Back to Journals » Neuropsychiatric Disease and Treatment » Volume 17
COP9 Signalosome Subunit 3 Restricts Neuroinflammatory Responses During Cerebral Ischemia/Reperfusion Injury Through Stabilizing Suppressor of Cytokine Signaling 3 Protein
Authors Liang E, Li X, Fu W, Zhao C, Yang B, Yang Z
Received 30 December 2020
Accepted for publication 22 March 2021
Published 22 April 2021 Volume 2021:17 Pages 1217—1227
DOI https://doi.org/10.2147/NDT.S298966
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Yu-Ping Ning
En Liang,1 Xiaojun Li,2 Wenjun Fu,2 Changtong Zhao,1 Baoying Yang,3 Zhonghua Yang2
1Department of Neurosurgery, The Affiliated Hexian Memorial Hospital of Southern Medical University, Guangzhou, People’s Republic of China; 2Centre for Integrative Medicine, School of Basic Medical Science, Guangzhou University of Chinese Medicine, Guangzhou, People’s Republic of China; 3Department of Neurosurgery, Guangdong Sanjiu Brain Hospital, Guangzhou, People’s Republic of China
Correspondence: Baoying Yang
Department of Neurosurgery, Guangdong Sanjiu Brain Hospital, No. 578 Shatai Nan Road, Baiyun District, Guangzhou, 510510, People’s Republic of China
Email [email protected]
Zhonghua Yang
Centre for Integrative Medicine, School of Basic Medical Science, Guangzhou University of Chinese Medicine, No. 232 Waihuan Dong Road, Panyun District, Guangzhou, 510006, People’s Republic of China
Email [email protected]
Background: The suppressor of cytokine signaling 3 (SOCS3) is a specific negative regulator of signal transducer and activator of transcription 3 (STAT3) signaling, which is predominantly activated to induce neuroinflammatory response in microglia and functions essential roles during cerebral ischemia-reperfusion (I/R) injury. Constitutive photomorphogenesis 9 (COP9) signalosome (CSN) is a signaling platform controlling protein stability by remodeling of cullin-RING ubiquitin ligases, which is recently reported to specifically recognize proteins with SOCS-box domains. However, whether SOCS3 is related to COP9 signalosome in neuroinflammation during cerebral I/R injury is completely unclear.
Methods: Mice subjected to transient middle cerebral artery occlusion (MCAO) and reperfusion, and BV2 microglia cells treated with oxygen-glucose deprivation and reoxygenation (OGD/R) were used to mimic cerebral I/R injury. Western blot, qRTPCR, immunofluorescence, and co-Immunoprecipitation assays were performed to explore the regulatory mechanism of SOCS3 on neuroinflammation and the relationship of SOCS3 and COP9 signalosome during cerebral I/R injury.
Results: SOCS3 expression is significantly upregulated in microglia during OGD/R treatment, and overexpression of SOCS3 suppresses OGD/R-induced STAT3 activation and inflammatory factor expression. Furthermore, we find that COP9 signalosome subunit 3 (CSN3) interacts with SOCS3 protein to enhance its stability, thereby resulting in restricting OGD/R-induced STAT3 activation and inflammatory response. Moreover, we find that knockdown of CSN3 evidently accelerates STAT3 activation, and aggravates cerebral I/R injury in vivo.
Conclusion: CSN3 restricts neuroinflammatory responses during cerebral I/R injury through stabilizing SOCS3 protein and indicates that CSN3 a potential therapeutic target for cerebral I/R injury.
Keywords: cerebral ischemia-reperfusion injury, neuroinflammation, suppressor of cytokine signaling 3, constitutive photomorphogenesis 9 signalosome, signal transducer and activator of transcription 3
Introduction
Ischemic stroke is a major cause of disability and death all over the world.1,2 Nowadays, thrombolysis treatment is a common therapeutic strategy for patients with acute cerebral ischemia in clinical, whereas recanalization of occluded arteries usually leads to a secondary injury, namely cerebral ischemia-reperfusion (I/R) injury.3,4 Many studies have reported that local excessive neuroinflammatory response in brain tissue is an essential pathogenesis of cerebral I/R.5–7 Thus, understanding the underlying mechanisms of neuroinflammation during cerebral I/R injury is important for developing new drugs for cerebral I/R therapy.
Microglia, as a resident macrophage in the brain, is activated during cerebral I/R process and produces many pro-inflammatory factors, such as tumor necrosis factor (TNFα) and interleukin-6 (IL-6), to aggravate brain injury8,9. The signal transducer and activator of transcription 3 (STAT3), an important transcriptional regulator of inflammatory gene, is phosphorylated and activated in macroglia by various stimuli, and induces pro-inflammatory immune reaction, such as IL-6, in response to various central nervous system insults, including cerebral I/R injury.10 Excreted IL-6 binds to the soluble IL-6 receptor on the cell membrane, subsequently resulting in STAT3 phosphorylation, and induction of IL-6 expression, which forms a positive feedback to intensify neuroinflammation,11 suggesting that STAT3 as a new therapeutic target for cerebral I/R injury. The suppressor of cytokine signaling (SOCS) 3 is found as a specific negative regulator of STAT3 signaling by inhibiting STAT3 phosphorylation.12 In addition, activation of SOCS3-STAT3 signaling by G14-humanin has been reported to have neuroprotective effects on cerebral I/R injury in rats.13 However, how SOCS3 is regulated in neuroinflammation during cerebral I/R injury is still not clear.
The constitutive photomorphogenesis 9 (COP9) signalosome (CSN) is an evolutionarily conserved multi-protein complex, consisting of eight subunits termed CSN1-CSN8, and is a signaling platform controlling the cellular ubiquitylation by remodeling cullin-RING ubiquitin ligases and interacting with deubiquitylating enzymes to rescue ubiquitylated proteins from degradation.14,15 COP9 signalosome is reported to be involved in regulating cell cycle DNA repair, and inflammatory gene expression through controlling protein degradation via the ubiquitin-proteasome system.16 For example, COP9 signalosome subunit 3 (CSN3), binds transcriptional factor interferon regulatory factor 5 to maintain its stability.17 CSN3 interacts with ATP-binding cassette transporters A1 (ABCA1) to mediate ABCA1 stability to promote cholesterol efflux.18 Recently, proteins with SOCS-box domains could be specifically recognized by cullin-RING ubiquitin ligases,19 which is regulated by COP9 signalosome.14 Thus, we suspected whether SOCS3 is associated with COP9 signalosome in neuroinflammation during cerebral I/R injury.
In this study, the regulatory mechanism of SOCS3 and the relationship between SOCS3 and COP9 signalosome in neuroinflammation during cerebral I/R injury were investigated using BV2 microglia cells treated with oxygen-glucose deprivation and reoxygenation (OGD/R) in vitro and mice subjected to focal cerebral I/R in vivo. This study firstly indicates CSN3 restricts neuroinflammatory responses during cerebral I/R injury through stabilizing SOCS3 protein.
Materials and Methods
Cell Culture and Oxygen–Glucose Deprivation/Reoxygenation (OGD/R) Treatment
Mouse microglial BV-2 cells were obtained from ATCC (Rockville, USA) and cultured in DMEM supplemented with 10% fetal bovine serum at 37°C and 5% CO2 in a humidified incubator. For OGD/R treatment, after 80% confluence, the cells were switched from a normal feeding medium containing serum and glucose to an oxygen-glucose-deprived medium, and incubated in a hypoxic chamber with CO2/N2 (5%/95%) for 3 h. The cells were then returned to the normal feeding medium and incubated under normal conditions for different hours as reperfusion.
Small Interfering RNA (siRNA) and Plasmid Transfection
siRNA against SOCS3 (si-SOCS3, 5ʹ-CAAGACCUUCAGCUCCAAGTT-3ʹ), and siRNA negative control (si-NC, 5ʹ-UUCUCCGAACGUGUCACGUTT-3ʹ) were synthesized by RiboBio (Guangzhou, China). pCMV3-N-FLAG-SOCS3, pCMV3-N-Myc-CSN3 plasmids were purchased from SiboBiological (Beijing, China). siRNAs or plasmids were transfected into BV2 cells with Lipo3000 (Invitrogen), as the manufacturers’ instructions described.
Immunofluorescence
After OGD/R-0h/24h treatment, BV2 cells were fixed with 4% paraformaldehyde and then permeabilized with Triton X-100. Subsequently, cells were incubated with anti-SOCS3 antibody (ab14939, Abcam) at 4°C overnight, and then incubated with secondary antibodies [goat anti-mouse Alexa Fluor 594 (A11032, Invitrogen)] at room temperature for 1 h. Nuclei were stained with DAPI (Roche, USA) for 10 min at room temperature. The cells were examined with a fluorescence microscope (Nikon TE300, Japan).
Quantitative Real-Time PCR (qPCR)
After treatment, total RNA of the BV-2 cells was isolated with TRIzol, and then was reverse-transcribed into cDNA using a reverse transcriptase kit (Promega, USA). qPCR was performed using SYBR green PCR Master Mix (Applied Biosystems, USA). The relative mRNA expressions were calculated by 2-ΔΔCt method. The qPCR primers were as following: IL-6, Forward5ʹ-GTACTCCAGAAGACCAGAGG-3ʹ, Reverse5ʹ-TGCTGGTG ACAACCACGGCC-3ʹ; SOCS3, Forward5ʹ-ATGGTCACCCACAGCAAGTTT-3ʹ, Reverse5ʹ-TCCAGTAGAATCCGCTCTCCT-3ʹ; Cyclin D1, Forward5ʹ-ATGGAAGGACCCTTGAGGC-3ʹ, Reverse5ʹ-CTTCACGGCTTGCTCGTTCT-3ʹ; myeloid cell leukemia sequence 1 (Mcl1), Forward5ʹ-AAAGGCGGCTGCATAAGTC-3ʹ, Reverse5ʹ-TGGCGGTATAGGTCGTCCTC-3ʹ; GAPDH, Forward 5ʹ-GCCAAGGCTGTGGGCAAGGT-3ʹ, Reverse5ʹ-TCTCCAGGCGGCACGTCAGA-3ʹ;
Protein Extraction and Western Blot
After treatments, BV2 cells were collected and lysed using NP40 lysis buffer (Beyotime Biotechnology, China) on ice. Lysates were quantified by the BCA kit (Beyotime Biotechnology, China), separated by 10% SDS-PAGE gel and then transferred onto polyvinylidene difluoride membranes. Membranes were blocked, incubated sequentially with primary and secondary antibodies. The immunoblots were detected using chemiluminescence (ECL Plus detection system). Bands were quantified using Image J software. The primary antibodies used were as follows: SOCS3 (1:1000, ab14939, Abcam), STAT3 (1:1000, ab68153, Abcam), phosphorylated STAT3 (pSTAT3, 1:1000, ab76315, Abcam), flag (1:1000, 80010-1-RR, ProteinTech), GAPDH (1:1000, 60004-1-Ig, ProteinTech), Myc (1:1000, 16286-1-AP, ProteinTech), CSN2 (1:1000, 10969-2-AP, ProteinTech), CSN3 (1:1000, ab229807, Abcam), CSN4 (1:1000, 10464-1-AP, ProteinTech), CSN5 (1:1000, 27511-1-AP, ProteinTech), CSN6 (1:1000, ab77299, Abcam), CSN7 (1:1000, ab133548, Abcam), CSN8 (1:1000, 10089-2-AP, ProteinTech). The secondary antibodies used in our work were as follows: goat anti-rabbit IgG-HRP (1:3000, SA00001-2, ProteinTech), goat anti-mouse IgG-HRP (1:3000, SA00001-1, ProteinTech).
Protein Stability Assay
After OGD/R-24h treatment, BV2 cells were treated with 50 μg/mL CHX (cycloheximide, Beyotime Biotechnology, China), 10 μM MG132 (Beyotime Biotechnology, Chin) or DMSO for 24 h, and then collected for the following experiments.
Co-Immunoprecipitation (Co-IP)
BV-2 cells were transfected with 4 μg pCMV3-N-FLAG-SOCS3 and pCMV3-N-Myc-CSN3 plasmids for 48 h. Cell lysates were incubated with an anti-Flag antibody overnight at 4°C, and then incubated with protein A/G-agarose beads for 4 h at 4°C. The immunoprecipitated proteins were separated by 10% SDS-PAGE and detected by Western blot with anti-Myc or anti-Flag antibodies. For endogenous immunoprecipitations, non-transfected cell lysates were incubated with anti-SOCS3 antibodies or IgG (control). Western blot was performed using anti-CSN2/3/4/5/6/7 antibodies.
Middle Cerebral Artery Occlusion (MCAO) Surgery
Experiments were performed under a project license (No.20200810001) granted by institutional ethics board of Guangzhou University of Chinese Medicine. MCAO surgery was performed as previous description.20 In short, mice were anesthetized with 4% isoflurane, and then the focal cerebral ischemia was produced by intraluminal occlusion of the right middle cerebral artery using a silicone coated nylon (6.0) monofilament. After 60 min, the occluding filament was withdrawn to allow blood reperfusion for 24 h. Then, the mice were used for further study.
Lentiviral Gene Transfer into Mouse Brains
Briefly, mice were kept immobile on a stereotactic apparatus under 4% isoflurane. Then, 3 µL of lentivirus solution (Lenti-shCSN3, or Lenti-NC 4.0×108 IU, obtained from Genechem) was directly injected (0.2 μL/min) into the right striatum at 2 mm lateral and 0.8 mm anterior to the bregma at a depth of 3.0 mm with a Stoelting injection system, as previously described.21 At 10 days after viral vector injection, the cerebral I/R operation was prepared using these mice.
Neurobehavioral Assessment
Neurological function was assessed by the modified neurological severity scores (mNSS), according to previous description.22 mNSS is graded on a scale of 0 to 18 (normal score, 0; maximal deficit score, 18). The higher score, the more severe is the injury.
Measurement of Infarct Size
Two percent of TTC (Sigma, USA) staining was conducted to evaluate the infarct volume, as previously described,23 and infarct volume was quantified using Image J.
Immunohistochemistry
Mice were anesthetized and intracardially perfused with 0.9% sodium chloride followed by 4% paraformaldehyde. Brains were removed and post-fixed in 4% PFA overnight at 4°C. Coronal brain sections (10 μm) were cut on a freezing microtome. The sections were stained overnight at 4°C using anti-CSN3 antibody (1:50, ab229807, Abcam) or anti-SOCS3 (1:50, ab14939, Abcam). Then, the sections were incubated using an appropriate secondary antibody for 1 h at room temperature, and the sections were imaged by microscope.
Statistical Analysis
Differences between two groups or multiple groups were analyzed by Student’s t-test or one-way analysis of variance (ANOVA), respectively. Statistical analyses were performed using R software (v 3.4.2) and GraphPad Prism software (v 8.00). P value <0.05 was considered statistically significant.
Results
SOCS3 Suppresses OGD/R-Induced STAT3 Activation and Inflammatory Factor Expression in BV-2 Microglia Cells
To explore the roles of SOCS3 in neuroinflammatory responses during cerebral I/R, we firstly detected SOCS3 protein expressions in BV-2 microglia cells under OGD/R treatment. As shown in Figure 1A, OGD/R treatment significantly increased SOCS3 protein expression in microglia cells. Besides, immunofluorescence analysis found that OGD/R treatment induced the nuclear distribution of SOCS3 in microglia cells (Figure 1B). Furthermore, downregulation of SOCS3 by transfecting siRNA-SOCS3 (si-SOCS3) significantly enhanced OGD/R-induced inflammatory factor, IL-6 expression, whereas, overexpression of SOCS3 evidently suppressed (Figure 1C and D). Given that SOCS3 is an important negative regulator of STAT3 signaling, which mediates the activation of inflammatory responses.12 Next, we examined the effect of SOCS3 on STAT3 signaling in microglia cells under OGD/R condition and found that overexpression of SOCS3 significantly suppressed OGD/R-induced STAT3 activation, and transcription of STAT3 downstream gene, Cyclin D1 and myeloid cell leukemia sequence 1 (Mcl1) (Figure 1E and F). Overall, these results indicate that SOCS3 suppresses OGD/R-induced STAT3 activation and inflammatory factor expression in BV-2 microglia cells.
 |
Figure 1 SOCS3 suppresses OGD/R-induced STAT3 activation and inflammatory factor expression in BV-2 microglial cells. (A) SOCS3 protein expression was examined by Western blot in OGD/R-treated BV2 microglia. GAPDH protein expression was used as loading control. (B) Cellular distribution of SOCS3 protein (Red) were detected by immunofluorescence in OGD/R-treated BV2 microglia. Nuclei were stained with DAPI (blue). (C) SOCS3 mRNA expression was examined by qPCR in BV2 microglia transfected with the indicated siRNAs or plasmids. (D) IL6 mRNA expression was examined by qPCR in BV2 microglia transfected with the indicated siRNAs or plasmids under OGD/R condition. (E) Protein expressions of STAT3 and phosphorylated STAT3 (p-STAT3) were examined by Western blot in BV2 microglia transfected with pCMV3-N-FLAG-SOCS3 plasmid under OGD/R condition. (F) mRNA expressions of Cyclin D1 and MCL1 were examined by qPCR in BV2 microglia transfected with pCMV3-N-FLAG-SOCS3 plasmid under OGD/R condition. Data are represented as means ± SEM (n=3; *Represents P < 0.05). |
COP9 Signalosome Complex Interacts with SOCS3 Protein and is Involved in Regulating SOCS3 Protein in Microglia During OGD/R Injury
To explore the underlying mechanism of OGD/R-induced upregulation of SOCS3 in microglia, we examined the SOCS3 mRNA expressions in microglia under OGD/R condition. As shown in Figure 2A, OGD/R treatment significantly increased SOCS3 mRNA expression in microglia. Furthermore, we used the protein synthesis inhibitor CHX and the proteasome inhibitor MG132 to further explore the underlying mechanism of SOCS3 upregulated expression under OGD/R condition. Compared to DMSO (as control) or CHX treatment, MG132 treatment significantly suppressed OGD/R-induced upregulated expression of SOCS3 protein in microglia (Figure 2B), indicating that degradation of SOCS3 protein may be inhibited during OGD/R injury. Given that COP9 signalosome has been reported to regulate deubiquitination and protect proteins from proteasome-mediated degradation,24 we further examined whether COP9 signalosome interacts with SOCS3 protein in microglia. Co-IP analysis showed that COP9 signalosome subunits CSN2/3/4/5/6/7/8, especially CSN3, could be obviously immunoprecipitated by anti-SOCS3 antibody (Figure 2C). Moreover, the interaction between exogenous SOCS3 and exogenous CSN3 was also confirmed in 293T cells by Co-IP analysis (Figure 2D), indicating that SOCS3 indeed interacts with COP9 signalosome. In addition, OGD/R treatment obviously enhanced SOCS3 interacting with CSN3 (Figure 2E). Overall, these results indicate that COP9 signalosome complex interacts with SOCS3 protein and is involved in regulating SOCS3 protein in microglia during OGD/R injury.
 |
Figure 2 COP9 signalosome complex interacts with SOCS3 protein and is involved in regulating SOCS3 protein in microglia during OGD/R injury. (A) SOCS3 mRNA expression was examined by qPCR in OGD/R-treated BV2 microglia. (B) SOCS3 protein expression was examined by Western blot in BV2 microglia treated with OGD/R and subsequently treated with 50 μg/mL CHX (cycloheximide), 10 μM MG132 or DMSO for 24 h. (C) The interaction between SOCS3 and CSNs by Co-IP in BV2 microglia. (D) The interaction between SOCS3 and CSN3 by Co-IP in 293T cells transfected with the indicated plasmids. (E) The interaction between SOCS3 and CSN3 by Co-IP in ODG/R treated with BV2 microglia. Data are represented as means ± SEM (n=3; *Represents P < 0.05). |
CSN3 Promotes SOCS3 Protein Stabilization in Microglia During OGD/R Injury
To further illuminate the molecular mechanism of COP9 signalosome complex regulating SOCS3 protein, we subsequently examined the effect of knockdown of COP9 signalosome subunit CSN3 on SOCS3 protein expression in microglia. As shown in Figure 3A and B, knockdown of CSN3 evidently decreased SOCS3 interacting with other subunits of COP9 signalosome, such as CSN4 and CSN5. Furthermore, we also found that knockdown of CSN3 not only obviously inhibited OGD/R-induced upregulation of SOCS3 protein in microglia but also decreased SOCS3 interacting with CSN4 and CSN5 (Figure 3C). To further confirm that CSN3 promotes SOCS3 protein stabilization through proteasome pathway, BV-2 microglia cells were transfected with si-CSN3, and then treated with CHX for different hours. Western blot analysis showed that under CHX treatment, SOCS3 protein level was significantly decreased in CSN3-knockdown BV-2 microglia cells, compared to control (Figure 3D). Overall, these results indicate that CSN3 promotes SOCS3 protein stabilization in microglia during OGD/R injury.
 |
Figure 3 CSN3 promotes SOCS3 protein stabilization in microglia during OGD/R injury. (A) SOCS3 mRNA expression was examined by qPCR in BV2 microglia transfected with the indicated siRNAs. (B) The interaction between SOCS3 and CSN4/5 by Co-IP in BV2 microglia. (C) The interaction between SOCS3 and CSN4/5 by Co-IP in BV2 microglia transfected with si-CSN3 and then treated with ODG/R. (D) SOCS3 protein expression was examined by Western blot in BV2 microglia transfected with si-CSN3 and subsequently treated with 10 μM MG132 for 24 h. Data are represented as means ± SEM (n=3; *Represents P < 0.05). |
CSN3 Restricting OGD/R-Induced STAT3 Activation and Inflammatory Responses Dependents on SOCS3
To further explore the role of CSN3 on inflammatory responses in microglia during OGD/R injury, we then investigated the effect of overexpression of CSN3 on inflammatory factor IL-6 expression. As shown in Figure 4A and B, overexpression of CSN3 did not evidently affect IL-6 expression in microglia under normal condition, whereas overexpression of CSN3 significantly inhibited OGD/R-induced IL-6 expression. Furthermore, we found that overexpression of CSN3 significantly suppressed OGD/R-induced STAT3 activation (Figure 4C). However, knockdown of SOCS3 evidently suppressed the effects of overexpression of CSN3 on STAT3 activation and transcription of STAT3 downstream genes, Cyclin D1 and Mcl1 in microglia under OGD/R condition (Figure 4D and E). Overall, these results indicate that CSN3 restricting OGD/R-induced STAT3 activation and inflammatory responses dependents on SOCS3.
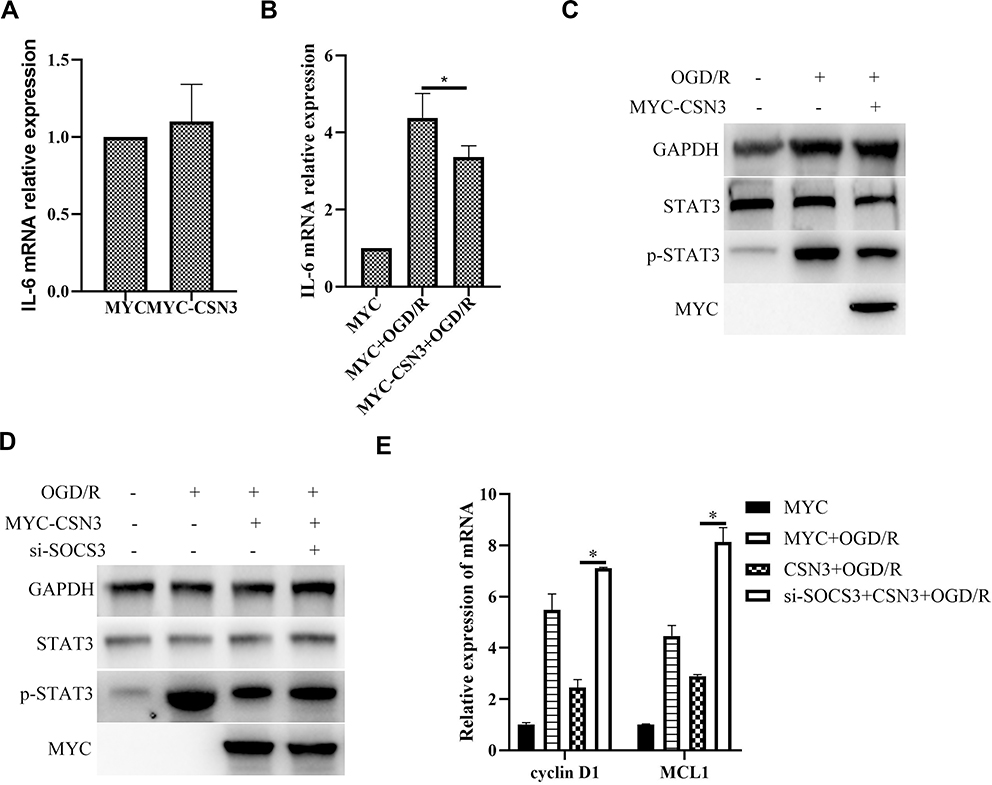 |
Figure 4 CSN3 restricting OGD/R-induced STAT3 activation and inflammatory responses dependents on SOCS3. (A) IL6 mRNA expression was examined by qPCR in BV2 microglia transfected with pCMV3-N-Myc-CSN3. (B) IL6 mRNA expression was examined by qPCR in BV2 microglia transfected with pCMV3-N-Myc-CSN3 and then treated with ODG/R. (C) Protein expressions of STAT3 and p-STAT3 were examined by Western blot in BV2 microglia transfected with pCMV3-N-Myc-CSN3 and then treated with ODG/R. (D) Protein expressions of STAT3 and p-STAT3 were examined by Western blot in BV2 microglia transfected with pCMV3-N-Myc-CSN3 and si-SOCS3, and then treated with ODG/R. (E) mRNA expressions of Cyclin D1 and MCL1 were examined by qPCR in BV2 microglia transfected with pCMV3-N-Myc-CSN3 and si-SOCS3, and then treated with ODG/R. Data are represented as means ± SEM (n=3; *Represents P < 0.05). |
Knockdown of CSN3 Aggravates Cerebral I/R Injury in vivo
We further examined the roles of CSN3 on cerebral I/R injury in vivo using a mice cerebral I/R model by occluding the middle cerebral artery (MCAO) for 1 h, and then reperfusing for 24 h. CSN3 expression in the brain was downregulated by injection of lentivirus-mediated shRNA-CSN3 (Lenti-shCSN3) into the lateral ventricle of mice before MCAO operation (Figure 5A). As expected, pre-injection of Lenti-shRNA-CSN3 obviously aggravated cerebral I/R-induced neurological deficit (Figure 5B) and increased cerebral infarction size (Figure 5C and D). Furthermore, we examined the STAT3 expression level in the ischemic boundary zone of brain tissue by immunohistochemistry and found that STAT3 expression was significantly increased and more STAT3 protein translocated into nucleus in the ischemic boundary zone of mice pre-injected with Lenti-shCSN3, compared to those mice pre-injected with Lenti-shNC (Figure 5E). Overall, knockdown of CSN3 aggravated cerebral I/R injury.
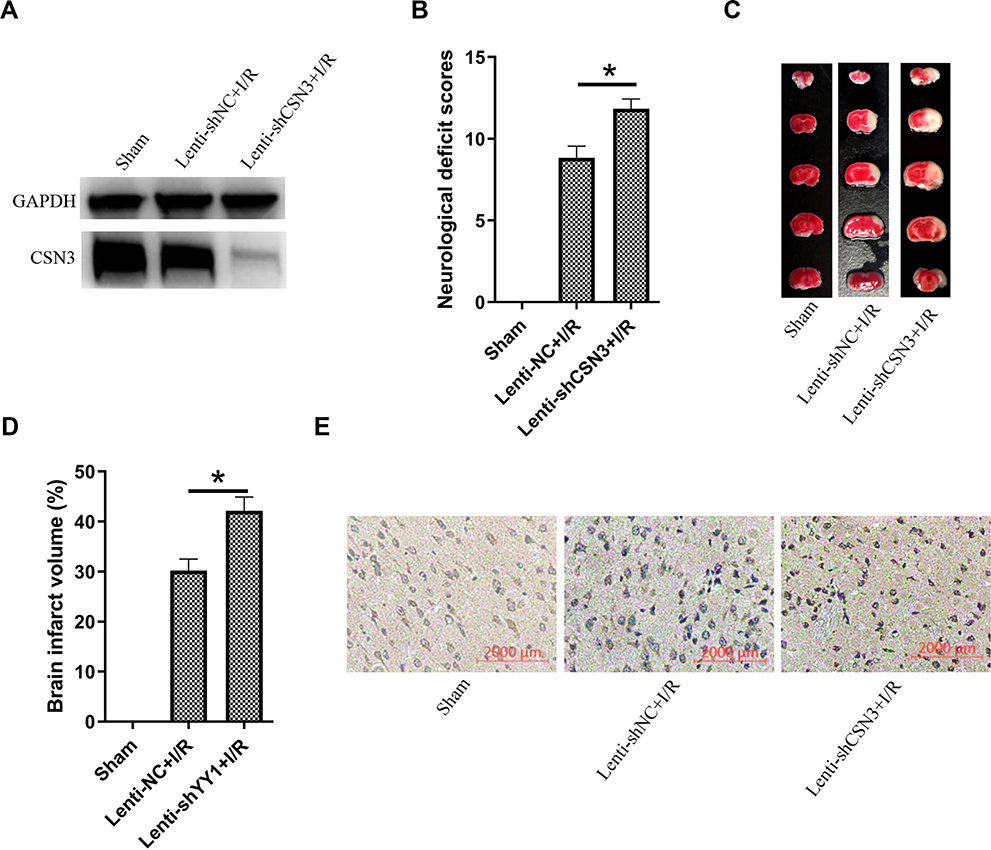 |
Figure 5 Knockdown of CSN3 aggravated cerebral I/R injury in vivo. (A) CSN3 protein expressions were examined by Western blot in the ischemic boundary zone of brain tissue of mice pre-infected with Lenti-shCSN3 or Lenti-shNC and subjected with MCAO/R. (B) Neurological scores were assessed at 24 h after cerebral I/R (n=6 per group, *represents P < 0.05). (C) Representative photographs of coronal brain sections stained with TTC. Pale areas represent infarction. (D) Quantification of infarct volume at 24 h after cerebral I/R (n=6 per group, *Represents P < 0.05). (E) Immunohistochemical staining for STAT3 in the periinfarct area. |
Discussion
SOCS3 is a well-known negative regulator of the Janus kinases-signal transducer and activator of transcription 3 (JAK/STAT3) signaling pathway,25 which has been reported to be activated to induce inflammatory responses and be involved in various diseases, such as cerebral I/R injury, myocardial cerebral I/R injury, and inflammatory bowel disease.26–28 Through binding tyrosine kinase receptor, primary receptor shared IL-6 receptor subunit gp130 and bindinJAK2, SOCS3 inhibits STAT3 phosphorylation to restrain inflammation.29 Here, we find that SOCS3 expression is gradually upregulated in microglia during I/R injury, and overexpression of SOCS3 evidently inhibits I/R-induced STAT3 phosphorylation and inflammatory factor expression, further suggesting SOCS3 functions protective effects in brain inflammation.
SOCS3 protein has two functional domains: a central SH2 domain, which binds the defined tyrosine-phosphorylated substrates on the cytokine receptor, and a SOCS-box domain at the C-terminus, which could be recognized as substrate by a Cullin-RING E3 ubiquitin-protein ligase complex,19,30,31 indicating that SOCS3 could also be regulated by deubiquitination. In this study, we explored the relationship between SOCS3 and COP9 signalosome which regulates the activity of Cullin-RING ubiquitin E3 ligase,32 during cerebral I/R injury. We find that SOCS3 interacts with COP9 subunit CSN3, which is essential for stabilize SOCS3 protein in microglia during cerebral I/R injury, thereby suppressing STAT3 activation and induction of pro-inflammatory factors. Besides, Nuclear factor kappa B (NF-κB) Signaling is also activated and essential for neuroinflammation during cerebral I/R injury.33 Generally, upon stimulation, the inhibitor of NF-κB, IκBα, is ubiquitinated and then degraded by the 26S proteasome, thereby leading to that NF-κB is released and translocated into the nucleus to induce expressions of pro-inflammatory factors.34,35 Recently, COP9 signalosome has been reported to function in the protection of IκBα from degradation by regulating the deneddylation of CRL1, resulting in reduction of NF-κB activation upon tumor necrosis factor (TNF) stimulation.36,37 Whether NF-кB signaling is also regulated by the same COP9 signalosome during cerebral I/R injury is still unknown and the relationship among SOCS3, NF-кB and COP9 signalosome complex needs to be investigated in the following studies. We also identify other subunits of COP9 signalosome interact with SOCS3 in microglia. Given that these CSNs cooperate with each other to form COP9 signalosome complex to function, and that the association between CSN4/5 and SOCS3 is decreased by CSN3 knockdown in microglia, it is probably that SOCS3 interacting with COP9 signalosome depends on CSN3, which needs further confirmation in the future studies. The detailed mechanism of COP9 signalosome stabilizing SOCS3 by deubiquitination during cerebral I/R injury is also worthy of further study. In addition, there are also some limitations in our present study. First, BV2 microglia cell line was used to explore the underlying mechanism of COP9 signalosome complex regulating SOCS3 protein during neuroinflammation in vitro. In the following studies, primary microglia should be used. Second, for the in vivo experiments, CSN3 knockout mice should be used in the following studies. Third, SOCS family has eight members: SOCS1-7 and the cytokine-inducible SH2-containing protein, which all negatively regulate the JAK-STAT signaling pathway.38 Except SOCS3, whether COP9 signalosome complex also regulating other SOCS members in neuroinflammation during cerebral I/R injury should be further investigated in the future studies.
Conclusion
This study firstly reports that CSN3 restricts neuroinflammatory responses during cerebral I/R injury through stabilizing STAT3 protein and identifies a protective role of COP9 signalosome in cerebral I/R. Our results contribute to deeply understanding the regulatory mechanism of SOCS3 in neuroinflammation and also providing potential therapeutic targets for cerebral I/R injury.
Availability of Supporting Data
The data will be available on request.
Ethical Approval
All animals were maintained according to the American Animal Protection Legislation. This study was approved by the Institutional Animal Care and Use Committee of the Guangzhou University of Chinese Medicine.
Funding
This study was supported by Project of Administration of Traditional Chinese Medicine of Guangdong Province of China (20201419) and ShenZhen Science and Technology Planning Project (JCYJ20190812163201666).
Disclosure
The authors reported no conflicts of interest for this work.
References
1. Zuo G, Zhang D, Mu R, et al. Resolvin D2 protects against cerebral ischemia/reperfusion injury in rats. Mol Brain. 2018;11(1):9. doi:10.1186/s13041-018-0351-1
2. Shvedova M, Anfinogenova Y, Atochina-Vasserman EN, Schepetkin IA, Atochin DN. c-Jun N-terminal Kinases (JNKs) in myocardial and cerebral ischemia/reperfusion injury. Front Pharmacol. 2018;9:715. doi:10.3389/fphar.2018.00715
3. Liu L, Chen H, Jin J, et al. Melatonin ameliorates cerebral ischemia/reperfusion injury through SIRT3 activation. Life Sci. 2019;239:117036. doi:10.1016/j.lfs.2019.117036
4. Hou S, Zhao MM, Shen PP, Liu XP, Sun Y, Feng JC. Neuroprotective effect of salvianolic acids against cerebral ischemia/reperfusion injury. Int J Mol Sci. 2016;17.
5. Amantea D, Bagetta G. Excitatory and inhibitory amino acid neurotransmitters in stroke: from neurotoxicity to ischemic tolerance. Curr Opin Pharmacol. 2017;35:111–119. doi:10.1016/j.coph.2017.07.014
6. Kishimoto M, Suenaga J, Takase H, et al. Oxidative stress-responsive apoptosis inducing protein (ORAIP) plays a critical role in cerebral ischemia/reperfusion injury. Sci Rep. 2019;9(1):13512. doi:10.1038/s41598-019-50073-8
7. Wu L, Xiong X, Wu X, et al. Targeting oxidative stress and inflammation to prevent ischemia-reperfusion injury. Front Mol Neurosci. 2020;13:28. doi:10.3389/fnmol.2020.00028
8. Gong Z, Pan J, Shen Q, Li M, Peng Y. Mitochondrial dysfunction induces NLRP3 inflammasome activation during cerebral ischemia/reperfusion injury. J Neuroinflammation. 2018;15(1):242. doi:10.1186/s12974-018-1282-6
9. Amani H, Shahbazi MA, D’Amico C, Fontana F, Abbaszadeh S, Santos HA. Microneedles for painless transdermal immunotherapeutic applications. J Control Release. 2021;330:185–217. doi:10.1016/j.jconrel.2020.12.019
10. Satriotomo I, Bowen KK, Vemuganti R. JAK2 and STAT3 activation contributes to neuronal damage following transient focal cerebral ischemia. J Neurochem. 2006;98(5):1353–1368. doi:10.1111/j.1471-4159.2006.04051.x
11. Mudter J, Neurath MF. Il-6 signaling in inflammatory bowel disease: pathophysiological role and clinical relevance. Inflamm Bowel Dis. 2007;13(8):1016–1023. doi:10.1002/ibd.20148
12. Groner B, Lucks P, Borghouts C. The function of Stat3 in tumor cells and their microenvironment. Semin Cell Dev Biol. 2008;19(4):341–350. doi:10.1016/j.semcdb.2008.06.005
13. Zhu Y, Bu Q, Liu X, Hu W, Wang Y. Neuroprotective effect of TAT-14-3-3epsilon fusion protein against cerebral ischemia/reperfusion injury in rats. PLoS One. 2014;9(3):e93334. doi:10.1371/journal.pone.0093334
14. Qin N, Xu D, Li J, Deng XW. COP9 signalosome: discovery, conservation, activity, and function. J Integr Plant Biol. 2020;62(1):90–103. doi:10.1111/jipb.12903
15. Glickman MH, Rubin DM, Coux O, et al. A subcomplex of the proteasome regulatory particle required for ubiquitin-conjugate degradation and related to the COP9-signalosome and eIF3. Cell. 1998;94(5):615–623. doi:10.1016/S0092-8674(00)81603-7
16. Milic J, Tian Y, Bernhagen J. Role of the COP9 Signalosome (CSN) in Cardiovascular Diseases. Biomolecules. 2019;9(6):9. doi:10.3390/biom9060217
17. Korczeniewska J, Barnes BJ. Corrected and Republished from: the COP9 signalosome interacts with and regulates interferon regulatory factor 5 protein stability. Mol Cell Biol. 2018;38(3):38. doi:10.1128/MCB.00493-17
18. Boro M, Govatati S, Kumar R, et al. Thrombin-Par1 signaling axis disrupts COP9 signalosome subunit 3-mediated ABCA1 stabilization in inducing foam cell formation and atherogenesis. Cell Death Differ. 2020.
19. Kamura T, Maenaka K, Kotoshiba S, et al. VHL-box and SOCS-box domains determine binding specificity for Cul2-Rbx1 and Cul5-Rbx2 modules of ubiquitin ligases. Genes Dev. 2004;18(24):3055–3065. doi:10.1101/gad.1252404
20. Chen Y, Zhang L, Ni J, et al. LLDT-8 protects against cerebral ischemia/reperfusion injury by suppressing post-stroke inflammation. J Pharmacol Sci. 2016;131(2):131–137. doi:10.1016/j.jphs.2016.05.003
21. Wang HJ, Tang XL, Huang G, et al. Long non-coding KCNQ1OT1 promotes oxygen-glucose-deprivation/reoxygenation-induced neurons injury through regulating MIR-153-3p/FOXO3 axis. J Stroke Cerebrovasc Dis. 2020;29(10):105126. doi:10.1016/j.jstrokecerebrovasdis.2020.105126
22. Chen J, Li Y, Wang L, et al. Therapeutic benefit of intravenous administration of bone marrow stromal cells after cerebral ischemia in rats. Stroke. 2001;32(4):1005–1011. doi:10.1161/01.STR.32.4.1005
23. Zhang XC, Gu AP, Zheng CY, et al. YY1/LncRNA GAS5 complex aggravates cerebral ischemia/reperfusion injury through enhancing neuronal glycolysis. Neuropharmacology. 2019;158:107682. doi:10.1016/j.neuropharm.2019.107682
24. Dubiel W, Chaithongyot S, Dubiel D, Naumann M. The COP9 signalosome: a multi-DUB complex. Biomolecules. 2020;10(7):10. doi:10.3390/biom10071082
25. Gao Y, Zhao H, Wang P, Wang J, Zou L. The roles of SOCS3 and STAT3 in bacterial infection and inflammatory diseases. Scand J Immunol. 2018;88(6):e12727. doi:10.1111/sji.12727
26. Wang R, Zhang S, Yang Z, et al. Mutant erythropoietin enhances white matter repair via the JAK2/STAT3 and C/EBPbeta pathway in middle-aged mice following cerebral ischemia and reperfusion. Exp Neurol. 2020;113553.
27. Wu L, Huang WQ, Yu CC, Li YF. Moderate hydrogen peroxide postconditioning ameliorates ischemia/reperfusion injury in cardiomyocytes via STAT3-induced calcium, ROS, and ATP homeostasis. Pharmacology. 2020;1–11. doi:10.1159/000511961
28. Yin Y, Liu W, Dai Y. SOCS3 and its role in associated diseases. Hum Immunol. 2015;76(10):775–780. doi:10.1016/j.humimm.2015.09.037
29. Carow B, Rottenberg ME. SOCS3, a major regulator of infection and inflammation. Front Immunol. 2014;5:58. doi:10.3389/fimmu.2014.00058
30. Piessevaux J, Lavens D, Peelman F, Tavernier J. The many faces of the SOCS box. Cytokine Growth Factor Rev. 2008;19(5–6):371–381. doi:10.1016/j.cytogfr.2008.08.006
31. Kazi JU, Kabir NN, Flores-Morales A, Ronnstrand L. SOCS proteins in regulation of receptor tyrosine kinase signaling. Cell Mol Life Sci. 2014;71(17):3297–3310. doi:10.1007/s00018-014-1619-y
32. Cavadini S, Fischer ES, Bunker RD, et al. Cullin-RING ubiquitin E3 ligase regulation by the COP9 signalosome. Nature. 2016;531(7596):598–603. doi:10.1038/nature17416
33. Zhang C, Chen S, Zhang Z, et al. Asiaticoside alleviates cerebral ischemia-reperfusion injury via NOD2/Mitogen-Activated Protein Kinase (MAPK)/Nuclear Factor kappa B (NF-kappaB) signaling pathway. Med Sci Monit. 2020;26:e920325.
34. Mitchell JP, Carmody RJ. NF-kappaB and the transcriptional control of inflammation. Int Rev Cell Mol Biol. 2018;335:41–84.
35. Hayden MS, Ghosh S. NF-kappaB in immunobiology. Cell Res. 2011;21(2):223–244. doi:10.1038/cr.2011.13
36. Lee JH, Yi L, Li J, et al. Crystal structure and versatile functional roles of the COP9 signalosome subunit 1. Proc Natl Acad Sci U S A. 2013;110(29):11845–11850. doi:10.1073/pnas.1302418110
37. Asare Y, Shagdarsuren E, Schmid JA, et al. Endothelial CSN5 impairs NF-kappaB activation and monocyte adhesion to endothelial cells and is highly expressed in human atherosclerotic lesions. Thromb Haemost. 2013;110(07):141–152. doi:10.1160/TH13-02-0155
38. McCormick SM, Heller NM. Regulation of macrophage, dendritic cell, and microglial phenotype and function by the SOCS Proteins. Front Immunol. 2015;6:549. doi:10.3389/fimmu.2015.00549
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.