurons, hemorrhage and edema, and contusion volumes were determined using an image analysis program (Konghai Co., Beijing, China) as described previously.16 Briefly, the normal hemisphere area of each slice (A1) was measured by an image analysis program, and the normal hemisphere volume (V1) was calculated: V1 = ΣA1 × D (mm3), where D represents the interval between two adjacent slices (0.6 mm). Then, the area of normal brain tissue of each slice (A2) in the injured hemisphere was measured, and the volume of normal brain tissue in the injured hemisphere (V2) was calculated: V2 = ΣA2 × D (mm3). The contusion volume = V1 – V2.
For the determination of white matter damage, 5 μm-thick coronal sections through the dorsal striatum were collected. The sections were stained with Luxol fast blue-periodic acid Schiff (LFB-PAS) and Bielschowsky’s silver impregnation to reveal myelin and axons, respectively.17,18
The slices stained with LFB-PAS or Bielschowsky’s silver stains were used to measure the optical densities (ODs) of myelin (LFB-PAS) and axons (Bielschowsky’s silver impregnation).18,19 The measured OD values reflect the stainability of white matter, and a decreased OD value indirectly reflects destruction of white matter because of loss of stainability. The OD values of corpus callosum in normal (left) side and damaged (right) side were measured by an image analysis program (Konghai Co., Beijing, China), and the results were expressed as a percentage of the normal side.
Sample Collection and Preparation
The tissues of right injured hemisphere were dissected at 4 °C, and samples were prepared as described previously20,21 with the following modifications. Briefly, rats were killed under anesthesia. The intact brain was collected, and the right (injured) hemisphere was dissected into three sections beginning 2 mm from the anterior tip of the frontal lobe and 2 mm from the posterior tip of the occipital lobe. The middle part tissue as traumatic brain tissue was collected. The traumatic brain tissues were homogenized in 10 volumes of homogenization buffer A (20 mM N-2-hydroxyethylpiperazine-N’-2ʹ-ethanesulfonic acid (HEPES), 250 mM sucrose, 10% glycerol, 10 mM KCl, 1.5 mM MgCl2, 1 mM EDTA, 1 mM EGTA, 1% Triton-X 100, and 1% protease inhibitor cocktail, pH 7.5). The homogenates were centrifuged at 1000 × g at 4 °C for 15 min to separate the homogenates into supernatant A and pellet A. Pellet A was washed with buffer A three times, and suspended in equal volumes of buffer B (20 mM HEPES, 20% glycerol, 1.5 mM MgCl2, 420 mM NaCl, 1 mM EDTA, 1 mM EGTA, 1 mM DTT, 1% Triton-X 100, and 10% protease inhibitor cocktail, pH 7.5) and vortexed at maximum speed for 15 s. The suspension was placed on ice for 30 min and then centrifuged at 14,000 × g for 30 min at 4 °C, and the supernatant containing the nuclear fraction was transferred and stored at −70 °C. Supernatant A was further centrifuged at 16,000 × g for 30 min at 4 °C, and the supernatant was used as the cytosolic fraction. The protein concentrations in the cytosolic and nuclear fractions were determined using the bicinchoninic acid (BCA) method.
Redox Status Assay
The redox status of the cortex after induction of CCI was assessed by assaying the level of lipid peroxidation and the levels of antioxidative ability in cytosolic fractions. The antioxidative ability was determined by the activities of SOD, the levels of GSH and total-anti-oxidizing-capability (T-AOC). The lipid peroxidation was determined by measuring the levels of malondialdehyde (MDA). The activities of SOD, the T-AOC and the levels of GSH and MDA were measured by kits (Nanjing Jiancheng Bioengineering Institute, Nanjing, China).
Calpain Activity Assay
Calpain activity in cytosolic fractions was assayed using a modification of a previously described method.22 Briefly, cleavage of the fluorogenic calpain substrate (Suc-LY-AMC; Calbiochem) to its fluorescent product was used to measure calpain activity. The cytosolic fractions (20 μL) were incubated with calpain reaction buffer (250 μL; 20 mM HEPES, pH 7.6; 1 mM EDTA; 50 mM NaCl; and 0.1% (v/v) 2-mercaptoethanol) containing 20 μM Suc-LY-AMC (Calbiochem), a fluorogenic calpain substrate, for 1 h at 37 °C. The free AMC released by proteolysis was assessed (excitation, 380 nm; emission, 460 nm) by a multifunctional microplate reader (BioTek Instruments, Inc., Winooski, VT, USA). Fluorescent arbitrary units were converted into micromoles of AMC released per hour and milligrams of protein using a standard curve of free AMC (Santa Cruz Biotech., CA, USA).
Western Blot
The protein levels of calpastatin, αII-spectrin, microtubule-associated protein 2 (MAP2), neurofilament heavy chain (NF-H), myelin basic protein (MBP), Nrf2, Keap1, thioredoxin interacting protein (TXNIP), HO-1, Trx 1 and β-actin in cytosolic fractions, and the levels of Nrf2, Keap1, TXNIP, Trx 1 and histone H3 in nuclear fractions were separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE). Briefly, equal masses of proteins (30 μg) were separated by SDS-PAGE, and molecular weight markers (Abcam Inc., MA, USA) were loaded onto each gel for protein band identification. The proteins on the gel were transferred to a PVDF membrane (Millipore, MA, USA) and blocked with Tris-buffered saline containing 1% Tween 20. The membrane was then probed with antibody which reacted with calpastatin (1:1000), MAP2 (1:500), Nrf2 (1:250), Keap1 (1:250), neurofilament heavy chain (NF-H; 1:500), or histone H3 (1:500, Abcam, Cambridge, United Kingdom), αII-spectrin (1:400), MBP (1:400, Millipore, MA, USA), TXNIP (1:250), Trx 1 (1:250, Cell Signaling Technology, MA, USA), HO-1 (1:500, R&D, Minneapolis, MN, USA), β-actin (1:2000, Sigma-Aldrich Corp., MO, USA) at 4 °C overnight and subsequently by horseradish peroxidase-conjugated secondary antibodies. Antibody binding was visualized by chemiluminescence (Millipore, MA, USA). β-actin and histone H3 were used as an internal control for the cytosolic and nuclear fractions, respectively. The results were expressed as percentages of the levels in sham-operated rats.
Statistical Analysis
Data are presented as mean ± SEM. The contusion volumes; the average optical densities (ODs) of Bielschowsky’s silver staining and LFB-PAS-hematoxylin impregnation, the activities of calpain and SOD; the T-AOC; and the levels of GSH, MDA, calpastatin, αII-spectrin, MAP2, NF-H, MBP, Nrf2, Keap1, TXNIP, HO-1, and Trx 1 were evaluated by one-way ANOVA with a post hoc LSD test. NSS was analyzed with a non-parametric Kruskal Wallis ANOVA with Dunn’s post hoc test. A P value of < 0.05 was defined as statistically significant.
Results
Effects of CPCGI on Neurobehavior and Contusion Volumes Due to CCI
Before the induction of CCI, all animals showed a normal neurological severity score (NSS) (score = 0). The vehicle-treated rats showed significant neurological deficits at 6 h, 24 h and 3 days after induction of CCI, and NSS values were 11.3 ± 0.2, 11.1 ± 0.2, and 10.6 ± 0.3, respectively. Treatment with 0.2 mL/kg CPCGI attenuated the neurological deficits, but the difference did not reach statistical significance. Treatment with 0.6 mL/kg CPCGI and 2 mL/kg CPCGI markedly reduced the neurological deficits compared with vehicle-treated rats (Figure 1A, 0.6 mL/kg: P < 0.05, 0.01 and 0.01 at 6 h, 24 h and 3 days post-trauma, respectively; 2 mL/kg: P < 0.05 and 0.01 at 24 h and 3 days post-trauma, respectively). In vehicle-treated rats, the contusion volume due to CCI was 16.4 ± 1.0 mm3. Compared with vehicle-treated rats, treatment with 0.2 mL/kg of CPCGI decreased the contusion volume, but the difference was not statically significant (Figure 1B and C). Treatment with 0.6 mL/kg and 2 mL/kg CPCGI significantly reduced the contusion volumes (P < 0.01 and 0.05 vs vehicle-treated rats, respectively). These data indicated the improvement of neurological functions and decreased contusion volumes in a dose-dependent manner by CPCGI after CCI.
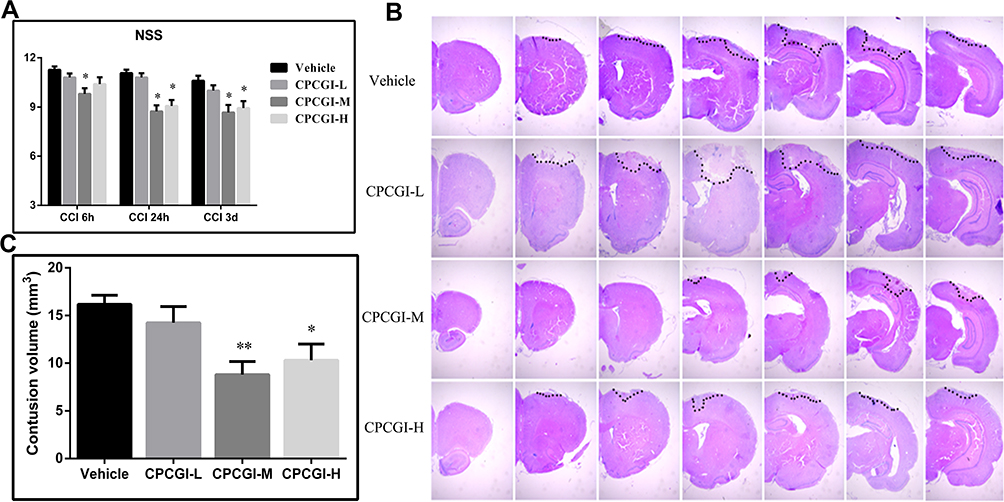 |
Figure 1 Effects of CPCGI on neurological deficits and contusion volumes in CCI rats (n=14/group). (A) Effects of CPCGI on NSS. (B) Representative photographs of selected H&E-stained coronal sections. (C) The bar graph presents the contusion volumes as determined by H&E staining in tissues from vehicle- or CPCGI-treated rats. *P < 0.05 and **P < 0.01 vs the vehicle group. |
Effects of CPCGI on Corpus Callosum Damage
The corpus callosum is mainly composed of myelinated nerve fibers. In the present study, the white matter damage was evaluated through investigating the corpus callosum morphology after induction of CCI. Axons and myelins were stained by Bielschowsky’s silver staining and LFB-PAS-hematoxylin, respectively. Typical photomicrographs of the right corpus callosum are shown in Figure 2. In the vehicle-treated rats, the axons and myelin sheaths showed significant damage. Axons showed irregular, twisted profiles and segmental fragmentation. The myelin sheaths lost their LFB-PAS stainability and showed empty spaces (vacuoles) separating myelin sheaths in the lesions of white matter. Overall statistical analysis demonstrated the axonal and myelin sheath damage in the corpus callosum in vehicle-treated rats (both P < 0.01 vs sham-operated rats). CPCGI at a dose of 0.2 mL/kg had no significant effect on the axons and myelin sheaths of the corpus callosum due to CCI. Treatment with 0.6 mL/kg and 2 mL/kg CPCGI markedly reduced the axonal and myelin sheath injury compared with vehicle-treated rats (axon: both P < 0.05; myelin sheath: P < 0.01 and 0.05, respectively). Our data showed dose-dependent protective effects of CPCGI on the white matter damage due to CCI.
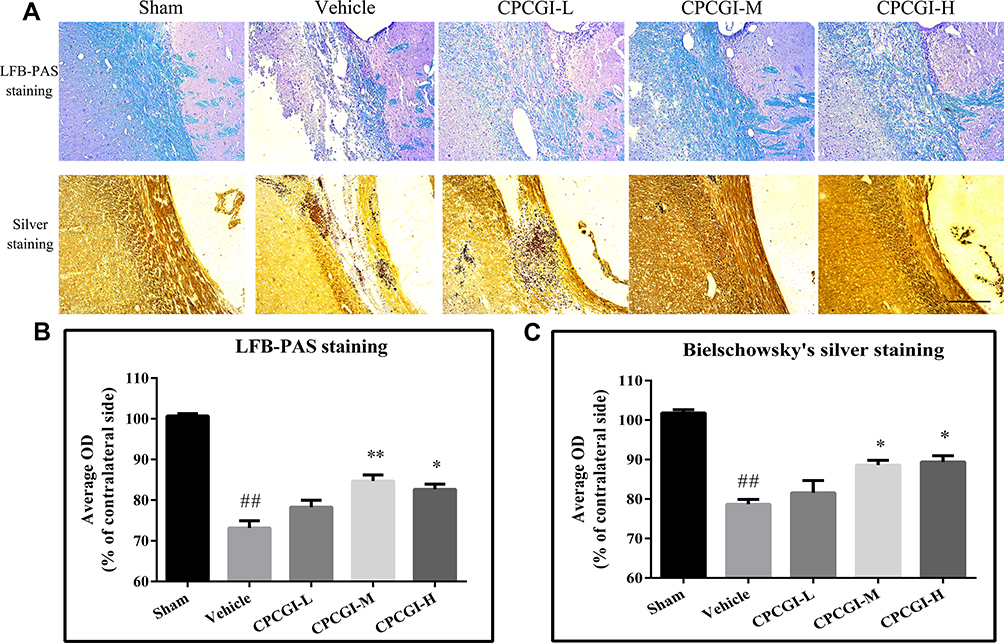 |
Figure 2 Effects of CPCGI on corpus callosum damage due to CCI. (A) Representative corpus callosum images from animals of all groups. Scale bar=100 μm. (B and C) The bar graph presents the myelin sheath and axonal damage in the corpus callosum from LFB-PAS staining and Bielschowsky’s silver staining in sham-operated and vehicle- or CPCGI-treated rats. The data are presented as the mean ± SEM (n=13/group). ##P < 0.01 vs sham-operated rats; *P < 0.05 and **P < 0.01 vs vehicle-treated rats. |
Effects of CPCGI on Calpain Activity and Calpastatin Levels
The results are shown in Figure 3. In vehicle-treated rats, the calpain activity was increased significantly (P < 0.01 vs sham-operated rats), and the calpastatin protein level was decreased significantly (P < 0.05 vs sham-operated rats). Treatment with CPCGI markedly reduced the calpain activity and enhanced the calpastatin protein level in traumatic tissues compared with vehicle-treated rats (both P < 0.05).
 |
Figure 3 Effects of CPCGI on calpain activity and calpastatin protein levels in traumatic brain tissues. (A) Calpain activity; (B) Western blot analysis with calpastatin antibody; (C) The bar graph presents the calpastatin protein levels, as determined by Western blot analysis, in sham-operated and vehicle- or CPCGI-treated rats. The data are presented as the mean ± SEM (n=6/group). #P < 0.05 and ##P < 0.01 vs sham-operated rats; *P < 0.05 vs vehicle-treated rats. |
Effects of CPCGI on the Levels of αII-Spectrin, MAP2, MBP and NF-H
αII-Spectrin, MAP2, NF-H and MBP are substrates for calpain. We investigated the effects of CPCGI treatment on the levels of αII-spectrin, MAP2, NF-H and MBP in traumatic brain tissues, and the results are shown in Figure 4. The protein levels of αII-spectrin, MAP2, NF-H and MBP were decreased significantly in traumatic brain tissues (P < 0.05, 0.01, 0.05 and 0.05 vs sham-operated rats, respectively). CPCGI treatment enhanced the protein levels of αII-spectrin, MAP2, NF-H and MBP in traumatic brain tissues (P < 0.05, 0.01, 0.05 and 0.05 vs vehicle-treated rats, respectively).
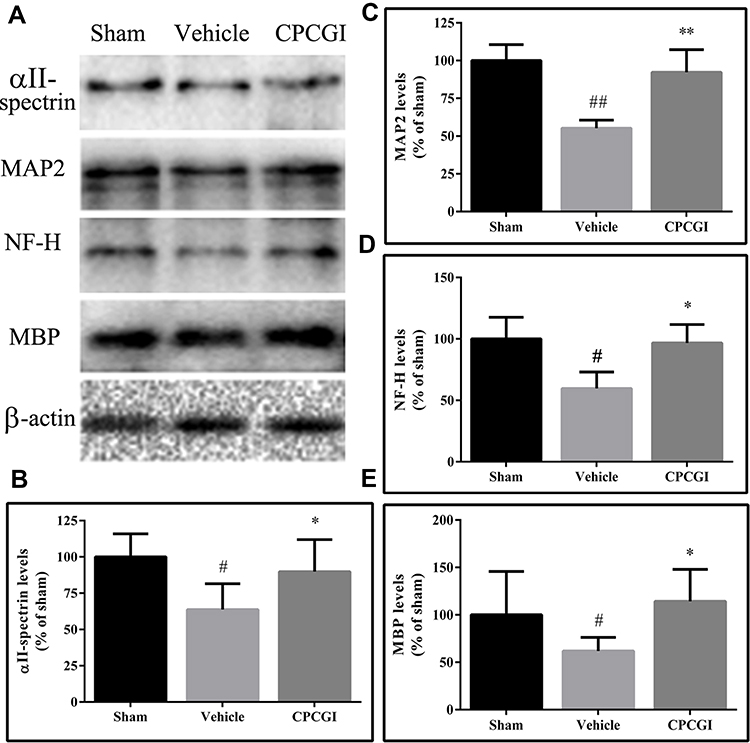 |
Figure 4 Effects of CPCGI on the protein levels of αII-spectrin, MAP2, NF-H and MBP in CCI tissues. The data are presented as the mean ± SEM (n=6/group). (A) Western blot analysis of αII-spectrin, MAP2, NF-H and MBP expression. (B–E) The bar graphs present the protein levels of αII-spectrin, MAP2, NF-H and MBP, as determined by Western blot analysis, in tissues from sham-operated and vehicle- or CPCGI-treated rats, respectively. #P < 0.05 and ##P < 0.01 vs sham-operated rats. *P < 0.05 and **P < 0.01 vs vehicle-treated rats. |
Effects of CPCGI on the Levels of Nrf2 and Keap1
To determine the Nrf2 activation, the protein levels of Nrf2 and Keap1 in cytosolic and nuclear were assessed. As shown in Figure 5, the protein levels of Nrf2 in the cytosol and nucleus were decreased significantly (both P < 0.05 vs sham-operated rats), whereas the protein levels of Keap1 in the cytosol and nucleus had no significant changes compared with that in sham-operated rats. Treatment with CPCGI markedly enhanced the protein levels of Nrf2 in the cytosol and nucleus (both P < 0.05 vs vehicle-treated rats) and significantly reduced the protein levels of Keap1 in the nucleus (P < 0.01 vs vehicle-treated rats), although it had no significant effect on the protein levels of Keap1 in the cytosol compared with that in vehicle-treated rats. Moreover, the Nrf2/Keap1 ratio in the cytosol in vehicle-treated rats was decreased significantly (P < 0.05 vs sham-operated rats), and that in the nucleus in vehicle-treated rats was increased compared with that in sham-operated rats, but the difference was not significant. Treatment with CPCGI enhanced the Nrf2/Keap1 ratios in the cytosol and nucleus (P < 0.05 and 0.01 vs sham-operated rats, respectively). These data indicated that CPCGI increased the expression and nuclear translocation of Nrf2 during traumatic brain injury.
 |
Figure 5 Effects of CPCGI on Nrf2/Keap1 signaling in TBI tissues after CCI. (A) Western blot analysis of Nrf2, Keap1, TXNIP and Trx 1 expression. (B and C) The bar graphs present the protein levels of Nrf2 in the cytosol and the nucleus, as determined by Western blot analysis. (D and E) The bar graphs present the protein levels of Keap1 in the cytosol and the nucleus, as determined by Western blot analysis. (F and G) The bar graphs present the Nrf2/Keap1 ratios in the cytosol and the nucleus. (H and I) The bar graphs present the protein levels of TXNIP in the cytosol and the nucleus, as determined by Western blot analysis. (J and K) The bar graphs present the protein levels of Trx 1 in the cytosol and the nucleus, as determined by Western blot analysis. (L and M) The bar graphs present the Trx 1/TXNIP ratios in the cytosol and the nucleus. The data are expressed as the mean ± SEM (n=6/group). #P <0.05 vs sham-operated rats; *P < 0.05 and **P < 0.01 vs vehicle-treated rats. |
Effects of CPCGI on the Levels of Trx 1 and TXNIP
Nrf2 is reported to drive the expression of Trx 1, while TXNIP is an endogenous inhibitory protein of Trx 1. The protein levels of Trx 1 and TXNIP in the cytosol and nucleus were investigated, and the results are shown in Figure 5. The protein levels of TXNIP in the cytosol in vehicle-treated rats were decreased significantly (P < 0.05 vs sham-operated rats), although that in the nucleus in vehicle-treated rats had no significant changes compared with that in sham-operated rats. Treatment with CPCGI had no significant effects on the protein levels of TXNIP in the cytosol, whereas it markedly reduced that in nucleus (P < 0.01 vs vehicle-treated rats).
The Trx 1 levels in cytosol in traumatic brain tissues were increased significantly (P < 0.05 vs sham-operated rats). Although the Trx 1 levels in the nucleus in traumatic rats were also increased compared with those in sham-operated rats, the difference did not reach statistical significance. CPCGI treatment enhanced Trx 1 levels in the cytosol (P < 0.05 vs vehicle-treated rats), although it had no significant effects on the levels in the nucleus compared with vehicle-treated rats. A significant increase in the Trx 1/TXNIP ratio in the cytosol was found (P < 0.05 vs sham-operated rats), and treatment with CPCGI markedly upregulated the Trx 1/TXNIP ratio in cytosol (P < 0.05 vs vehicle-treated rats). In the nucleus, the Trx 1/TXNIP ratio in vehicle-treated rats had no significant change compared with that in sham operated rats, and CPCGI treatment increased the Trx 1/TXNIP ratio compared with that in vehicle-treated rats, but the difference did not reach statistical significance.
Effects of CPCGI on the Levels of HO-1
HO-1 is one of the target genes of Nrf2. In the present study, we found that the protein levels of HO-1 in traumatic brain tissues were increased compared with those in sham-operated rats, but the difference did not reach statistical significance (Figure 6A). CPCGI treatment markedly increased the protein levels of HO-1 in traumatic brain tissues (P < 0.05 vs vehicle-treated rats).
 |
Figure 6 Effects of CPCGI on HO-1 levels and redox status in TBI tissues after CCI. (A) Western blot analysis of HO-1 expression. (B) The bar graph presents the protein levels of HO-1, as determined by Western blot analysis, in tissues from sham-operated and vehicle- or CPCGI-treated rats. (C) SOD activities. (D) GSH levels. (E) T-AOC. (F) MDA levels. The data are presented as the mean ± SEM (n=6/group). # P < 0.05 and ## P < 0.01 vs sham-operated rats. *P < 0.05 and **P < 0.01 vs vehicle-treated rats. |
Effects of CPCGI on Redox Status
The redox status in traumatic brain tissues was determined through assaying the SOD activities, T-AOC, and the levels of GSH and MDA, and the results are shown in Figure 6B–F. Compared with sham-operated rats, the levels of GSH in traumatic brain tissues had no significant changes, but the activities of SOD and the T-AOC were markedly reduced (both P < 0.05), and the levels of MDA were markedly increased (P < 0.01). Treatment with CPCGI increased the activities of SOD and the levels of GSH and T-AOC, and reduced the levels of MDA in traumatic brain tissues (P < 0.05, 0.01, 0.05 and 0.01 vs vehicle-treated rats, respectively). Our data indicated the reduction of anti-oxidative capacity and aggravation of oxidative damage in CCI rats, and CPCGI enhanced the anti-oxidative capacity and significantly decreased oxidative damage.
Discussion
The present study primarily investigated the protective effects of CPCGI against the brain injury due to CCI. Our results showed that CPCGI reduced the neurological deficits, attenuated corpus callosum injury, and decreased the contusion volumes in a dose-dependent manner. CPCGI had no effects on Keap1 and TXNIP levels, but it enhanced Nrf2 and Trx 1 levels, Nrf2/Keap1 and Trx 1/TXNIP ratios in cytosol. CPCGI reduced Keap1 and TXNIP levels, and enhanced Nrf2 levels and Nrf2/Keap1 ratio, but it had no effects on Trx 1 levels and the Trx 1/TXNIP ratio in the nucleus. Furthermore, CPCGI increased the HO-1 levels, the SOD activities and the GSH levels and total anti-oxidative capacities and decreased the MDA levels in traumatic brain tissues. Moreover, it increased the calpastatin levels, reduced the calpain activities, and attenuated the degradation of αII-spectrin, MAP2, NF-H and MBP in traumatic brain tissues. These data confirmed the neuroprotective effects of CPCGI against gray and white matter damage due to CCI, and suggest that the activation of Nrf2 signaling and reduction of oxidative stress-mediated calpain over-activation could be one mechanism of CPCGI against brain trauma.
Accumulating evidence demonstrates the detrimental role of oxidative stress in brain trauma. The formation of reactive oxygen species (ROS) is increased after TBI. These ROS react with proteins, DNA and polyunsaturated fatty acids in membrane phospholipids, leading to cell damage. Moreover, decreased activities of SOD and GSH peroxidase and increased levels of MDA are reported after TBI. In addition, antioxidants and ROS scavengers protect the brain against experimental TBI.3,23 Our data further demonstrated the contribution of ROS to secondary damage due to TBI and suggest that restoring antioxidative defense systems and reducing oxidative stress may be one mechanism by which CPCGI protects against TBI.
It is well known that Nrf2 signaling plays a key role in endogenous protection against oxidative stress, which regulates the intracellular redox status by driving the gene transcription of some antioxidants and phase II detoxifying enzymes. Keap1 is an endogenous inhibitory protein of Nrf2. Under normal conditions, it binds with Nrf2 to form the Nrf2/Keap1 complex, which sequesters Nrf2 in the cytoplasm and promotes Nrf2 degradation via ubiquitination, thereby maintaining the balance of cellular redox homeostasis.4,24,25 The thioredoxin (Trx) system, a major cellular thiol-reducing and antioxidant system, is composed of NADPH, Trx reductase, and Trx. TXNIP is an endogenous inhibitor of the Trx system that binds with the reduced form of Trx and blocks its reducing potential.26 It is reported that Nrf2 drives the gene expression of Trx 1 and suppresses the gene expression of TXNIP under oxidative stress.27 During TBI, the beneficial roles of Nrf2 signaling are supported by pharmacological and genetic approaches.4,28,29 In the present study, the levels of Nrf2 in the cytosol and nucleus and the ratio of Nrf2/Keap 1 in the cytosol were decreased significantly after traumatic brain injury. Meanwhile, the ratio of Nrf2/Keap 1 in the nucleus was increased, although it did not reach statistical significance. In addition, the levels of Trx 1 and the ratio of Trx 1/TXNIP in cytosol was increased significantly, while the levels of Trx 1 and the ratio of Trx 1/TXNIP in nucleus had no significant changes. These data suggest that Nrf2 signaling could be activated and that the Trx system plays more important roles in the cytosol than in the nucleus during traumatic brain injury. CPCGI treatment enhanced Nrf2 and Trx 1 levels and Nrf2/Keap1 and Trx 1/TXNIP ratios in cytosol, reduced Keap1 and TXNIP levels in the nucleus, and enhanced Nrf2 levels and the Nrf2/Keap1 ratio in the nucleus, although it had no significant effects on Keap1 and TXNIP levels in the cytosol or on Trx 1 levels and the Trx 1/TXNIP ratio in the nucleus. Moreover, CPCGI enhanced cytosolic HO-1 levels. These data suggest that CPCGI could repress oxidative stress through prompting the activation of anti-oxidative Nrf2 signaling and increasing the expression of Nrf2-driven genes during TBI.
Calpains, a family of cysteine proteases, are activated by calcium, and regulated reversibly by calcium and calpastatin, an endogenous calpain inhibitor.8 Calpains participate in the pathophysiology of TBI, which is supported by the degradation of substrates and the protective effects of calpain inhibition after induction of TBI.9,30 Oxidative stress is reported to participate in calpain activation by elevating intracellular calcium under pathological conditions.31 In rodent models of TBI, suppression of oxidative stress restores mouse cortical mitochondrial bioenergetics and calcium buffering and reduces calpain-mediated cytoskeletal damage.32,33 Moreover, Nrf2 activation reduces lipid and protein peroxidation, preserves mitochondrial respiratory function, and attenuates calpain-mediated cytoskeletal breakdown.29 In addition, calpains in neurons and axons are activated and involved in neuronal and axonal damage.34,35 These data suggest that the disruption of intracellular calcium homeostasis due to oxidative stress could result in the activation of calpain and the degradation of structural proteins, leading to the axonal and neuronal damage during TBI. Our results showed that CPCGI activated Nrf2 signaling, promoted the expression of the target genes, and reduced the oxidative damage during TBI. Moreover, CPCGI enhanced calpastatin levels, reduced calpain activities, and attenuated substrate degradation. These data suggest that CPCGI protects against TBI through promoting antioxidative capacity and suppressing oxidative stress-induced overactivation of calpain.
αII-Spectrin, a major structural component of cortical membrane cytoskeleton, is located in axons, presynaptic terminals and neuronal bodies. Microtubules consist of polymers of α- and β-tubulin heterodimers, and MAP2 is reported to be involved in microtubule assembly, stability, and elongation. Neurofilaments are present in perikarya and dendrites, particularly abundant in axons, which participate in the assembly of neuronal intermediate filaments. MBP, the major protein component in the myelin sheath that encases axons, is central to maintaining the structural homeostasis of the myelin sheath. It is well known that αII-spectrin, NF-H, MAP2, and MBP are involved in the generation of the neuronal and axonal cytoarchitecture through crosstalking with other cytoskeletal and membrane proteins, such as tubulins, neurofilament light and middle chain, β-spectrin, actins, and ankyrins, which play critical roles in maintaining normal neuronal structure and functions, including neuronal shape, radial growth of axons, axon caliber, and nerve conduction.36–40 It is reasonable that the degradation of αII-spectrin, NF-H, MAP2, and MBP would result in the gray and white matter damage in the brain. αII-spectrin, NF-H, MAP2, and MBP are substrates for calpain, and their degradation after TBI is reported.9,30 These data combined with our results suggest that TBI-induced activation of calpain could result in the degradation of αII-spectrin, MAP2, NF-H and MBP, leading to neuronal and axonal dysfunction and neuronal cell death. The inhibition of CCI-induced activation of calpain by CPCGI could protect the brain against TBI through suppressing the degradation of cytoskeletal proteins and MBP, reducing the damage to gray and white matter, and promoting the recovery of neurological function.
CPCGI is composed of polypeptides, gangliosides and hypoxanthine, which is extracted from porcine brain tissues and rabbit muscle tissues. CPCGI is used to treat acute and chronic brain trauma in China, but it lacks the support of experimental studies. Cerebrolysin, extracted from porcine brain tissue, consists of peptides and amino acids. A large amount of data has shown the neuroprotective effect of cerebrolysin against brain ischemia and brain trauma, which contributes to its neuroprotective and neurotrophic properties, such as its ability to reduce excitotoxicity, lessen oxidative stress, attenuate neuroinflammation, and improve survival and neurogenesis.41,42 Therefore, it is reasonable that CPCGI may exert neuroprotective effects through mechanisms similar to cerebrolysin. Moreover, gangliosides protect the brain from experimental brain trauma,43,44 and hypoxanthine reduces hypoxic-ischemic brain injury.45,46 These data suggest the beneficial role of CPCGI treatment in brain trauma. Our data showed that CPCGI reduced the neurological deficits, attenuated corpus callosum injury, and decreased the contusion volumes in a dose-dependent manner, suggesting the neuroprotective effects of CPCGI against brain injury due to TBI. Figure 7 summarizes the possible mechanisms of CPCGI against traumatic brain injury.
 |
Figure 7 Overview of neuroprotection of CPCGI against traumatic brain injury. (-) Negative effects; (+) Positive effects. |
Conclusion
In summary, these data confirm the protective effects of CPCGI against white matter and gray matter damage induced by TBI and suggest that stimulating Nrf2 signaling, and suppressing oxidative stress-mediated calpain overactivation could be one mechanism by which CPCGI protects against TBI.
Abbreviations
TBI, traumatic brain injury; Nrf2, nuclear factor erythroid-2-related factor 2; Keap1, Kelch-like ECH-associated protein 1; ARE, antioxidant response element; SOD, superoxide dismutase; HO-1, heme oxygenase 1; Trx, thioredoxin; ROS, reactive oxygen species; CPCGI, compound porcine cerebroside and ganglioside injection; CCI, controlled cortical impact; NSS, neurological severity score; NS, normal saline; H&E, hematoxylin and eosin; LFB-PAS, Luxol fast blue–periodic acid Schiff; ODs, optical densities; HEPES, N-2-hydroxyethylpiperazine-N’-2ʹ-ethanesulfonic acid; T-AOC, total-antioxidizing capability; MDA, malondialdehyde; MAP2, microtubule-associated protein 2; NF-H, neurofilament heavy chain; MBP, myelin basic protein; TXNIP, thioredoxin interacting protein; SDS-PAGE, sodium dodecyl sulfate-polyacrylamide gel electrophoresis.
Data Sharing Statement
All data are published in this article and materials can be obtained from the corresponding author upon reasonable request.
Disclosure
The authors report no conflicts of interest for this work.
References
1. Maas AIR, Menon DK, Adelson PD, et al. Traumatic brain injury: integrated approaches to improve prevention, clinical care, and research. Lancet Neurol. 2017;16(12):987–1048. doi:10.1016/S1474-4422(17)30371-X
2. Kaur P, Sharma S. Recent advances in pathophysiology of traumatic brain injury. Curr Neuropharmacol. 2018;16(8):1224–1238. doi:10.2174/1570159X15666170613083606
3. Ng SY, Lee AYW. Traumatic brain injuries: pathophysiology and potential therapeutic targets. Front Cell Neurosci. 2019;13:528. doi:10.3389/fncel.2019.00528
4. Zhang L, Wang H. Targeting the NF-E2-related factor 2 pathway: a novel strategy for traumatic brain injury. Mol Neurobiol. 2018;55(2):1773–1785. doi:10.1007/s12035-017-0456-z
5. Trushina E, McMurray CT. Oxidative stress and mitochondrial dysfunction in neurodegenerative diseases. Neuroscience. 2007;145(4):1233–1248. doi:10.1016/j.neuroscience.2006.10.056
6. Chen X, Wang H, Zhou M, et al. Valproic acid attenuates traumatic brain injury-induced inflammation in vivo: involvement of autophagy and the Nrf2/ARE Signaling Pathway. Front Mol Neurosci. 2018;11:117. doi:10.3389/fnmol.2018.00117
7. Zhang M, Wu J, Ding H, Wu W, Xiao G. Progesterone provides the pleiotropic neuroprotective effect on traumatic brain injury through the Nrf2/ARE signaling pathway. Neurocrit Care. 2017;26(2):292–300. doi:10.1007/s12028-016-0342-y
8. Goll DE, Thompson VF, Li H, Wei W, Cong J. The calpain system. Physiol Rev. 2003;83(3):731–801. doi:10.1152/physrev.00029.2002
9. Saatman KE, Creed J, Raghupathi R. Calpain as a therapeutic target in traumatic brain injury. Neurotherapeutics. 2010;7(1):31–42. doi:10.1016/j.nurt.2009.11.002
10. Wang M, Zhang Y, Feng L, et al. Compound porcine cerebroside and ganglioside injection attenuates cerebral ischemia-reperfusion injury in rats by targeting multiple cellular processes. Neuropsychiatr Dis Treat. 2017;13:927–935. doi:10.2147/NDT.S129522
11. Chen X, Hua X, Sun D, Fan W. [Compound porcine cerebroside and ganglioside relieves brain injury and promotes expression of cerebellin 4 in neonatal mice with intrauterine hypoxia]. Xi Bao Yu Fen Zi Mian Yi Xue Za Zhi = Chin J Cell Mol Immunol. 2019;35(8):721–726 Chinese.
12. Wang X, Zhao J. Neuroprotective effect of CPCGI on Alzheimer’s disease and its mechanism. Mol Med Rep. 2020;21(1):115–122. doi:10.3892/mmr.2019.10835
13. Miao Y, Wang R, Wu H, Yang S, Qiu Y. CPCGI confers neuroprotection by enhancing blood circulation and neurological function in cerebral ischemia/reperfusion rats. Mol Med Rep. 2019;20(3):2365–2372. doi:10.3892/mmr.2019.10472
14. Zweckberger K, Hackenberg K, Jung CS, et al. Cerebral metabolism after early decompression craniotomy following controlled cortical impact injury in rats. Neurol Res. 2011;33(8):875–880. doi:10.1179/1743132811Y.0000000017
15. Chen J, Sanberg PR, Li Y, et al. Intravenous administration of human umbilical cord blood reduces behavioral deficits after stroke in rats. Stroke. 2001;32(11):2682–2688. doi:10.1161/hs1101.098367
16. Satchell MA, Zhang X, Kochanek PM, et al. A dual role for poly-ADP-ribosylation in spatial memory acquisition after traumatic brain injury in mice involving NAD+ depletion and ribosylation of 14-3-3gamma. J Neurochem. 2003;85(3):697–708. doi:10.1046/j.1471-4159.2003.01707.x
17. Pantoni L, Garcia JH, Gutierrez JA. Cerebral white matter is highly vulnerable to ischemia. Stroke. 1996;27(9):
18. Schabitz WR, Li F, Fisher M. The N-methyl-D-aspartate antagonist CNS 1102 protects cerebral gray and white matter from ischemic injury following temporary focal ischemia in rats. Stroke. 2000;31(7):1709–1714. doi:10.1161/01.STR.31.7.1709
19. Gu Y, Zhao Y, Qian K, Sun M. Taurine attenuates hippocampal and corpus callosum damage, and enhances neurological recovery after closed head injury in rats. Neuroscience. 2015;291:331–340. doi:10.1016/j.neuroscience.2014.09.073
20. Pei DS, Wang XT, Liu Y, et al. Neuroprotection against ischaemic brain injury by a GluR6-9c peptide containing the TAT protein transduction sequence. Brain. 2006;129(Pt 2):465–479. doi:10.1093/brain/awh700
21. Solaroglu I, Tsubokawa T, Cahill J, Zhang JH. Anti-apoptotic effect of granulocyte-colony stimulating factor after focal cerebral ischemia in the rat. Neuroscience. 2006;143(4):965–974. doi:10.1016/j.neuroscience.2006.09.014
22. Vosler PS, Sun D, Wang S, et al. Calcium dysregulation induces apoptosis-inducing factor release: cross-talk between PARP-1- and calpain-signaling pathways. Exp Neurol. 2009;218(2):213–220. doi:10.1016/j.expneurol.2009.04.032
23. Cornelius C, Crupi R, Calabrese V, et al. Traumatic brain injury: oxidative stress and neuroprotection. Antioxid Redox Signal. 2013;19(8):836–853. doi:10.1089/ars.2012.4981
24. Schmidlin CJ, Dodson MB, Madhavan L, Zhang DD. Redox regulation by NRF2 in aging and disease. Free Radic Biol Med. 2019;134:702–707. doi:10.1016/j.freeradbiomed.2019.01.016
25. Kopacz A, Kloska D, Forman HJ, Jozkowicz A, Grochot-Przeczek A. Beyond repression of Nrf2: an update on Keap1. Free Radic Biol Med. 2020. doi:10.1016/j.freeradbiomed.2020.03.023
26. Lu J, Holmgren A. The thioredoxin antioxidant system. Free Radic Biol Med. 2014;66:75–87. doi:10.1016/j.freeradbiomed.2013.07.036
27. Tonelli C, Chio IIC, Tuveson DA. Transcriptional regulation by Nrf2. Antioxid Redox Signal. 2018;29(17):1727–1745. doi:10.1089/ars.2017.7342
28. Lu XY, Wang HD, Xu JG, Ding K, Li T. Deletion of Nrf2 exacerbates oxidative stress after traumatic brain injury in mice. Cell Mol Neurobiol. 2015;35(5):713–721. doi:10.1007/s10571-015-0167-9
29. Miller DM, Singh IN, Wang JA, Hall ED. Nrf2-ARE activator carnosic acid decreases mitochondrial dysfunction, oxidative damage and neuronal cytoskeletal degradation following traumatic brain injury in mice. Exp Neurol. 2015;264:103–110. doi:10.1016/j.expneurol.2014.11.008
30. Ma M. Role of calpains in the injury-induced dysfunction and degeneration of the mammalian axon. Neurobiol Dis. 2013;60:61–79. doi:10.1016/j.nbd.2013.08.010
31. Starkov AA, Chinopoulos C, Fiskum G. Mitochondrial calcium and oxidative stress as mediators of ischemic brain injury. Cell Calcium. 2004;36(3–4):257–264. doi:10.1016/j.ceca.2004.02.012
32. Hill RL, Singh IN, Wang JA, Hall ED. Time courses of post-injury mitochondrial oxidative damage and respiratory dysfunction and neuronal cytoskeletal degradation in a rat model of focal traumatic brain injury. Neurochem Int. 2017;111:45–56. doi:10.1016/j.neuint.2017.03.015
33. Mustafa AG, Wang JA, Carrico KM, Hall ED. Pharmacological inhibition of lipid peroxidation attenuates calpain-mediated cytoskeletal degradation after traumatic brain injury. J Neurochem. 2011;117(3):579–588. doi:10.1111/j.1471-4159.2011.07228.x
34. Ai J, Liu E, Wang J, Chen Y, Yu J, Baker AJ. Calpain inhibitor MDL-28170 reduces the functional and structural deterioration of corpus callosum following fluid percussion injury. J Neurotrauma. 2007;24(6):960–978. doi:10.1089/neu.2006.0224
35. Buki A, Farkas O, Doczi T, Povlishock JT. Preinjury administration of the calpain inhibitor MDL-28170 attenuates traumatically induced axonal injury. J Neurotrauma. 2003;20(3):261–268. doi:10.1089/089771503321532842
36. Goodman SR, Zimmer WE, Clark MB, Zagon IS, Barker JE, Bloom ML. Brain spectrin: of mice and men. Brain Res Bull. 1995;36(6):593–606. doi:10.1016/0361-9230(94)00264-2
37. Huang CY, Zhang C, Zollinger DR, Leterrier C, Rasband MN. An alphaII spectrin-based cytoskeleton protects large-diameter myelinated axons from degeneration. J Neurosci. 2017;37(47):11323–11334. doi:10.1523/JNEUROSCI.2113-17.2017
38. Bodakuntla S, Jijumon AS, Villablanca C, Gonzalez-Billault C, Janke C. Microtubule-associated proteins: structuring the cytoskeleton. Trends Cell Biol. 2019;29(10):804–819. doi:10.1016/j.tcb.2019.07.004
39. Laser-Azogui A, Kornreich M, Malka-Gibor E, Beck R. Neurofilament assembly and function during neuronal development. Curr Opin Cell Biol. 2015;32:92–101. doi:10.1016/j.ceb.2015.01.003
40. Boggs JM. Myelin basic protein: a multifunctional protein. Cell Mol Life Sci. 2006;63(17):1945–1961. doi:10.1007/s00018-006-6094-7
41. Bornstein N, Poon WS. Accelerated recovery from acute brain injuries: clinical efficacy of neurotrophic treatment in stroke and traumatic brain injuries. Drugs Today. 2012;48 Suppl A:43–61. doi:10.1358/dot.2012.48(Suppl.A).1739723
42. Brainin M. Cerebrolysin: a multi-target drug for recovery after stroke. Expert Rev Neurother. 2018;18(8):681–687. doi:10.1080/14737175.2018.1500459
43. Benady A, Freidin D, Pick CG, Rubovitch V. GM1 ganglioside prevents axonal regeneration inhibition and cognitive deficits in a mouse model of traumatic brain injury. Sci Rep. 2018;8(1):13340. doi:10.1038/s41598-018-31623-y
44. Rubovitch V, Zilberstein Y, Chapman J, Schreiber S, Pick CG. Restoring GM1 ganglioside expression ameliorates axonal outgrowth inhibition and cognitive impairments induced by blast traumatic brain injury. Sci Rep. 2017;7:41269. doi:10.1038/srep41269
45. Mink RB, Dutka AJ, Kumaroo KK, Hallenbeck JM. No conversion of xanthine dehydrogenase to oxidase in canine cerebral ischemia. Am J Physiol. 1990;259(6 Pt 2):H1655–1659. doi:10.1152/ajpheart.1990.259.6.H1655
46. Mink R, Johnston J. The effect of infusing hypoxanthine or xanthine on hypoxic-ischemic brain injury in rabbits. Brain Res. 2007;1147:256–264. doi:10.1016/j.brainres.2007.02.004
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.