")
Back to Journals » International Journal of Chronic Obstructive Pulmonary Disease » Volume 14
Copper-Heparin Inhalation Therapy To Repair Emphysema: A Scientific Rationale
Authors Janssen R , Wouters EFM, Janssens W , Daamen WF , Hagedoorn P, de Wit HAJM, Serré J , Gayan-Ramirez G, Franssen FME , Reynaert NL, von der Thüsen JH , Frijlink HW
Received 24 August 2019
Accepted for publication 31 October 2019
Published 25 November 2019 Volume 2019:14 Pages 2587—2602
DOI https://doi.org/10.2147/COPD.S228411
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Richard Russell
Rob Janssen,1 Emiel FM Wouters,2 Wim Janssens,3 Willeke F Daamen,4 Paul Hagedoorn,5 Hugo AJM de Wit,6 Jef Serré,3 Ghislaine Gayan-Ramirez,3 Frits ME Franssen,2 Niki L Reynaert,2 Jan H von der Thüsen,7 Henderik W Frijlink5
1Department of Pulmonary Medicine, Canisius-Wilhelmina Hospital, Nijmegen, The Netherlands; 2Department of Respiratory Medicine, Maastricht University Medical Center+, Maastricht, The Netherlands; 3Laboratory of Respiratory Diseases, Department of Chronic Diseases, Metabolism and Ageing (CHROMETA), KU Leuven, Leuven, Belgium; 4Department of Biochemistry, Radboud Institute for Molecular Life Sciences, Radboud University Medical Center, Nijmegen, The Netherlands; 5Department of Pharmaceutical Technology and Biopharmacy, Groningen Research Institute of Pharmacy, Groningen, University of Groningen, Groningen, The Netherlands; 6Department of Clinical Pharmacy, Canisius-Wilhelmina Hospital, Nijmegen, The Netherlands; 7Department of Pathology, Erasmus Medical Center, Rotterdam, The Netherlands
Correspondence: Rob Janssen
Department of Pulmonary Medicine, Canisius-Wilhelmina Hospital, Weg Door Jonkerbos 100, Nijmegen, SZ 6532, The Netherlands
Email [email protected]
Abstract: Current pharmacotherapy of chronic obstructive pulmonary disease (COPD) aims at reducing respiratory symptoms and exacerbation frequency. Effective therapies to reduce disease progression, however, are still lacking. Furthermore, COPD medications showed less favorable effects in emphysema than in other COPD phenotypes. Elastin fibers are reduced and disrupted, whereas collagen levels are increased in emphysematous lungs. Protease/antiprotease imbalance has historically been regarded as the sole cause of emphysema. However, it is nowadays appreciated that emphysema may also be provoked by perturbations in the sequential repair steps following elastolysis. Essentiality of fibulin-5 and lysyl oxidase-like 1 in the elastin restoration process is discussed, and it is argued that copper deficiency is a plausible reason for failing elastin repair in emphysema patients. Since copper-dependent lysyl oxidases crosslink elastin as well as collagen fibers, copper supplementation stimulates accumulation of both proteins in the extracellular matrix. Restoration of abnormal elastin fibers in emphysematous lungs is favorable, whereas stimulating pulmonary fibrosis formation by further increasing collagen concentrations and organization is detrimental. Heparin inhibits collagen crosslinking while stimulating elastin repair and might therefore be the ideal companion of copper for emphysema patients. Efficacy and safety considerations may lead to a preference of pulmonary administration of copper-heparin over systemic administration.
Keywords: collagen, copper, desmosine, elastin, emphysema, fibulin-5, heparin, lysyl oxidase, lysyl oxidase-like protein 1, tropoelastin
Introduction
Emphysema is defined as dilatation and/or destruction of airspaces distal to terminal bronchioles.1 Chronic obstructive pulmonary disease (COPD), on the other hand, is a condition characterized by persistent respiratory symptoms in combination with airflow limitation at spirometry.2 Although emphysema is considered to be a subtype of COPD,2 emphysematous lesions on computed tomography (CT) may also be detected in nonobstructive subjects.3
Inhaled bronchodilators, whether or not combined with corticosteroids, form the mainstay of pharmacotherapy for COPD.2 These drugs are primarily intended to relieve symptoms and reduce exacerbation frequency with little if any effect on attenuation of disease progression.2 Furthermore, current therapy seems to be less effective in patients with emphysema than in those with other COPD phenotypes.4,5 The objective of this manuscript is to provide a theoretical rationale for a potentially novel disease-modifying therapy for emphysema patients to decelerate disease progression and reverse structural changes.
Given the low perfusion/ventilation ratio of the usually most damaged upper lung zones,6 inhalation therapy may be preferred over systemic routes of drug administration to optimize delivery to emphysematous areas. Another advantage of local therapy is that it requires lower doses of medicine, which reduces the risk of systemic side-effects.
Causes Of Emphysema
A long time after the discovery that genetic deficiency of the antiprotease α-1 antitrypsin has the ability to induce premature emphysema, the protease/antiprotease imbalance remains the prevailing paradigm as the monocausal explanation for all forms of emphysema. Chronic inhalation of tobacco smoke is the most prevalent risk factor of emphysema, which may negatively modulate the aforementioned equilibrium by both upregulating protease and downregulating antiprotease activity.7,8 Various lines of evidence suggest that disturbances of repair mechanisms in the lungs may also be causes of emphysema.9–12
Elastin And Collagen
The pulmonary parenchyma is that portion of the lungs involved in gas transfer and consists of alveoli, respiratory bronchioles and capillaries. Lung parenchymal cells are encompassed by a three-dimensional network of extracellular matrix. Fibrillar collagens (type I, II, III, V and XI) and elastin are the most abundant proteins in the pulmonary extracellular matrix.13,14 Elastin has unique elastic properties,14 facilitating deformability of the pulmonary compartment during the breathing cycle, and collagen provides lungs with tensile strength.13 Elastin fibers are abnormal in pulmonary parenchyma of patients with emphysema due to a combination of proteolytic degradation and inefficacious repair. Most studies found decreased absolute elastin concentrations in emphysematous lungs.15–18 Some early reports did not find such a reduction;19,20 however, validity of their methods to quantify elastin has been questioned.17 Collagen levels are increased in emphysematous alveolar walls of patients with moderate COPD, which is further enhanced in those with very severe airflow limitation.15 However, the organization/maturation level of collagen type I is reduced in both airways and parenchyma of patients with COPD.21–23
Emphysematous remodeling of the extracellular matrix may have detrimental physiological consequences, i.e., airflow limitation as well as static and dynamic hyperinflation, leading to shortness of breath and exercise limitation.6 Elastin content in alveolar walls positively relates to spirometric values,16 whereas the reverse is true with regard to alveolar collagen.15
Modulatory treatments for patients with emphysema should suppress collagen accumulation and stimulate repair of disrupted elastin fibers. Development of completely new elastin fibers may not always be advantageous. This is illustrated by pulmonary elastin levels that are increased in patients with idiopathic pulmonary fibrosis (IPF) but also inversely associated with both lung function tests and survival.24
Elastogenesis And Elastin Repair
Elastic fiber formation in the lungs is usually restricted to early life.25 The largest and main part of elastic fibers consists of elastin, which organizes itself into a network of crosslinked polymers through a series of intricate sequential steps (Figure 1).14 In short, (a) elastin’s precursor tropoelastin is produced within mesenchymal cells and secreted into the extracellular matrix, (b) tropoelastin proteins then undergo a process of spontaneous self-aggregation (i.e., coacervation), (c) tropoelastin polymers are subsequently attached to a pre-deposited scaffold of microfibrils and (d) are finally crosslinked with other tropoelastin polymers into mature elastic fibers.
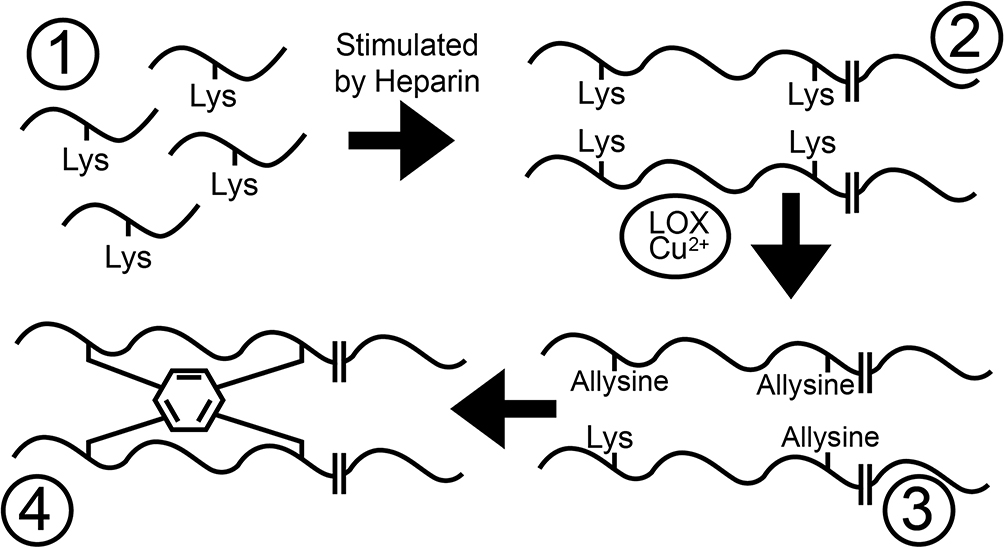 |
Figure 1 Tropoelastin monomers (1) aggregate into polymers (2). Lysine is transformed by copper-dependent lysyl oxidase (LOX-Cu2+) into highly reactive allysine (3). Three allysines and one untransformed lysine on adjacent fibers spontaneously condense into one (iso)desmosine amino acid (depicted as a hexagon; 4). |
Although there are some important differences, repair steps in response to proteolytic elastolysis are rather similar to those of initial elastogenesis.10,11,26,27
Lysyl Oxidase And Lysyl Oxidase-Like Proteins
Prototype lysyl oxidase (LOX) and LOX-like (LOXL) proteins 1 to 4 form a superfamily of metalloenzymes produced by fibroblasts.28 They catalyze the process of elastin crosslinking by oxidizing lysine amino acids into highly reactive allysine residues (Figure 1). Subsequently, three allysines and one untransformed lysine on adjacent tropoelastin polymers spontaneously react into stable end-products, e.g., desmosine and isodesmosine, that are exclusively present in elastic fibers.29
LOX enzymes are also involved in the crosslinking process of collagen, which gives rise to various crosslinks that can also be found in elastic fibers.30
Fibulins
Fibulins form a seven-membered group of secreted proteins that facilitate interactions between extracellular matrix proteins and components of basal membranes.31 Fibulin-4 and fibulin-5, in particular, play nonredundant roles in multiple steps of elastic tissue formation. Although their actions seem to be rather comparable, fibulin-4 and fibulin-5 are unable to replace or compensate each other.10 The preferential binding between prototype LOX and fibulin-4 is crucial for elastin crosslinking in the initial phase of elastogenesis,26,27 whereas LOXL1/fibulin-5 interactions have similar pivotal functions during elastin restoration later in life.10,11
Convergence And Divergence Between Fibulin-5 And Lysyl Oxidase-Like Protein 1 During Elastin Repair
Protease activity does not lead to permanent loss of pulmonary elastic fibers and emphysema formation as long as damage is instantly and properly repaired.32 Numerous genes coding for proteins involved in elastin restoration processes, such as elastin and fibulin-5, are among the strongest upregulated loci in lungs of patients with COPD.33,34 Intratracheal administration of elastases produces an initial rapid formation of emphysema, followed by a phase of elastin repair in which pulmonary expression of fibulin-5 and elastin is enhanced.35,36 These studies suggest that both humans and animals try to repair damaged elastin fibers after proteolytic insults in an attempt to prevent irreversible emphysematous destruction.33–36
The relevance of fibulin-5 in the pathogenesis of emphysema, through its role in the elastin repair process, has been illustrated in various studies.10,35 For instance, fibulin-5 knock-out mice survive to adulthood but not without developing severe lung emphysema.10 Furthermore, individuals with loss of function mutations in the fibulin-5 gene develop cutis laxa,37 a disease that is characterized by inelastic skin and with emphysema as a frequent manifestation.38
Successful elastin restoration also depends on proper function of LOX enzymes and particularly of LOXL1. Administration of a pan-LOX inhibitor has been shown to exacerbate elastase-induced emphysema formation,9 and mice in which LOXL1 was knocked-out developed a comparable emphysematous phenotype as when switching off fibulin-5.10,11 These data underscore the importance of LOXL1 in emphysema formation. However, in contrast to fibulin-5,33 LOXL1 activity is not upregulated but downregulated in patients with emphysema.39 The reason for this difference may well lay in the fact that LOXL1, unlike fibulin-5, depends on a cofactor, i.e., copper.40 Whereas synthesis of repair proteins can be autonomically upregulated following proteolytic insults, tissues are dependent on exogenous supply to provide them with extra copper.
Animal Models Of Copper Deficiency-Induced Emphysema
Low pulmonary copper levels have been acknowledged for more than forty years to possess emphysema-inducing potency in animals.41–45 Copper deficiency may lead to both developmental41,43 and acquired emphysema.44
Protease levels are not significantly elevated in models of copper deficiency;44 however, histologic appearance of emphysematous lesions in copper-deficient animals is similar to those provoked by intratracheal administration of elastases.42,46 These findings illustrate that emphysema in copper deficiency is mainly caused by failing elastogenesis and/or elastin repair mechanisms and not because of protease/antiprotease imbalance.
Emphysema Induced By Copper-Deficient Diets In Rodents
Pregnant rats and hamsters were separated into groups that were made copper-deficient through dietary means and control arms that received regular nutrition.41,43 The mothers’ diet was also fed to newborns after weaning.
Postmortem examination revealed that extensive bullous emphysematous changes had developed in lungs of copper-deficient rodents.41,43 Alveolar internal surface area was reduced as a reflection of damage to alveoli, and airspaces in the remaining fairly normal-appearing lung areas were enlarged. Pressure–volume curves of copper-deficient animals shifted upwards and leftwards due to, respectively, lung hyperinflation and increased compliance.
Changes In Pulmonary Extracellular Matrix Of Copper-Deficient Rats
Elastin concentration in the lungs was significantly reduced by 17%, whereas collagen levels remained stable.41
After a period of being fed a copper-deficient diet, some 5–10 weeks old rats were repleted with copper to assess reversibility of emphysematous lesions. Pulmonary elastin density and ultrastructure returned to near normal. However, no effect of repletion was found on alveolar size. This suggests that copper-induced developmental emphysema is partially but not fully reversible.
Changes In Pulmonary Extracellular Matrix Of Copper-Deficient Hamsters
Pulmonary (iso)desmosine levels were slightly, though significantly, increased in copper-deficient compared to copper-sufficient hamsters.43 This finding is not only opposite to what would have been anticipated but is also contradictory to the decreased desmosine levels that were found in copper-deficient chicks.47 A reason for this lack of decreased elastin-crosslinks in experimental hamsters could have a methodological basis, since (iso)desmosine in biological samples can only be reliably quantified with liquid chromatography-tandem mass spectrometry (LC-MS/MS).29 For example, capillary electrophoresis combined with laser-induced fluorescence – which is a more sophisticated method to assess (iso)desmosine than the amino acid analyzer method used by Soskel et al43 – measured about 100-times higher desmosine levels in plasma of patients with emphysema induced by α1-antitrypsin deficiency than LC-MS/MS.48,49 This illustrates the problem that techniques other than LC-MS/MS lack the specificity to differentiate (iso)desmosine-crosslinks from other elastin-derived peptides.29
LOX activity was assessed, and although no significant difference (i.e., p = 0.125) was found between groups, LOX activity in relation to the total weight of proteins tended to be lower in copper-deficient (269 cpm/mg protein) than in copper-sufficient (335 cpm/mg protein) hamsters. Before discarding this statistically nonsignificant decrease as irrelevant, one should realize that concentrations of maintenance proteins other than LOX are not lower but higher in emphysematous lungs.33 Decreased expression of LOX mRNA was found in a rat model of copper deficiency-induced emphysema, whereas fibulin-5 mRNA concentrations were increased.44 This pattern of gene expression in copper-deficient rats reminisces to upregulation of fibulin-5 and downregulation of LOX enzymes in human COPD lungs.33,39
Given that copper is also the essential cofactor of the cytosolic enzyme superoxide dismutase (SOD), activity of this protein was quantified as well.43 In contrast to LOX, SOD activity was significantly diminished in the copper-deficient lungs. Soskel et al therefore explained the paradox of low pulmonary copper levels but not significantly decreased LOX activity by ascribing emphysema formation in hamsters to reduced activity of SOD rather than of LOX.
We argue that it would have been more appropriate if LOXL1 would have been specifically quantified, as hamsters were sacrificed at the age of 8 to 10 weeks.43 Initial elastogenesis in rodent lungs – in which LOX plays a major role – is practically completed within the first two weeks after birth.50 Subsequent pulmonary elastin metabolism progresses slowly in the ensuing months50 and involves LOXL1 rather than LOX.11 Whether the LOX-activity assay that was applied by Soskel et al reflects either only prototype or pan-LOX activity has never been assessed.43,51,52
Contrary to in hamsters,43 LOX activity was significantly reduced in lungs of copper-deficient chicks.45 A potential reason for the more pronounced difference in LOX activity between copper-deficient and control chicks might be that the birds were, unlike the rodents,43 still in a developmental state (i.e. 23-days-old) in which LOX rather than LOXL1 played a pivotal role.45
Given issues with the methods that were used, this hamster model43 should ideally be repeated in order to quantify (iso)desmosine as well as LOXL1 protein, mRNA and activity levels with contemporary technologies.
Emphysema Induced By Copper-Deficient/Zinc-Supplemented Diet In Porcine
Whereas copper is LOX’s essential cofactor, zinc has an inhibitory effect on the enzyme’s activity.53 Pigs were therefore raised on a copper-deficient/zinc-supplemented diet in order to induce emphysema.42
Perforated alveolar walls were observed in lungs of animals who had received the copper-deficient diet. The lungs demonstrated features of emphysema such as airspace enlargement, decrease in alveolar surface areas and increased compliance.
Elastin levels in the lungs remained similar between copper-deficient/zinc-supplemented and control pigs. The degree of elastin crosslinking was not assessed; however, we suspect a reduction of (iso)desmosine in lungs of copper-deficient/zinc-supplemented pigs. In contrast to elastin, collagen was significantly increased in lung tissues from the experimental animals.42 A plausible explanation for this divergent reaction of collagen and elastin to copper deficiency might be that collagen’s resistance to proteolytic degradation is less dependent on LOX-mediated crosslinking than elastin. This is probably because non-enzymatic crosslinks from glycated amino acid residues are more prevalent in collagen than elastic fibers,54 which also confer some protection against proteases,55 whereas tropoelastin monomers are instantaneously degraded if they are not enzymatically crosslinked by LOX.56
Cellular Aspects Of Emphysema Induced By Copper-Deficient Diet Combined With Copper Chelator
Male rats received a copper-deficient diet in combination with administration of a copper chelator (i.e., tetrathiomolybdate).44 The microscopic examination of lung sections showed emphysematous destruction with enlargement of the alveoli.
Based on the pulmonary downregulation of both LOX and vascular endothelial growth factor (VEGF) gene expression, it was suggested that copper-deficient rats not only acquire emphysema due to extracellular processes but also via cellular mechanisms. The relevance of VEGF-downregulation in the pathogenesis of emphysema has been established in animal studies demonstrating that lowering either expression or activity of this growth factor induces emphysematous changes.57,58 Mizuno et al hypothesized that copper depletion causes detachment of alveolar cells from their supporting matrix, which may result in a form of programmed cell death named anoikis.44,59 The authors subsequently sought evidence for this theory and found support by demonstrating reduced FAK, AKT and ERK1/2 phosphorylation patterns as well as enhanced expression of caspase-8 and p53 genes, which can all be regarded as indicative for anoikis activity.44 This specific form of apoptosis may potentially form a mechanistic link between copper-induced failure of maintenance processes in the extracellular matrix and reduced cellularity of emphysematous lungs.
Copper Deficiency Causes Emphysema, But Does Emphysema Induce Copper Deficiency?
Models using various animal species have unequivocally demonstrated that copper deficiency has the potential to cause emphysema.41–45 However, there is also an animal model to suggest that this mechanism may also work the other way around, namely that emphysema formation induces pulmonary copper deficit.60
Transgenic mice with lung-specific TNF-α overexpression were used as a model for emphysema.60 A variety of metal and micronutrient concentrations were measured in various tissues. Pulmonary zinc and selenium concentrations did not significantly vary between mice with normal and overexpressed TNF-α. However, a 75% decline of copper concentrations was found in lungs of TNF-α overexpressing compared to age-matched wild-type mice, whereas copper levels remained fairly stable in blood and other organs. Importantly, this pattern of isolated pulmonary copper deficiency seems to be also present in human COPD.61,62
mRNA levels of LOXL1 – as an enzyme that is more important for elastin maintenance than prototype LOX11 – were unfortunately not quantified. However, expression of the gene coding for LOX was significantly downregulated in TNF-α overexpressing animals. We suspect that the following sequential steps may have led to pulmonary copper deficiency in these transgenic animals (Figure 2). High pulmonary TNF-α levels attract macrophages and neutrophils to the lungs, which produce proteolytic enzymes that accelerate elastolysis. Damage to pulmonary elastic fibers increases the need for elastin repairing proteins as well as for copper to activate additional LOX enzymes. Contrary to animal models of emphysema induced by copper-deficient diets,41–45 copper supply to the lungs will not be reduced in TNF-α overexpressing mice.60 Normal pulmonary copper levels, however, may be insufficient when massive elastin repair is required. Such increased demand for copper may induce copper deficiency and reduce levels of activated LOX(L), hampering elastin restoration. Since partially degraded elastin has enhanced vulnerability to further proteolytic degradation,63 failing elastin restoration due to copper deficiency may provoke a vicious circle of elastolysis (Figure 2).
 |
Figure 2 Smoking induces inflammation, which is associated with enhanced proteolytic activity leading to accelerated elastin degradation. Damage to elastic fibers upregulates synthesis of elastin repair proteins and increases copper demand to activate additional LOXL1 enzymes. This may deplete copper stores, which may lead to decreased LOXL1 activity and impaired elastin repair. Cigarette smoke also has direct inhibitory effects on LOXL1 activity. A vicious circle of elastolysis and copper depletion may arise (bold arrows), as partially degraded elastin is more accessible to proteases than intact fibers. Copper supplementation may provide an escape from the vicious circle by activating LOXL1 and facilitating efficacious elastin repair (dashed arrows). Green=positive effect, yellow=neutral effect and red=negative effect. |
The concept of expenditure-induced deficit of cofactors, rather than primary deficiency, has been demonstrated in other pathological circumstances. Administration of vitamin D, for example, promotes vascular mineralization as well as synthesis of the vitamin K-dependent calcification inhibitor matrix Gla protein.64,65 The need for vitamin K to activate surplus matrix Gla protein consequently increases, which will induce vitamin K-deficiency if demand outstrips supply. Similar to vitamin K’s ability to prevent vitamin D-induced calcification, we suggest that copper supplementation may interrupt the vicious circle of emphysema formation (Figure 2).
There might be an alternative explanation for reduced LOX activation yet sufficient circulating levels of copper in emphysematous lungs. The import of copper from the extracellular matrix into cells is facilitated by copper transporter 1 (Ctr1),66 which occurs as either full-length or truncated protein.67 The latter exhibits about 50% reduced capacity for copper uptake compared to the full-length variant.67 Truncated Ctr1 is generated through cleavage of full-length Ctr1 by cathepsin proteases.68 Since activity of these proteolytic enzymes is upregulated in patients with COPD,69 it is plausible that cleavage of Ctr1 is enhanced in emphysematous lungs leading to reduced intracellular copper availability for LOX activation.
The most relevant remaining questions that need to be answered in future animal studies are whether smoking-induced emphysema induces copper deficiency and whether pulmonary copper deficiency amplifies the development of smoking-induced emphysema.
Human Data Suggesting That Copper Deficiency Induces Emphysema
Whether deficiency of copper relates to the development of emphysema in humans is currently unknown; however, some data may provide a rationale to explore this possibility.39,62
Copper In Exhaled Breath Condensate
Collecting exhaled breath condensate (EBC) is a noninvasive method to sample the airways. EBC of stable COPD patients contains lower copper levels than of controls, and copper levels in EBC are positively related to forced expiratory volume in one second (FEV1) in individuals with COPD.62
It is tempting, though highly speculative, to suggest that low copper levels in EBC – as a reflection of pulmonary copper status – may lead to reduced spirometric values through prevention of LOX(L) activation.
Importantly, no differences were found in EBC copper levels between different FEV1-based stages of disease severity.62 It might be that low pulmonary copper levels play a role in the initiation rather than the progression of COPD.
Emphysema As A Potential Complication Of Menkes Disease
Menkes disease is a lethal genetic disorder characterized by neurological deterioration in combination with connective tissue abnormalities. Emphysema may be a complication of Menkes disease.70,71 The syndrome is caused by mutations in Menkes P-type ATPase (ATP7A) gene coding for an intracellular copper-transporter.72 Inherited dysfunction of ATP7A is also the reason why so-called Blotchy mice develop early emphysema.73 Cells with mutant proteins on their surfaces are unable to take up copper in cells, which precludes activation of LOX by copper leading to inefficacious elastin development and restoration processes. Copper supplementation therapy cannot negate this,70 since copper is intracellularly incorporated into LOX proteins.74
A patient with Menkes disease has been described who developed respiratory distress from about 9 months of age.70 CT revealed bilateral emphysematous blebs, and chest X-rays demonstrated lung hyperinflation which progressed over several months. He died at 14 months of age. Postmortem examination of the lungs demonstrated severe and diffuse panlobular emphysema in combination with various-sized bullae.
Although the link between disrupted copper metabolism and emphysema formation in Menkes disease is interesting from a pathogenic standpoint, it is important to realize that there are fundamental differences between this genetic disorder and smoking-related emphysema.
Lysyl Oxidase Enzymes In Nongenetic Human Emphysema
Levels of copper-activated LOX, LOXL1 and LOXL2 were decreased in emphysematous compared to nonemphysematous lung samples obtained from COPD patients who had undergone surgical lung cancer resection.39 Postoperative pathological examination confirmed the diagnosis of lung cancer in 87.5% of the cases. It is important to appreciate that LOX enzymes seem to be also involved in tumorigenesis.72 No data are currently available to answer the question whether LOX activity is also decreased in emphysematous lungs without concomitant lung cancer.
Copper metabolism domain containing-1 (COMMD1) is a protein that regulates cellular copper concentration through regulation of export from cells to the extracellular matrix,75 and its levels appear to be reduced in human emphysematous lung tissue.39 The authors attributed their findings of low activated LOX levels to decreased copper concentrations in the pulmonary extracellular matrix caused by depletion of COMMD1.39 However, this seems unlikely since copper incorporation into LOX is not an extracellular but intracellular process72 and COMMD1 is not a regulator of cellular copper uptake. Reduced levels of activated LOX, LOXL1 and LOXL2 proteins in affected areas39 may nevertheless suggest local copper deficiency in lungs of patients with emphysema, however, via other mechanisms than reduction in the level of COMMD1.
Smoking, Copper And Inhibition Of Lysyl Oxidase
Cigarette smoke is the single most important risk factor for the development of emphysema.76 Smokers usually first develop emphysematous lesions in the middle portion of secondary pulmonary lobules (i.e., centrilobular emphysema),77 probably because that is where inhaled particles deposit. Tobacco-derived toxins may interfere with LOX crosslinking, and consequently with elastin repair, through several mechanisms.
Increased Copper Demand
The smoking-induced inflammatory response associates with increased recruitment of protease-producing leukocytes to the lungs, resulting in acceleration of elastin degradation. Increased elastolysis subsequently upregulates restoration processes and thereby the need for copper to activate additional LOX enzymes. Tobacco inhalation may potentially deplete pulmonary copper ions through this mechanism (Figure 2).60
Inhibition Of Lysyl Oxidase By Nicotine And/Or Cadmium
Another reason why inhalation of cigarette smoke may hamper LOX crosslinking is by its dose-dependent decreasing effect on LOX levels and activity.78 Tobacco contains a myriad of harmful chemicals, of which nicotine and cadmium are the most likely candidates to be responsible for the inhibitory effect of smoke condensate on LOX enzymes.79–81
Nicotine seems to have the capacity to chelate metals, which is exemplified by its reducing effect on copper levels in senile plaques.82 Exposure of pregnant rats to nicotine results in the development of panlobular emphysema in their offspring,80 which is largely undone by maternal copper supplementation.79 These results suggest that a chelating effect of nicotine exposure leads to inadequate copper concentrations to activate sufficient LOX enzymes for elastogenesis and that this effect can be reversed by administrating additional copper.
Whereas cadmium is undetectable in human tissues at birth, its concentrations gradually increase over time due to intake levels that exceed levels of excretion.83 Inhaled tobacco constitutes a significant source of cadmium exposure for smoking individuals.84 Postmortem examination demonstrated about three times higher hepatic cadmium concentrations in subjects with COPD compared to nonobstructive controls.85 Pulmonary cadmium levels are also elevated in emphysema patients compared to individuals with healthy lungs.86 Nebulizing rats with cadmium chloride results in dilation, distortion and destruction of alveoli in such a pattern that lesions resemble human centrilobular emphysema.81 Remarkably, short-term exposure to cadmium induces upregulation of LOX, whereas chronic exposure reduces LOX levels.87,88 Similar to nicotine-mediated LOX-inhibition,79 copper supplementation also neutralizes cadmium-mediated LOX-inhibition.89,90
The influence of cigarette smoke on LOX is rather complex as tobacco leaves also contain copper,91,92 which may potentially stimulate pulmonary LOX activity.93,94 Although plasma copper levels are elevated in smokers compared to nonsmokers,95 the net effect of cigarette smoke extract on LOX is – as discussed in the above – inhibitory.78
Expert Opinion
We consider the gene expression study of Brandsma et al as a landmark study since it was the first to demonstrate that repair mechanisms are strongly upregulated in lungs of regular COPD patients.33 What we are bound to ask is why this effort is not successful, as the answer to this question may be key to establish modulatory pharmacotherapy for patients with emphysema in order to efficaciously restore pulmonary proteolytic damage.
Rationale For Copper Inhalation Therapy In Emphysema
Based on presence of pulmonary copper deficiency yet normal circulating copper levels in TNF-α overexpressing transgenic mice60 and the suggestion that lung-specific deficiency of copper is also present in patients with COPD,61,62 the possibility of using topical lung supplementation of copper was proposed as a novel treatment strategy for individuals with emphysema in order to restore pulmonary copper levels and thereby local elastin repair capacity.60 Although we agree that this concept should be further explored, we also foresee major limitations as various animal models have demonstrated adverse side effects of inhaled copper monotherapy.96–99
It is to be kept in mind that copper-dependent LOX enzymes are not only elastin but also collagen crosslinkers. The formation of crosslinks promotes organization, stabilization and accumulation of collagen, which are hallmarks of fibrosis. Whereas the organization of collagen fibrils is reduced in COPD lungs,21–23 collagen maturation is enhanced in lung parenchyma of patients with IPF.100 This difference in collagen organization may well be due to the level of enzymatic crosslinking by LOX enzymes. Both animal and human data suggest that particularly the degree of pulmonary LOXL1 activity is decisive for whether or not fibrotic lung disease develops. LOXL1 knock-out mice are protected against pulmonary fibrosis induced by overexpression of the profibrotic cytokine transforming growth factor-β1,101 whereas levels of LOXL1 are increased in lungs of patients with IPF (Figure 3) and associate with the thickness of collagen fibers.102 The effects of inhaled copper oxide nanoparticles were studied in mice, and it was found that they stimulated pulmonary collagen accumulation culminating in lung fibrosis.97 The fact that copper-chelator tetrathiomolybdate proved protective against bleomycin-induced lung fibrosis corroborates the existence of mechanistic links between high pulmonary copper levels, collagen accumulation and fibrosis formation.96 We expect that inhalation therapy with copper in emphysema patients would cause a shift from emphysematous towards fibrotic parenchymal lung disease.
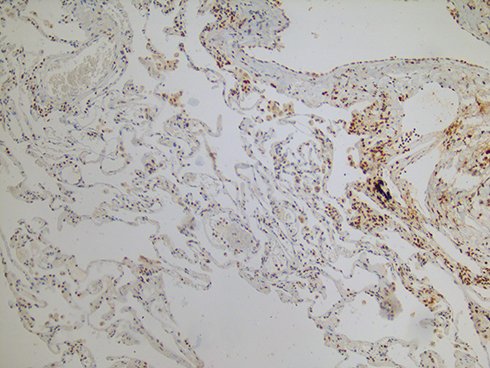 |
Figure 3 Immunohistochemistry image of an explant lung from a patient with combined pulmonary fibrosis and emphysema showing increased lysyl oxidase-like protein 1 (LOXL1) levels in fibrotic areas and decreased LOXL1 levels in emphysematous areas (brown=LOXL1; blue=nucleus; pink=cytoplasm). |
Heparin Added To Inhalation Formulations With Copper
Heparin is a glycosaminoglycan and has the highest negative charge density per disaccharide of any known biological molecule. We propose that heparin should be added to inhalation formulations with copper for various reasons (Table 1; Figure 4).
 |
Table 1 Reasons For Adding Heparin To Copper Inhalation Formulations |
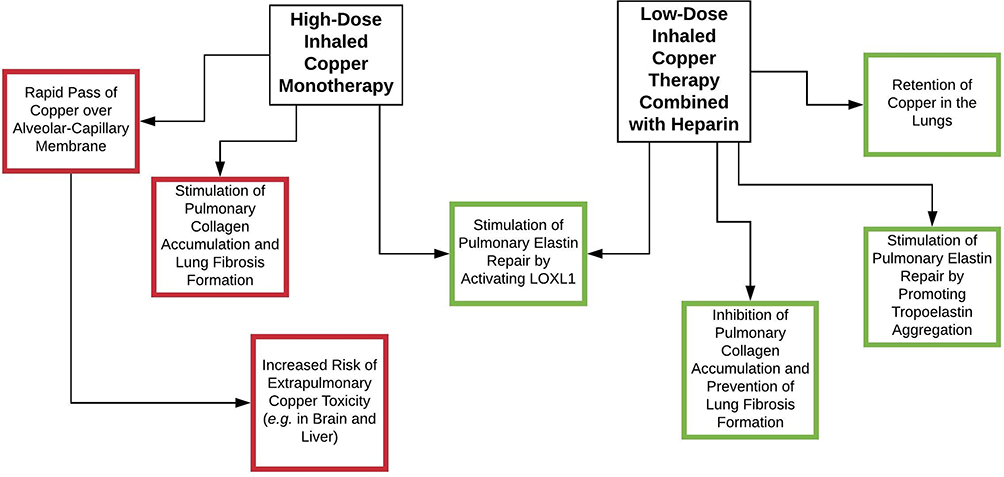 |
Figure 4 Inhalation therapy with copper stimulates elastin repair by activating LOXL1. Inhaled copper monotherapy, however, also stimulates lung fibrosis formation by stimulating collagen crosslinking, stabilization and accumulation. Furthermore, high-dose copper is needed when administered as monotherapy, since unbound copper rapidly passes the alveolar-capillary membrane. This increases the risk of extrapulmonary copper toxicity. Addition of heparin retains copper in the pulmonary compartment, which makes that lower copper doses suffice. Heparin also has an inhibitory effect on collagen crosslinking while stimulating elastin repair by promoting aggregation of tropoelastin. Green=positive effect, yellow=neutral effect and red=negative effect. |
Heparin As Specific Inhibitor Of Collagen Crosslinking And Fibrosis Formation
Heparin is a ligand of both collagen and elastin, however, its effect on LOX-mediated crosslinking is fundamentally different for both proteins. In vitro experiments demonstrate that heparin – when present prior to collagen fibrillogenesis – strongly inhibits collagen crosslinking, which reduces collagen stabilization and thereby the occurrence of fibrosis.103 The antifibrotic effect of heparin also seems to be relevant in lungs of mammals as experiments show that aerosolized heparin prevents bleomycin-induced fibrosis in rabbits by reducing pulmonary collagen and hydroxyproline-crosslink contents.104 We therefore propose that heparin is the ideal companion of copper in inhalation formulations to prevent copper-induced maturation and further accumulation of collagen fibrils in emphysematous lungs.
Heparin As Promotor Of Tropoelastin Polymerization And Elastin Repair
The second step of elastogenesis is the aggregation of tropoelastin monomers into continuous layers of polymerized fibers (Figure 1).14 Heparin, as a strong anionic molecule, has the ability to facilitate this process of self-assembly by neutralizing positively charged amino acids of tropoelastin proteins and to thereby undo the repellent effect that similar charged tropoelastin proteins have on each other.105 Calculations suggest that the negative charge of heparin is approximately in balance with the positive charge of tropoelastin.105
Heparin As Ideal Copper-Carrier With Regard To Elastin Repair
Heparin may not only be the ideal companion of copper in inhalation formulations because of its inhibitory properties on collagen crosslinking and synergistic effect on elastin repair but may have more potential advantageous properties.
A study assessing the ability of copper to induce angiogenesis in the cornea suggested that copper ions are in need for a carrier to reduce toxicity and potentiate activity.106 Both copper chloride and copper sulfate monotherapies were only angiogenic at dosages that induced inflammatory responses, whereas copper bound to either ceruloplasmin or heparin was effective at nontoxic doses. The fact that unbound copper ions do not only have such adverse side effects in eyes but also in lungs was suggested by studies in which inhaled copper oxide upregulated pulmonary expression of major proinflammatory genes,97,98 as well as the number of macrophages, neutrophils and lymphocytes in bronchoalveolar lavage fluid (BALF).99
Although ceruloplasmin is the natural copper-carrier in the blood, heparin might be the ideal chaperone for copper with regard to pulmonary elastin repair. Heparin, at least in vitro, preferentially binds to positively charged lysine residues in tropoelastin proteins.105 It may thereby deliver copper ions to the sites where they are needed most, such as free margins of proteolytically destructed alveoli, where elastogenesis is particularly taking place.36 Exchange of copper for other cations, from heparin molecules, may subsequently provide local LOXL1-producing cells with copper.
Heparin As A Retainer Of Copper In The Lungs
Copper ions do not remain in the pulmonary compartment for extended periods of time following inhalation but rapidly pass the alveolar-capillary membrane to enter the circulation.97 The fast clearance from the lungs implies that relatively high copper doses are needed to achieve the intended local therapeutic effect. This is problematic given that the fibrotic effect of copper on lung tissue is dose-dependent.97 High-dose copper inhalation therapy will also have a significant incremental effect on circulating levels of copper, which is unwanted as it increases the risk for cerebral and hepatic copper toxicity.107,108
In contrast to copper, heparin is retained in the lungs for days after inhaled administrations,109,110 which makes that up to 400,000 international units (I.U.) of heparin (resulting in a delivery dose of 32,000 I.U. to the lower respiratory tract) can be safely nebulized,111 whereas 5000 I.U. are intravenously used as a bolus to initiate anticoagulation therapy. Almost 40% of the inhaled heparin dose that initially deposits in the lower respiratory tract remains in the lungs after 24 hrs,109 and a single nebulization session with heparin (moderately) affects systemic coagulation in human volunteers for up to two weeks.110 Heparin is taken up and stored by macrophages without being degraded, and it is only released upon death of the storage cell.110 Furthermore, it is to be expected that inhaled heparin will make interactions with various positively charged components of the airways and airspaces, such as lysine amino acid residues in elastic fibers,105 which may provide another mechanism to prevent heparin molecules from directly entering the systemic circulation.
The addition of heparin to copper inhalation formulations may be the key to retain copper within lungs and thereby achieve efficacious LOXL activation with minimal risk of copper toxicity.
Tolerability Of Copper
Copper is an essential nutrient and it has been estimated that a daily intake of 0.75 mg would maintain copper balance in a stable 70-kg adult.112 A double-blinded study assessed effects of 10 mg copper per day, as copper gluconate, and found no change on either copper concentrations in serum, urine or hair.113 Levels of hematocrit, triglycerides, cholesterol and liver function tests were also unaltered.
The safe upper limits of population mean intakes have been set on 10 mg/day for women and 12 mg/day for men.114 Significantly lower doses may be sufficient for activation of LOX enzymes by means of pulmonary copper-heparin administration.
Copper-Heparin Inhalation Therapy
Given that copper ions carry double positive charges (Cu2+), copper-heparin ionic interactions will arise when they are combined with sodium-heparin in a solution.115 A maximum of 23 to 24 copper ions can be bound to one heparin molecule.115 Copper ions display initial site-specific coordinated binding to heparin,116 which is distinct from connections that heparin makes with other cations.117 Electrostatic interaction with copper ions induces subtle conformational changes to heparin loosening its helical structure.115,117 We assessed whether varying heparin and copper concentrations affect nebulization characteristics of aerosolized copper-heparin solutions, which does not appear to be the case (Table 2). We therefore assume that it is acceptable to extrapolate published 99mtechnetium-labeled sodium-heparin (99mTc-heparin) data to predict pulmonary deposition distribution of copper-heparin solutions.
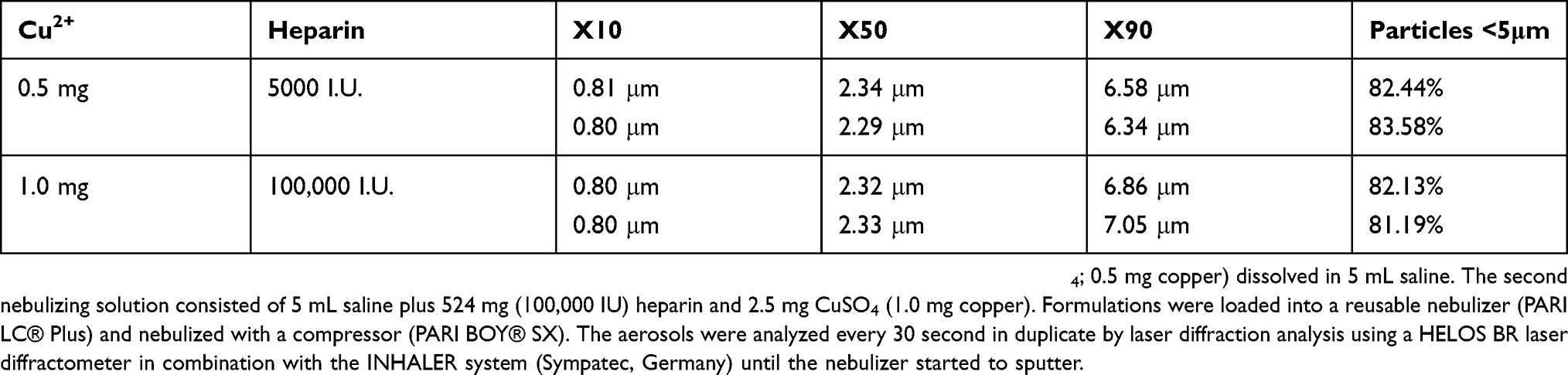 |
Table 2 Particle Size Distribution Of Nebulized Copper-Heparin Solutions |
Deposition of inhaled 99mTc-heparin has been quantified in the airways and airspaces of healthy subjects after a single nebulization.109 Of note, 99mTc-labeling does not have a modulatory effect on the deposition pattern of nebulized heparin. Less than 15% of the nebulizer charge reaches the mouth and 7.6% reaches the lower respiratory tract. The level of nebulized heparin that deposits in the lungs seems to be remarkably low; however, it is comparable to deposition rates after nebulization of β2-agonists with a jet nebulizer.118
Airspaces in the lung apices are typically most severely affected in patients with smoking-induced emphysema.77 Nearly 50% of the heparin dose that arrives at the pulmonary compartment deposits in the upper lung zones and about one-third in the periphery, which suggests that nebulization is a suitable method to reach emphysematous lung areas. Based on 99mTc-heparin distribution proportions, combined with the fact that one mole copper is needed for the activation of one mole LOX, we are able to estimate nebulizer-charging doses to deliver required amounts of copper-heparin in order to activate certain quantities of LOX enzymes in target zones.
It has to be taken into account that a large proportion of nebulized copper-heparin solution will deposit in the small airways <2 mm in internal diameter, which are affected in the early stages of COPD.119–121 Copper-heparin might potentially also have advantageous effects in this part of the lungs, given that levels of collagen and elastin are, respectively, increased and decreased in both airways and parenchyma of patients with COPD.15,16
Five-Year View
The effects of inhalation therapy with copper-heparin should first be tested in animal models of TNF-α overexpression-, elastase- and smoking-induced emphysema, where mean linear intercept and alveolar surface area should be used as endpoints. Micro CT-scanning could also be applied as a sophisticated technique to quantify emphysema and fibrosis formation. Furthermore, pulmonary levels of copper, (tropo)elastin, collagen, (iso)desmosine, hydroxyproline and gene expression of maintenance proteins should be assessed as well as LOX(L) protein and activity levels. Copper should also be measured in serum, liver and brains to assess the effect of copper-heparin inhalation therapy on extrapulmonary copper concentrations.
Next, the effect of a single copper-heparin nebulization on systemic copper and coagulation levels should be studied in healthy volunteers to assess safety and tolerability. High-dose inhalation therapy with heparin has been proven safe in healthy volunteers as well as in COPD and IPF patients,111,122,123 and the intended copper dose (i.e., somewhere between 0.1 and 1.0 mg/day) is far beneath the safe upper limit of daily intake.114 Adverse side-effects related to inhaled copper-heparin are therefore not expected.
If proven safe and tolerable, a clinical trial of a few weeks should be conducted in emphysema patients to assess the effects of copper-heparin nebulization on (tropo)elastin, (iso)desmosine, hydroxyproline and LOXL1 levels in BALF. Desmosines only occur in elastic fibers from which they can be released by proteases.124 Their levels in BALF therefore specifically reflect the rate of elastolysis in the lungs. As partially degraded elastic fibers have increased susceptibility for proteolytic degradation63 and inhaled copper supplementation is supposed to enhance elastin repair in the lungs by activating pulmonary LOXL1,94 we would expect a decreasing effect of the proposed treatment on BALF (iso)desmosine levels.
If inhalation treatment with nebulized copper-heparin formulations proves efficacious in animal and human studies, more convenient and efficacious (i.e., lower loading doses to achieve equal concentrations in the lower respiratory tract) copper-heparin inhalation forms, such as dry-powder formulations, should be developed.
We hope that within five years copper-heparin inhalation therapy has been explored as modulatory treatment for patients with emphysema to repair proteolytic lung damage.
Conclusions
Emphysema is a very common respiratory disorder for which no regenerating therapy is currently available. Besides protease/antiprotease imbalance, disturbances in the maintenance of elastin may also cause emphysema. LOXL1 is a copper-dependent enzyme that plays a crucial step in the restoration of proteolytic damage to elastic fibers. We speculate that local availability of copper is the rate-limiting step in the pulmonary elastin repair process. Copper inhalation therapy may be a valid therapeutic option for patients with emphysema; however, inhaled copper monotherapy is associated with stimulation of collagen accumulation and fibrosis formation. Heparin may be the ideal companion of copper for various reasons: i.e., (a) stimulating elastin repair, (b) inhibiting collagen crosslinking and (c) reducing copper toxicity by extending copper retention time in the lungs and thereby reducing the required copper dose in inhalation formulations.
Disclosure
Dr Rob Janssen reports a grant from Stichting Astma Bestrijding to study the effect of copper/heparin on emphysema formation in a mouse model, is one of the owners of Desmosine.com, and has a patent WO 2019/139479; Mr Paul Hagedoorn and Prof. Dr. Henderik W. Frijlink report that their employer has a license agreement with AstraZeneca on the Genuair inhaler and with PureIMS on the Twincer and Cyclops inhalers; Dr Frits M.E. Franssen report grants, personal fees from AstraZeneca, personal fees from Boehringer Ingelheim, personal fees from Chiesi, personal fees from GlaxoSmithKline, grants, personal fees from Novartis, personal fees from TEVA, outside the submitted work. The authors report no other conflicts of interest in this work.
References
1. The definition of emphysema. Report of a national heart, lung, and blood institute, division of lung diseases workshop. Am Rev Respir Dis. 1985;132:182–185. doi:10.1164/arrd.1985.132.1.182
2. Vogelmeier CF, Criner GJ, Martinez FJ, et al. Global strategy for the diagnosis, management, and prevention of chronic obstructive lung disease 2017 report: GOLD executive summary. Am J Respir Crit Care Med. 2017;195:557–582. doi:10.1164/rccm.201701-0218PP
3. Regan EA, Lynch DA, Curran-Everett D, et al. Clinical and radiologic disease in smokers with normal spirometry. JAMA Intern Med. 2015;175:1539–1549. doi:10.1001/jamainternmed.2015.2735
4. Al-Kassimi FA, Alhamad EH, Al-Hajjaj MS, et al. Can computed tomography and carbon monoxide transfer coefficient diagnose an asthma-like phenotype in COPD? Respirology. 2017;22:322–328. doi:10.1111/resp.12902
5. Papi A, Vestbo J, Fabbri L, et al. Extrafine inhaled triple therapy versus dual bronchodilator therapy in chronic obstructive pulmonary disease (TRIBUTE): a double-blind, parallel group, randomised controlled trial. Lancet. 2018;391:1076–1084. doi:10.1016/S0140-6736(18)30206-X
6. West JB, Luks AM. West’s Pulmonary Pathophysiology: The Essentials. Philadelphia, PA: Wolters Kluwer; 2017.
7. Betsuyaku T, Nishimura M, Yoshioka A, Takeyabu K, Miyamoto K, Kawakami Y. [Neutrophil elastase and elastin-derived peptides in BAL fluid and emphysematous changes on CT scans]. Nihon Kyobu Shikkan Gakkai Zasshi. 1996;34 Suppl:69–74.
8. Janoff A, Carp H. Possible mechanisms of emphysema in smokers: cigarette smoke condensate suppresses protease inhibition in vitro. Am Rev Respir Dis. 1977;116:65–72. doi:10.1164/arrd.1977.116.1.65
9. Kuhn C
10. Nakamura T, Lozano PR, Ikeda Y, et al. Fibulin-5/DANCE is essential for elastogenesis in vivo. Nature. 2002;415:171–175. doi:10.1038/415171a
11. Liu X, Zhao Y, Gao J, et al. Elastic fiber homeostasis requires lysyl oxidase-like 1 protein. Nat Genet. 2004;36:178–182. doi:10.1038/ng1297
12. Niewoehner DE, Hoidal JR. Lung fibrosis and emphysema: divergent responses to a common injury? Science. 1982;217:359–360. doi:10.1126/science.7089570
13. van der Rest M, Garrone R. Collagen family of proteins. Faseb J. 1991;5:2814–2823. doi:10.1096/fasebj.5.13.1916105
14. Mithieux SM, Weiss AS. Elastin. Adv Protein Chem. 2005;70:437–461. doi:10.1016/S0065-3233(05)70013-9
15. Eurlings IM, Dentener MA, Cleutjens JP, et al. Similar matrix alterations in alveolar and small airway walls of COPD patients. BMC Pulm Med. 2014;14:90. doi:10.1186/1471-2466-14-90
16. Black PN, Ching PS, Beaumont B, Ranasinghe S, Taylor G, Merrilees MJ. Changes in elastic fibres in the small airways and alveoli in COPD. Eur Respir J. 2008;31:998–1004. doi:10.1183/09031936.00017207
17. Chrzanowski P, Keller S, Cerreta J, Mandl I, Turino GM. Elastin content of normal and emphysematous lung parenchyma. Am J Med. 1980;69:351–359. doi:10.1016/0002-9343(80)90004-2
18. Cardoso WV, Sekhon HS, Hyde DM, Thurlbeck WM. Collagen and elastin in human pulmonary emphysema. Am Rev Respir Dis. 1993;147:975–981. doi:10.1164/ajrccm/147.4.975
19. Pierce JA, Hocott JB, Ebert RV. The collagen and elastin content of the lung in emphysema. Ann Intern Med. 1961;55:210–222. doi:10.7326/0003-4819-55-2-210
20. Fitzpatrick M. Studies of human pulmonary connective tissue. III. Chemical changes in structural proteins with emphysema. Am Rev Respir Dis. 1967;96:254–265. doi:10.1164/arrd.1967.96.2.254
21. Tjin G, Xu P, Kable SH, Kable EP, Burgess JK. Quantification of collagen I in airway tissues using second harmonic generation. J Biomed Opt. 2014;19:36005. doi:10.1117/1.JBO.19.3.036005
22. Abraham T, Hogg J. Extracellular matrix remodeling of lung alveolar walls in three dimensional space identified using second harmonic generation and multiphoton excitation fluorescence. J Struct Biol. 2010;171:189–196. doi:10.1016/j.jsb.2010.04.006
23. Abraham T, Hirota JA, Wadsworth S, Knight DA. Minimally invasive multiphoton and harmonic generation imaging of extracellular matrix structures in lung airway and related diseases. Pulm Pharmacol Ther. 2011;24:487–496. doi:10.1016/j.pupt.2011.03.008
24. Enomoto N, Suda T, Kono M, et al. Amount of elastic fibers predicts prognosis of idiopathic pulmonary fibrosis. Respir Med. 2013;107:1608–1616. doi:10.1016/j.rmed.2013.08.008
25. Shapiro SD, Endicott SK, Province MA, Pierce JA, Campbell EJ. Marked longevity of human lung parenchymal elastic fibers deduced from prevalence of D-aspartate and nuclear weapons-related radiocarbon. J Clin Invest. 1991;87:1828–1834. doi:10.1172/JCI115204
26. Mäki JM, Räsänen J, Tikkanen H, et al. Inactivation of the lysyl oxidase gene Lox leads to aortic aneurysms, cardiovascular dysfunction, and perinatal death in mice. Circulation. 2002;106:2503–2509. doi:10.1161/01.cir.0000038109.84500.1e
27. McLaughlin PJ, Chen Q, Horiguchi M, et al. Targeted disruption of fibulin-4 abolishes elastogenesis and causes perinatal lethality in mice. Mol Cell Biol. 2006;26:1700–1709. doi:10.1128/MCB.26.5.1700-1709.2006
28. Grau-Bové X, Ruiz-Trillo I, Rodriguez-Pascual F. Origin and evolution of lysyl oxidases. Sci Rep. 2015;5:10568. doi:10.1038/srep10568
29. Turino GM, Lin YY, He J, Cantor JO, Ma S. Elastin degradation: an effective biomarker in COPD. COPD. 2012;9:435–438. doi:10.3109/15412555.2012.697753
30. Harris ED, Rayton JK, Balthrop JE, DiSilvestro RA, Garcia-de-Quevedo M. Copper and the synthesis of elastin and collagen. Ciba Found Symp. 1980;79:163–182. doi:10.1002/9780470720622.ch9
31. Papke CL, Yanagisawa H. Fibulin-4 and fibulin-5 in elastogenesis and beyond: insights from mouse and human studies. Matrix Biol. 2014;37:142–149. doi:10.1016/j.matbio.2014.02.004
32. Tuder RM, Yoshida T, Fijalkowka I, Biswal S, Petrache I. Role of lung maintenance program in the heterogeneity of lung destruction in emphysema. Proc Am Thorac Soc. 2006;3:673–679. doi:10.1513/pats.200605-124SF
33. Brandsma CA, van den Berge M, Postma DS, et al. A large lung gene expression study identifying fibulin-5 as a novel player in tissue repair in COPD. Thorax. 2015;70:21–32. doi:10.1136/thoraxjnl-2014-205091
34. Deslee G, Woods JC, Moore CM, et al. Elastin expression in very severe human COPD. Eur Respir J. 2009;34:324–331. doi:10.1183/09031936.00123008
35. Kuang PP, Goldstein RH, Liu Y, Rishikof DC, Jean JC, Joyce-Brady M. Coordinate expression of fibulin-5/DANCE and elastin during lung injury repair. Am J Physiol Lung Cell Mol Physiol. 2003;285:L1147–L1152. doi:10.1152/ajplung.00098.2003
36. Lucey EC, Goldstein RH, Stone PJ, Snider GL. Remodeling of alveolar walls after elastase treatment of hamsters. Results of elastin and collagen mRNA in situ hybridization. Am J Respir Crit Care Med. 1998;158:555–564. doi:10.1164/ajrccm.158.2.9705021
37. Loeys B, Van Maldergem L, Mortier G, et al. Homozygosity for a missense mutation in fibulin-5 (FBLN5) results in a severe form of cutis laxa. Hum Mol Genet. 2002;11:2113–2118. doi:10.1093/hmg/11.18.2113
38. Urban Z, Davis EC. Cutis laxa: intersection of elastic fiber biogenesis, TGFβ signaling, the secretory pathway and metabolism. Matrix Biol. 2014;33:16–22. doi:10.1016/j.matbio.2013.07.006
39. Besiktepe N, Kayalar O, Ersen E, Oztay F. The copper dependent-lysyl oxidases contribute to the pathogenesis of pulmonary emphysema in chronic obstructive pulmonary disease patients. J Trace Elem Med Biol. 2017;44:247–255. doi:10.1016/j.jtemb.2017.08.011
40. Molnar J, Fong KS, He QP, et al. Structural and functional diversity of lysyl oxidase and the LOX-like proteins. Biochim Biophys Acta. 2003;1647:220–224. doi:10.1016/s1570-9639(03)00053-0
41. O’Dell BL, Kilburn KH, McKenzie WN, Thurston RJ. The lung of the copper-deficient rat. A model for developmental pulmonary emphysema. Am J Pathol. 1978;91:413–432.
42. Soskel NT, Watanabe S, Hammond E, Sandberg LB, Renzetti AD
43. Soskel NT, Watanabe S, Sandberg LB. Mechanisms of lung injury in the copper-deficient hamster model of emphysema. Chest. 1984;85:70S–73S. doi:10.1378/chest.85.6_supplement.70s
44. Mizuno S, Yasuo M, Bogaard HJ, et al. Copper deficiency induced emphysema is associated with focal adhesion kinase inactivation. PLoS One. 2012;7:e30678. doi:10.1371/journal.pone.0030678
45. Harris ED, Biochemical defect in chick lung resulting from copper deficiency. J Nutr. 1986;116:252–258. doi:10.1093/jn/116.2.252
46. Kuhn C, Yu SY, Chraplyvy M, Linder HE, Senior RM. The induction of emphysema with elastase. II. Changes in connective tissue. Lab Invest. 1976;34:372–380.
47. Lefevre M, Heng H, Rucker RB. Dietary cadmium, zinc and copper: effects on chick lung morphology and elastin cross-linking. J Nutr. 1982;112:1344–1352. doi:10.1093/jn/112.7.1344
48. Fregonese L, Ferrari F, Fumagalli M, Luisetti M, Stolk J, Iadarola P. Long-term variability of desmosine/isodesmosine as biomarker in alpha-1-antitrypsin deficiency-related COPD. COPD. 2011;8:329–333. doi:10.3109/15412555.2011.589871
49. Ma S, Lin YY, Cantor JO, et al. The effect of alpha-1 proteinase inhibitor on biomarkers of elastin degradation in alpha-1 antitrypsin deficiency: an analysis of the RAPID/RAPID extension trials. Chronic Obstr Pulm Dis. 2016;4:34–44. doi:10.15326/jcopdf.4.1.2016.0156
50. Dubick MA, Rucker RB, Cross CE, Last JA. Elastin metabolism in rodent lung. Biochim Biophys Acta. 1981;672:303–306. doi:10.1016/0304-4165(81)90297-x
51. Pinnell SR, Martin GR. The cross-linking of collagen and elastin: enzymatic conversion of lysine in peptide linkage to alpha-aminoadipic-delta-semialdehyde (allysine) by an extract from bone. Proc Natl Acad Sci USA. 1968;61:708–716. doi:10.1073/pnas.61.2.708
52. Misiorowski RL, Ulreich JB, Chvapil M. A microassay for lysyl oxidase activity. Anal Biochem. 1976;71:186–192. doi:10.1016/0003-2697(76)90026-9
53. Chvapil M, Misiorowski R. In vivo inhibition of lysyl oxidase by high dose of zinc. Proc Soc Exp Biol Med. 1980;164:137–141. doi:10.3181/00379727-164-40836
54. Reiser K, McCormick RJ, Rucker RB. Enzymatic and nonenzymatic cross-linking of collagen and elastin. Faseb J. 1992;6:2439–2449. doi:10.1096/fasebj.6.7.1348714
55. Nowotny K, Grune T. Degradation of oxidized and glycoxidized collagen: role of collagen cross-linking. Arch Biochem Biophys. 2014;542:56–64. doi:10.1016/j.abb.2013.12.007
56. Tinker D, Geller J, Romero N, Cross CE, Rucker RB. Tropoelastin production and tropoelastin messenger RNA activity. Relationship to copper and elastin cross-linking in chick aorta. Biochem J. 1986;237:17–23. doi:10.1042/bj2370017
57. Kasahara Y, Tuder RM, Cool CD, Lynch DA, Flores SC, Voelkel NF. Endothelial cell death and decreased expression of vascular endothelial growth factor and vascular endothelial growth factor receptor 2 in emphysema. Am J Respir Crit Care Med. 2001;13:737–744. doi:10.1164/ajrccm.163.3.2002117
58. Kasahara Y, Tuder RM, Taraseviciene-Stewart L, et al. Inhibition of VEGF receptors causes lung cell apoptosis and emphysema. J Clin Invest. 2000;106:1311–1319. doi:10.1172/JCI10259
59. Gilmore AP. Anoikis. Cell Death Differ. 2005;12(Suppl 2):1473–1477. doi:10.1038/sj.cdd.4401723
60. Liu L, Geng X, McDermott J, et al. Copper Deficiency in the Lungs of TNF-α Transgenic Mice. Front Physiol. 2016;7:234. doi:10.3389/fphys.2016.00234
61. Karul AB, Karadag F, Yensel N, Altinisik M, Altun C, Cildag O. Should chronic obstructive pulmonary disease outpatients be routinely evaluated for trace elements? Biol Trace Elem Res. 2003;94:41–48. doi:10.1385/BTER:94:1:41
62. Mutti A, Corradi M, Goldoni M, Vettori MV, Bernard A, Apostoli P. Exhaled metallic elements and serum pneumoproteins in asymptomatic smokers and patients with COPD or asthma. Chest. 2006;129:1288–1297. doi:10.1378/chest.129.5.1288
63. Bostancioglu K, Mecham RP, Wallach JM. Elastolysis of normal and partially cross-linked elastin. Biochem Int. 1987;15:263–269.
64. Price PA, Buckley JR, Williamson MK. The amino bisphosphonate ibandronate prevents vitamin D toxicity and inhibits vitamin D-induced calcification of arteries, cartilage, lungs and kidneys in rats. J Nutr. 2001;131:2910–2915. doi:10.1093/jn/131.11.2910
65. Fraser JD, Otawara Y, Price PA. 1,25-Dihydroxyvitamin D3 stimulates the synthesis of matrix gamma-carboxyglutamic acid protein by osteosarcoma cells. Mutually exclusive expression of vitamin K-dependent bone proteins by clonal osteoblastic cell lines. J Biol Chem. 1988;263:911–916.
66. Dancis A, Haile D, Yuan DS, Klausner RD. The Saccharomyces cerevisiae copper transport protein (Ctr1p). Biochemical characterization, regulation by copper, and physiologic role in copper uptake. J Biol Chem. 1994;269:25660–25667.
67. Maryon EB, Molloy SA, Kaplan JH. O-linked glycosylation at threonine 27 protects the copper transporter hCTR1 from proteolytic cleavage in mammalian cells. J Biol Chem. 2007;282:20376–20387. doi:10.1074/jbc.M701806200
68. Öhrvik H, Logeman B, Turk B, Reinheckel T, Thiele DJ. Cathepsin protease controls copper and cisplatin accumulation via cleavage of the ctr1 metal-binding ectodomain. J Biol Chem. 2016;291:13905–13916. doi:10.1074/jbc.M116.731281
69. Lesser M, Padilla ML, Cardozo C. Induction of emphysema in hamsters by intratracheal instillation of cathepsin B. Am Rev Respir Dis. 1992;145:661–668. doi:10.1164/ajrccm/145.3.661
70. Daish P, Wheeler EM, Roberts PF, Jones RD. Menkes’s syndrome. Report of a patient treated from 21 days of age with parenteral copper. Arch Dis Child. 1978;53:956–958. doi:10.1136/adc.53.12.956
71. Grange DK, Kaler SG, Albers GM, Petterchak JA, Thorpe CM, DeMello DE. Severe bilateral panlobular emphysema and pulmonary arterial hypoplasia: unusual manifestations of Menkes disease. Am J Med Genet A. 2005;139A:151–155. doi:10.1002/ajmg.a.31001
72. Shanbhag V, Jasmer-McDonald K, Zhu S, et al. ATP7A delivers copper to the lysyl oxidase family of enzymes and promotes tumorigenesis and metastasis. Proc Natl Acad Sci USA. 2019;116:6836–6841. doi:10.1073/pnas.1817473116
73. Hunt DM. Primary defect in copper transport underlies mottled mutants in the mouse. Nature. 1974;249:852–854. doi:10.1038/249852a0
74. Kosonen T, Uriu-Hare JY, Clegg MS, Keen CL, Rucker RB. Incorporation of copper into lysyl oxidase. Biochem J. 1997;327:283–289. doi:10.1042/bj3270283
75. McDonald FJ. COMMD1 and ion transport proteins: what is the COMMection? Focus on “COMMD1 interacts with the COOH terminus of NKCC1 in Calu-3 airway epithelial cells to modulate NKCC1 ubiquitination”. Am J Physiol Cell Physiol. 2013;305:C129–C130. doi:10.1152/ajpcell.00128.2013
76. Foster WL
77. Sweet HC, Wyatt JP, Fritsch AJ, et al. Panlobular and centrilobular emphysema: correlation of clinical findings with pathologic patterns. Ann Intern Med. 1961;55:565–581. doi:10.7326/0003-4819-54-5-1060_3
78. Chen LJ, Zhao Y, Gao S, et al. Downregulation of lysyl oxidase and upregulation of cellular thiols in rat fetal lung fibroblasts treated with cigarette smoke condensate. Toxicol Sci. 2005;83:372–379. doi:10.1093/toxsci/kfi019
79. Maritz GS, Windvogel S. Is maternal copper supplementation during alveolarization protecting the developing rat lung against the adverse effects of maternal nicotine exposure? A morphometric study. Exp Lung Res. 2003;29:243–260. doi:10.1080/01902140303785
80. Maritz GS, Windvogel S. Chronic maternal nicotine exposure during gestation and lactation and the development of the lung parenchyma in the offspring. Response to nicotine withdrawal. Pathophysiology. 2003;10:69–75. doi:10.1016/j.pathophys.2003.10.001
81. Snider GL, Hayes JA, Korthy AL, Lewis GP. Centrilobular emphysema experimentally induced by cadmium chloride aerosol. Am Rev Respir Dis. 1973;108:40–48. doi:10.1164/arrd.1973.108.1.40
82. Zhang J, Liu Q, Chen Q, et al. Nicotine attenuates beta-amyloid-induced neurotoxicity by regulating metal homeostasis. Faseb J. 2006;20:1212–1214. doi:10.1096/fj.05-5214fje
83. Koch HJ
84. Scherer G, Barkemeyer H. Cadmium concentrations in tobacco and tobacco smoke. Ecotoxicol Environ Saf. 1983;7:71–78. doi:10.1016/0147-6513(83)90050-7
85. Lewis GP, Lyle H, Miller S. Association between elevated hepatic water-soluble protein-bound cadmium levels and chronic bronchitis and-or emphysema. Lancet. 1969;2:1330–1333. doi:10.1016/s0140-6736(69)90866-6
86. Pääkkö P, Kokkonen P, Anttila S, Kalliomäki PL. Cadmium and chromium as markers of smoking in human lung tissue. Environ Res. 1989;49:197–207. doi:10.1016/S0013-9351(89)80065-9
87. Almassian B, Trackman PC, Iguchi H, Boak A, Calvaresi D, Kagan HM. Induction of lung lysyl oxidase activity and lysyl oxidase protein by exposure of rats to cadmium chloride: properties of the induced enzyme. Connect Tissue Res. 1991;25:197–208. doi:10.3109/03008209109029156
88. Sampson CE, Chichester CO, Hayes JA, Kagan HM. Alterations in collagen biosynthesis and in metallothionein in lungs of rats acutely or repeatedly exposed to cadmium chloride aerosol. Am Rev Respir Dis. 1984;129:619–624.
89. Li W, Chou IN, Boak A, Kagan HM. Downregulation of lysyl oxidase in cadmium-resistant fibroblasts. Am J Respir Cell Mol Biol. 1995;13:418–425. doi:10.1165/ajrcmb.13.4.7546771
90. Zhao Y, Gao S, Chou IN, Toselli P, Stone P, Li W. Inhibition of the expression of lysyl oxidase and its substrates in cadmium-resistant rat fetal lung fibroblasts. Toxicol Sci. 2006;90:478–489. doi:10.1093/toxsci/kfj112
91. Dias Fde S, Bonsucesso JS, Oliveira LC, Dos Santos WN. Preconcentration and determination of copper in tobacco leaves samples by using a minicolumn of sisal fiber (Agave sisalana) loaded with Alizarin fluorine blue by FAAS. Talanta. 2012;89:276–279. doi:10.1016/j.talanta.2011.12.027
92. Pelit FO, Demirdöğen RE, Henden E. Investigation of heavy metal content of Turkish tobacco leaves, cigarette butt, ash, and smoke. Environ Monit Assess. 2013;185:9471–9479. doi:10.1007/s10661-013-3266-4
93. Rucker RB, Romero-Chapman N, Wong T, et al. Modulation of lysyl oxidase by dietary copper in rats. J Nutr. 1996;126:51–60. doi:10.1093/jn/126.1.51
94. Werman MJ, Bhathena SJ, Turnlund JR. Dietary copper intake influences skin lysyl oxidase in young men. Nutritional Biochem. 1997;8:201–204. doi:10.1016/S0955-2863(97)00004-1
95. Kocyigit A, Erel O, Gur S. Effects of tobacco smoking on plasma selenium, zinc, copper and iron concentrations and related antioxidative enzyme activities. Clin Biochem. 2001;34:629–633. doi:10.1016/s0009-9120(01)00271-5
96. Ovet H, Oztay F. The copper chelator tetrathiomolybdate regressed bleomycin-induced pulmonary fibrosis in mice, by reducing lysyl oxidase expressions. Biol Trace Elem Res. 2014;162:189–199. doi:10.1007/s12011-014-0142-1
97. Lai X, Zhao H, Zhang Y, et al. Intranasal delivery of copper oxide nanoparticles induces pulmonary toxicity and fibrosis in C57BL/6 mice. Sci Rep. 2018;8:4499. doi:10.1038/s41598-018-22556-7
98. Costa PM, Gosens I, Williams A, et al. Transcriptional profiling reveals gene expression changes associated with inflammation and cell proliferation following short-term inhalation exposure to copper oxide nanoparticles. J Appl Toxicol. 2018;38:385–397. doi:10.1002/jat.3548
99. Gosens I, Cassee FR, Zanella M, et al. Organ burden and pulmonary toxicity of nano-sized copper (II) oxide particles after short-term inhalation exposure. Nanotoxicology. 2016;10:1084–1095. doi:10.3109/17435390.2016.1172678
100. Pena AM, Fabre A, Débarre D, et al. Three-dimensional investigation and scoring of extracellular matrix remodeling during lung fibrosis using multiphoton microscopy. Microsc Res Tech. 2007;70:162–170. doi:10.1002/jemt.20400
101. Bellaye PS, Shimbori C, Upagupta C, et al. Lysyl oxidase-like 1 protein deficiency protects mice from adenoviral transforming growth factor-β1-induced pulmonary fibrosis. Am J Respir Cell Mol Biol. 2018;58:461–470. doi:10.1165/rcmb.2017-0252OC
102. Tjin G, White ES, Faiz A, et al. Lysyl oxidases regulate fibrillar collagen remodelling in idiopathic pulmonary fibrosis. Dis Model Mech. 2017;10:1301–1312. doi:10.1242/dmm.030114
103. Gavriel P, Kagan HM. Inhibition by heparin of the oxidation of lysine in collagen by lysyl oxidase. Biochemistry. 1988;27:2811–2815. doi:10.1021/bi00408a022
104. Günther A, Lübke N, Ermert M, et al. Prevention of bleomycin-induced lung fibrosis by aerosolization of heparin or urokinase in rabbits. Am J Respir Crit Care Med. 2003;168:1358–1365. doi:10.1164/rccm.2201082
105. Wu WJ, Vrhovski B, Weiss AS. Glycosaminoglycans mediate the coacervation of human tropoelastin through dominant charge interactions involving lysine side chains. J Biol Chem. 1999;274:21719–21724. doi:10.1074/jbc.274.31.21719
106. Raju KS, Alessandri G, Ziche M, Gullino PM. Ceruloplasmin, copper ions, and angiogenesis. J Natl Cancer Inst. 1982;69:1183–1188. doi:10.1093/jnci/69.5.1183
107. Li DD, Zhang W, Wang ZY, Zhao P. Serum copper, zinc, and iron levels in patients with alzheimer’s disease: a meta-analysis of case-control studies. Front Aging Neurosci. 2017;9:300. doi:10.3389/fnagi.2017.00300
108. Müller T, Müller W, Feichtinger H. Idiopathic copper toxicosis. Am J Clin Nutr. 1998;67:1082S–1086S. doi:10.1093/ajcn/67.5.1082S
109. Bendstrup KE, Chambers CB, Jensen JI, Newhouse MT. Lung deposition and clearance of inhaled (99m)Tc-heparin in healthy volunteers. Am J Respir Crit Care Med. 1999;160:1653–1658. doi:10.1164/ajrccm.160.5.9809123
110. Jaques LB, Mahadoo J, Kavanagh LW. Intrapulmonary heparin. A new procedure for anticoagulant therapy. Lancet. 1976;2:1157–1161. doi:10.1016/s0140-6736(76)91679-2
111. Bendstrup KE, Gram J, Jensen JI. Effect of inhaled heparin on lung function and coagulation in healthy volunteers. Eur Respir J. 2002;19:606–610. doi:10.1183/09031936.02.00105202
112. Shike M, Roulet M, Kurian R, Whitwell J, Stewart S, Jeejeebhoy KN. Copper metabolism and requirements in total parenteral nutrition. Gastroenterology. 1981;81:290–297. doi:10.1016/S0016-5085(81)80060-1
113. Pratt WB, Omdahl JL, Sorenson JR. Lack of effects of copper gluconate supplementation. Am J Clin Nutr. 1985;42:681–682. doi:10.1093/ajcn/42.4.681
114. Institute of Medicine (US) Panel on Micronutrients. Dietary Reference Intakes for Vitamin A, Vitamin K, Arsenic, Boron, Chromium, Copper, Iodine, Iron, Manganese, Molybdenum, Nickel, Silicon, Vanadium and Zinc. Washington, DC: National Academies Press (US); 2001.
115. Lages B, Stivala SS. Interaction of the polyelectrolyte heparin with copper(II) and calcium. Biopolymers. 1973;12:127–143. doi:10.1002/bip.1973.360120112
116. Chung MC, Ellerton NF. Viscosity at low shear and circular dichroism studies of heparin. Biopolymers. 1976;15:1409–1423. doi:10.1002/bip.1976.360150713
117. Rudd TR, Skidmore MA, Guimond SE, et al. Site-specific interactions of copper(II) ions with heparin revealed with complementary (SRCD, NMR, FTIR and EPR) spectroscopic techniques. Carbohydr Res. 2008;343:2184–2193. doi:10.1016/j.carres.2007.12.019
118. Marshall LM, Francis PW, Khafagi FA. Aerosol deposition in cystic fibrosis using an aerosol conservation device and a conventional jet nebulizer. J Paediatr Child Health. 1994;30:65–67. doi:10.1111/j.1440-1754.1994.tb00569.x
119. Hogg JC, Chu F, Utokaparch S, et al. The nature of small-airway obstruction in chronic obstructive pulmonary disease. N Engl J Med. 2004;350:2645–2653. doi:10.1056/NEJMoa032158
120. McDonough JE, Yuan R, Suzuki M, et al. Small-airway obstruction and emphysema in chronic obstructive pulmonary disease. N Engl J Med. 2011;365:1567–1575. doi:10.1056/NEJMoa1106955
121. Koo HK, Vasilescu DM, Booth S, et al. Small airways disease in mild and moderate chronic obstructive pulmonary disease: a cross-sectional study. Lancet Respir Med. 2018;6:591–602. doi:10.1016/S2213-2600(18)30196-6
122. Shute JK, Calzetta L, Cardaci V, Di Toro S, Page CP, Cazzola M. Inhaled nebulised unfractionated heparin improves lung function in moderate to very severe COPD: a pilot study. Pulm Pharmacol Ther. 2018;48:88–96. doi:10.1016/j.pupt.2017.10.001
123. Markart P, Nass R, Ruppert C, et al. Safety and tolerability of inhaled heparin in idiopathic pulmonary fibrosis. J Aerosol Med Pulm Drug Deliv. 2010;23:161–172. doi:10.1089/jamp.2009.0780
124. Umeda H, Aikawa M, Libby P. Liberation of desmosine and isodesmosine as amino acids from insoluble elastin by elastolytic proteases. Biochem Biophys Res Commun. 2011;411:281–286. doi:10.1016/j.bbrc.2011.06.124
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.