Back to Journals » Vascular Health and Risk Management » Volume 17
COVID-19 Sepsis: Pathogenesis and Endothelial Molecular Mechanisms Based on “Two-Path Unifying Theory” of Hemostasis and Endotheliopathy-Associated Vascular Microthrombotic Disease, and Proposed Therapeutic Approach with Antimicrothrombotic Therapy
Authors Chang JC ![]()
Received 27 December 2020
Accepted for publication 24 March 2021
Published 1 June 2021 Volume 2021:17 Pages 273—298
DOI https://doi.org/10.2147/VHRM.S299357
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Konstantinos Tziomalos
Jae C Chang
Department of Medicine, University of California Irvine School of Medicine, Irvine, CA, USA
Correspondence: Jae C Chang 6 Via Castilla Unit B, Laguna Woods, CA, 92637, USA
Tel +1-949-943-9988
Email [email protected]
Abstract: COVID-19 sepsis is characterized by acute respiratory distress syndrome (ARDS) as a consequence of pulmonary tropism of the virus and endothelial heterogeneity of the host. ARDS is a phenotype among patients with multiorgan dysfunction syndrome (MODS) due to disseminated vascular microthrombotic disease (VMTD). In response to the viral septicemia, the host activates the complement system which produces terminal complement complex C5b-9 to neutralize pathogen. C5b-9 causes pore formation on the membrane of host endothelial cells (ECs) if CD59 is underexpressed. Also, viral S protein attraction to endothelial ACE2 receptor damages ECs. Both affect ECs and provoke endotheliopathy. Disseminated endotheliopathy activates two molecular pathways: inflammatory and microthrombotic. The former releases inflammatory cytokines from ECs, which lead to inflammation. The latter initiates endothelial exocytosis of unusually large von Willebrand factor (ULVWF) multimers and FVIII from Weibel–Palade bodies. If ADAMTS13 is insufficient, ULVWF multimers activate intravascular hemostasis of ULVWF path. In activated ULVWF path, ULVWF multimers anchored to damaged endothelial cells recruit circulating platelets and trigger microthrombogenesis. This process produces “microthrombi strings” composed of platelet-ULVWF complexes, leading to endotheliopathy-associated VMTD (EA-VMTD). In COVID-19, microthrombosis initially affects the lungs per tropism causing ARDS, but EA-VMTD may orchestrate more complex clinical phenotypes, including thrombotic thrombocytopenic purpura (TTP)-like syndrome, hepatic coagulopathy, MODS and combined micro-macrothrombotic syndrome. In this pandemic, ARDS and pulmonary thromboembolism (PTE) have often coexisted. The analysis based on two hemostatic theories supports ARDS caused by activated ULVWF path is EA-VMTD and PTE caused by activated ULVWF and TF paths is macrothrombosis. The thrombotic disorder of COVID-19 sepsis is consistent with the notion that ARDS is virus-induced disseminated EA-VMTD and PTE is in-hospital vascular injury-related macrothrombosis which is not directly related to viral pathogenesis. The pathogenesis-based therapeutic approach is discussed for the treatment of EA-VMTD with antimicrothrombotic regimen and the potential need of anticoagulation therapy for coinciding macrothrombosis in comprehensive COVID-19 care.
Keywords: acute respiratory distress syndrome; ARDS, ADAMTS13, endotheliopathy, macrothrombosis; microthrombosis, multiorgan dysfunction syndrome; MODS
Introduction
Coronaviruses are a large family of viruses that usually cause mild to moderate upper-respiratory tract illnesses. Three major coronaviruses have emerged from animal reservoirs over the past two decades and have caused serious and widespread illnesses and claimed human lives according to National Institute of Allergy and Infectious Disease.1 The coronaviruses causing severe acute respiratory syndrome (SARS-CoV) had significant outbreaks in China in 2002 and Middle East respiratory syndrome (MERS-CoV) in South Korea in 2015. Now, COVID-19 due to SARS-CoV-2 has sparked pandemic since its outbreak in late 2019 from Wuhan, China. The origin of the viral spread to human is undetermined at this time.2,3 COVID-19 pandemic has created unprecedented political, economic and societal dislocation worldwide and claimed over two million lives as of early months of 2021.
Coronaviral infection typically begins with constitutional flu-like and mild respiratory symptoms, and in severe cases progresses to inflammation, pneumonia and acute respiratory distress syndrome (ARDS).4–6 The clinical manifestations of COVID-19 have been similar to previous outbreaks of SARS and MERS. However, the transmission is faster and clinical symptoms more extensive. The outcome has been worse with higher morbidity and mortality.7–9 In the beginning, particular concern was incompletely understood pathogenesis of ARDS. Later, complex hemostatic phenotypes of micro and macrothrombotic disorders and multiorgan dysfunction syndrome (MODS) had become apparent, including inexplicable gangrene involving the extremities, and digits in particular.10–14 Like other pathogens, severe COVID-19 sepsis was found to be associated with complement activation,15–17 endotheliopathy,18,19 microvascular injury and thrombosis,16 and pathologic hemostasis.20,21 Previously, these mechanisms were affirmed to be involved in other pathogen-induced sepsis and identified as the pathogenesis causing ARDS, MODS, thrombotic disease and coagulopathy via endotheliopathy-associated vascular microthrombotic disease (EA-VMTD).6,22
In this focused review, hemostatic nature of COVID-19 sepsis, hematologic phenotypes and thrombotic and coagulation findings will be analyzed from published clinical literatures. The pathogenesis of the sepsis will be constructed utilizing two novel hemostatic mechanisms: “two-path unifying theory” and “two-activation theory of the endothelium”. These theories have already established the unique concept of EA-VMTD that is associated with activated unusually large von Willebrand factor (ULVWF) path of hemostasis, which is different from macrothrombosis and coagulopathy occurring as a result of combined activation of ULVWF and tissue factor (TF) paths.23 The pathogenetic mechanism of macrothrombosis typified by pulmonary thromboembolism (PTE) and deep vein thrombosis (DVT) coexisting with ARDS in COVID-19 will be discussed from the concept of the hemostatic fundamentals. In the end, theory-based therapeutic approach will be proposed. Further, the management of coexisting ARDS and macrothrombotic disorder (i.e., PTE) will be separated since this is a very important practical issue in COVID-19 sepsis.
Perspective on New Therapeutic Direction Based on Theory
The clinical course of each viral sepsis is influenced by the combined expression of infectivity and virulence of the pathogen, and immune competence and response mechanism of the host. In early pandemic stage, the management of COVID-19 was centered on the efforts of identifying effective antiviral agents to eradicate the pathogen with significant and modest success. After one year of the pandemic, preventive measure is prioritized to offer adaptive immunity for world’s populace via virus specific vaccines. Coordinated vaccination program has been activated and is in progress.
When pathogen intrudes into the blood stream, two nature-endowed biological mechanisms are activated to protect and maintain proper homeostasis of the body and to overcome septicemia and prevent sepsis. One is defensive physiological response through activated innate and adaptive immune system to neutralize the pathogen, and the other is healthy hemostatic system to protect the endothelial integrity and prevent destructive endothelial responses leading to sepsis,22 as summarized in Figure 1. Since COVID-19 sepsis is characterized by complement activated endotheliopathy, the therapy can target the pathogenetic mechanism involving the endothelial dysfunction.
 |
Figure 1 Physiological and pathological response mechanisms in sepsis. Notes: In sepsis, host response is characterized by two biological mechanisms. One is physiologic defensive mechanism through immune system, and the other is pathologic destructive mechanism through endothelial system. The mechanism of physiologic and pathologic responses is summarized. It is known the complement system protecting host through innate immune system could trigger harmful endothelial molecular pathogenesis. This dual role of the complement system must be nature’s rule just like normal hemostasis, which protects human lives in external bodily injury, but also may harm human lives in intravascular injury through thrombogenesis. Reproduced from Chang JC. Sepsis and septic shock: endothelial molecular pathogenesis associated with vascular microthrombotic disease. Thromb J. 2019;17:10.22Abbreviations: APC, antigen presenting cell; DIC, disseminated intravascular coagulation; DIT, disseminated intravascular microthrombosis; EA-VMTD, endotheliopathy-associated vascular microthrombotic disease; MAHA, microangiopathic hemolytic anemia; MODS, multiorgan dysfunction syndrome; MOF, multiorgan failure; NO, nitric oxide; IF, interferon; IL, interleukin; LPS, lipopolysaccharide; TNF, tumor necrosis factor; TTP, thrombotic thrombocytopenic purpura. |
The main pathology of COVID-19 sepsis is microthrombosis, and its clinical phenotype is EA-VMTD, which primary manifestation is ARDS. Since ARDS is the organ phenotype of disseminated microthrombosis in the pulmonary vasculature, the treatment can be directed to counteract the formation of microthrombi.6 Microthrombi are produced by the activated ULVWF path of hemostasis after release of the multimers from endothelial cells (ECs) and activation of platelets as shown in Figure 2.22 Theoretically, antimicrothrombotic therapy can prevent microthrombogenesis and also resolve microthrombi formed from the molecular endothelial pathogenesis as illustrated in Figure 3.
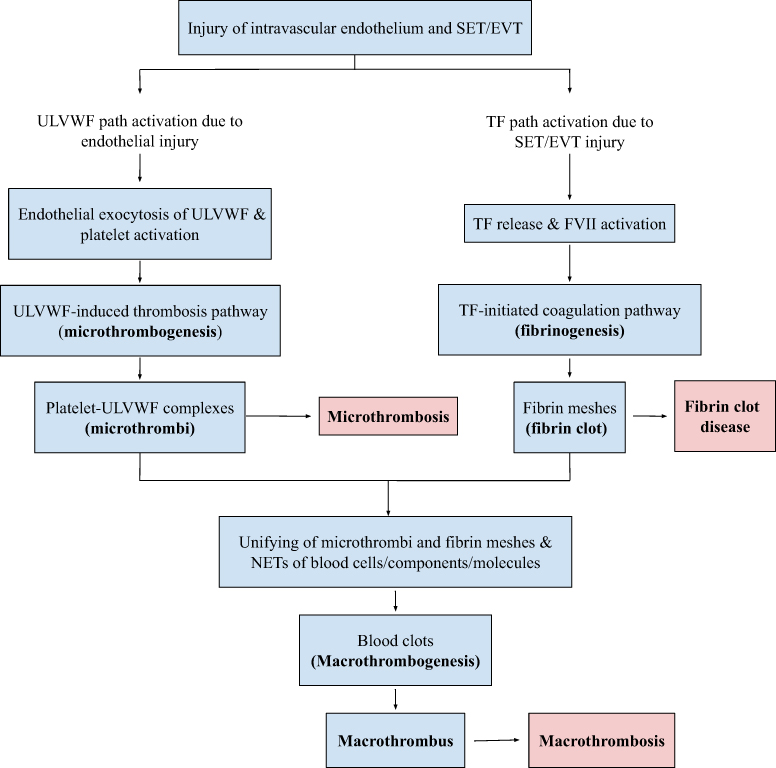 |
Figure 2 Normal in vivo hemostasis based on “two-path unifying theory.” Notes: Following avascular injury, invivo hemostatic system triggers the activation of two independent sub-hemostatic paths: microthrombotic (ULVWF) and fibrinogenetic (TF). Both are initiated by the damage of ECs and SET/EVT due to external bodily injury and intravascular injury. In activated ULVWF path from ECs damage, released ULVWF multimers recruit platelets and produce microthrombi strings via microthrombogenesis, but in activated TF path from SET/EVT damage, released TF activates FVII and produces fibrin meshes via extrinsic coagulation cascade. The final path of invivo hemostasis is macrothrombogenesis, in which microthrombi strings and fibrin meshes become unified together with incorporation of NETs, including red blood cells, neutrophils, DNAs and histones. This unifying event called macrothrombogenesis promotes “hemostatic plug” and wound healing in external bodily injury and produces “macrothrombus” in intravascular injury. Reproduced from Chang JC. Sepsis and septic shock: endothelial molecular pathogenesis associated with vascular microthrombotic disease. Thromb J. 2019;17:10.22Abbreviations: EA-VMTD, endotheliopathy-associated vascular microthrombotic disease; EVT, extravascular tissue; NETs, neutrophil extracellular traps; SET, subendothelial tissue; TF, tissue factor; ULVWF, unusually large von Willebrand factor multimers. |
 |
Figure 3 Proposed endothelial pathogenesis of SARS-CoV-2 viral sepsis based on “two-activation theory of the endothelium.” Notes: Endothelial molecular pathogenesis of ARDS as one organ phenotype among MODS is illustrated. The underlying pathologic nature of ARDS is ahemostatic disease caused by endotheliopathy due to complement activation and viral Sprotein-endothelial receptor ACE2 interaction that promotes the activation of two molecular pathways. One is inflammatory pathway, which releases cytokines and provokes inflammation, including inflammatory fever, malaise and myalgia. The other is microthrombotic pathway, which promotes exocytosis of ULVWF and platelet activation and triggers much more deadly DIT via microthrombogenesis, leading to EA-VMTD. It orchestrates consumptive thrombocytopenia, MAHA, TTP-like syndrome and MODS. Abbreviations: ACE2, angiotensin converting enzyme 2; ARDS, acute respiratory distress syndrome; DIT, disseminated intravascular thrombosis; ECs, endothelial cells; IL, interleukin; MODS, multiorgan dysfunction syndrome; SARS, severe acute respiratory syndrome; TCIP, thrombocytopenia in critically ill patients; TNF, tumor necrosis factor; TTP, thrombotic thrombocytopenic purpura; EA-VMTD, endotheliopathy-associated vascular microthrombotic disease; ULVWF, unusually large von Willebrand factor. |
Thrombotic Disorders and Thrombotic Syndromes
Soon after declaration of pandemic, ARDS has been found to be caused by disseminated microthrombosis in associated with endotheliopathy, which is entirely consistent with EA-VMTD as previously predicted pathogenesis of ARDS occurring in every sepsis and critical illnesses.6 Later, in some patients, ARDS coincided with macrothrombosis such as PTE and DVT. Therefore, COVID-19 has been convinced to be a complex hemostatic disease, culminating to the composite of variable thrombotic phenotypes.
Contemporary concept of thrombotic disorder is based on the theory that all the thrombotic disorders are caused by the same coagulation process initiated by activated TF pathway following intravascular injury. To date, ARDS due to microthrombosis and PTE due to macrothrombosis are considered as the same diseases but variable expression due to involvement in different caliber and size of the vessel. I questioned this simplified concept of activated TF pathway-induced thrombogenesis. It was convinced that microthrombosis and macrothrombosis must be two different thrombotic disorders originating from two different sub-hemostatic paths as a result of different level (depth) of intravascular injury.6,22 Microthrombosis in sepsis occurs due to ECs injury, but macrothrombosis in vascular trauma (e.g., surgery) develops due to combined ECs and subendothelial tissue (SET) injury of the blood vessel wall. These hemostatic fundamental principles are detailed in Table 1 and vascular wall physiology based hemostatic mechanism is illustrated in Figure 2. Succinctly, the damage from virus-induced endotheliopathy initiating ARDS is confined to ECs and is disseminated, but the damage from in-hospital-related vascular injury initiating PTE extends to from ECs to SET of blood vessel wall and is localized at injury site.
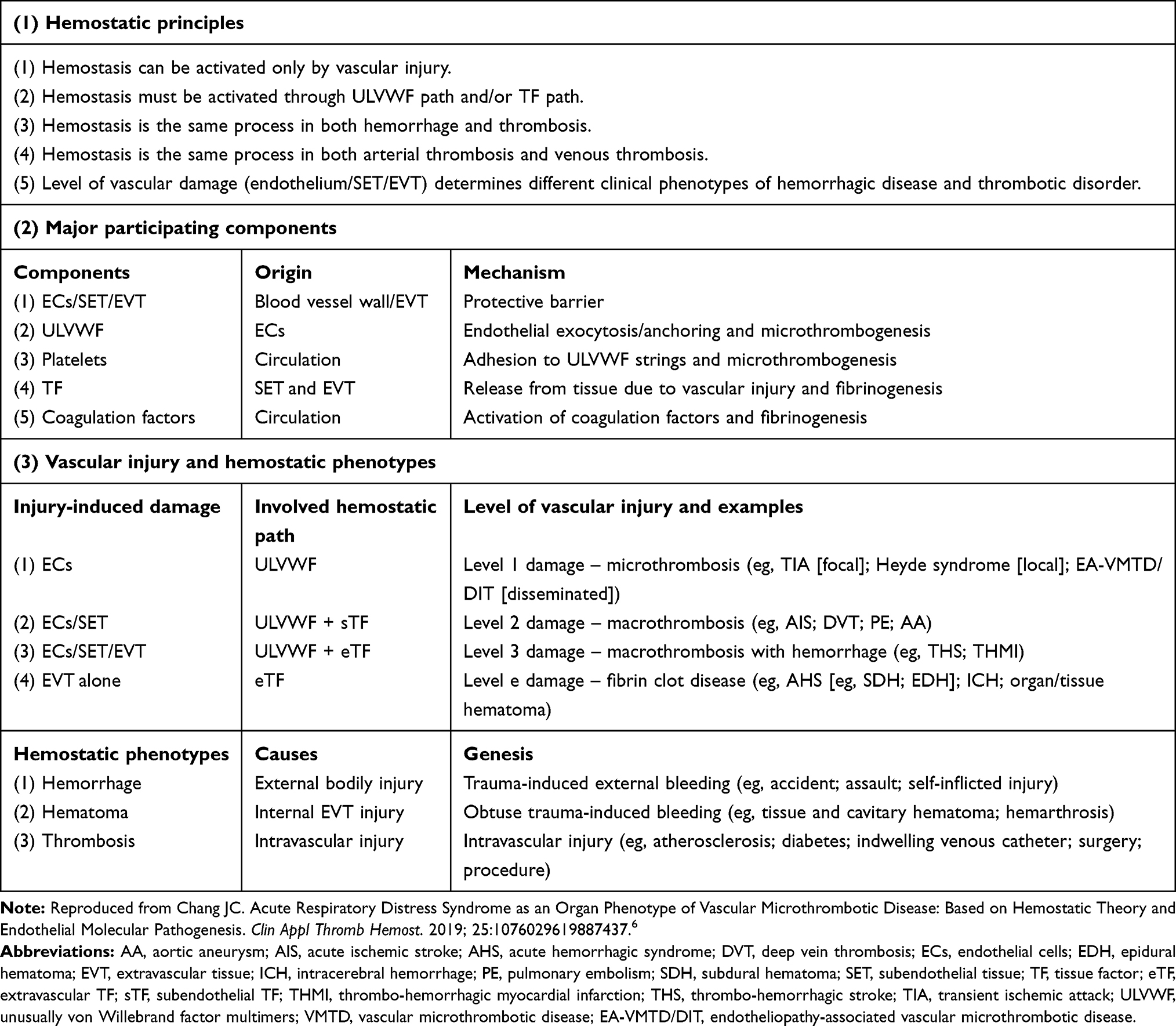 |
Table 1 Three Essentials in Normal Hemostasis |
In COVID-19, some patients had coexisting microthrombosis and macrothrombosis, which suggested two different thrombogenetic mechanisms were involved and produced two entirely different thrombi via different levels of vascular wall injury. The genesis and pathobiological feature between ARDS and PTE are summarized in Table 2. It is important to understand two phenotypes of the thrombotic disorders require different therapeutic approaches.
 |
Table 2 Biological and Hemostatic Characteristics Between Microthrombosis and Macrothrombosis Observed in COVID-19 Sepsis |
Microthrombotic Syndromes in COVID-19
VMTD includes every microthrombosis-associated disease occurring in any vascular system, which could be generalized, regional, or local/focal, and be hereditary or acquired, and also be arterial or venous disease. Microthrombi are formed of multiple “microthrombi strings” composed of platelets-ULVWF multimer complexes and tend to anchor to the endothelial membrane tethering to the direction of blood flow in circulation, typically at the terminal microvascular tree. The microthrombi strings partially obstruct the vascular lumen and slow down the blood flow within arterioles and capillaries, exposing the organ and tissue to hypoxia. The classification of VMTD is presented in Table 3 to assist the reader for comprehension of VMTD in the understanding of COVID-19 sepsis. Microthrombi are the underlying pathology of disseminated EA-VMTD and can affect the microvasculature of every organ, which may lead to TTP-like syndrome and hypoxic MODS.
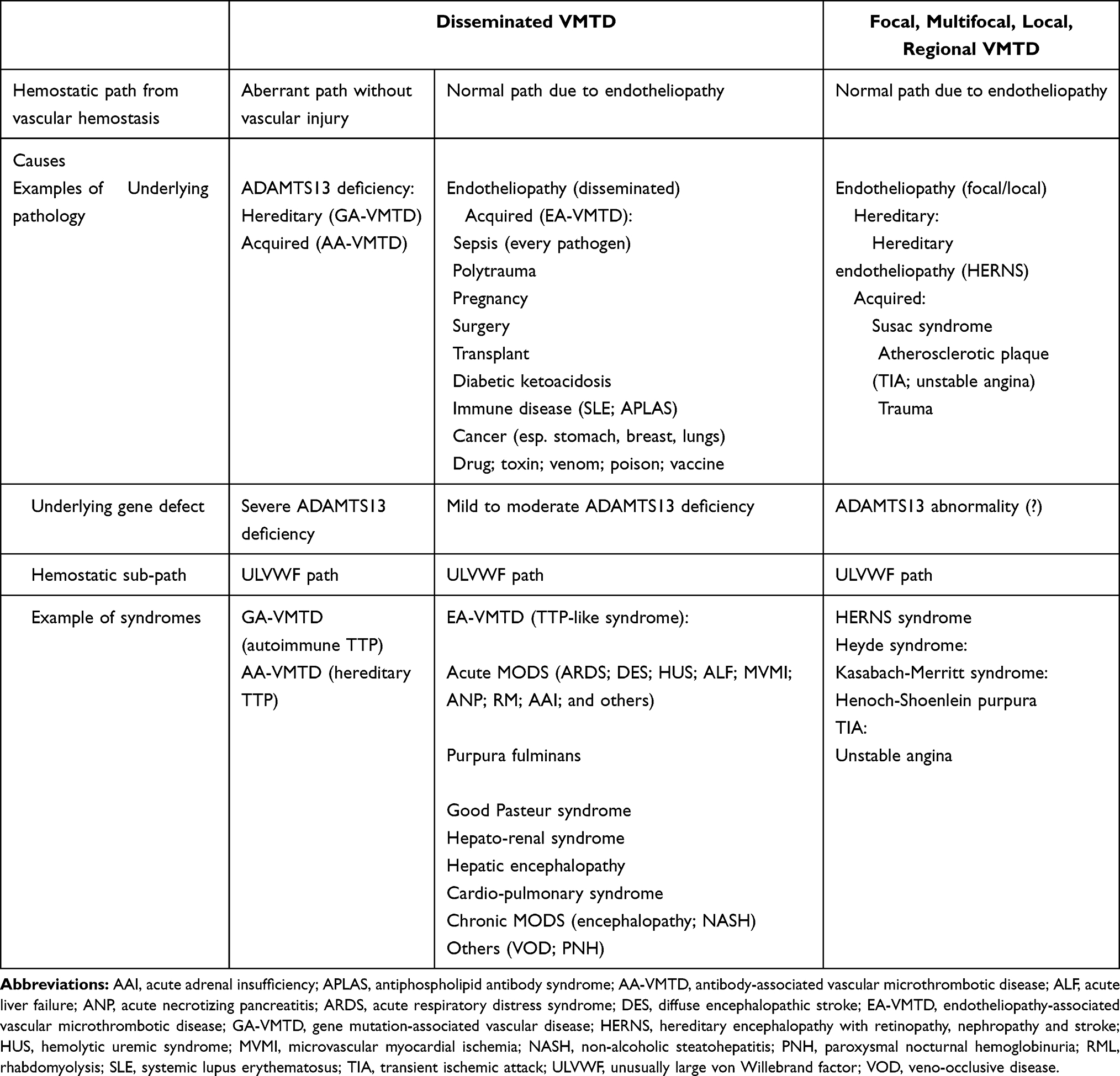 |
Table 3 Classification of Vascular Microthrombotic Disease |
Acute Respiratory Distress Syndrome
The lungs are the most common organ affected by VMTD in coronaviral sepsis. ARDS is the major prototype of organ dysfunction syndrome affected by EA-VMTD. In COVID-19, two main manifestations are inflammation and respiratory distress due to disseminated microthrombosis in the pulmonary vasculature.16,18 The character term of “microthrombi strings” representing platelet-ULVWF multimer complexes has been designated6 after the concept derived from the insightful works of the group of scientists who demonstrated the endothelial cell-bound ULVWF multimers aggregate platelets to form platelet-ULVWF complexes ex vivo.24 These complexes are the same to the theoretical microthrombi strings proposed for ULVWF path in “two path unifying theory” of hemostasis22 shown in Figure 2. The mechanism of microthrombogenesis will be discussed after introduction of “two-activation theory of the endothelium” (Figure 3).
The spike (S) protein of SARS-CoV-2 is attracted to the angiotensin converting enzyme 2 (ACE2) on ECs25 and compromises endothelial function. In addition, similar to other pathogens, COVID-19 sepsis activates complement system leading to formation of terminal complement complex C5b-9.15−17 C5b-9 neutralizes virus, but also may cause the channel (pore) formation on the endothelial membrane of the host if CD59 known as a glycoprotein protecting ECs membrane is underexpressed,26,27 Subsequent endothelial activation and dysfunction result in inflammation and exocytosis of ULVWF multimers from damaged ECs. The inflammation is due to released inflammatory cytokines, and pulmonary vascular microthrombosis is due to ULVWF multimers forming microthrombi with platelets.
The molecular mechanism of how the tropism and endothelial heterogeneity work in ARDS is a partially understood mystery beyond the interaction between S protein and the receptor ACE2. The characteristic feature of susceptibility of the lungs producing ARDS in COVID-19 should encourage the research on the mechanisms of interfacing between human and nature as well as gene and environment that could explain other organ syndromes such as adrenal insufficiency in meningococcus, hemolytic-uremic syndrome due to Shiga toxin of Escherichia coli O157:H7 and acute liver failure associated with hepatitis B virus infection, and others.
TTP-Like Syndrome
Thrombotic thrombocytopenic purpura (TTP) is a classical hematologic disease representing VMTD. It is caused by severe deficiency of ULVWF cleaving protease ADAMTS13 and is characterized by triad of 1) thrombocytopenia; 2) microangiopathic hemolytic anemia (MAHA); and 3) one or more organ dysfunction syndrome, commonly in the brain and kidneys. On the other hand, TTP-like syndrome caused by EA-VMTD is a hemostatic disease with exactly the same triad and is often associated with mild to moderate deficiency of ADAMTS13, about 25–75% of normal, and occurs with critical illnesses such as sepsis. The analysis of clinical features of TTP-like syndrome identified it was associated with sepsis and other illnesses that are promoting complement activation and endotheliopathy,28 TTP-like syndrome typically is accompanied by thrombocytopenia as a result of consumption of platelets, and characterized by two endothelial markers, which are overexpression of von Willebrand factor (VWF) antigen and increased FVIII activity. All of the disseminated microthrombosis-associated disorders belong to the umbrella group of VMTD, which includes GA-VMTD for gene mutation-associated VMTD, AA-VMTD for antibody-associated VMTD, and EA-VMTD.28 The critical difference between TTP and TTP-like syndrome is the fact that TTP occurs due to severe ADAMTS13 deficiency, but TTP-like syndrome occurs as a hemostatic disorder due to endotheliopathy.
When COVID-19 pandemic was declared in early 2020, one of the serious concerns was a life-threatening ARDS could further progress to severe hematologic and coagulation disorders, including severe thrombocytopenia, MAHA, TTP-like syndrome, and thrombo-hemorrhagic syndrome that, in the past, claimed very high morbidity and mortality in bacterial sepsis. As presaged, ARDS was characterized by microthrombotic disease primarily affecting the lungs, sometimes with MODS. Fortunately, even in critically ill patients, thrombocytopenia has been mild to modest. Schistocytosis and MAHA were uncommonly reported, and TTP-like syndrome was diagnosed rarely although well-documented cases were described.29–32 Deadly thrombo-hemorrhagic coagulopathy, which has been known as acute disseminated intravascular coagulation (“DIC”) has not been a significant issue in COVID-19 to date. The fear perceived at the beginning of the pandemic has been eased.
Our understanding of EA-VMTD was conceptualized from clinical cases of TTP-like syndrome which retrospectively was recognized as a hemostatic disease initiated by complement activation and endotheliopathy.28 Thus, I initially assumed a good number of patients with disseminated VMTD would be associated with some degree of TTP-like syndrome. However, this pandemic has enlightened us that, depending upon the pathogenicity of each pathogen and host response, EA-VMTD can be manifested as a wide spectrum of clinical phenotypes from uncomplicated EA-VMTD to complicated EA-VMTD associated with combined micro-macrothrombotic syndrome, including severe thrombocytopenia, TTP-like syndrome, MODS, thrombo-hemorrhagic syndrome. Certainly, although MODS was reported commonly in COVID-19, TTP-like syndrome and thrombo-hemorrhagic syndrome have occurred but rarely.
My interpretation is the mildness of thrombocytopenia could have been due to coinciding reactive thrombocytosis when the lungs were involved by microthrombosis leading to ARDS. The extramedullary megakaryopoiesis in the lungs has been well-known mechanism.33–35 Also, uncommon MAHA and fewer cases with schistocytes might have been related to less shear stress of blood flow in pulmonary circulation because arterial blood pressure in the pulmonary vasculature is normally a lot lower than systemic blood pressure at 8–20 mm Hg at rest.36 Therefore, EA-VMTD in organ dysfunction involving primarily in the lungs was less vulnerable to intravascular hemolysis. Additionally, at molecular level in ARDS, lower degree complement activation of C3a, C3c and C5b-9 was apparent when compared to pathogen-induced sepsis,15 which also could have contributed to less hemolytic complication.
Despite of uncommon hematologic and hemostatic complications, ARDS has been a life-threatening clinical phenotype of EA-VMTD. A retrospective propensity matched control study showed improved survival of serious COVID-19 patients with therapeutic plasma exchange (TPE),37 which supports the benefit from indirect supply of the protease ADAMTS13 even without TTP-like syndrome. Considering these findings and data in COVID-19, “EA-VMTD” should stand as the diagnostic term of choice representing endotheliopathy with or without TTP-like syndrome.
Multiorgan Dysfunction Syndrome (MODS)
Sepsis-associated MODS can be defined as organ specific dysfunction of two or more organs due to hypoxia-induced physical and/or biochemical abnormalities caused by EA-VMTD in the patient with complement-activated endotheliopathy.28 It may be simultaneous or sequential in organ involvement. The medical literatures of COVID-19 have recorded many cases of MODS involving in the lungs, liver, heart, brain, kidneys, muscle, pancreas, adrenals, and nerve, and others,38–43 which phenotypes are summarized in Figure 4 with the mechanism of its pathogenesis. MODS often occurs with inflammation.15,18,37,44,45 Sometimes it is called cytokine storm in severe case. This additional activation of inflammatory pathway had been predicted to occur according to “two-activation theory of the endothelium“ as shown in Figure 3.22
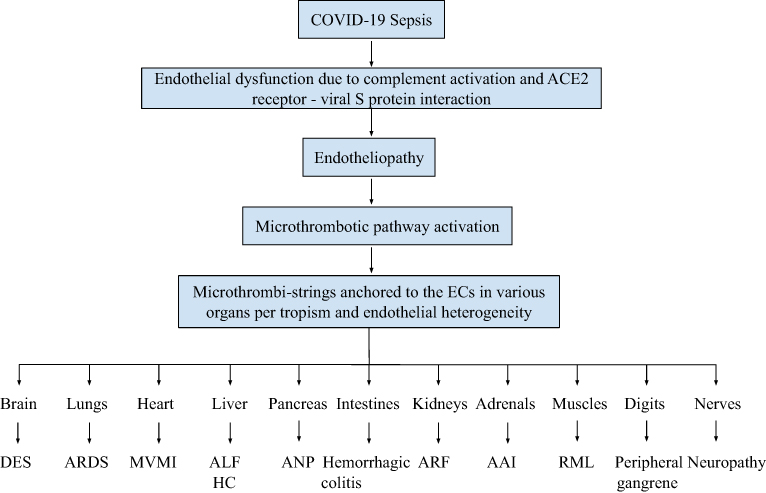 |
Figure 4 Pathogenesis of multiorgan dysfunction syndrome in COVID-19. Notes: The pathogenesis of MODS occurring in COVID-19 is summarized. Just like other sepsis, any organ can be involved by VMTD caused by the SARS viruses. However, MODS occurs much more commonly in the lungs as ARDS resulting from the viral tropism and host’s endothelial heterogeneity. The illustration is self-explanatory. Abbreviations: AAI, acute adrenal insufficiency; ALF, acute liver failure; ANP, acute necrotizing pancreatitis; ARDS, acute respiratory distress syndrome; ARF, acute renal failure; DES, diffuse encephalopathic stroke; HC, hepatic coagulopathy; MVMI, microvascular myocardial infarction; RML, rhabdomyolysis. |
The clinical organ syndromes, in addition to ARDS, have been termed: acute liver failure/fulminant hepatic failure, diffuse myocardial ischemia, encephalopathy/diffuse encephalopathic stroke, acute renal failure/hemolytic-uremic syndrome, acute necrotizing pancreatitis/hypoxic pancreatitis, rhabdomyolysis, adrenal insufficiency, and peripheral neuropathy. All of these clinical syndromes have been associated with TTP-like syndrome. Any organ syndrome in sepsis should alert the potential of underlying EA-VMTD, especially when associated with thrombocytopenia. Therefore, proper diagnostic surveillance for additional hemolytic anemia is recommended in every organ dysfunction syndrome of critically ill patients. Some case reports of organ dysfunction syndrome due to sepsis and now COVID-19 interpreted that; acute pancreatitis triggered TTP-like syndrome, acute liver failure was extrapulmonary manifestation of ARDS, or hepatic encephalopathy was the result of acute liver failure causing metabolic encephalopathy. Also, some reports have thought coexisting organ syndromes were the result of cross-talk between or among organs. Now, we can understand that the concept of underling EA-VMTD has placed every organ in equal footing for the potential of causing organ dysfunction syndrome rather than one causing the other(s).
To date, the prevailing pathogenic mechanism for organ syndrome has been direct invasion of pathogen and/or toxin into the organs, but pathological findings of microthrombosis within the organs and laboratory changes of increased expression of VWF antigen and increased activity of FVIII supporting endothelial pathogenesis have further confirmed in COVID-19 that the major mechanism causing organ syndrome is disseminated intravascular microthrombosis. Since microthrombi strings partially obstruct the microvasculature causing hypoxia of the affected organ, organ dysfunction is reversible if EA-VMTD is diagnosed in a timely manner and treated with TPE. Even septic patient with delirium and coma still can recover from hypoxic encephalopathy. In general, the development of MODS indicates advancing disease and portends poor prognosis.
Macrothrombosis
Although the primary disorder of COVID-19 was ARDS, serious macrothrombosis, especially PTE and DVT, was commonly superimposed and coexisted in the same patient.46–53 Localized acute ischemic stroke and acute myocardial infarction were also observed, but could not be blamed to microthrombosis of EA-VMTD due to their macrothrombotic nature. Because ARDS coexistence with PTE in the lungs, COVID-19 has been considered to cause a complex thrombotic disorder represented by both microthrombosis and macrothrombosis. In this pandemic, the majority of physicians has managed this complex thrombosis with traditional TF path inhibiting anticoagulation because ARDS and PTE/DVT were different expression of the same disease. In past several decades, numerous therapeutic trials for sepsis-associated coagulopathy (e.g., “DIC”) had failed to show any benefit from anticoagulants relied on the pathogenesis of activated TF path. In retrospect, the failure can be recognized as the result of distinctively different two pathogeneses between microthrombosis of ARDS and macrothrombosis of PTE/VTE. This conception of the sameness of all thrombi has been persisted in medical community so long, and some pathologists have been persuaded to call microthrombi in pathologic specimens as platelet-fibrin thrombi, fibrin rich microthrombi, or fibrin deposits35,54,55 even though microthrombi strings contain no fibrin components. This issue of the different character between microthrombi and macrothrombus should be clarified through an appropriate discussion forum. The answer should come from the understanding of true mechanism of hemostasis in vivo.
Interesting questions are; why do microthrombi and macrothrombus each occur in different-sized vessels? how can two different thrombi be produced from the same TF-FVIIa complex activated coagulation cascade mechanism? what are the differences in their character between microthrombi and macrothrombus? It is logical to conclude microthrombi of ARDS and macrothrombus of PTE are two different blood clots not only in size, location and genesis, but also in their intrinsic character of the thrombi. We know microthrombi are composed mostly of platelet-ULVWF complexes,24 and macrothrombus is partly made of platelet, fibrin clot and extracellular traps, Assuredly, microthrombi have to be formed from another path different from TF path-initiated hemostasis. No wonder, why anticoagulation therapy failed for the treatment of EA-VMTD of sepsis-associated coagulopathy. I have wrestled with this conceptual mystery of two different thromboses and “fibrin clot disease” occurring in acute promyelocytic leukemia. Finally, a novel “two-path unifying theory” of hemostasis was proposed and updated as shown in Figure 2.23
Figure 5 schematically illustrates cross section of blood vessel histology and hemostatic components, which explains the physiologic consequences from intravascular damage limited to ECs compared to that from combined ECs and SET damage.56 Disseminated endotheliopathy limited to ECs damage (e.g., sepsis) activates lone ULVWF path of hemostasis that produces microthrombosis in the microvasculature, but localized vascular injury (e.g., surgery) in the large vessel activates combined ULVWF path and TF path that produces macrothrombosis at the damage site. The difference is summarized in Table 2. Indeed, microthrombosis of ARDS is unrelated to coexisting macrothrombosis occurring in some cases of COVID-19. Since without vascular injury hemostasis cannot not be initiated and without hemostasis thrombus cannot be formed,57 a localized vascular injury leading to macrothrombosis must have occurred as a result of vascular trauma from the risk factor such as “hospitalization”.
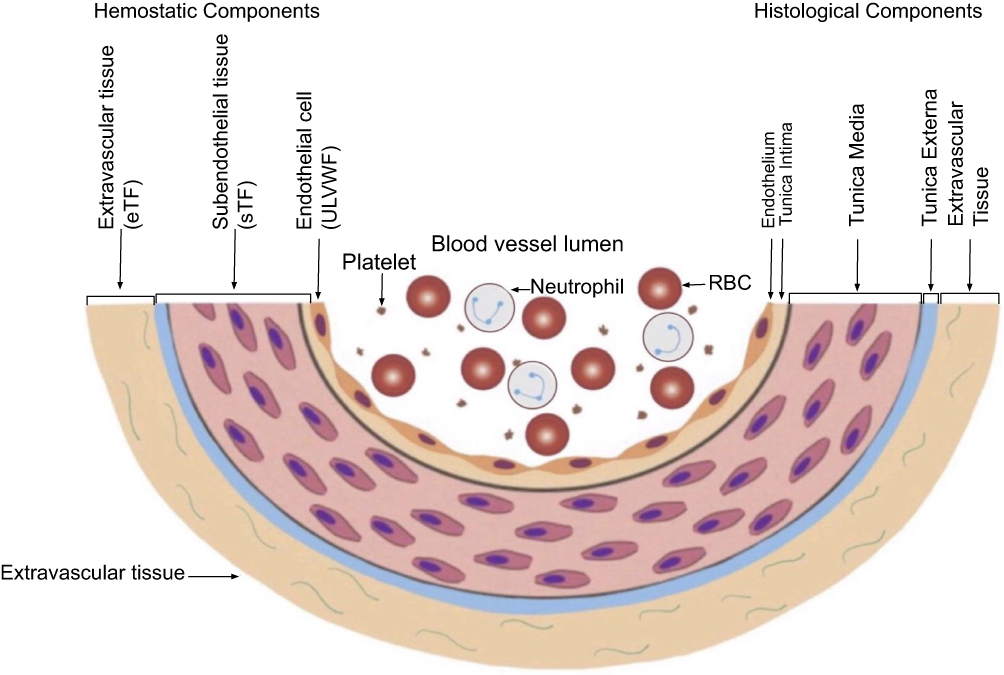 |
Figure 5 Schematic illustration of cross section of blood vessel histology and hemostatic components. Notes: The blood vessel wall is the site of hemostasis (coagulation) to produce the hemostatic plug in external bodily vascular injury and to stop hemorrhage. It is also the site of hemostasis (thrombogenesis) to produce intravascular blood clots (thrombus) in intravascular injury to cause thrombosis. Its histologic components are divided into the endothelium, tunica intima, tunica media and tunica externa, and each component has its function contributing to molecular hemostasis. As shown in the illustration, ECs damage triggers exocytosis of ULVWF and SET damage promotes the release of sTF from tunica intima, tunica media and tunica externa. EVT damage releases eTF from the outside of blood vessel wall. This depth of blood vessel injury contributes to the genesis of different thrombotic disorders such as microthrombosis, macrothrombosis, fibrin clot disease, hematoma and thrombo-hemorrhagic clots. This concept is especially important in the understanding of different phenotypes of stroke and heart attack. Reproduced from Chang JC. Acute Respiratory Distress Syndrome as an Organ Phenotype of Vascular Microthrombotic Disease: Based on Hemostatic Theory and Endothelial Molecular Pathogenesis. Clin Appl Thromb Hemost. 2019; 25:1076029619887437.6Abbreviations: ECs, endothelial cells; EVT, extravascular tissue; SET, subendothelial tissue; TF, tissue factor; eTF, extravascular TF; sTF, subendothelial tissue factor; ULVWF, unusually large von Willebrand factor. |
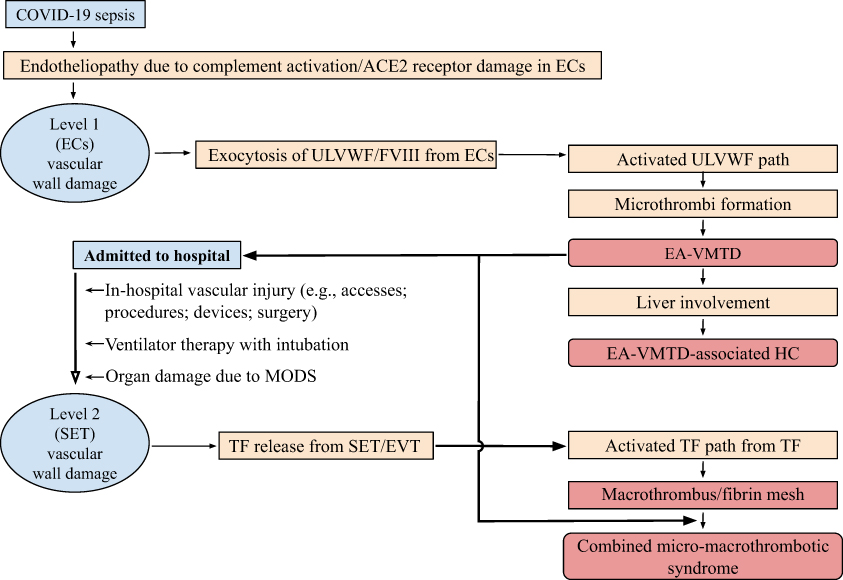 |
Figure 6 Proposed pathogenesis of thrombotic and coagulopathic syndromes observed in COVID-19. Notes: In COVID-19 viral sepsis, disseminated endotheliopathy promotes inflammatory and microthrombotic pathways. The latter pathway is identical to ULVWF path due to Level 1 (ECs) damage-induced hemostasis in localized vascular injury. Disseminated endothelial molecular pathogenesis of activated ULVWF path triggers the formation of microthrombi and causes EA-VMTD, which orchestrates several hemostatic phenotypes of ARDS and MODS. If microthrombosis involves in the liver and leads to hepatic necrosis, especially in an underling disease such as liver cirrhosis. This unique coagulopathy can be termed “EA-VMTD with HC” (it can be recognized as acute “DIC”).23 Also, ICU admission increases the risk of additional serious complication of macrothrombosis from in-hospital vascular injury, ventilator therapy with intubation and perhaps also from MODS. The Level 2 vascular wall damage of SET/EVT from vascular injury unrelated to viral sepsis could release of TF that activates TF path and produce macrothrombosis such as PTE and DVT (Table 2). Further, the interaction of microthrombi strings of EA-VMTD formed from microthrombogenesis and fibrin meshes formed by thrombin following activated TF path from in-hospital vascular damage could cause “combined micro-macrothrombotic syndrome” presenting with peripheral or limb gangrene (see text for discussion). This proposed mechanism is derived from “two-path unifying theory” of hemostasis and endothelial molecular pathogenesis. Abbreviations: “DIC”, false disseminated intravascular coagulation; EA-VMTD, endotheliopathy-associated vascular microthrombotic disease; DVT, deep vein thrombosis; ECs, endothelial cells; EVT, extravascular tissue; ICU, intensive care unit; HC, hepatic coagulopathy; MODS, multiorgan dysfunction syndrome; PTE, pulmonary thromboembolism; SET, subendothelial tissue; TF, tissue factor; ULVWF, unusually large von Willebrand factor multimers. |
Macrothrombosis is common complication from vascular injury after admission to the hospital, especially in intensive care unit which is a high-risk environment due to numerous vascular interventions in septic and non-septic conditions.49,50,58–62 The simple fact is the vascular events (e.g., surgery, accesses, procedures, devices and ventilators) may cause local vascular wall damage which releases ULVWF and TF and initiate thrombogenesis, leading to macrothrombosis at the injury site and spreading to other sites. In addition, distinguished from this macrothrombosis, inexplicable macrothrombotic disorder in COVID-19 characterized by “multiple” small macrothromboses mixed with microthrombotic disorder was observed in the lungs in the same patient,52,53,63,64 and also occurred in the digits with gangrene12–14 and brain with acute ischemic stroke and cerebral venous sinus thrombosis (CVST), which pathogenesis is unidentified yet. This “multiple” small macrothrombosis typically tends to occur in severe ARDS patients.
Because the reports in the literature were not from cooperative studies providing the patient care, it was difficult to know how commonly macrothrombosis coincided with COVID-19. My impression is that the significance of macrothrombosis was overemphasized in patients with ARDS. The incidence of macrothrombosis is estimated between 5% and 10% of critically ill patients with significantly higher rate in the intensive care unit. However, general perception has been the macrothrombotic complication occurred more commonly in COVID-19 than other pathogen-associated ARDS. Could this have been due to overzealous vascular intervention for monitoring and more aggressive usage of controlled respiratory therapy in this hyped pandemic? The prevalent reports of coexisting PTE and relatively mild thrombocytopenia with evidence of active megakaryopoiesis in the lungs of COVID-1935,65,66 may suggest locally produced platelets from reactive thrombocytosis could also have contributed to the formation of “multiple” small macrothrombi in the compromised vasculature of the lungs. Further, could it have been due to combined micro-macrothrombotic syndrome in the lungs similar to multiple peripheral digital gangrene?
VMTD with Coagulopathy
In early stage of COVID-19, initial evidence of potential coagulopathy was elevated fibrinogen, increased FVIII activity, and overexpressed ULVWF/VWF/VWF antigen with thrombocytopenia. Since fibrinogen is synthesized in the liver, hyperfibrinogenemia could have been caused by mild dysfunction of the highly vascularized liver in early stage of microthrombosis, especially with preexisting liver disease. However, in advanced stage of COVID-19, hypofibrinogenemia was common, which was attributed to decreased synthesis of fibrinogen due to liver failure. Overexpression of ULVWF/VWF/VWF antigen and increased FVIII activity were due to the release from Weibel–Palade bodies in endotheliopathy, which have become the best endothelial markers that can be used along with mild to moderate decrease of ADAMTS13 activity in early diagnosis of endothelial dysfunction.6
Unlike in viral hemorrhagic fever occurring in Eboli sepsis, coagulopathy presenting with hemorrhagic disease has been uncommon in COVID-19 even though mildly prolonged prothrombin time (PT) and activated partial thromboplastin time (aPTT), and increased fibrin degradation products (FDPs)/D-dimer have been encountered commonly.67,68 Significant hepatic coagulopathy resulting from complication of EA-VMTD, which is similar to contemporary concept of acute “DIC,” was uncommon.68,69 It could have been due to relatively lower levels of activated complement factors15 and/or possibly insignificant tropism of the viral molecules to the liver. Undetermined factors have lowered the incidence of serious thrombo-hemorrhagic phenotype, and by chance spared tragic loss of many lives in pandemic.
Combined Micro-Macrothrombotic Syndrome with Gangrene
This inexplicable thrombotic syndrome has rarely occurred but commonly been reported in the COVID-19 literature because of its oddity.14,70–76 However, this gangrene had been described enough in sepsis of the non-COVID-19 literature22,77–80 because of its mysterious nature and serious consequences with high morbidity and sometimes death. The diagnosis of combined micro-macrothrombotic syndrome is affirmed logically from two facts in every patient: 1) underlying disseminated microthrombosis (i.e., EA-VMTD); and 2) gangrene formation which never occurs without macrothrombosis. In the past, the pathogenesis could not be identified because of our shortcomings on the complete picture of hemostasis. Now, armed with novel “two-path unifying theory” of hemostasis (Figure 2) and “two-activation theory of the endothelium” (Figure 3) as well as the “three essentials of hemostasis” (Table 1), the pathogenesis of this serious life-threatening disorder can be established as shown in (Figure 6).
Combined micro-macrothrombotic syndrome presenting with several different phenotypes is characterized by the gangrene presenting with single or multiple, small or large, often symmetrical and peripheral, sometimes as ischemic (acrocyanosis) or hemorrhagic, with either a large isolated or disseminated lesion(s), involving commonly digits or sometimes exposed areas of the limb, skin and subcutaneous tissue as well-demarcated gangrene. Often, it occurs as “symmetrical peripheral gangrene” involving the terminal parts of digits. These gangrene phenotypes typically occur in association with sepsis in hospitalized patients coinciding with underlying disseminated microthrombosis (i.e., EA-VMTD) following surgery or other vascular events.
It should be emphasized that “gangrene” is the dead and altered tissue lesion beyond necrosis or infarction. It is caused by “multiple” small arterial macrothrombi due to complete cutoff of blood supply to the distal parts without collateral circulation. Unlike “necrosis” or” infarction,” “gangrene” is the dead, dried and shrunken tissue lesion discolored to black without much pain or inflammation, indicating the all of the microvasculature in the area is out of service supplying of oxygen and destroying the entire tissue. Gangrene is created by denatured hemoglobin molecules. Previously, I theorized that peripheral gangrene was triggered by “multiple” small macrothrombi formed from ongoing microthrombi of activated ULVWF path in sepsis unifying – according to “ two-path unifying theory” – with “fibrin meshes” from activated TF path following additional vascular injury due to surgery or vascular devices.22,77 These “multiple” small macrothrombi are suspected to be “microthrombi strings-fibrin meshes” complexes, completely shutting off blood supply to the distal part of circulation. This combined micro-macrothrombotic syndrome can explain the pathogenesis of purpura fulminans73,80 that is associated with protein C deficiency and sepsis complicated by vascular injury, and also diabetic gangrene, Fournier’s gangrene, necrotizing fasciitis, and other gangrene disorders. Some have called this syndrome was a gangrene type of “DIC.”75
In COVID-19 sepsis, for example, “microthrombi strings” are formed from activated ULVWF path due to endotheliopathy (i.e., sepsis) and “fibrin meshes” are produced from activated TF path due to vascular injury (e.g., femoral or subclavian artery device). Fibrin meshes in circulation travel to downstream arterial microvascular trees and encounter microthrombi strings localized in the microvasculature of every involved digits. These two different thrombi would unify to form “multiple” small macrothrombi composed of “microthrombi strings-fibrin meshes” complexes based on the hemostatic theory. These “multiple” small (minute) macrothrombi could completely block circulation at every similar-sized branches of small vascular trees of the digits via macrothrombogenesis. This pathogenesis is similar to “vascular access steal syndrome” seen kidney dialysis patients. The hemoglobin of red blood cells trapped within the small arteries distally would be completely deprived of oxygen supply to distal areas of the tissue without any collateral circulation. The hemoglobin would be denatured into dark organic compounds and/or inorganic molecules such as methemoglobin and/or ferric disulfide, which be deposited into surrounding dead tissues to produce dry black gangrene. This interaction between COVID-19 septic endotheliopathy and vascular access-induced injury explain the unique “combined micro-macrothrombotic syndrome.”22
Symmetrical peripheral gangrene,79 peripheral digit ischemic syndrome,77 limb hemorrhagic gangrene,70 limb ischemia,71 acrocyanosis,14 purpura fulminans,73,75,76,80 and perhaps coumadin-induced gangrene and diabetic leg gangrene have been associated with sepsis-associated microthrombosis, sometimes with underlying thrombophilia. These gangrene syndromes represent variable phenotypes of combined micro-macrothrombotic syndrome observed in COVID-19. The experience in COVID-19 pandemic has offered an opportunity for us to reexamine the complexity of microthrombogenesis, fibrinogenesis and macrothrombogenesis, and their interactions as well as true hemostasis in vivo.
Cytokine Storm
Endotheliopathy is manifested by both inflammation and EA-VMTD. Inflammation is common in COVID-19 due to endothelial release of various cytokines, including interleukin (IL)-1, IL-2, Il-6, tumor necrosis factors and interferons. If inflammation is severe, it is called cytokine storm. The cross-talk mechanism between inflammation and coagulation was proposed to explain frequent association of two conditions in sepsis-associated coagulopathy, but cytokine syndrome is neither the cause nor result of microthrombotic disorder. Some immunologists and coagulation specialists have thought that cytokine storm would have a major impact on the morbidity and mortality of COVID-19. However, in ARDS with severe COVID-19, circulating cytokine levels were significantly lower compared to those with bacterial sepsis, which suggests cytokine storm was not so serious feature of COVID-19.81 The finding of relatively low level of cytokines in spite of severe ARDS tends to support EA-VMTD and inflammation are independent processes occurring in endotheliopathy, which implies anticytokine therapy would not be an effective regimen treating COVID-19.
Further, even though inflammatory response may cause toxic state with fever, headache, malaise, myalgia, and gastrointestinal symptoms, cytokine storm is not a disease, but is only a symptom complex which is transient and reversible. On the contrary, disseminated EA-VMTD is a structural disease that may cause TTP-like syndrome and lead to organ hypoxia and MODS. Eventually, organ dysfunction, if prolonged and not reversed, organ failure directly contributes to the demise of the patient. TPE was employed for the treatment of COVID-19 and had shown improved overall survival in a good retrospective case control study.37 The benefit was claimed to be the result of removal of cytokines, but the decreased mortality was more likely from the result of replenished ADAMTS13 that had counteracted microthrombogenesis.
Hemostatic, Hematologic and Coagulation Laboratory Findings
Adequate coagulation laboratory results are available from the clinical and research literatures. The data fully support previously proposed endothelial pathogenesis of coronaviral sepsis.6 The underlying pathology of COVID-19 sepsis has been confirmed to be EA-VMTD via complement activation, endotheliopathy, activated ULVWF path of hemostasis, microthrombogenesis and ADAMTS13 insufficiency, and could lead to complex thrombo-coagulopathic syndromes.15,18,19,30,68,82–91 This is the same endothelial mechanism causing septic syndrome in every pathogen-induced septicemia, eventually leading to various phenotypes of EA-VMTD.
As anticipated from endothelial molecular mechanism, decreased activity of ADAMTS13 was very important findings in the tested patients.82,83,90 The result is consistent with key role of the enzyme in the pathogenesis of microthrombogenesis as proposed.6,22,23 The laboratory findings have firmly supported the concept that COVID-19 sepsis is a hemostatic disease based on the following laboratory data.
- Consistently significant findings
- Normal or mild to moderate thrombocytopenia
- Uncommon but enough reported cases with MAHA
- Markedly increased ULVWF/VWF/VWF antigen expression19,83,88–90,92,93
- Markedly increased FVIII activity19,90,92–95
- Activated complement factors9,15,16
- Mild to moderately decreased activity of ADAMTS13 (25–75% of normal)82,83,90,96–98
- Increased fibrinogen in early stage of EA-VMTD or markedly decreased in advanced stage30,68,82,84–86,88,89
- Increased FDP(s)/D-dimer68,83–91,93
- Sometimes, significant abnormal findings
Hemostatic Pathogenesis of COVID-Induced Endotheliopathy
How Does Endotheliopathy Activate Hemostasis of ULVWF Path?
Hemostasis in vivo can be explained best by “two-path unifying theory” initiated by activated ULVWF path and TF path. The hemostatic mechanism was derived from the physiologic syllogism of vascular wall injury as illustrated in Figure 2. This theory represents true hemostasis in vivo99 and has been updated.6,22,23
The hemostatic components of blood vessel walls are consisted of two major coagulation factors: ULVWF multimers from ECs and TF from SET/extravascular tissue (EVT) shown in Figure 5. A vascular wall injury to ECs releases ULVWF multimers and the injury to SET/EVT releases TF. In external bodily injury (e.g., physical assault), bleeding occurs externally with release of ULVWF and TF, but internal vascular injury (e.g., dissecting aneurysm) releases ULVWF multimers and TF into circulation, and sometimes bleeding into EVT. ULVWF multimers recruit platelet to activate ULVWF path, and TF activates FVII to initiate TF path. The former produces “microthrombi strings” composed of platelet-ULVWF complexes via microthrombogenesis. The latter produces “fibrin meshes” via fibrinogenesis. In the last step, microthrombi strings and fibrin meshes unify together via macrothrombogenesis to form “hemostatic plug” in external bodily injury to stop bleeding, and to form “macrothrombus” in intravascular injury to cause macrothrombosis.
Sepsis is an intravascular disease involving the endothelium breached by septicemia. It is mediated by endotheliopathy which damage is limited ECs. Endotheliopathy in sepsis leads to exocytosis of ULVWF multimers and activates platelets. The released ULVWF multimers into intravascular space should be cleaved by the protease ADAMTS13 in normal person. However, if mild to moderate insufficiency of ADAMTS13 is present due to heterozygous mutation of the gene or excessive release of ULVWF multimers over the capacity handling of ADAMTS13, uncleaved ULVWF multimers become anchored to the damaged endothelial membrane and recruit platelets to form microthrombi strings via microthrombogenesis. These strings are microthrombi that partially obstruct the microvasculature.
Apart from normal hemostasis, released ULVWF in sepsis-induced endotheliopathy activates only partial hemostasis, which is called ULVWF (microthrombotic) path of hemostasis, but is disseminated in the entire microvascular system and leads to EA-VMTD. Endotheliopathy does not activate TF (fibrinogenetic) path because SET of vessel wall is intact in sepsis. Thus, coagulation factors (ie, FVII, FV, FX, FII and fibrinogen) are not involved in the formation of microthrombosis, and macrothrombus is not formed. Succinctly speaking, sepsis is caused by lone activation of ULVWF path in disseminated endotheliopathy, which results in partial hemostasis that causes pathologic microthrombosis.
ULVWF multimers are very large multimeric glycoproteins synthesized in the endothelium and stored within the Weibel–Palade bodies with FVIII.100 Because of their close relationship each other, the exocytosis of ULVWF simultaneously occurs with release of FVIII in endotheliopathy. Therefore, increased level of VWF in plasma is always associated with increased activity of FVIII. These increased VWF and FVIII are the best diagnostic endothelial markers for EA-VMTD along with thrombocytopenia and decreased activity of ADAMTS13 (approximately 25–75% of normal).28
The theory of hemostasis was derived from the same character of microthrombi composed of platelet-ULVWF complexes in TTP.99,101 Severe deficiency of the protease ADAMTS13 typically less than 5% of normal occurs in TTP due to either gene mutation or due to autoantibody production. However, endotheliopathy leads to EA-VMTD/TTP-like syndrome when it is associated with mild to modest ADAMTS13 insufficiency.28,56 The formation of microthrombi takes place on the endothelium. Some cases of EA-VMTD could cause the triad of thrombocytopenia, MAHA and organ dysfunction syndrome. In such case, it is called TTP-like syndrome. The theory of in vivo hemostasis is consistent with the works of the coagulation scientists who observed the characteristic platelet-ULVWF strings anchored to the ECs ex vivo and in vivo.102−105
This vascular model of hemostasis can easily define the concept of phenotypes of variable thrombotic disorders via two important elements in vascular injury, which are: 1) depth of vascular wall damage; and 2) extent of involvement of vascular tree system. Now, the different clinical phenotypes of thrombotic disorders (e.g., ARDS and PTE) in COVID-19 can be understood from their different causes and pathogeneses.56
How Does Endotheliopathy Orchestrate Molecular Pathogenesis?
In intravascular injury, when ULVWF path is activated due to focal or local detachment of a small atherosclerotic ECs plaque(s), some microthrombi strings could be produced, which clinical phenotype is focal microthrombotic syndrome such as transient ischemic attack or angina pectoris without systemic implication.56 In contrast to this focal phenotype of endothelial injury, disseminated endotheliopathy produces a disseminated phenotype of EA-VMTD with a spectrum of clinical syndromes from consumptive thrombocytopenia to severe combined micro-macrothrombotic syndrome and diverse microthrombotic syndromes such as thrombocytopenia in critically ill patients, TTP-like syndrome, MODS, and hepatic coagulopathy in-between. This complexity cannot be reconciled by “two-path unifying theory” of hemostasis alone. Figure 3 showing “two-activation theory of the endothelium” answers the rest of pathophysiological mechanism.57 Therefore, microthrombotic pathway displayed in endothelial molecular pathogenesis is the extended version of ULVWF path of hemostasis applicable to the pathogenesis of disseminated endotheliopathy.
In COVID-19 sepsis, when terminal complement complex C5b-9 and S protein of SARS-CoV-2 attack ECs and promote endotheliopathy,19 similar to other pathogen, two important molecular events occur: 1) release of inflammatory cytokines, including various interleukins, tumor necrosis factors, and interferons;44,81 and 2) platelet activation and exocytosis of ULVWF multimers.9,19,90,92 The former promotes inflammation, which mechanism is called “activation of inflammatory pathway,” and the latter mediates microthrombogenesis, which is triggered by “activation of microthrombotic pathway.” These two independent pathways were proposed in the framework of “two-activation theory of the endothelium.”57 Many scientists have opined inflammatory pathway plays a major role on the pathogenesis of COVID-19. But the experience from COVID-19 pandemic, activated microthrombotic pathway contributes to more deadly pathogenesis leading to ARDS of EA-VMTD, which orchestrates consumptive thrombocytopenia, MAHA, TTP-like syndrome, MODS, thrombo-coagulopathic syndrome, and combined micro-macrothrombotic syndrome.
Role of ADAMTS13 as a Modulator of ULVWF Path
ADAMTS13, which is a zinc containing metalloproteinase cleaving ULVWF multimers to smaller VWF and modulating of thrombogenesis in intravascular injury, was predicted to be decreased in ARDS based on two hemostatic theories.6 This protease was found to be insufficient when tested in severe COVID-19 patients.82,83,90,96,98 This important proteolytic enzyme is needed to cleave the excess ULVWF multimers that are released from damaged ECs and to prevent microthrombogenesis.
The role of ADAMTS13 preventing TTP and TTP-like syndrome (i.e., EA-VMTD) is well established.101 This enzyme also contributes to downregulating macrothrombogenesis of stroke,106–108 myocardial infarction,109,110 and DVT111 via ULVWF path perhaps before unifying mechanism prior to form macrothrombus. Therefore, ADAMTS13 deficiency is a “thrombophilia” modulating ULVWF path of hemostasis in contrast to protein C deficiency which is a “thrombophilia” modulating TF path. It is predicted “purpura fulminans” occurring in sepsis is likely associated with combined deficiency of ADAMTS13 of ULVWF path and protein C deficiency of TF path, leading to disseminated form of combined micro-macrothrombotic syndrome according to two hemostatic theories. Over the last few months, cases of purpura fulminans were reported in COVID-19.73,76
Both ADAMTS13 and ABO blood group genes are closely linked at the same chromosome 9q34.2 location, and non-O blood group population has been associated with increased susceptibility and poorer prognosis compared to O blood group in COVID-19.112–114 Therefore, non-O blood group individuals are expected to have decreased ADAMTS13 activity that can cause more severe phenotypes of EA-VMTD and poorer prognosis. The relationship in the triangle of ADAMTS13 activity, ABO blood group antigen expression and severity of EA-VMTD in patient with COVID-19 and other sepsis would be an interesting epidemiologic study, which could identify the more vulnerable population to sepsis and septic progression.
Interpretation for Laboratory Findings and Diagnostic Approach
The followings are the hematologic and coagulation abnormalities observed in COVID-19. Their interpretation is summarized based on endothelial pathogenesis.
- Mild to moderate thrombocytopenia → likely due to consumption during microthrombogenesis, but with partial compensation from extramedullary megakaryopoiesis in the lungs35,65,66
- Rare cases with schistocytes in blood film and hemolysis → likely due to: 1) uncommon hemolysis in ARDS of COVID-19 secondary to less production of C5b-9 than in viral sepsis;15 and 2) less shear stress of blood flow at the pulmonary vasculature35
- Prolonged PT, if present → due to decreased FVII, FX, FV, FII and fibrinogen in hepatic coagulopathy
- Prolonged aPTT, if present → due to decreased FX, FV, FIX, FII and fibrinogen in hepatic coagulopathy
- Overexpression of ULVWF/VWF/VWF antigen and increased FVIII activity → due to endothelial exocytosis
- Decreased ADAMTS13 activity → due to: 1) heterozygous gene mutation or polymorphism; and/or 2) excessive release of ULVWF multimers creating an imbalance between the enzyme and substrate multimers
- Abnormal fibrinogen levels → increased likely due to early transient liver dysfunction and decreased due to advanced hepatic necrosis resulting from microthrombosis
- D-dimer → positive due to “fibrinolysis” in MODS and coexisting macrothrombosis with on-going EA-VMTD, but negative in “fibrinogenolysis” without MODS even with on-going EA-VMTD
- Soluble fibrin monomer (SFM) → positive due to increased “fibrinogenesis”
- FDP(s) → positive due to fibrinolysis and/or fibrinogenolysis
In EA-VMTD, if PT and aPTT are prolonged, it would be helpful to determine the activity of FVII, perhaps with other liver dependent factors (FII, FV, FIX, and FX) to confirm hepatic coagulopathy. If combined micro-macrothrombotic syndrome with gangrene occurs in sepsis, in addition to ADAMTS13 activity, protein C and protein S activities, and the test for FV-Leiden and others would be needed to exclude potential underlying congenital or acquired thrombophilia. Previously, insufficient coagulation tests had precluded identifying the conceptual difference between acute “DIC” and hepatic coagulopathy in sepsis-associated coagulopathy, which is now summarized in Table 4. If hypofibrinogenemia, and prolonged PT and aPTT occur with thrombocytopenia, increased activity of VWF and FVIII, and decreased FVII in sepsis, the diagnosis of EA-VMTD associated hepatic coagulopathy can be established. Since serious hemorrhagic syndrome has not been a major issue in this pandemic, COVID19 sepsis is not considered to be a hemorrhagic disorder.
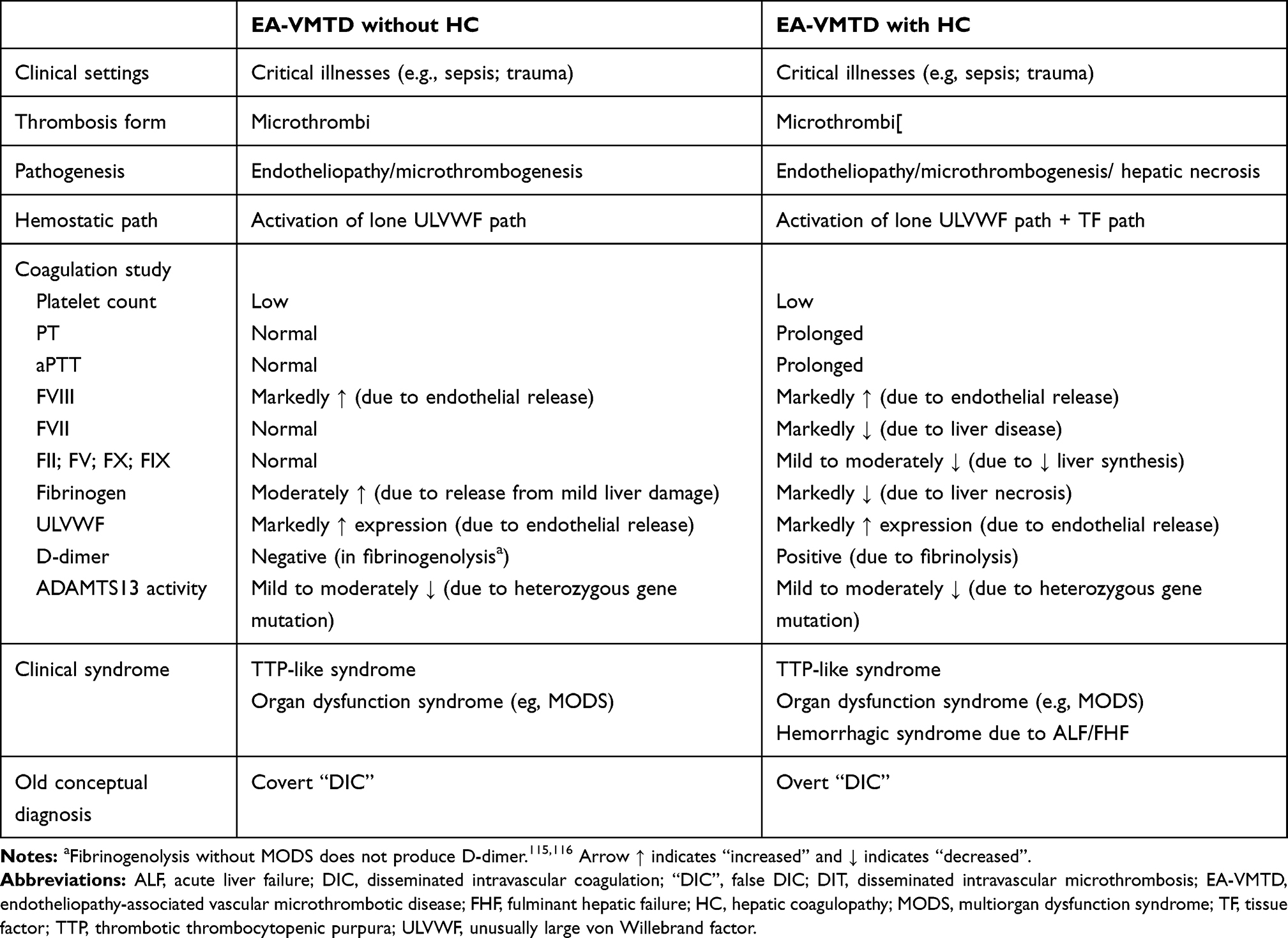 |
Table 4 Hemostatic Characteristics Between EA-VMTD without HC and EA-VMTD with HC |
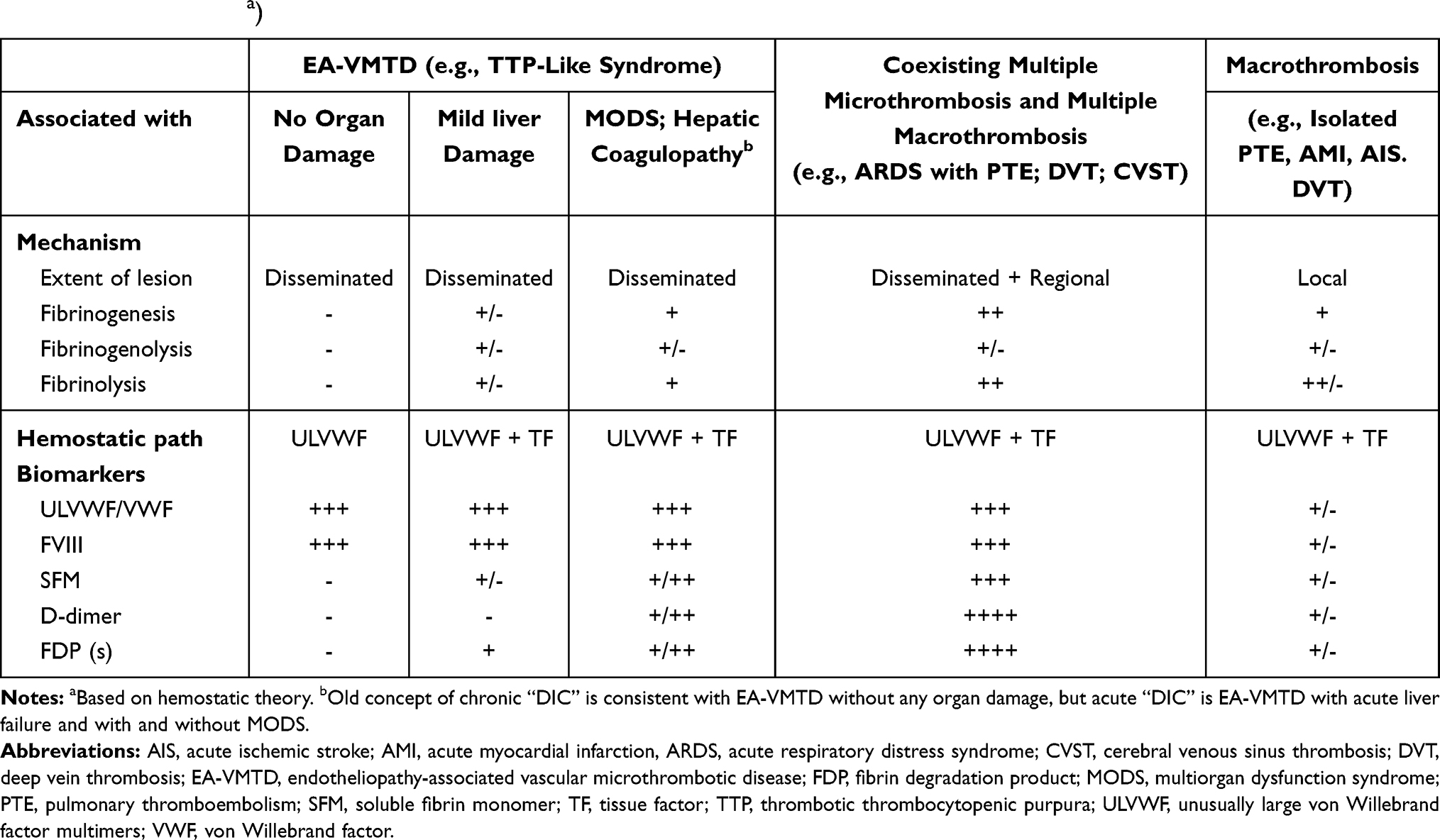 |
Table 5 Expected Biomarker Changes in Thrombo-Coagulopathic Disorders Based on “Two-Path Unifying Theory” of in vivo Hemostasis (Estimateda) |
In regard to interpreting D-dimer, SFM and FDP(s), when increased fibrinogen is cleaved by plasmin, fragments X, Y, Dg and E but no D-dimer are produced, however, when cross-linked fibrin clots are cleaved by plasmin, several FDP(s) and D-dimer are produced.115 If fibrinogen is catalyzed by thrombin following TF path activation, SFM is formed. Thus, in EA-VMTD, D-dimer should be negative unless organ damage leading to TF expression has occurred due to MODS or macrothrombosis is coexisted, but SFM becomes positive following fibrinogenesis and fibrinolysis according to the hemostatic theory. For this reason, D-dimer is negative in EA-VMTD without organ damage, but is positive in EA-VMTD with organ damage,116 including hepatic coagulopathy. Macrothrombosis such as multiple PTE in COVID-19 with EA-VMTD causes strongly positive D-dimer because vascular injury releasing TF due to SET/EVT damage of the organ leads to fibrinogenesis and fibrinolysis. Generally, in sepsis, significantly positive D-dimer is an important marker indicative of advancing MODS116,117 or concurrent macrothrombosis.
The theoretical diagnostic utility of D-dimer and FDP(s) in thrombotic disorders is approximated in Table 5. D-dimer is a complex marker to interpret in coexisting microthrombosis, macrothrombosis and coagulopathy as well as MODS because the value also varies depending upon not only microthrombosis with/without liver involvement, with/without MODS, and with/without additional macrothrombosis, but also the severity of thrombotic disorders. Theoretically, negative D-dimer infers uncomplicated EA-VMTD in the absence of activated TF path, but positive D-dimer in EA-VMTD may suggest the damage of SET of the vessel wall leading to activated TF path (i.e., MODS) and portends poorer prognostic outcome.
Treatment Options Based on in vivo Hemostatic Theory
Since beginning of pandemic, the use of anticoagulation and thromboprophylaxis with drugs such as TF path inhibitors (e.g., low molecular weight heparin), antiplatelet agents, and fibrinolytic therapy118–120 was advocated for “comprehensive” treatment and prevention of both microthrombosis and macrothrombosis. Anticytokine therapy also has been attempted and debated.121–124 Anticoagulation and anti-inflammatory regimens have shown no measurable benefit on mortality of ARDS and uncertain effect on developed macrothrombotic disorders. The lack of success could have been presaged because traditional anticoagulants counteracting TF path is theoretically ineffective for microthrombosis (e.g., ARDS)6 and was not beneficial in numerous clinical trials for sepsis-associated coagulopathy. Anticytokine therapy suppressed inflammation but did not reduced mortality.121 The proposition is the demise of the patient does not occur due to activated inflammatory pathway but due to activated ULVWF (microthrombotic) pathway. The main culprit of therapeutic failure is due to our lack of comprehension on different pathogeneses between ARDS in sepsis and PTE in virus-unrelated vascular injury.
Targeting EA-VMTD
The primary goal treating ARDS and associated MODS caused by EA-VMTD is resolving and preventing pathologic microthrombosis that is resulting from activation of ULVWF path. The logical approach counteracting the pathogenesis is to prevent and remove the excess of ULVWF multimers. The following is theoretically effective antimicrothrombotic regimens.
- Proteolytic therapy with ULVWF-cleaving protease ADAMTS13 to inhibit ULVWF path of hemostasis (i.e., recombinant (r) ADAMTS13)
- Surrogate therapy using plasma exchange to supply ADAMTS13 (i.e., TPE)
- Anticomplement therapy directed against activated complement components to prevent endotheliopathy (e.g., eculizumab)
- Disulfide bond reducing mucolytic therapy to inhibit ULVWF path (i.e., N-acetylcysteine)
Recombinant ADAMTS13
Recombinant (r) ADAMTS13 has been available for clinical trials in GA-VMTD due to severe ADAMTS13 deficiency (ie, hereditary TTP). In animal model, its prophylactic administration protected ADAMTS13 knockout mice from developing TTP-like syndrome, and therapeutic dose reduced the incidence and severity of TTP findings.125 It has not been used yet for conceptually established diagnosis of EA-VMTD in the clinical setting. Recent studies and review showed correlation between decreased level of ADAMTS13 and poor outcomes of sepsis in mice and in humans.126,127 Furthermore, ADAMTS13‐deficient mice were partly rescued from widespread microthrombosis in staphylococcus aureus sepsis by the administration of rADAMTS13.128 Theoretically, rADAMTS13 is the best regimen for the treatment of VMTD, but it has not been utilized in EA-VMTD or TTP-like syndrome. The hemostatic nature of VMTD caused by the endothelial molecular pathogenesis should encourage therapeutic trials for COVID-19.6,22 This regimen is expected to become a standard therapeutic agent for the prevention and treatment of the entire clinical spectrum of EA-VMTD and perhaps antibody-associated (AA)-VMTD.
Therapeutic Plasma Exchange
TPE as a surrogate for ADAMTS13 has been the best option in the treatment of TTP and TTP-like syndrome with safety and good to excellent results if treated in early stage of EA-VMTD with an appropriate safeguard. It has shown promising result in case series, and clinical trials are in progress for COVID-19.37,129–134 Since ARDS is caused by microthrombosis and often advances to various organ phenotypes due to disseminated microthrombosis, TPE may become a valuable therapeutic regimen in most of patients with early stage of ARDS and other organ syndromes. The main drawback is that TPE may not be readily available in many patient care centers, and is very time-consuming and labor-intensive procedure. For this reason, TPE may not be therapeutic solution for the large number of COVID-19 patients in pandemic.
Complement Inhibitors
Since endotheliopathy in COVID-19 sepsis is promoted by complement-mediated damage to the endothelium of the host,9,15–17 which triggers microthrombogenesis,28 the complement inhibitor eculizumab has been suggested135 to use as an indirect antimicrothrombotic agent to suppress endotheliopathy. Complement activation inhibitor eculizumab is a long-acting monoclonal antibody approved for the treatment of paroxysmal nocturnal hemoglobinuria. This antibody targets complement component C5 and prevents its cleavage into C5a and C5b, thereby inhibiting the formation of C5b-9. Considering eculizumab downregulates C5b-9, it could inhibit on-going endothelial dysfunction and prevent both exocytosis of ULVWF and inflammation. In view of this theoretical ground6,22,135,136 and initial promising clinical experience from complement inhibition,137–139 several clinical trials are currently underway to evaluate anticomplement agents for the treatment of COVID-19. The complement inhibitors seem to have a potential role, but should be employed with special care in immunologically safer stage of sepsis140 since it might interfere with the innate immune system that protects the host from septicemia and undefined pneumonia.
Disulfide Bond Reducing Mucolytic Therapy
N-acetylcysteine (NAC) is N-acetyl derivative of the amino acid L-cysteine. It is a precursor in the formation of antioxidant glutathione in the body and is a disulfide bond reducing agent with mucolytic activity. The sulfhydryl group confers antioxidant effects and is able to reduce free radicals. This drug has been used in acetaminophen overdose and toxicity, cystic fibrosis and chronic obstructive lung disease, and symptomatic inhalation treatment as a mucolytic agent for thick mucus. Its oral form is over-the-counter medicine with very little side effects. It is found to possess potential antimicrothrombotic activity,141 inhibiting the ULVWF path of hemostasis.
The hemostatic function of human ULVWF depends on the normal assembly of disulfide-linked multimers from approximately 250-kDa subunits. Subunits initially form dimers through disulfide bonds near the COOH terminus. Dimers then form multimers through disulfide bonds closely at the NH2 terminus of each subunit.142 ULVWF multimers released from ECs are intrinsically active in binding platelets102,103 and are suspected the essential component promoting microthrombogensis in MODS of sepsis.22 ADAMTS13 exerts a disulfide bond reducing activity that primarily targets the bonds located in the domain of A2 in plasma ULVWF multimers.143 Therefore, it is theorized that rADAMTS13 inhibit microthrombogenesis in intravascular space by cleaving ULVWF in the patient with TTP and also proteolyze ULVWF bound to microthrombi strings anchored to the damaged endothelium in EA-VMTD.
Similar to ADAMTS13, NAC exerts analogous effect of proteolytic activity on ULVWF by reducing disulfide bonds. In addition to being involved in the antioxidant mechanism, NAC has disulfide breaking activity on the intracellular bond located in platelet binding A1 domain of ULVWF. It also inhibits VWF-dependent platelet aggregation and collagen binding.141 The same process may explain the mucolytic action of NAC which is due to its effect in reducing heavily cross-linked mucoproteins.144,145 A few clinical reports and animal models have suggested potential benefit of NAC in disseminated intravascular microthrombosis.146–148 NAC is inexpensive and readily available drug with a very high safety profile for oral use.
NAC has been investigated in ARDS as an antioxidant agent without the knowledge of underlying pathophysiological mechanism caused by molecular endothelial pathogenesis.149–152 The benefit as antioxidant agent was not consistently demonstrated in limited clinical trials, but in a meta-analysis some positive results had encouraged further study.153 In my opinion, first, the case number in the clinical trials was insufficient for statistical analysis. Second, the dosage of NAC might have been too low to draw a valid conclusion on efficacy. Third, inclusion of the patients on ventilator support with tracheal intubation might have influenced the outcome due to additional hemostatic complications such as PTE related to vascular injury and skewed its interpretation.154–156 These limitations could have precluded fair assessment of NAC response. In one limited, but controlled, study Suter et al149 concluded that NAC showed improvement of oxygenation but resulted in no beneficial effect on mortality of their study patients. However, a second look at the study suggests that NAC had a beneficial effect on ARDS. This benefit on oxygenation could be interpreted as positive effect on pulmonary vascular microthrombosis even though the study used a low dose at 40 mg/kg/day and that was treated for a short duration of 3 days.
Currently, with an evidence review, clinical therapeutic trials with NAC for the prevention and treatment for ARDS in COVID-19 are in progress.157 Clinical trials utilizing the targeted agent rADAMTS13 which are based on the “two-path unifying theory” of hemostasis and “two-activation theory of the endothelium” should be able to determine its potential benefit not only for COVID-19, but also for the entire spectrum of pathogen-induced sepsis, including ARDS and MODS and TTP-like syndrome as well as EA-VMTD. Theoretically and for practicality, NAC, if rADAMTS13 cannot be available for one reason or another for clinical trials, may become an acceptable therapeutic agent in pandemic. The treatment may be given parenterally to allow an effective dose comparable to that used in acetaminophen poisoning depending upon severity of EA-VMTD. If parenteral therapy of NAC is found to be effective, this therapeutic dosage may be estimated and calculated for an oral regimen.
Targeting Macrothrombosis in the Intensive Care Setting
In addition to ARDS caused by microthrombosis, macrothrombosis with clinical phenotypes of multiple DVT, PTE and cerebral venous sinus thrombosis, acute ischemic stroke and acute myocardial infarction is an important issue because it should be managed differently. Considering the former is from endotheliopathy and the latter is from localized vascular damage, rational management is as follows.
- Prevention by limiting in-hospital related vascular accesses as much as possible
- Thromboprophylaxis prior to significant vascular intervention
- Anticoagulation for developed macrothrombosis
For the prevention of macrothrombosis, a two-pronged approach should be considered; one is to limit the risk factors by minimizing the vascular damage from vascular accesses, procedures, endovascular devices and surgery, and prudent decision on ventilator support with practice guideline and surveillance; the other is a rational decision for a short term thromboprophylaxis when needed. For the treatment of developed macrothrombosis, therapeutic anticoagulation counteracting TF path of hemostasis such as low molecular weight heparin should be a standard treatment. Albeit anticoagulant therapy superimposed to antimicrothrombotic regimen is anticipated to be safe and effective, close monitoring for potential bleeding complication is warranted.
Conclusion
The COVID-19 pandemic is a 21st century challenge to humanity and civilization. With organized efforts of medical community worldwide, the pathophysiological mechanisms of the viral sepsis are being uncovered. The underlying pathogenesis is found to be generalized endotheliopathy triggered by complement activation and subsequent endothelial molecular dysfunction, leading to inflammatory syndrome and VMTD. The endothelial molecular pathogenesis has been unequivocally established based on hemostatic evaluation, and clinical and pathologic findings. Theoretically, targeted antimicrothrombotic regimen is expected to be effective against EA-VMTD and its complications, including TTP-like syndrome and MODS. Additional anticoagulant therapy would be needed if macrothrombosis due to the virus-unrelated events in hospital coincides with ARDS in COVID-19.
Abbreviations
AIS, acute ischemic stroke; ANP, acute necrotizing pancreatitis; ARDS, acute respiratory distress syndrome; aPTT, activated partial thromboplastin time; CVST, cerebral venous sinus thrombosis; DIC, disseminated intravascular coagulation; “DIC”, false disseminated intravascular coagulation; DIT, disseminated intravascular microthrombosis; DES, diffuse encephalopathic stroke; DVT, deep vein thrombosis; ECs, endothelial cells; EVT, extravascular tissue; HUS, hemolytic-uremic syndrome; FDP(s), fibrin degradation products; IL, interleukin; MAHA, microangiopathic hemolytic anemia; MERS, middle east respiratory syndrome; MODS, multi-organ dysfunction syndrome; MVMI, microvascular myocardial ischemia; NAC, N-acetylcysteine; NETs, neutrophil extracellular traps; PT, prothrombin time; PTE, pulmonary thromboembolism; SARS, severe acute respiratory syndrome; SET, subendothelial tissue; SFM, soluble fibrin monomer; TF, tissue factor; TPE, therapeutic plasma exchange; TTP, thrombotic thrombocytopenic purpura; ULVWF, unusually large von Willebrand factor multimers; VMTD, vascular microthrombotic disease; AA-VMTD, antibody-associated VMTD; EA-VMTD, endotheliopathy-associated VMTD; GA-VMTD, gene mutation associated VMTD.
Data Sharing Statement
Data sharing not applicable to this article as no datasets were generated or analyzed during the current study.
Acknowledgments
The author expresses sincere appreciation to Miss Emma Nichole Zebrowski for her drawing on the structure of the blood vessel wall in relation to hemostasis and the illustrative art works of Figures. Also, the author express thanks to Dr. H. Bradford Hawley, M.D. for his review of the manuscript and critical suggestions in infectious aspects of COVID-19 pandemic.
Funding
There is no support of funding in research, preparation and publication of this article.
Disclosure
The author reports no conflicts of interest in this work.
References
1. NIH. Information for Researchers: Coronaviruses. National Institute of Allergy and Infectious Disease. Available from: https://www.niaid.nih.gov/diseases-conditions/coronaviruses.
2. Morens DM, Breman JG, Calisher CH, et al. The Origin of COVID-19 and why it matters. Am J Trop Med Hyg. 2020;103:955–959.
3. Bolsen T, Palm R, Kingsland JT. Framing the origins of COVID-19. Sci Commun. 2020;1075547020953603.
4. Jih TK. Acute respiratory distress syndrome (ARDS) and severe acute respiratory syndrome (SARS): are we speaking different languages? J Chin Med Assoc. 2005;68:1–3.
5. Li X, Ma X. Acute respiratory failure in COVID-19: is it “typical” ARDS? Crit Care. 2020;24:198.
6. Chang JC. Acute respiratory distress syndrome as an organ phenotype of vascular microthrombotic disease: based on hemostatic theory and endothelial molecular pathogenesis. Clin Appl Thromb Hemost. 2019;25:1076029619887437.
7. Romagnoli S, Peris A, De Gaudio AR, et al. SARS-CoV-2 and COVID-19: from the bench to the bedside. Physiol Rev. 2020;100(4):1455–1466.
8. Felsenstein S, Herbert JA, McNamara PS, Hedrich CM. COVID-19: immunology and treatment options. Clin Immunol. 2020;215:108448.
9. Fletcher-Sandersjöö A, Bellander BM. Is COVID-19 associated thrombosis caused by overactivation of the complement cascade? A literature review. Thromb Res. 2020;194:36–41.
10. Lodigiani C, Iapichino G, Carenzo L, et al. Venous and arterial thromboembolic complications in COVID-19 patients admitted to an academic hospital in Milan, Italy. Thromb Res. 2020;191:9–14. doi:10.1016/j.thromres.2020.04.024
11. Bompard F, Monnier H, Saab I, et al. Pulmonary embolism in patients with COVID-19 pneumonia. Eur Respir J. 2020;56:2001365.
12. Schultz K, Wolf JM. Digital ischemia in COVID-19 patients: case report. J Hand Surg Am. 2020;45:518–522.
13. Kanitakis J, Lesort C, Danset M, et al. Chilblain-like acral lesions during the COVID-19 pandemic (“COVID toes”): histologic, immunofluorescence, and immunohistochemical study of 17 cases. J Am Acad Dermatol. 2020;83:870–875.
14. Pourdowlat G, Naderi Z, et al. Acrocyanosis and digital necrosis are associated with poor prognosis in COVID-19. Clin Case Rep. 2020;8:2769–2772.
15. de Nooijer AH, Grondman I, Janssen NAF, et al. Complement activation in the disease course of COVID-19 and its effects on clinical outcomes. J Infect Dis. 2021;223:214–224.
16. Magro C, Mulvey JJ, Berlin D, et al. Complement associated microvascular injury and thrombosis in the pathogenesis of severe COVID-19 infection: a report of five cases. Transl Res. 2020;220:1–13.
17. Cugno M, Meroni PL, Gualtierotti R, et al. Complement activation in patients with COVID-19: a novel therapeutic target. J Allergy Clin Immunol. 2020;146:215–217.
18. Ackermann M, Verleden SE, Kuehnel M, et al. Pulmonary vascular endothelialitis, thrombosis, and angiogenesis in Covid-19. N Engl J Med. 2020;383:120–128.
19. Goshua G, Pine AB, Meizlish ML, et al. Endotheliopathy in COVID-19-associated coagulopathy: evidence from a single-centre, cross-sectional study. Lancet Haematol. 2020;7:e575–e582.
20. Al-Samkari H, Karp Leaf RS, Dzik WH, et al. COVID-19 and coagulation: bleeding and thrombotic manifestations of SARS-CoV-2 infection. Blood. 2020;136:489–500.
21. Panigada M, Bottino N, Tagliabue P, et al. Hypercoagulability of COVID-19 patients in intensive care unit: a report of thromboelastography findings and other parameters of hemostasis. J Thromb Haemost. 2020;18:1738–1742.
22. Chang JC. Sepsis and septic shock: endothelial molecular pathogenesis associated with vascular microthrombotic disease. Thromb J. 2019;17:10.
23. Chang JC. Disseminated intravascular coagulation: new identity as endotheliopathy-associated vascular microthrombotic disease based on in vivo hemostasis and endothelial molecular pathogenesis. Thrombosis J. 2020;18:25.
24. Bernardo A, Ball C, Nolasco L, et al. Platelets adhered to endothelial cell-bound ultra-large von Willebrand factor strings support leukocyte tethering and rolling under high shear stress. J Thromb Haemost. 2005;3:562–570.
25. Ali A, Vijayan R. Dynamics of the ACE2-SARS-CoV-2/SARS-CoV spike protein interface reveal unique mechanisms. Sci Rep. 2020;10:14214.
26. Farkas I, Baranyi L, Ishikawa Y, et al. CD59 blocks not only the insertion of C9 into MAC but inhibits ion channel formation by homologous C5b-8 as well as C5b-9. J Physiol. 2002;539(Pt 2):537–545.
27. Kumar A, Narayan RK, Kumari C, et al. SARS-CoV-2 cell entry receptor ACE2 mediated endothelial dysfunction leads to vascular thrombosis in COVID-19 patients. Med Hypotheses. 2020;145:110320.
28. Chang JC. TTP-like syndrome: novel concept and molecular pathogenesis of endotheliopathy-associated vascular microthrombotic disease. Thromb J. 2018;16:20.
29. Capecchi M, Mocellin C, Abbruzzese C, Mancini I, Prati D, Peyvandi F. Dramatic presentation of acquired thombotic thrombocytopenic purpura associated with COVID-19. Haematologica. 2020;105:e540.
30. Hindilerden F, Yonal-Hindilerden I, Akar E, et al. Covid-19 associated autoimmune thrombotic thrombocytopenic purpura: report of a case. Thromb Res. 2020;195:136–138.
31. Jhaveri KD, Meir LR, Flores chang BS, et al. Thrombotic microangiopathy in a patient with COVID-19. Kidney Int. 2020;98:509–512.
32. Gavriilaki E, Brodsky RA. Severe COVID-19 infection and thrombotic microangiopathy: success does not come easily. Br J Haematol. 2020;189:e227–e30.
33. Lefrançais E, Ortiz-Muñoz G, Caudrillier A, et al. The lung is a site of platelet biogenesis and a reservoir for haematopoietic progenitors. Nature. 2017;544(7648):105–109.
34. Kroll MH, Afshar-Kharghan V. Platelets in pulmonary vascular physiology and pathology. Pulm Circ. 2012;2:291–308.
35. Rapkiewicz AV, Mai X, Carsons SE, et al. Megakaryocytes and platelet-fibrin thrombi characterize multi-organ thrombosis at autopsy in COVID-19: a case series. EClinicalMedicine. 2020;24:100434.
36. Chemla D, Castelain V, Provencher S, et al. Evaluation of various empirical formulas for estimating mean pulmonary artery pressure by using systolic pulmonary artery pressure in adults. Chest. 2009;135:760–768.
37. Kamran SM, Mirza ZE, Naseem A, et al. plasma exchange for coronavirus disease-2019 triggered cytokine release syndrome; a retrospective propensity matched control study. PLoS One. 2021;16:e0244853.
38. Zaim S, Chong JH, Sankaranarayanan V, et al. COVID-19 and multiorgan response. Curr Probl Cardiol. 2020;45:100618.
39. Chen T, Wu D, Chen H, et al. Clinical characteristics of 113 deceased patients with coronavirus disease 2019: retrospective study [published correction appears in BMJ. 2020 Mar 31;368:m1295]. BMJ. 2020;368:m1091.
40. Robba C, Battaglini D, Pelosi P, et al. Multiple organ dysfunction in SARS-CoV-2: MODS-CoV-2. Expert Rev Respir Med. 2020;14:865–868.
41. Wang L, Zhang Y, Zhang S. Cardiovascular impairment in COVID-19: learning from current options for cardiovascular anti-inflammatory therapy. Front Cardiovasc Med. 2020;7:78.
42. Elsoukkary SS, Mostyka M, Dillard A, et al. Autopsy findings in 32 patients with COVID-19: a single-institution experience. Pathobiology. 2021;88:56–68.
43. Mokhtari T, Hassani F, Ghaffari N, et al. COVID-19 and multiorgan failure: a narrative review on potential mechanisms. J Mol Histol. 2020;51:613–628.
44. Wang J, Jiang M, Chen X, et al. Cytokine storm and leukocyte changes in mild versus severe SARS-CoV-2 infection: review of 3939 COVID-19 patients in China and emerging pathogenesis and therapy concepts. J Leukoc Biol. 2020;108:17–41.
45. Simpson JM, Newburger JW. Multisystem inflammatory syndrome in children in association with COVID-19. Circulation. 2020;142:437–440.
46. Ortega-Paz L, Capodanno D, Montalescot G, et al. Coronavirus disease 2019-associated thrombosis and coagulopathy: review of the pathophysiological characteristics and implications for antithrombotic management. J Am Heart Assoc. 2021;10:e019650.
47. Mondal S, Quintili AL, Karamchandani K, et al. Thromboembolic disease in COVID-19 patients: a brief narrative review. J Intensive Care. 2020;8:70.
48. Hajra A, Mathai SV, Ball S, et al. Management of thrombotic complications in COVID-19: an update. Drugs. 2020;80:1553–1562.
49. Zhang R, Ni L, Di X, et al. Systematic review and meta-analysis of the prevalence of venous thromboembolic events in novel coronavirus disease-2019 patients. J Vasc Surg Venous Lymphat Disord. 2021;9:289–298.e5).
50. Boonyawat K, Chantrathammachart P, Numthavaj P, et al. Incidence of thromboembolism in patients with COVID-19: a systematic review and meta-analysis. Thrombosis J. 2020;18:34.
51. Merrill JT, Erkan D, Winakur J, et al. Emerging evidence of a COVID-19 thrombotic syndrome has treatment implications. Nat Rev Rheumatol. 2020;16:581–589.
52. Wichmann D, Sperhake JP, Lütgehetmann M, et al. Autopsy findings and venous thromboembolism in patients with COVID-19: a prospective cohort study. Ann Intern Med. 2020;173:268–277.
53. Lax SF, Skok K, Zechner P, et al. Pulmonary arterial thrombosis in COVID-19 with fatal outcome: results from a prospective, single-center, clinicopathologic case series. Ann Intern Med. 2020;173:350–361.
54. Bois MC, Boire NA, Layman AJ, et al. COVID-19-associated nonocclusive fibrin microthrombi in the heart. Circulation. 2021;143:230–243.
55. Whyte CS, Morrow GB, Mitchell JL, Chowdary P, Mutch NJ. Fibrinolytic abnormalities in acute respiratory distress syndrome (ARDS) and versatility of thrombolytic drugs to treat COVID-19. J Thromb Haemost. 2020;18:1548–1555.
56. Chang JC. Stroke classification: critical role of unusually large von willebrand factor multimers and tissue factor on clinical phenotypes based on novel “two-path unifying theory” of hemostasis. Clin Appl Thromb Hemost. 2020;26:1076029620913634.
57. Chang JC. Thrombogenesis and thrombotic disorders based on ‘two-path unifying theory of hemostasis’: philosophical, physiological, and phenotypical interpretation. Blood Coagul Fibrinolysis. 2018;29:585–595.
58. Minet C, Potton L, Bonadona A, et al. Venous thromboembolism in the ICU: main characteristics, diagnosis and thromboprophylaxis. Crit Care. 2015;19:287.
59. Marietta M, Coluccio V, Luppi M. COVID-19, coagulopathy and venous thromboembolism: more questions than answers. Intern Emerg Med. 2020;15:1375–1387.
60. Zhang C, Zhang Z, Mi J, et al. The cumulative venous thromboembolism incidence and risk factors in intensive care patients receiving the guideline-recommended thromboprophylaxis. Medicine (Baltimore). 2019;98:e15833.
61. Geerts W, Selby R. Prevention of venous thromboembolism in the ICU. Chest. 2003;124(6 Suppl):357S–363S.
62. Hamada SR, Espina C, Guedj T, et al. High level of venous thromboembolism in critically ill trauma patients despite early and well-driven thromboprophylaxis protocol. Ann Intensive Care. 2017;7:97.
63. Borczuk AC, Salvatore SP, Seshan SV, et al. COVID-19 pulmonary pathology: a multi-institutional autopsy cohort from Italy and New York City. Mod Pathol. 2020;33:2156–2168.
64. Carsana L, Sonzogni A, Nasr A, et al. Pulmonary post-mortem findings in a series of COVID-19 cases from northern Italy: a two-centre descriptive study. Lancet Infect Dis. 2020;20:1135–1140.
65. Valdivia-Mazeyra MF, Salas C, Nieves-Alonso JM, et al. Increased number of pulmonary megakaryocytes in COVID-19 patients with diffuse alveolar damage: an autopsy study with clinical correlation and review of the literature. Virchows Arch. 2020;1–10.
66. Thachil J, Lisman T. Pulmonary Megakaryocytes in Coronavirus Disease 2019 (COVID-19): roles in thrombi and fibrosis. Semin Thromb Hemost. 2020;46:831–834.
67. Palumbo D, Guazzarotti G, De Cobelli F. Spontaneous major hemorrhage in COVID-19 patients: another brick in the wall of SARS-CoV-2-associated coagulation disorders? J Vasc Interv Radiol. 2020;31:1494–1496.
68. Hardy M, Lecompte T, Douxfils J, et al. Management of the thrombotic risk associated with COVID-19: guidance for the hemostasis laboratory. Thrombosis J. 2020;18:17.
69. Tsutsumi T, Saito M, Nagai H, et al. Association of coagulopathy with liver dysfunction in patients with COVID-19. Hepatol Res. 2021;51:227–232.
70. Li T, Lu H, Zhang W. Clinical observation and management of COVID-19 patients. Emerg Microbes Infect. 2020;9:687–690.
71. Qian SZ, Pan JY. COVID-19 with limb ischemic necrosis. J Cardiothorac Vascul Anesth COVID-19. 2020;34:2846–2847.
72. Suarez-Valle A, Fernandez-Nieto D, Diaz-Guimaraens B, et al. Acro-ischaemia in hospitalized COVID-19 patients. J Eur Acad Dermatol Venereol. 2020;34:e455–e457.
73. Khan IA, Karmakar S, Chakraborty U, et al. Purpura fulminans as the presenting manifestation of COVID-19. Postgrad Med J. 2021.
74. Marzano AV, Cassano N, Genovese G, et al. Cutaneous manifestations in patients with COVID-19: a preliminary review of an emerging issue. Br J Dermatol. 2020;183:431–442.
75. Novara E, Molinaro E, Benedetti I, et al. Severe acute dried gangrene in COVID-19 infection: a case report. Eur Rev Med Pharmacol Sci. 2020;24(10):5769–5771.
76. Del Giudice P, Boudoumi D, Le Guen B, et al. Catastrophic acute bilateral lower limbs necrosis associated with COVID-19 as a likely consequence of both vasculitis and coagulopathy. J Eur Acad Dermatol Venereol. 2020;34(11):e679–e680.
77. Chang JC, Ikhlaque N. Peripheral digit ischemic syndrome can be a manifestation of postoperative thrombotic thrombocytopenic purpura. Ther Apher Dial. 2004;8:413–418.
78. Warkentin TE. Microvascular thrombosis and ischaemic limb losses in critically Ill patients. Hamostaseologie. 2019;39:6–19.
79. Shimbo K, Yokota K, Miyamoto J, et al. Symmetrical peripheral gangrene caused by septic shock. Case Rep Plast Surg Hand Surg. 2015;2:53–56.
80. Davis MD, Dy KM, Nelson S. Presentation and outcome of purpura fulminans associated with peripheral gangrene in 12 patients at Mayo Clinic. J Am Acad Dermatol. 2007;57:944–956.
81. Kox M, Waalders NJB, Kooistra EJ, et al. Cytokine levels in critically ill patients with COVID-19 and other conditions. JAMA. 2020;324:1565–1567.
82. Martinelli N, Montagnana M, Pizzolo F, et al. A relative ADAMTS13 deficiency supports the presence of a secondary microangiopathy in COVID 19. Thrombo Res. 2020;193:170–172.
83. Mancini I, Baronciani L, Artoni A, et al. The ADAMTS13-von Willebrand factor axis in COVID-19 patients. J Thromb Haemost. 2021;19:513–521.
84. Fogarty H, Townsend L, Cheallaigh CN, et al. COVID-19 coagulopathy in Caucasian patients. Br J Haematol. 2020;189:1044–1049.
85. McGonagle D, O’Donnell JS, Sharif K, et al. Immune mechanisms of pulmonary intravascular coagulopathy in COVID-19 pneumonia. Lancet Rheumatol. 2020;2:e437–45.
86. Zhang Y, Xiao M, Zhang S, et al. Coagulopathy and antiphospholipid antibodies in patients with Covid-19. N Engl J Med. 2020;382:e38.
87. Marchandot B, Sattler L, Jesel L, et al. COVID-19 related coagulopathy: a distinct entity? J Clin Med. 2020;9:E1651.
88. Becker RC. COVID-19 update: covid-19-associated coagulopathy. J Thromb Thrombolysis. 2020;50:54–67.
89. Levi M, Thachil J, Iba T, et al. Coagulation abnormalities and thrombosis in patients with COVID-19. Lancet Haematol. 2020;7:e438–e440.
90. Escher R, Breakey N, Lämmle B. Severe COVID-19 infection associated with endothelial activation. Thromb Res. 2020;190:62.
91. Sridharan M, Ashrani A, Chen D. et al. Evaluation of soluble fibrin monomer complex in patients with Sars-Cov-2 COVID-19. Infection Blood. 2020;136(Supplement 1):27–28.
92. Ladikou EE, Sivaloganathan H, Milne KM, et al. Von Willebrand factor (vWF): marker of endothelial damage and thrombotic risk in COVID-19? Clin Med (Lond). 2020;20:e178–e182.
93. Joly BS, Siguret V, Veyradier A. Understanding pathophysiology of hemostasis disorders in critically ill patients with COVID-19. Intensive Care Med. 2020;46:1603–1606.
94. Cipolloni L, Sessa F, Bertozzi G, et al. Preliminary Post-Mortem COVID-19 evidence of endothelial injury and factor VIII hyperexpression. Diagnostics (Basel). 2020;10:575.
95. Rauch A, Labreuche J, Lassalle F, et al. Coagulation biomarkers are independent predictors of increased oxygen requirements in COVID-19. J Thromb Haemost. 2020;18:2942–2953.
96. Huisman A, Beun R, Sikma M, et al. Involvement of ADAMTS13 and von Willebrand factor in thromboembolic events in patients infected with SARS-CoV-2. Int J Lab Hematol. 2020;42:e211–e212.
97. Bazzan M, Montaruli B, Sciascia S, et al. Low ADAMTS 13 plasma levels are predictors of mortality in COVID-19 patients. Intern Emerg Med. 2020;15:861–863.
98. Madeeva DV, Christian J, Goshua G, et al. VWF/ADAMTS13 ratios are potential markers of immunothrombotic complications in patients with COVID-19: a cross-sectional study. Blood. 2020;136(Supplement 1):34–35.
99. Chang JC. Hemostasis based on a novel ‘two-path unifying theory’ and classification of hemostatic disorders. Blood Coagul Fibrinolysis. 2018;29:573–584.
100. Turner NA, Moake JL. Factor VIII is synthesized in human endothelial cells, packaged in Weibel-Palade bodies and secreted bound to ULVWF strings. PLoS One. 2015;10:e0140740.
101. Tsai HM. Pathophysiology of thrombotic thrombocytopenic purpura. Int J Hematol. 2010;91(1):1–19.
102. De Ceunynck K, De Meyer SF, Vanhoorelbeke K. Unwinding the von Willebrand factor strings puzzle. Blood. 2013;121:270–277.
103. Mourik MJ, Valentijn JA, Voorberg J, et al. von Willebrand factor remodeling during exocytosis from vascular endothelial cells. J Thromb Haemost. 2013;11:2009–2019.
104. Huang J, Roth R, Heuser JE, et al. Integrin αvβ3 on human endothelialcells binds von Willebrand factor strings under fluid shear stress. Blood. 2009;113:1589–1597.
105. Chauhan AK, Goerge T, Schneider SW, et al. Formation of platelet strings and microthrombi in the presence of ADAMTS-13 inhibitor does not require P-selectin or beta3 integrin. J Thromb Haemost. 2007;5:583–589.
106. Chen X, Cheng X, Zhang S, et al. ADAMTS13: an emerging target in stroke therapy. Front Neurol. 2019;10:772.
107. Sonneveld MA, de Maat MP, Portegies ML, et al. Low ADAMTS13 activity is associated with an increased risk of ischemic stroke. Blood. 2015;126:2739–2746.
108. Denorme F, Kraft P, Pareyn I, et al. Reduced ADAMTS13 levels in patients with acute and chronic cerebrovascular disease. PLoS One. 2017;12:e0179258.
109. Eerenberg ES, Teunissen PF, van den Born BJ, et al. The role of ADAMTS13 in acute myocardial infarction: cause or consequence? Cardiovasc Res. 2016;111:194–203.
110. Horii M, Uemura S, Uemura M, et al. Acute myocardial infarction as a systemic prothrombotic condition evidenced by increased von Willebrand factor protein over ADAMTS13 activity in coronary and systemic circulation. Heart Vessels. 2008;23:301–307.
111. Pagliari MT, Boscarino M, Cairo A, et al. ADAMTS13 activity, high VWF and FVIII levels in the pathogenesis of deep vein thrombosis. Thromb Res. 2021;197:132–137.
112. Zhao J, Yang Y, Huang H, et al. Relationship between the ABO blood group and the coronavirus disease 2019 (COVID-19) Susceptibility. Clin Infect Dis. 2020;ciaa1150.
113. Wu Y, Feng Z, Li P, et al. Relationship between ABO blood group distribution and clinical characteristics in patients with COVID-19. Clin Chim Acta. 2020;509:220–223.
114. Li J, Wang X, Chen J, et al. Association between ABO blood groups and risk of SARS‐CoV‐2 pneumonia. Br J Haematol. 2020;2020(190):24–27.
115. Gaffney PJ. Distinction between fibrinogen and fibrin degradation products in plasma. Clin Chim Acta. 1975;65:109–115.
116. Shorr AF, Thomas SJ, Alkins SA, et al. D-dimer correlates with proinflammatory cytokine levels and outcomes in critically ill patients. Chest. 2002;121:1262–1268.
117. Goebel PJ, Williams JB, Gerhardt RT. A pilot study of the performance characteristics of the D-dimer in presumed sepsis. West J Emerg Med. 2010;11:173–179.
118. Miesbach W, Makris M. COVID-19: coagulopathy, risk of thrombosis, and the rationale for anticoagulation. Clin Appl Thromb Hemost. 2020;26:1076029620938149.
119. Maldonado E, Tao D, Mackey K. Antithrombotic therapies in COVID-19 disease: a systematic review. J Gen Intern Med. 2020;17:1–9.
120. Wang J, Hajizadeh N, Moore EE, et al. Tissue plasminogen activator (tPA) treatment for COVID-19 associated acute respiratory distress syndrome (ARDS): a case series. J Thromb Haemost. 2020;18:1752–1755.
121. Salama C, Han J, Yau L, et al. Tocilizumab in patients hospitalized with Covid-19 pneumonia. N Engl J Med. 2021;384:20–30.
122. Stone JH, Frigault MJ, Serling-Boyd NJ, et al. Efficacy of tocilizumab in patients hospitalized with Covid-19. N Engl J Med. 2020;383:2333–2344.
123. FitzGerald GA. Misguided drug advice for COVID-19. Science. 2020;367(6485):1434.
124. Rizk JG, Kalantar-Zadeh K, Mehra M, et al. Pharmaco-immunomodulatory therapy in COVID-19. Drugs. 2020;80:1267–1292.
125. Schiviz A, Wuersch K, Piskernik C, et al. A new mouse model mimicking thrombotic thrombocytopenic purpura: correction of symptoms by recombinant human ADAMTS13. Blood. 2012;119:6128–6135.
126. Azfar MF, Khan MF, Habib SS, et al. Prognostic value of ADAMTS13 in patients with severe sepsis and septic shock. Clin Invest Med. 2017;40:E49–E58.
127. Levi M, Scully M, Singer M. The role of ADAMTS-13 in the coagulopathy of sepsis. J Thromb Haemost. 2018;16:646–651.
128. Peetermans M, Meyers S, Liesenborghs L, et al. Von Willebrand factor and ADAMTS13 impact on the outcome of Staphylococcus aureus sepsis. J Thromb Haemost. 2020;18:722–731.
129. Khamis F, Al-Zakwani I, Al Hashmi S, et al. Therapeutic plasma exchange in adults with severe COVID-19 infection. Int J Infect Dis. 2020;99:214–218.
130. Dogan L, Kaya D, Sarikaya T, et al. Plasmapheresis treatment in COVID-19-related autoimmune meningoencephalitis: case series. Brain Behav Immun. 2020;87:155–158.
131. Faqihi F, Alharthy, A, Alodat, Met al. A pilot study of therapeutic plasma exchange for serious SARS CoV-2 disease (COVID-19): a structured summary of a randomized controlled trial study protocol. Trials. 2020;21:506.
132. Keith P, Day M, Choe C, et al. The successful use of therapeutic plasma exchange for severe COVID-19 acute respiratory distress syndrome with multiple organ failure. SAGE Open Med Case Rep. 2020;8:2050313X20933473.
133. Shi H, Zhou C, He P, et al. Successful treatment with plasma exchange followed by intravenous immunoglobulin in a critically ill patient with COVID-19. Int J Antimicrob Agents. 2020;56:105974.
134. Adeli SH, Asghari A, Tabarraii R, et al. Therapeutic plasma exchange as a rescue therapy in patients with coronavirus disease 2019: a case series. Pol Arch Intern Med. 2020;130:455–458.
135. Valga F, Vega- Díaz N, Macia M, et al. Targeting complement in severe Coronavirus disease 2019 to address microthrombosis. Clin Kidney J. 2020;13:477–479.
136. Stahel PF, Barnum SR. Complement inhibition in coronavirus disease (COVID)-19: a neglected therapeutic option. Front Immunol. 2020;11:1661.
137. Annane D, Heming N, Grimaldi-Bensouda L, et al. Eculizumab as an emergency treatment for adult patients with severe COVID-19 in the intensive care unit: a proof-of-concept study. EClinicalMedicine. 2020;28:100590.
138. Diurno F, Numis FG, Porta G, et al. Eculizumab treatment in patients with COVID-19: preliminary results from real life ASL Napoli 2 Nord experience. Eur Rev Med Pharmacol Sci. 2020;24:
139. Mastaglio S, Ruggeri A, Risitano AM, et al. The first case of COVID-19 treated with the complement C3 inhibitor AMY-101. Clin Immunol. 2020;215:108450.
140. Java A, Apicelli AJ, Liszewski MK, et al. The complement system in COVID-19: friend and foe? JCI Insight. 2020;5:e140711.
141. Chen J, Reheman A, Gushiken FC, et al. N-acetylcysteine reduces the size and activity of von Willebrand factor in human plasma and mice. J Clin Invest. 2011;121:593–603.
142. Dong Z, Thoma RS, Crimmins DL, et al. Disulfide bonds required to assemble functional von Willebrand factor multimers. J Biol Chem. 1994;269:6753–6758.
143. Yeh HC, Zhou Z, Choi H, et al. Disulfide bond reduction of von Willebrand factor by ADAMTS-13. J Thromb Haemost. 2010;8:2778–2788.
144. Aldini G, Altomare A, Baron G, et al. N-acetylcysteine as an antioxidant and disulphide breaking agent: the reasons why. Free Radic Res. 2018;52:751–762.
145. Samuni Y, Goldstein S, Dean OM, et al. The chemistry and biological activities of N-acetylcysteine. Biochim Biophys Acta. 2013;1830:4117–4129.
146. Cabanillas G, Popescu-Martinez A. N-acetylcysteine for relapsing thrombotic thrombocytopenic purpura: more evidence of a promising drug. Am J Ther. 2016;23:e1277–1279.
147. Rottenstreich A, Hochberg-Klein S, Rund D, et al. The role of N-acetylcysteine in the treatment of thrombotic thrombocytopenic purpura. J Thromb Thrombolysis. 2016;41:678–683.
148. Tersteeg C, Roodt J, Van Rensburg WJ, et al. N-acetylcysteine in preclinical mouse and baboon models of thrombotic thrombocytopenic purpura. Blood. 2017;129:1030–1038.
149. Suter PM, Domenighetti G, Schaller MD, et al. N-acetylcysteine enhances recovery from acute lung injury in man. A randomized, double-blind, placebo-controlled clinical study. Chest. 1994;105:190–194.
150. Domenighetti G, Suter PM, Schaller MD, et al. Treatment with N-acetylcysteine during acute respiratory distress syndrome: a randomized, double-blind, placebo-controlled clinical study. J Crit Care. 1997;12:177–182.
151. Bernard GR, Wheeler AP, Arons MM, et al. A trial of antioxidants N-acetylcysteine and procysteine in ARDS. The Antioxidant in ARDS Study Group. Chest. 1997;112:164–172.
152. Moradi M, Mojtahedzadeh M, Mandegari A, et al. The role of glutathione-S-transferase polymorphisms on clinical outcome of ALI/ARDS patient treated with N-acetylcysteine. Respir Med. 2009;103:434–441.
153. Zhang Y, Ding S, Li C, et al. Effects of N-acetylcysteine treatment in acute respiratory distress syndrome: a meta-analysis. Exp Ther Med. 2017;14:2863–2868.
154. Divatia JV, Khan PU, Myatra SN. Tracheal intubation in the ICU: life-saving or life threatening? Indian J Anaesth. 2011;55:470–475.
155. Spieth PM, de Abreu MG. Pulmonary embolism in mechanically ventilated patients: what the eye doesn’t see, the heart can still grieve over. Crit Care Med. 2012;40:3320–3321.
156. Guglielmetti G, Quaglia M, Sainaghi PP, et al. “War to the knife” against thromboinflammation to protect endothelial function of COVID-19 patients. Crit Care. 2020;24:365.
157. Shi Z, Puyo CA. N-Acetylcysteine to Combat COVID-19: an evidence review. Ther Clin Risk Manag. 2020;16:1047–1055.
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.