Back to Journals » International Journal of Chronic Obstructive Pulmonary Disease » Volume 11 » Issue 1
COPD disease severity and innate immune response to pathogen-associated molecular patterns
Authors Fan V ![]() , Gharib S, Martin T, Wurfel M
, Gharib S, Martin T, Wurfel M
Received 14 August 2015
Accepted for publication 7 November 2015
Published 4 March 2016 Volume 2016:11(1) Pages 467—477
DOI https://doi.org/10.2147/COPD.S94410
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Prof. Dr. Richard Russell
Vincent S Fan,1,2 Sina A Gharib,2,3 Thomas R Martin,1,2 Mark M Wurfel2
1VA Puget Sound Health Care System, Seattle, WA, USA; 2Division of Pulmonary & Critical Care Medicine, Department of Medicine, University of Washington, Seattle WA, USA; 3Computational Medicine Core, Center for Lung Biology, University of Washington, Seattle, WA, USA
Abstract: The airways of COPD patients are often colonized with bacteria leading to increased airway inflammation. This study sought to determine whether systemic cytokine responses to microbial pathogen-associated molecular patterns (PAMPs) are increased among subjects with severe COPD. In an observational cross-sectional study of COPD subjects, PAMP-induced cytokine responses were measured in whole blood ex vivo. We used PAMPs derived from microbial products recognized by toll-like receptors 1, 2, 4, 5, 6, 7, and 8. Patterns of cytokine response to PAMPs were assessed using hierarchical clustering. One-sided Student’s t-tests were used to compare PAMP-induced cytokine levels in blood from patients with and without severe COPD, and for subjects with and without chronic bronchitis. Of 28 male patients, 12 had moderate COPD (FEV1 50%–80%) and 16 severe COPD (FEV1 <50%); 27 participants provided data on self-reported chronic bronchitis, of which 15 endorsed chronic bronchitis symptoms and 12 did not. Cytokine responses to PAMPs in severe COPD were generally lower than in subjects with milder COPD. This finding was particularly strong for PAMP-induced interleukin (IL)-10, granulocyte colony stimulating factor, and IL-1β. Subjects with chronic bronchitis showed higher PAMP-induced IL-1RA responses to most of the PAMPs evaluated. COPD patients with more severe disease demonstrated a diminished cytokine response to PAMPs, suggesting that chronic colonization with bacteria may dampen the systemic innate immune response.
Keywords: chronic obstructive pulmonary disease, innate immunity, inflammation, chronic bronchitis, toll-like receptors
Introduction
Bacterial colonization of the lower airways is common in patients with stable COPD, but the role of the host immune response to infectious organisms in COPD is poorly understood. Toll-like receptors (TLRs) constitute a family of eleven transmembrane proteins that differentially recognize microbial products containing highly conserved motifs known as pathogen-associated molecular patterns (PAMPs).1 PAMP recognition through TLRs results in secretion of several cytokines including interleukin (IL)-8, MCP-1, MIP1α, tumor necrosis factor (TNF)α, and IL-1β.2,3 TLR-mediated responses are critical for innate immunity and modulation of the adaptive immune response.4
COPD patients have increased airway inflammation5–8 as well as increased systemic inflammation compared to those without COPD9 with elevation in levels of C-reactive protein, fibrinogen, circulating lymphocytes, and TNFα.10 There is evidence that the innate immune response in the airways and lung in COPD may be impaired;11,12 however, studies of innate immune responses in circulating peripheral leukocytes have shown mixed results. For example, peripheral blood monocytes from COPD patients secrete higher amounts of peptidoglycan-induced, but not lipopolysaccharide (LPS)-induced, TNFα and IL-6 ex vivo compared with controls.13 In addition, peripheral blood neutrophils from patients with COPD secrete slightly higher amounts of LPS-induced IL-8.14 There are no published studies linking innate immune responses mediated by TLRs in peripheral leukocytes and COPD severity.
We sought to better understand the relationship between COPD severity and innate immune responses to selected PAMPs likely to be present in airways of COPD patients. The primary aim of this pilot study was to determine whether PAMP-induced cytokine production in peripheral whole blood increases with disease severity.
Materials and methods
Study subjects
Subjects with a clinical history of COPD were recruited from a pulmonary clinic at an academic medical center in Seattle, WA, USA, between September 2006 and January 2008. Inclusion criteria were: 1) postbronchodilator FEV1/FVC <0.70; 2) FEV1 <80% predicted; 3) age 50 to 89 years; 4) >10 pack-years cigarette smoking; and 5) no respiratory illnesses and no treatment with prednisone or antibiotics in the past 4 weeks. Exclusion criteria were: 1) primary diagnosis of asthma; 2) >15% postbronchodilator change in FEV1; 3) chronic inflammatory or infectious disease; 4) cancer; 5) autoimmune disease; 6) chronic renal failure with a serum creatinine >1.5, 7) chronic liver disease; and 8) chronic antibiotic use.
Because of difficulty recruiting, after six subjects we modified the exclusion criteria to allow subjects with a bronchodilator response >15%, since bronchodilator response does not differentiate between COPD and asthma.15 In addition, we allowed those with chronic renal failure not requiring dialysis and nonmetastatic cancer (eg, prostate cancer) provided there was no history of lung cancer.
This study was approved by the VA Puget Sound Health Care System Human Studies Committee (ID: 01439) and has been registered with ClinicalTrials.gov. Identifier: NCT02637219. All subjects provided written informed consent.
Data collection
Spirometry
If no results were available in the last 12 months, spirometry was performed before and after administration of a short-acting bronchodilator.
Clinical factors
COPD disease severity was defined based on the Global Initiative for Chronic Obstructive Lung Disease (GOLD) categorization of lung function using spirometry, grouping participants into moderate (GOLD 2: 50%≤ FEV1 <80% predicted) and severe (GOLD 3–4: FEV1 <50% predicted).16 Subjects completed a questionnaire regarding smoking history, exacerbations, and COPD-related symptoms including chronic bronchitis (cough productive of sputum for at least 3 months for 2 consecutive years). COPD medications, COPD-related health care utilization, and comorbidities were abstracted from the medical record.
Whole blood stimulation assay
Subjects were asked to avoid vigorous exercise,17 alcohol,18,19 and nonsteroidal anti-inflammatory medications20,21 in the 24 hours preceding phlebotomy, which was performed between 8 am and 10 am to minimize circadian variations in cytokine responses.17,22 Whole blood (30 mL) was drawn into sterile-buffered citrate (0.2 M, pH 7.3) for anticoagulation, and transported to the analysis laboratory within 3 hours.
The assay of PAMP-induced inflammatory responses simultaneously measured whole blood responses to various PAMPs in a 96-well format as previously described.23 Seven different TLR agonists were tested (Table S1): 1) pam3CSK4 (N-palmitoyl-S-[2,3-bis(palmitoyl-oxy)-(2RS)-propyl]-[R]-cysteinyl-[S]-seryl-[S]-lysyl-[S]-lysyl-[S]-lysyl-[S]-lysine), a TLR1/2 agonist; 2) zymosan derived from Saccharomyces cerevisiae, a TLR6/2 agonist; 3) FSL-1 derived from Mycoplasma salivarium, a TLR6/2 agonist; 4) both smooth form LPS (O111:B4) from Escherichia coli and rough form LPS (Re595) from Salmonella Minnesota, TLR4 agonists; 5) flagellin from Salmonella typhimurium, a TLR5 agonist, and 6) R848, an imidazoquinoline compound that is a TLR7/TLR8 agonist.24,25
Whole blood samples were mixed 1:1 with RPMI-1640 medium (Thermo Fisher Scientific, Waltham, MA, USA), and applied to a 96-well plate containing PAMP dilutions within 60 minutes of phlebotomy. The plates were incubated at 37°C for 6 hours in a 5% CO2 incubator, supernatants collected, and the concentrations of inflammatory (IL-1β, IL-6, IL-8, TNFα, MCP-1, granulocyte colony stimulating factor) and anti-inflammatory (IL-1RA, IL-10) cytokines and chemokines were measured using a multiplex cytometric polystyrene bead-based immunoassay system as dictated by the manufacturer (R&D Luminex, Austin, TX, USA).26,27 For immunoassay values that fell below the limits of detection, we substituted the value of the lower limit of detection minus 10%. For values that were above the upper limit of detection, we substituted the value of the upper limit of detection plus 10%.
Analysis
Cytokine measurements were done in duplicate and average values were calculated. Unless otherwise specified, analyses were performed using STATA (StataCorp LP, College Station, TX, USA). Cytokine levels were nonnormally distributed and therefore displayed as medians with inter-quartile range.
To identify patterns of cytokine response to PAMPs using a nonbiased approach, we used cluster analysis to map the cytokine level data for every cytokine–PAMP combination by individual subject.28 Cytokine levels were first log2-transformed, and then data for each cytokine-PAMP combination were normalized and analyzed using two-dimensional hierarchical clustering using Euclidean distance metric.
To examine whether cytokine responses to each PAMP differed based on clinical characteristics (eg, disease severity or history of chronic bronchitis), we used Student’s t-tests to compare each cytokine–PAMP response after log-transformation of cytokine levels. To test our hypothesis that the leukocytes of patients with more severe disease would in general produce more PAMP-induced cytokines, we used one-sided statistical tests to determine the significance of our results.
To visualize patterns in associations between PAMP-induced cytokine responses and COPD patient characteristics, we used the P-values generated by the one-sided Student’s t-tests to produce heatmaps in which the strength of the association is represented by a different color. We created a heatmap for the hypothesis that patients with severe COPD would have higher PAMP-induced cytokine levels, and another complementary heatmap for the alternate hypothesis that severe COPD would be associated with lower cytokine levels. Given the small sample size, no adjusted analyses were performed. To explore whether the association between disease severity and cytokine response differed by inhaled steroid use, we performed stratified analyses by inhaled steroid use.
Because of the large number of hypothesis testing for cytokine–PAMP combinations, we applied false discovery rate analysis using the Q-value,29 which is preferred over Bonferroni-type adjustments for controlling false positives in experimental designs including multiple partially correlated measures.30 We used a bootstrapping approach to calculate Q-values based on P-values from the 256 comparisons in both upper and lower one-sided Student’s t-tests for FEV1 and chronic bronchitis models. We selected a false discovery rate (Q-value) cut-off value of <10% to designate significance.
Results
Subject characteristics
The mean age of participants was 69.3 years, and 57% had severe disease (FEV1 <50% predicted) (Table 1). In all, 75% were using a corticosteroid inhaler, with no difference in inhaled steroid use between those with moderate and severe COPD (75% vs 81%, P=0.69).
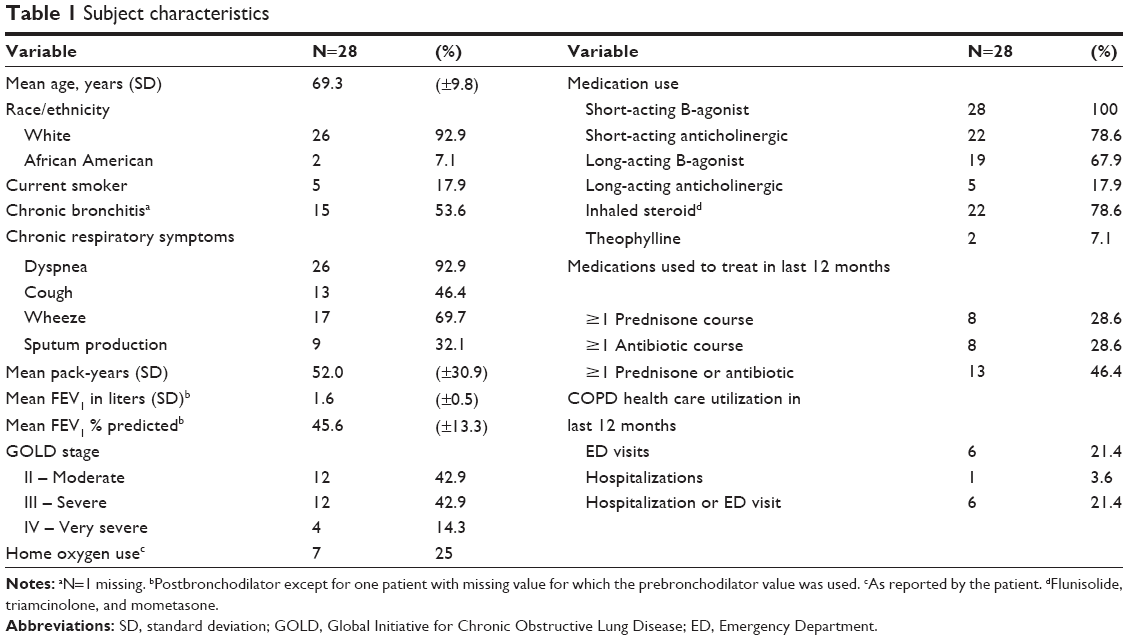 | Table 1 Subject characteristics |
Disease severity and cytokine response of whole blood to stimulation by PAMPs
In an unbiased analysis using two-way hierarchical clustering of all cytokine–PAMP pairs across subjects, many patients with severe COPD (ten out of 12 subjects to the left of Figure 1) had decreased cytokine production in response to PAMPs. The exception was production of the anti-inflammatory cytokine IL-1RA, in which the response was more heterogeneous in severe COPD. We did not observe segregation of subjects based on their PAMP–cytokine response when they were classified according to their chronic bronchitis status, with the exception of IL-1RA levels which were generally increased in patients with chronic bronchitis. Of note, the clustering algorithm tended to group responses by cytokine responses rather than by the stimulating PAMP, suggesting that agonists for different TLRs induced similar patterns of cytokine production in whole blood from COPD subjects.
 | Figure 1 Hierarchical clustering of cytokine-PAMP combinations by subject. |
We then examined the strength of the statistical association between COPD severity and each cytokine–PAMP combination. Heatmaps were generated showing the pattern of associations for two alternative hypotheses: 1) severe disease is associated with increased cytokine production; and 2) severe COPD is associated with reduced cytokine production (Figure 2A). The one-sided Student’s t-test results showed that the majority of PAMP–cytokine combinations in severe COPD had decreased cytokine production in response to stimulation with most PAMPs when compared to patients with less severe COPD. Severe COPD was associated with decreased IL-10 levels in response to zymosan, FSL-1, 0111:B4, and Re595UP, and decreased granulocyte colony stimulating factor and IL-1β production in response to all PAMPs except flagellin and R848. There was no difference in PAMP-induced IL-1RA production by disease severity. Unstimulated whole blood levels of TNFα and IL-6 (labeled as media only) were reduced in patients with more severe COPD, whereas unstimulated IL-6 was increased in those with severe COPD. Tables 2–4 show the magnitude of the nontransformed differences observed between severe and moderate COPD.
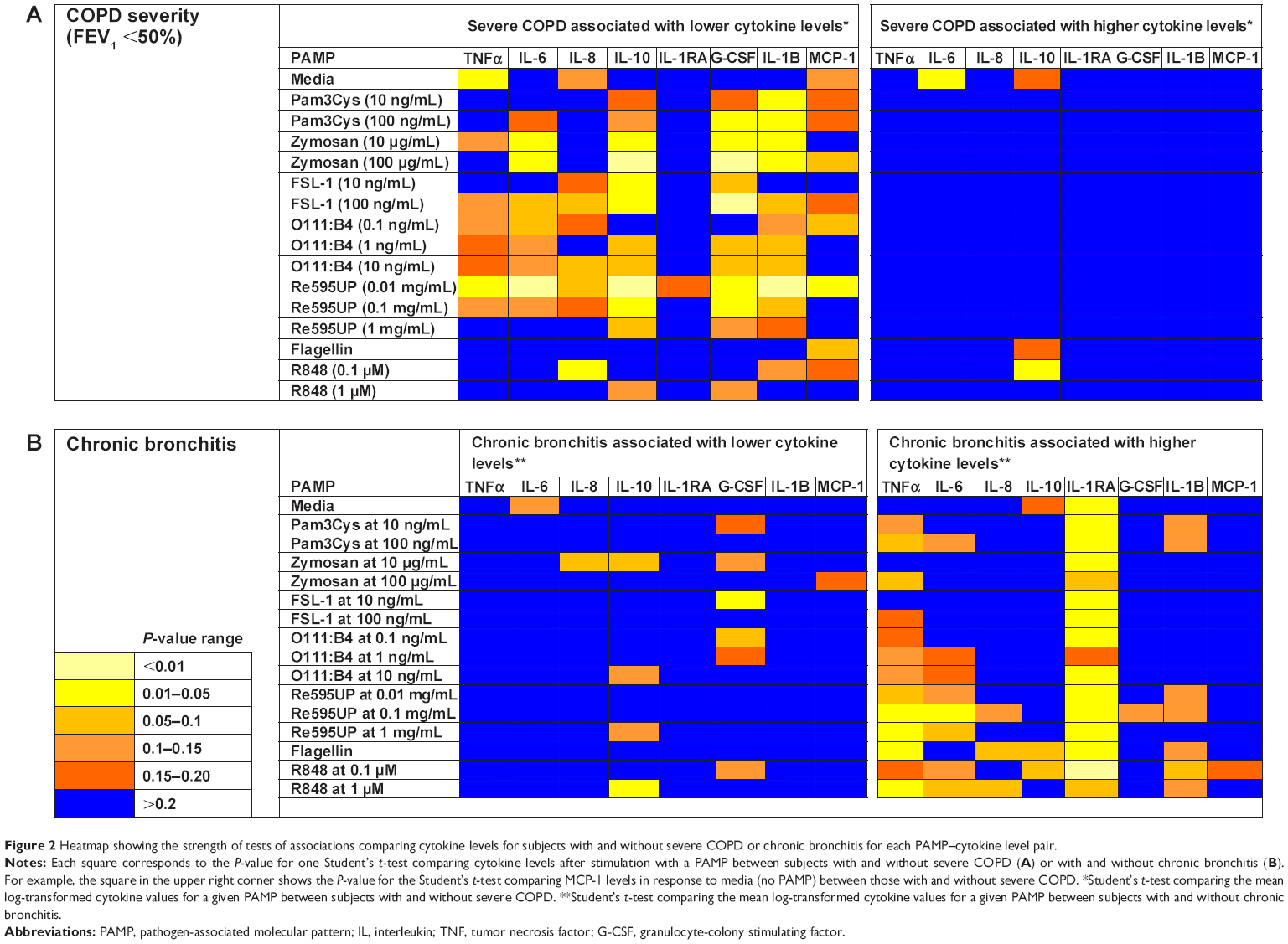 | Figure 2 Heatmap showing the strength of tests of associations comparing cytokine levels for subjects with and without severe COPD or chronic bronchitis for each PAMP–cytokine level pair. |
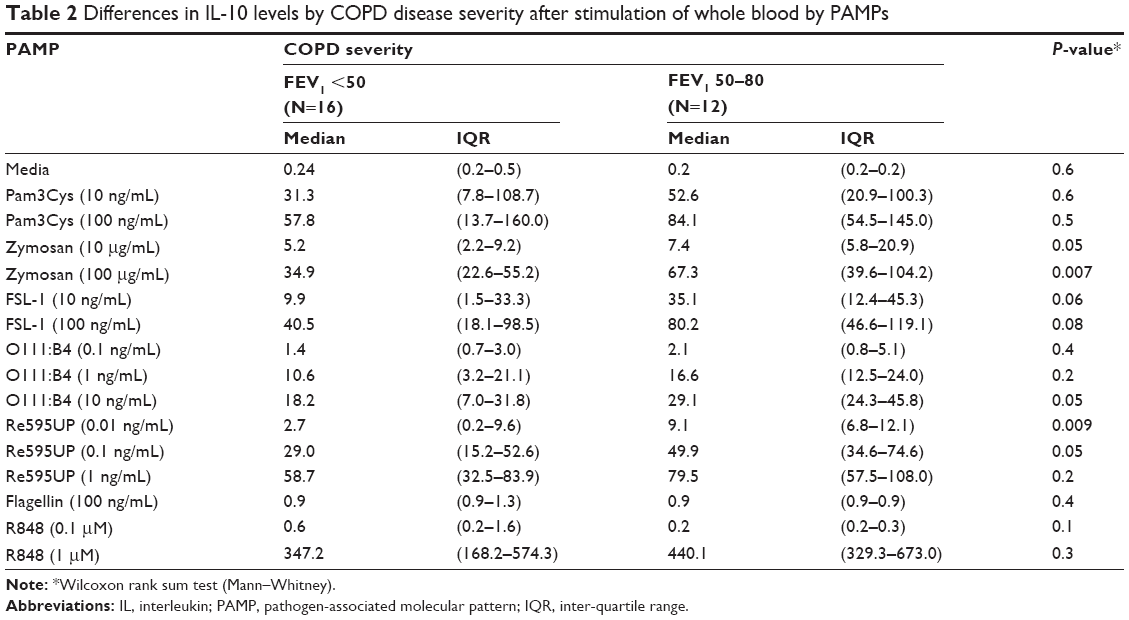 | Table 2 Differences in IL-10 levels by COPD disease severity after stimulation of whole blood by PAMPs |
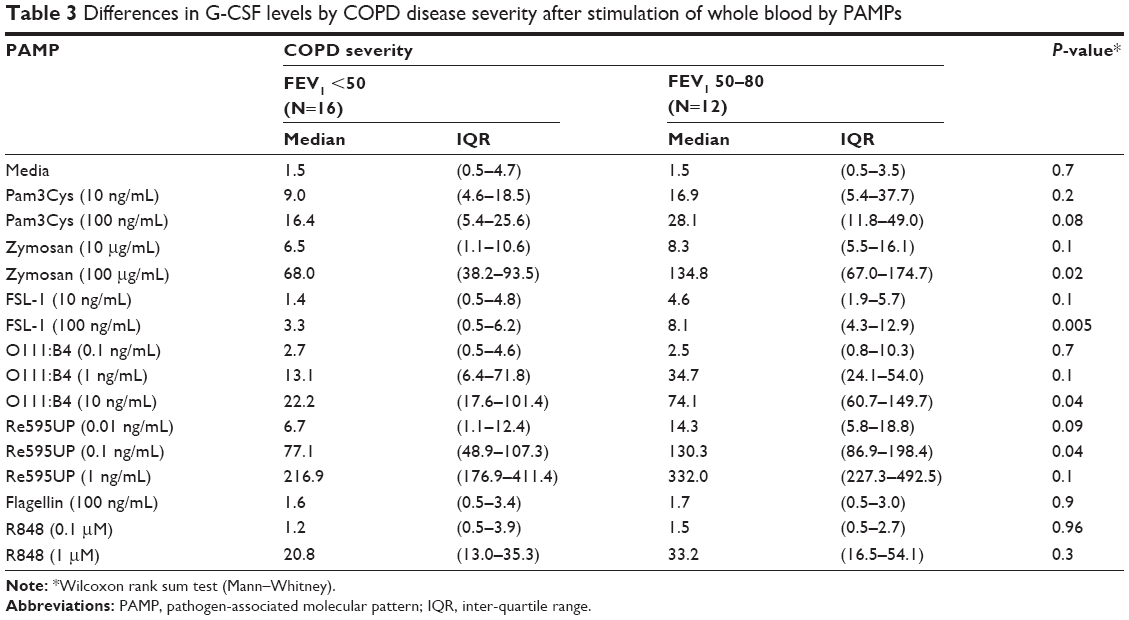 | Table 3 Differences in G-CSF levels by COPD disease severity after stimulation of whole blood by PAMPs |
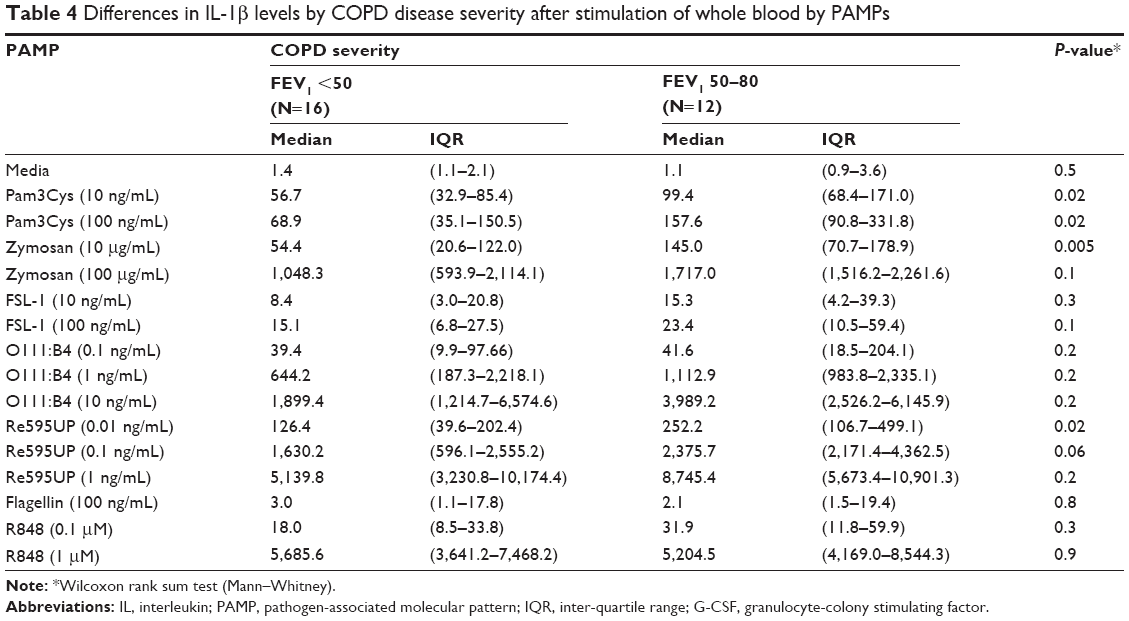 | Table 4 Differences in IL-1β levels by COPD disease severity after stimulation of whole blood by PAMPs |
Self-reported symptoms of chronic bronchitis were associated with increased cytokine production in whole blood for several PAMP–cytokine combinations compared to those without chronic bronchitis (Figure 2B). IL-1RA levels were increased in both unstimulated (media only) and PAMP-stimulated conditions in those with chronic bronchitis relative to those without bronchitis (Table 5 for nontransformed differences). Levels of TNFα were also elevated in response to flagellin, Re595UP, zymosan, and R848.
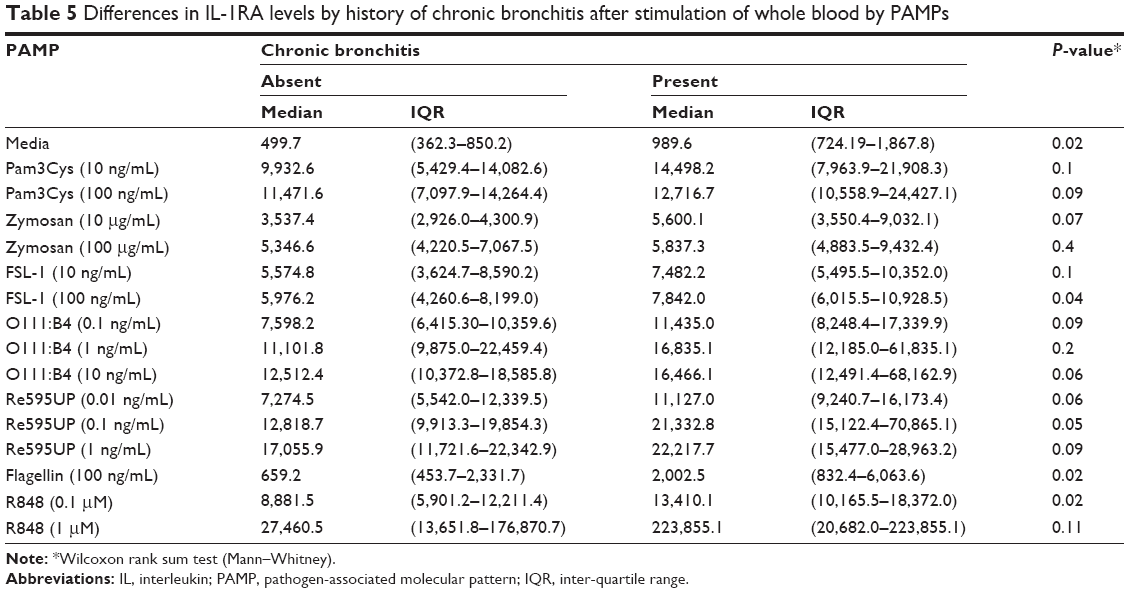 | Table 5 Differences in IL-1RA levels by history of chronic bronchitis after stimulation of whole blood by PAMPs |
To address multiple comparisons we calculated Q-values for each Student’s t-test comparing cytokine levels between subjects with and without severe COPD and chronic bronchitis. We identified 26 tests with results meeting a threshold false discovery rate (Q-value) ≤10% when testing the hypothesis that increased COPD severity is associated with decreased inflammation, and 19 tests for the hypothesis that chronic bronchitis is associated with increased inflammation. Therefore, most of the heatmap patterns observed using one-sided P-values in Figure 2 remained unaltered even after adjustment for multiple testing (Figure S1).
Because inhaled steroids may reduce systemic inflammation in COPD, we performed exploratory stratified analyses to see if the relationship between disease severity and cytokine levels differed between users and nonusers of inhaled steroids. Most patients, regardless of disease severity, were using inhaled steroids, which were associated with reduced PAMP-induced cytokine production (Figure S2).
However, when stratifying the analysis by inhaled steroid user, there was a similar pattern of reduction in PAMP-induced cytokine production among the 22 inhaled steroid users (Figure S3A). Among the six patients not using inhaled steroids (Figure S3B), more cytokines levels were reduced in the three subjects with severe COPD than in the three with less severe COPD; however, results should be interpreted with caution due to the small sample size.
Discussion
In a small sample of well-characterized COPD subjects, we found that those with severe COPD have decreased whole blood leukocyte responsiveness to PAMPs ex vivo as compared to subjects with less severe COPD. This suggests that the receptor and/or signaling mechanisms that mediate PAMP-induced responses are downregulated or suppressed in severe COPD. A potential cause of this downward modulation could be chronic stimulation by bacteria colonizing the airways of patients with COPD. In contrast to higher COPD severity, we found that subjects with chronic bronchitis have a tendency toward higher PAMP-induced cytokine responses, with significant elevations seen in PAMP-stimulated IL-1RA.
Chronic bacterial colonization of the airways is common in COPD with increasing prevalence as disease severity worsens: 25% with moderate COPD and more than 40% with severe disease have airways colonized with bacteria.31 Chronic colonization is associated with worse airway inflammation and more frequent exacerbations,3 with more neutrophils and higher levels of IL-8 and MMP-9.32,33 Another potential consequence of bacterial colonization of the airways in COPD is impairment of the host innate immune system resulting from repeated exposure to bacterial PAMPs.3 Our findings demonstrating reduced PAMP-induced responses in patients with more severe COPD are consistent with this possibility. Further support for PAMP-induced downregulation of innate immune responses was shown in a recent study comparing responses of alveolar macrophages (AM) obtained from bronchoalveolar lavage from COPD patients with frequent or infrequent exacerbations,34 in terms of LPS and pam3CSK4-induced IL-8 and TNFα production. This study showed reduced AM responses among frequent exacerbators who also had significantly reduced FEV1 compared with infrequent exacerbators. Also, AM isolated from COPD patients showed reduced LPS-induced expression of the TLR2 gene that was similar to smokers without COPD when compared with AM from nonsmokers.35 Thus, our findings in whole blood are consistent with an increasing body of literature demonstrating reduced innate immune responses in COPD patients with more severe lung disease.
How do our findings fit in the context of prior studies demonstrating increased levels of circulating inflammatory biomarkers in COPD? It is postulated that airway inflammation in COPD may lead to systemic inflammation. One theory suggests that airway inflammation “spills over” from the lung to the systemic circulation in COPD.9 In addition, bacterial products could also get access to blood in pulmonary microcirculation, affecting the systemic immune response. Notably, most prior studies of systemic inflammation in COPD have compared subjects with lung disease to control subjects without lung disease and have not looked at differences in inflammation across COPD subtypes.10 More recently, researchers using data from the ECLIPSE study found that increasing disease severity is associated with a small but significantly increased risk of persistent systemic inflammation.36
In contrast, during admission for community acquired pneumonia, patients with COPD demonstrate lower systemic inflammation than non-COPD community acquired pneumonia controls.37 This suggests that even during community acquired pneumonia when the patient is exposed to PAMPs released by the infecting pathogens, patients with COPD demonstrate a blunted innate immune response. Our results are more in line with this model in which leukocytes in patients with more severe COPD have an attenuated innate immune response.
We found decreased cytokine production with more severe COPD in response to bacterial PAMPs that bind to TLR-1/2, TLR-2/6, and TLR-4, but not those binding TLR5 or TLR7/8. Bacteria identified from COPD patients via bronchoalveolar lavage include Haemophilus influenza, Streptococcus pneumoniae, Moraxella catarrhalis, Enterobacteriaceae, and less commonly Pseudomonas aeruginosa, or Staphylococcus aureus.33 Most of these pathogenic bacteria produce PAMPs that would bind TLR-1/2 or TLR-2/6 and all gram-negative species have LPS that would bind TLR4.38 Although P. aeruginosa is flagellated, it is a fairly uncommon bacterial species to colonize the airways of COPD patients, and in our study the cytokine response to flagellin did not appear related to disease severity. Similarly, viral colonization is less likely in stable COPD, which may explain why responsiveness to TLR-7/8 did not differ by COPD severity.
We also found that the presence of chronic bronchitis is associated with increased PAMP-induced responses for some cytokines. COPD patients who produce sputum are more likely to have bacterial colonization (60%), with increased inflammation and significantly more neutrophils and higher levels of cytokines in the airways (eg, MCP-1, IL-6, TNFα) than nonsputum producers.39 Our results suggest that patients with chronic bronchitis represent a subset of patients with COPD with a distinct immune phenotype. This is consistent with the findings that distinct phenotypic groups characterized by airway inflammation or airflow obstruction can be identified among COPD patients.40
Our results show that patients with severe COPD exhibit a broad downregulation of the innate immune response to bacterial PAMPs. This phenomenon has been referred to as “microbial tolerance”, which may have the adverse consequence of limiting the inflammatory response during an actual infection.41 A similar immune tolerance has been observed in patients with cystic fibrosis whose peripheral blood monocytes had decreased inflammatory responses to LPS compared to health volunteers.42 The mechanisms underlying this downregulation or tolerance are not known but could involve the modulation of intracellular signaling molecules and/or negative feedback through autocrine effects of type 1 interferons.43,44
Our study has several limitations. The sample size was modest, and to ensure adequate recruitment in a sick older population with many comorbidities, we included patients with medical conditions that may be associated with systemic inflammation such as renal failure or nonmetastatic cancer. In addition, the majority of subjects (~80%) were using inhaled steroids which may have impacted the systemic innate immune response. Inhaled steroid use was similar between subjects with moderate and severe COPD, reducing the likelihood that it confounded our disease severity results. When restricting the analysis to inhaled steroid users, we saw a very similar pattern of reduced cytokine response to PAMP stimulation, suggesting that this diminished cytokine response occurs above and beyond the anti-inflammatory effects of inhaled steroids. Also, among the six patients not using inhaled steroids, cytokine responses to PAMPs still tended to be reduced among those with severe COPD compared to less severe COPD. A small proportion of the subjects (N=5) were active smokers, which may also have affected the results. We excluded other conditions that were likely to confound a relationship with severity including any exacerbation or worsening respiratory symptoms in the 4-week period prior to the study visit. Although no participant had a clinical diagnosis of asthma, we did include three patients with a bronchodilator response >15% and >400 cc improvement in FEV1 who may have had asthma–COPD overlap syndrome, which may have affected our results. It is important to note that this study may not be generalizable to women. Due to the small sample size, we could not perform adjusted analyses. Finally, the PAMP-induced response assays were performed with whole blood, not purified cell lines (ie, monocytes, neutrophils, lymphocytes, etc), so we cannot identify the cells responsible for cytokine production measured or the extent to which these responses are reflective of the alveolar and airway responses. However, there may be advantages to use whole-blood stimulation assays since they may more accurately reflect the innate immune inflammatory response to PAMP stimuli.45
Our study has several strengths. We used a well-characterized sample of patients with COPD. The blood samples were obtained and the stimulation assays performed under highly controlled conditions. Finally, the simultaneous measurement of the production of nine cytokines/chemokines in response to 15 different PAMP preparations allowed for a comprehensive assessment of peripheral blood innate immune responses.
Conclusion
We found that patients with more severe COPD had decreased innate immune response to bacterial PAMPs and that chronic sputum producers had elevated levels of the anti-inflammatory cytokine IL-1RA in response to bacterial PAMP stimulation. Overall, our findings raise the intriguing possibility that although COPD pathogenesis may be due to chronic innate immune activation, a paradoxical impairment of innate immunity characterizes those with more severe disease. Deficient immune response may then lead to increased susceptibility to airway infections and repeated exacerbations in severe COPD. Future studies with a larger sample size are needed to confirm these results and delineate mechanistic details and evaluate the clinical consequences of these observations.
Acknowledgments
Angela Horn assisted in recruitment, study visits, and data management for the study. This work was supported by Career Development Award, Award # 02-170-2, from the United States Department of Veterans Affairs Health Services Research and Development Program, as well as a grant by the National Institute of Allergy and Infectious Diseases (Award # U54 AI057141). The views expressed in this article are those of the authors and do not necessarily represent the views of the Department of Veterans Affairs.
Disclosure
The authors report no conflicts of interest in this work.
References
Bezemer GF, Sagar S, van Bergenhenegouwen J, et al. Dual role of Toll-like receptors in asthma and chronic obstructive pulmonary disease. Pharmacol Rev. 2012;64(2):337–358. | ||
Iwasaki A, Medzhitov R. Toll-like receptor control of the adaptive immune responses. Nat Immunol. 2004;5(10):987–995. | ||
Opitz B, van Laak V, Eitel J, Suttorp N. Innate immune recognition in infectious and noninfectious diseases of the lung. Am J Respir Crit Care Med. 2010;181(12):1294–1309. | ||
Kawai T, Akira S. The role of pattern-recognition receptors in innate immunity: update on Toll-like receptors. Nat Immunol. 2010;11(5):373–384. | ||
Vernooy JH, Kucukaycan M, Jacobs JA, et al. Local and systemic inflammation in patients with chronic obstructive pulmonary disease: soluble tumor necrosis factor receptors are increased in sputum. Am J Respir Crit Care Med. 2002;166(9):1218–1224. | ||
Yamamoto C, Yoneda T, Yoshikawa M, et al. Airway inflammation in COPD assessed by sputum levels of interleukin-8. Chest. 1997;112(2):505–510. | ||
Pesci A, Balbi B, Majori M, et al. Inflammatory cells and mediators in bronchial lavage of patients with chronic obstructive pulmonary disease. Eur Respir J. 1998;12(2):380–386. | ||
Riise GC, Ahlstedt S, Larsson S, et al. Bronchial inflammation in chronic bronchitis assessed by measurement of cell products in bronchial lavage fluid. Thorax. 1995;50(4):360–365. | ||
Agusti A. Systemic effects of chronic obstructive pulmonary disease: what we know and what we don’t know (but should). Proc Am Thorac Soc. 2007;4(7):522–525. | ||
Gan WQ, Man SF, Senthilselvan A, Sin DD. Association between chronic obstructive pulmonary disease and systemic inflammation: a systematic review and a meta-analysis. Thorax. 2004;59(7):574–580. | ||
Thorley AJ, Tetley TD. Pulmonary epithelium, cigarette smoke, and chronic obstructive pulmonary disease. Int J Chron Obstruct Pulmon Dis. 2007;2(4):409–428. | ||
Shaykhiev R, Crystal RG. Innate immunity and chronic obstructive pulmonary disease: a mini-review. Gerontology. 2013;59(6):481–489. | ||
Pons J, Sauleda J, Regueiro V, et al. Expression of Toll-like receptor 2 is up-regulated in monocytes from patients with chronic obstructive pulmonary disease. Respir Res. 2006;7:64. | ||
Baines KJ, Simpson JL, Gibson PG. Innate immune responses are increased in chronic obstructive pulmonary disease. PLoS one. 2011;6(3):e18426. | ||
Tashkin DP, Celli B, Decramer M, et al. Bronchodilator responsiveness in patients with COPD. Eur Respir J. 2008;31(4):742–750. | ||
Global Strategy for the Diagnosis, Management and Prevention of COPD, Global Initiative for Chronic Obstructive Lung Disease (GOLD): Updated 2015. 2015. | ||
DeRijk R, Michelson D, Karp B, et al. Exercise and circadian rhythm-induced variations in plasma cortisol differentially regulate interleukin-1 beta (IL-1 beta), IL-6, and tumor necrosis factor-alpha (TNF alpha) production in humans: high sensitivity of TNF alpha and resistance of IL-6. J Clin Endocrinol Metab. 1997;82(7):2182–2191. | ||
Nelson S, Bagby GJ, Bainton BG, Summer WR. The effects of acute and chronic alcoholism on tumor necrosis factor and the inflammatory response. J Infect Dis. 1989;160(3):422–429. | ||
Standiford TJ, Danforth JM. Ethanol feeding inhibits proinflammatory cytokine expression from murine alveolar macrophages ex vivo. Alcohol Clin Exp Res. 1997;21(7):1212–1217. | ||
Lozanski G, Ballou SP, Kushner I. Effect of flurbiprofen on cytokine production by human monocytes and U-937 and THP-1 cell lines. J Rheumatol. 1992;19(6):921–926. | ||
Housby JN, Cahill CM, Chu B, et al. Non-steroidal anti-inflammatory drugs inhibit the expression of cytokines and induce HSP70 in human monocytes. Cytokine. 1999;11(5):347–358. | ||
Entzian P, Linnemann K, Schlaak M, Zabel P. Obstructive sleep apnea syndrome and circadian rhythms of hormones and cytokines. Am J Respir Crit Care Med. 1996;153(3):1080–1086. | ||
Wurfel MM, Gordon AC, Holden TD, et al. Toll-like receptor 1 polymorphisms affect innate immune responses and outcomes in sepsis. Am J Respir Crit Care Med. 2008;178(7):710–720. | ||
Hemmi H, Kaisho T, Takeuchi O, et al. Small anti-viral compounds activate immune cells via the TLR7 MyD88-dependent signaling pathway. Nat Immunol. 2002;3(2):196–200. | ||
Jurk M, Heil F, Vollmer J, et al. Human TLR7 or TLR8 independently confer responsiveness to the antiviral compound R-848. Nat Immunol. 2002;3(6):499. | ||
Prabhakar U, Eirikis E, Davis HM. Simultaneous quantification of proinflammatory cytokines in human plasma using the LabMAP assay. J Immunol Methods. 2002;260(1–2):207–218. | ||
Khan SS, Smith MS, Reda D, Suffredini AF, McCoy JP Jr. Multiplex bead array assays for detection of soluble cytokines: comparisons of sensitivity and quantitative values among kits from multiple manufacturers. Cytometry. 2004;61B(1):35–39. | ||
Saeed AI, Sharov V, White J, et al. TM4: a free, open-source system for microarray data management and analysis. Biotechniques. 2003;34(2):374–378. | ||
Storey JD. A direct approach to false discovery rates. J R Statist Soc B. 2002;64, Part 3:479–498. | ||
Glickman ME, Rao SR, Schultz MR. False discovery rate control is a recommended alternative to Bonferroni-type adjustments in health studies. J Clin Epidemiol. 2014;67(8):850–857. | ||
Rosell A, Monso E, Soler N, et al. Microbiologic determinants of exacerbation in chronic obstructive pulmonary disease. Arch Intern Med. 2005;165(8):891–897. | ||
Sethi S, Maloney J, Grove L, Wrona C, Berenson CS. Airway inflammation and bronchial bacterial colonization in chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2006;173(9):991–998. | ||
Soler N, Ewig S, Torres A, Filella X, Gonzalez J, Zaubet A. Airway inflammation and bronchial microbial patterns in patients with stable chronic obstructive pulmonary disease. Eur Respir J. 1999;14(5):1015–1022. | ||
Berenson CS, Kruzel RL, Eberhardt E, et al. Impaired innate immune alveolar macrophage response and the predilection for COPD exacerbations. Thorax. 2014;69(9):811–818. | ||
Droemann D, Goldmann T, Tiedje T, Zabel P, Dalhoff K, Schaaf B. Toll-like receptor 2 expression is decreased on alveolar macrophages in cigarette smokers and COPD patients. Respir Res. 2005;6:68. | ||
Agusti A, Edwards LD, Rennard SI, et al. Persistent systemic inflammation is associated with poor clinical outcomes in COPD: a novel phenotype. PLoS One. 2012;7(5):e37483. | ||
Crisafulli E, Menendez R, Huerta A, et al. Systemic inflammatory pattern of patients with community-acquired pneumonia with and without COPD. Chest. 2013;143(4):1009–1017. | ||
Baral P, Batra S, Zemans RL, Downey GP, Jeyaseelan S. Divergent functions of Toll-like receptors during bacterial lung infections. Am J Respir Crit Care Med. 2014;190(7):722–732. | ||
Khurana S, Ravi A, Sutula J, et al. Clinical characteristics and airway inflammation profile of COPD persistent sputum producers. Respir Med. 2014;108(12):1761–1770. | ||
Lapperre TS, Snoeck-Stroband JB, Gosman MM, et al. Dissociation of lung function and airway inflammation in chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2004;170(5):499–504. | ||
Medvedev AE, Sabroe I, Hasday JD, Vogel SN. Tolerance to microbial TLR ligands: molecular mechanisms and relevance to disease. J Endotoxin Res. 2006;12(3):133–150. | ||
del Fresno C, Gomez-Pina V, Lores V, et al. Monocytes from cystic fibrosis patients are locked in an LPS tolerance state: down-regulation of TREM-1 as putative underlying mechanism. PLoS One. 2008;3(7):e2667. | ||
Bode JG, Ehlting C, Haussinger D. The macrophage response towards LPS and its control through the p38(MAPK)-STAT3 axis. Cell Signal. 2012;24(6):1185–1194. | ||
Biswas SK, Lopez-Collazo E. Endotoxin tolerance: new mechanisms, molecules and clinical significance. Trends Immunol. 2009;30(10):475–487. | ||
Duffy D, Rouilly V, Libri V, et al. Functional analysis via standardized whole-blood stimulation systems defines the boundaries of a healthy immune response to complex stimuli. Immunity. 2014;40(3):436–450. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.