")
Back to Journals » International Journal of General Medicine » Volume 16
Construction of circRNA–miRNA–mRNA Network Reveal Functional circRNAs and Key Genes in Acute Myeloid Leukemia
Authors Hu L, Zheng B, Yang Y, Chen C, Hu M
Received 25 December 2022
Accepted for publication 4 April 2023
Published 24 April 2023 Volume 2023:16 Pages 1491—1504
DOI https://doi.org/10.2147/IJGM.S402671
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Scott Fraser
Lianbo Hu,1 Bingrong Zheng,2 Yang Yang,2 Chunmei Chen,2 Meiwei Hu2
1Physical Examination Center, Affiliated Hangzhou First People’s Hospital, Zhejiang University School of Medicine, Hangzhou, Zhejiang, People’s Republic of China; 2Department of Hematology, the Second Affiliated Hospital of Zhejiang Chinese Medical University, Hangzhou, Zhejiang, People’s Republic of China
Correspondence: Meiwei Hu, Department of Hematology, the Second Affiliated Hospital of Zhejiang Chinese Medical University, No. 318 Chaowang Road, Hangzhou, Zhejiang, 310014, People’s Republic of China, Email [email protected]
Introduction: CircRNA is closely correlated with a wide variety of processes of acute myeloid leukemia (AML), whereas the novel circRNAs, their molecular mechanism and the specific function they played in AML should be explored in depth.
Methods: The microarray chip data of AML patients and normal samples in the Gene Expression Omnibus (GEO) database were selected to differentially expressed (DE) circRNA, miRNA, and mRNA genes. The miRNA gene was the intersection of the circRNA target gene predicted using CSCD and the miRNA gene screened from AML patients, while the mRNA gene was the intersection of the target gene mRNA of miRNA predicted using miRanda and miRTarBase software and the mRNA gene screened from AML patients. The hub mRNAs related to survival were further screened through Cox proportional hazard regression. CircRNA/miRNA/mRNA interaction network was constructed by using Cytoscape software.10 circRNAs and 6 miRNAs in bone marrow mononuclear cells (BMMNCs) of AML patients (n=43) and healthy controls (n=35) were determined by RT-qPCR. Correlations between them were analyzed by Pearson correlation coefficient.
Results: 10 circRNAs, 6 miRNAs, and 33 mRNAs were identified. Subsequently, the network of circRNAs, miRNAs, and hub genes was built using Cystoscope. Four key circRNAs, seven hub genes and their regulatory pathways were identified. The result of RT-qPCRs showed that hsa_circ_0009581 and hsa_circ_0005273 were significantly upregulated in AML patients while hsa_circ_0000497 and hsa_circ_0001947 were significantly downregulated. Hsa-miR-150-5p was significantly downregulated; hsa-miR-454-3p was upregulated in AML patients. Hsa_circ_0009581 and hsa-miR-150-5p; hsa_ circ_0001947 and hsa-miR-454-3p were inversely correlated using Pearson’s correlation coefficient.
Conclusion: This study suggests that differentially expressed circRNAs take on a critical significance to AML development and may be the effective therapeutic targets. We suppose that hsa_circ_0009581 promotes leukemia development through hsa-miR-150-5p and hsa_circ_0001947 through hsa-miR-454-3p. hsa_circ_0001947 and hsa_circ_0009581 may provide new directions in the pathogenesis of AML.
Keywords: acute myeloid leukemia, circRNA, pathogenesis, bioinformation analysis
Introduction
Acute myeloid leukemia (AML) is a group of highly aggressive hematological malignancies with abnormal proliferation and differentiation of hematopoietic stem cells.1 Abnormal blood cells proliferate in a considerable number and inhibit normal hematopoiesis, with the clinical manifestations of anemia, bleeding, hepatosplenomegaly and other organ infiltration. It is one of the most common hematologic malignancies, and the mortality rate is the highest compared with other types of leukemia.2 The treatment is based on conventional chemotherapy and hematopoietic stem cell transplantation, and molecular targeted therapy and cellular immunotherapy are promising for treating AML patients. However, they are still at risk for drug resistance and disease recurrence, thus resulting in shorter survival and poorer quality of life.3 Studying the molecular mechanism of AML development takes on a great significance in finding novel therapeutic targets and facilitating AML prognosis.
Circular RNAs (circRNAs) are a special type of closed-loop non-coding RNA molecule without 5’-caps or 3’ -polyadenylate tails. Existing research has suggested that circRNAs exhibit complex biological functions in the body.4 An increasing number of studies have suggested that circRNAs play a vital role in the development of tumors.5 Existing research has also suggested that circRNA is correlated with the emergence of drug-resistance in osteosarcoma,6 breast cancer,7 as well as chronic myeloid leukemia.8 CircRNA is stably expressed and is present at high level in body fluids, making it an ideal candidate for noninvasive liquid biopsy biomarkers in tumors.9 Existing research has suggested that circRNAs are capable of playing a role in the post-transcriptional regulation of AML by binding miRNAs, activating downstream signaling cascades, and regulating the expression of related genes.10 CircRNA is closely correlated with a wide variety of processes of AML and may serve as a novel target for AML diagnosis and treatment. With the development of bioinformatics technology, there have been increasing circRNA-based bioinformatics studies, whereas the molecular mechanism and the specific function of the novel circRNAs in AML should be explored in depth.
In this study, the microarray chip data in AML were analyzed and mined from Gene Expression Omnibus (GEO) database, differentially expressed circRNAs (DEcircRNAs), differentially expressed miRNAs (DEmiRNAs), and differentially expressed mRNAs (DEmRNAs) were selected using R software. circRNA/miRNA/mRNA interaction networks were built using Cytoscape. Moreover, the main functional pathways were evaluated through gene ontology (GO) enrichment analysis and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analysis. Lastly, validation and survival analysis of hub genes were conducted using The Cancer Genome Atlas (TCGA) database and genotype-tissue expression (GTEx) database. Subsequently, we performed real-time quantitative polymerase chain reaction (RT-qPCR) to confirm that these circRNAs and miRNAs are expressed differently in AML and healthy controls. The above results provide new directions for the pathogenesis and treatment of AML.
Materials and Methods
Data Acquisition and Identification of DEcircRNAs
Gene Expression Omnibus (GEO) database contains high-throughput gene expression data submitted by research institutions around the world. Microarray chip data from AML patients (GSE94591) in the GEO database were selected. R language was used to screen differential genes between AML patients and healthy controls for significant differential circRNAs (P<0.05, logFC absolute value > 2).
Prediction of miRNA Response Elements (MREs)
Cancer-Specific CircRNA Database (CSCD) explores the function of circRNAs by predicting the miRNA response elements (MREs) loci. We predicted the target genes of circRNAs by CSCD. The microarray data from AML patients in the GEO database (GSE142700-GPL26945) were selected, and the R language was employed to screen for meaningful miRNAs (P<0.05, logFC absolute value>1). By intersecting the circRNA-predicted target miRNAs with them, we selected 8 overlapping miRNAs for further mRNA prediction.
Forecast of miRNA Targeted Genes, Collection of AML Differentially Expressed Genes (DEG) and Obtaining Overlapping Genes
MiRanda was developed by researchers at the prestigious Memorial Sloan-Kettering Cancer Research Center to predict the binding sites of miRNAs interacting with their target genes. MiRTarBase is a manually collected database of experimentally verified miRNA target genes. For each miRNA target gene record, a unique miRNA-target interaction (referred to as MTs) number will be assigned. The target gene mRNA of miRNA was predicted by miRanda and miRTarBase software. Similarly, the microarray data from AML patients in the GEO database (GSE142700-GPL19956) were selected, and the R language was employed to screen for meaningful mRNAs (P<0.05, logFC absolute value>1). The genes between the DEGs and predicted miRNA target genes were obtained through the Venn diagram.
Functional Enrichment Analysis of Overlapped Genes
DAVID V6.8 (https://david.abcc.ncifcrf.gov/), an online bioinformatics resource, provides tools for functional interpretation of a large number of genes, which was used for Gene Ontology (GO) analysis and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analysis of overlapping genes. The setting was adopted to perform the R package, with p<0.05 and the count is>2.
Overall Survival (OS) Analysis of the Hub Genes
GEPIA 2 (Gene Expression Profiling Interactive Analysis) mainly focuses on The Cancer Genome Atlas (TCGA) database and Genotype-Tissue Expression (GTEx) project. Analysis includes tumor/normal differential expression profiling, expression analysis, survival analysis, similar genes, gene expression correlation and dimensionality reduction analysis. Overall survival (OS) analysis was used to explore the effect of differentially expressed hub genes on OS between healthy controls and AML patients.
Construction of circRNA–miRNA–mRNA Network
A circRNA-miRNA-mRNA regulatory network was constructed by mapping the regulatory networks using Cytoscape software (Figure 1).
 |
Figure 1 Flowchart of this study about constructing a circRNA/miRNA/mRNA network. |
AML Patients and Healthy Controls
We enrolled 43 patients with confirmed AML and 35 healthy controls who were admitted to the Second Affiliated Hospital of Zhejiang Chinese Medical University from Jan 2021 to Sep 2022. There was no significant difference in gender and age between patients with AML and healthy controls. Bone marrow aspirate was performed on two groups to collect BM cells. Bone marrow mononuclear cells (BMMNCs) were isolated using lymphocyte isolation medium (TBD Science).
RNA Extraction and Quantitative Real-Time PCR Validation
Total RNA were extracted from BMMNCs of AML patients and controls with TRIzol reagent (Life Technologies, CA, USA). The quantitative analysis of RNAs was performed by NanoDrop 2000 Spectrophotometer (Thermo Scientific, Wilmington, DE, USA). For circRNA, cDNA was synthesized using the PrimeScript™ RT reagent kit (Takara, Tokyo, Japan) under the guidance of the manufacturer’s instructions. Realtime-qPCR was performed using the TB Green® Premix Ex Taq™ II kit (Takara, Tokyo, Japan). PCR primers are listed in Table 1. For microRNA, cDNA was synthesized using the Mir-X miRNA First-Strand Synthesis Kit (Takara, Tokyo, Japan). The special sequences of hsa-miRNAs were amplified with the forward primer in Table 1. The universal reverse primer and U6 primers in Mir-X miRNA qRT-PCR TB Green Kit (Takara, Tokyo, Japan). Indicated genes were normalized to β-actin or U6. Thereafter, the folding change of genes was calculated by the 2-ΔΔCt analytic method.
 |
Table 1 Sequences of Primers for RT-qPCR and miRNA Related Sequence |
Ethical Statement
This study were approved by the Ethics Committee of The Second Affiliated Hospital of Zhejiang Chinese Medical University (the Institutional Review Board approval number: 2021-KL-125-01) and were carried out in accordance with the ethical principles of the Declaration of Helsinki. All patients signed the informed consent form.
Statistical Analysis
Bioinformatic statistical analyses were performed using R software (v.3.6.1) and the above software packages. Other statistical analyses were done using GraphPad Prism software. Correlations were analyzed by Pearson’s correlation coefficient. P < 0.05 was statistically significant.
Result
Acquiring 10 DEcircRNAs in Acute Myeloid Leukemia
To explore the function of circRNAs and construct a circRNA-miRNA-mRNA interaction network, we obtained the microarray dataset GSE94591 from the GEO database to identify the differentially expressed circRNAs in bone marrow specimens from healthy controls and AML patients. The gene chip was obtained from the Agilent-069978 Arraystar Human CircRNA microarray V1 platform. Using the Limma package, we obtained 4520 differentially expressed CircRNAs (Figure 2a). After screening at P<0.05 and differential fold > 4, we finally found that 72 CircRNAs matched and used the top 50 CircRNAs with the most significant differences for the heat map (as shown in Figure 2b). The top 5 circRNAs with the most significant rise and fall were used for the follow-up study. 10 circ RNAs were differentially expressed in bone marrow specimens from healthy controls and AML patients (Figure 2). The basic characteristics of the 10 circRNAs are shown in Table 2.
 |
Table 2 Basic Characteristics of These 10 circRNAs |
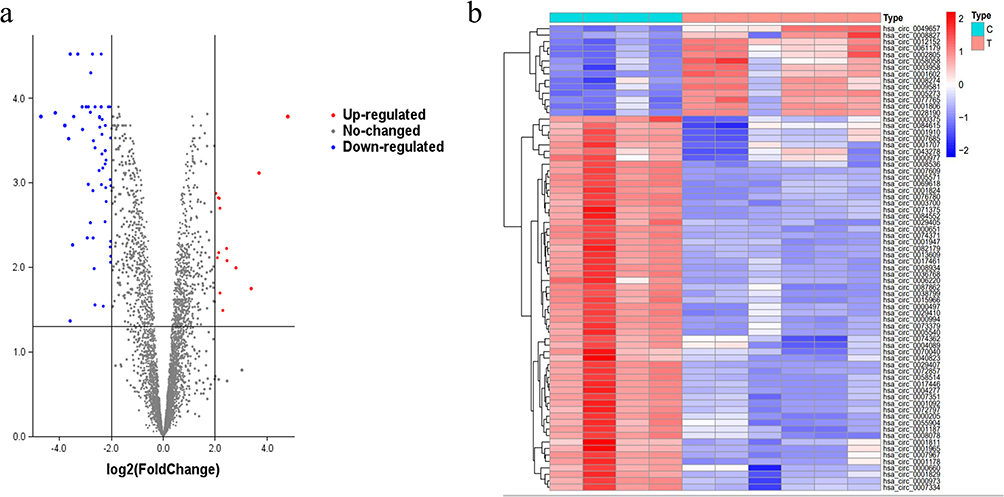 |
Figure 2 Acquired DEcircRNAs of AML. (a) Volcano plots of DEcircRNAs, the blue dots and red dots represent down- and up-expressed circRNAs. (b) Heatmap of circRNAs. Type: (C) healthy controls. (T) AML patients. |
Identification of 10 circRNA-miRNA Interactions
To explore circRNA-miRNA interactions, we previously analyzed the structures of 10 circRNAs (Figure 3a) which have the structures of MRE, RBP and ORF. We also predicted the target miRNAs to which CSCD binds. Meanwhile, we identified differentially expressed miRNAs between healthy controls and AML patients. The microarray dataset GSE142700 was obtained from the GEO database. Gene chips were obtained from NanoString nCounter Human miRNA platform. Using the Limma package, 777 differential circRNAs were screened (Figure 3b). After screening with P<0.05 and the differential fold > 2-fold, 54 miRNAs were finally found to be concordant, and the top 50 miRNAs with the most significant differences were mapped to a heat map (Figure 3c). circRNA-predicted target genes intersected with miRNAs screened by the AML patient microarray (Figure 3d), and 6 miRNAs intersections were identified, namely: hsa-miR-382-5p, hsa-miR-3190-3p, hsa-miR-150-5p, hsa-miR-1193, hsa-miR-3613-5p, hsa-miR-454-3p. Six miRNAs were used as follow-up studies.
 |
Figure 3 Differentially expressed miRNAs in AML patients. (a) Basic structures of the 10 circRNAs. (b) Volcano plots for differentially expressed miRNAs, the blue and red dots represent down- and up-expressed miRNAs. (c) Heatmap of differentially expressed miRNAs. Type: (C) healthy controls. (T) AML patients. (d) Venn graph showing the 6 overlapped genes by intersecting circRNA targeted genes and miRNA response elements from AML patients. |
Identification of 6 miRNA-mRNA Interactions
We used the miRanda and miRTarBase tools to predict the target mRNAs of the 6 miRNAs in order to investigate miRNA-mRNA interactions. At the same time, differentially expressed mRNAs between healthy controls and AML patients were identified. The microarray dataset GSE142700 was obtained from the GEO database. The gene chip was obtained from the NanoString nCounter human PanCancer Pathways Panel platform. Using the Limma package, 779 differential mRNAs were screened (as shown in Figure 4a). After screening with P<0.05 and the differential fold >2, 137 matched mRNAs were finally found, with the top 50 mRNAs with the most significant differences used to plot a heat map (Figure 4b). The target genes predicted by miRNA were intersected with the mRNAs screened by the AML patient microarray 33 mRNAs were found to intersect (Figure 4c) and used as follow-up studies.
 |
Figure 4 Differentially expressed mRNAs in AML patients. (a) Volcano plots of differentially expressed mRNAs, the blue and red dots represent down- and up-expressed miRNAs. (b) Heatmap for differentially expressed miRNAs. Type: (C) healthy controls. (T) AML patients. (c) Venn graph showing the 33 overlapped genes by intersecting the miRNA targeted genes and mRNA targets from AML patients. |
Function Enrichment Analyses
For the potential functions and biological roles of the 33 overlapping genes, we performed GO analysis, including biological process (BP), molecular function (MF) and cellular component (CC) and KEGG signaling pathway enrichment analysis. GO analysis showed that, in terms of BP, these overlapping genes were mainly enriched in response to oxygen levels, regulation of cell-cell adhesion, response to alcohol, regulation of leukocyte cell-cell adhesion, and cellular response to the abiotic stimulus. For MF, overlapping genes are mainly enriched in cytokine receptor binding, protein serine/threonine kinase activity, phosphatase binding, kinase regulator activity and transforming growth factor beta receptor binding. KEGG signaling pathway enrichment analysis revealed that it was mainly enriched in PI3K-Akt signaling pathway, Pancreatic cancer, colorectal cancer, AGE-RAGE signaling pathway in diabetic complications, hepatocellular carcinoma and other pathways (Figure 5).
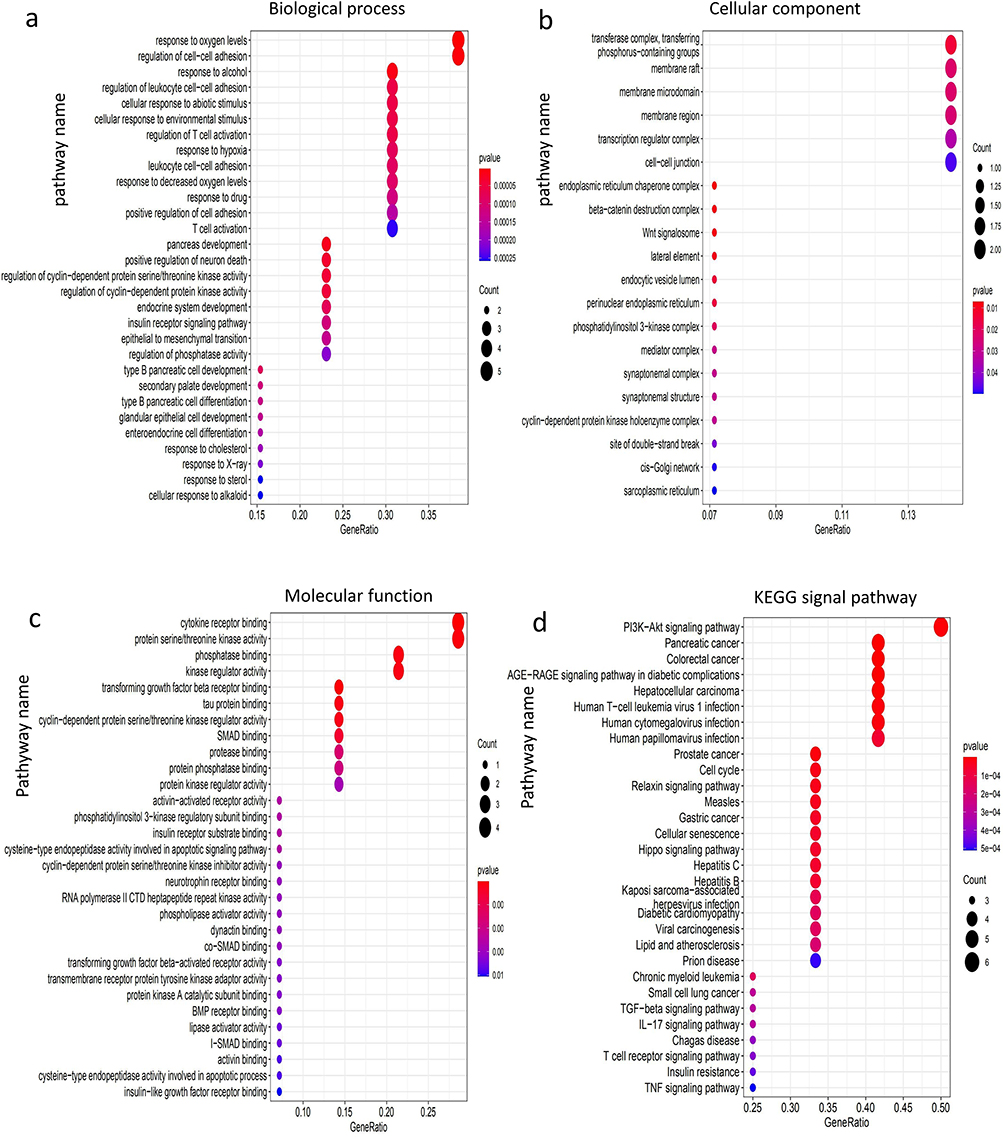 |
Figure 5 Dot plot of function enrichment analysis. (a) Biological process. (b) Cellular component. (c) Molecular function. (d) KEGG pathway. |
CircRNA/miRNA/mRNA Competing for Endogenous RNA (ceRNA) Network Construction
Based on the circRNAs, miRNAs, and mRNAs screened in the previous stage, an association network between circRNA, miRNA and hub genes were established by Cytoscape (Figure 6). circRNA/miRNA/mRNA regulatory axis, including hsa_circ_0009581/hsa-miR-150-5p/RAD51 axis, hsa_circ_0009581/hsa-miR-150-5p/CDK4 axis, hsa_circ_0009581/hsa-miR-150-5p/PKMYT1 axis, hsa_circ_0009581/hsa-miR-150-5p/MYB axis, hsa_circ_0009581/hsa-miR-150-5p/HSP90B1axis, hsa_circ_0009581/hsa-miR-150-5p/ZNF346 axis, hsa_circ_0009581/hsa-miR-150-5p/PIK3R1 axis, hsa_circ_0001947/hsa-miR-454-3p/CREB5 axis, hsa_circ_0001947/hsa-miR-454-3p/BMP6 axis, hsa_circ_0001947/hsa-miR-454-3p/TGFBR2 axis, hsa_circ_0007765/hsa-miR-382-5p/CASP3 axis, hsa_circ_0004277/hsa-miR-3190-3p/GSK3B axis, hsa_circ_0004277/hsa-miR-3190-3p/SETBP1axis, hsa_circ_0004277/hsa-miR-3190-3p/SMAD2 axis, were extracted from this network.
 |
Figure 6 ceRNA network. circRNAs, and miRNAs, hub-genes are shown in purple, yellow, and red respectively. circRNAs: dark purple: rising, light purple: falling; miRNAs: dark yellow: rising, light yellow: falling; hub-genes: dark red: rising, light red: falling. |
Effects of Hub-Genes on Overall Survival
Hub genes have been discovered. The overall survival (OS) time of patients with AML will now be shown, which is the biggest concern for all doctors. As shown in Figure 7a, the hub genes, MYB, HSP90B1, PIK3R1, SMAD2, PKMYT1, RAD51 and TGFBR2 were significantly differentially expressed between normal controls and AML patients from TCGA database and GTEx project. The overall survival of RAD51 is shown in Figure 7b, and higher expression of RAD51 is associated with poor overall survival of AML patients (HR = 2.2, P = 0.044).
 |
Figure 7 Survival analysis and validation of the 7 genes. (a) mRNA expression levels of 7 hub genes.*p<0.05. (b) The overall survival analysis of RAD51 related to AML patients. P < 0.05. |
Validation of Differentially Expressed circRNAs and miRNAs by qPCR
To investigate the expression of circRNA and miRNA from bioinformatic analysis and determine whether they may be important to leukemia, we used RT-qPCR to confirm that some circRNA and miRNA are expressed differently in AML and healthy controls. The different expression levels of 10 circRNAs and 6 miRNAs from AML patients (n=43) and healthy controls (n=35) were analyzed using RT-qPCRs. The results showed that hsa_circ_0009581 and hsa_circ_0005273 were significantly upregulated in AML patients; hsa_circ_0000497 and hsa_circ_0001947 were significantly downregulated in AML patients compared to that in the controls (Figure 8a, p <0.05). Hsa-miR-150-5p was significantly downregulated; hsa-miR-454-3p was upregulated in AML patients (Figure 8b, p<0.05). To explore the relationship between circ RNAs and miRNAs, hsa_circ_0009581 and hsa-miR-150-5p; hsa_ circ_0001947 and hsa-miR-454-3p were inversely correlated using Pearson’s correlation coefficient (Figure 8c). Hence, they might be regarded as prognostic indicators, but also could be designed as therapeutic targets of AML.
 |
Figure 8 Quantitative real-time PCR results of circRNAs and miRNAs expression. (a): RT-qPCR of 10 circRNAs (b): RT-qPCR of 6 miRNAs. (c): Relationships of circRNAs and miRNAs. **p<0.05. |
Discussion
AML has been confirmed as a highly heterogeneous malignant hematologic tumor in cytogenetics and molecular biology, which has been precisely diagnosed by MIMC (ie, morphology, immunology, molecular biology, and cytogenetics). Molecular mechanism research of AML has facilitated the development of several novel molecular targeted therapies over the past few years,11 whereas most AML patients will relapse, and the overall prognosis remains disappointing. CircRNAs are novel non-coding RNA family members with characteristic covalent closed loops. As high-throughput sequencing technology has been leaping forward, circRNAs, a novel class of RNA molecules, has been extensively investigated over the past few years. In AML, Li et al12 analyzed circRNA microarray from 10 samples (four healthy controls and six newly diagnosed AML patients included) and suggested that hsa_circ_0004277 may be a promising diagnostic marker and treatment target in AML. Chen et al13 have suggested that circ-ANAPC7 up-regulated significantly in AML, will be a potential biomarker for AML. Circ-ANAPC7 may play a certain role in AML pathogenesis by serving as a sponge for the miR-181 family. The above two studies involved few samples and used a single chip, so the mentioned results may not be ideally validated in large sample data. With the rapid advance of gene sequencing technology, there have been increasing biological data, and some research has employed multiple microarrays for analysis. For instance, Zhang et al14 adopted GSE116617 and GSE94591 to obtain the microarray data of circRNA, employed GSE142699 to obtain the microarray data of miRNA, and used GSE114868 to obtain the microarray data of mRNA. They built a circRNA-miRNA-hub gene subnetwork and revealed four key circRNAs (including hsa_circ_0074371, hsa_circ_0029407, hsa_circ_0001394, and hsa_circ_0000994) involved in the pathogenesis of AML. With one of the GEO microarrays consistent with to ours, this study is a better addition to Zhang et al study. The reason for the better addition is that the rest of the microarray data were different, such that the results obtained by overlapping were different.
In the current study, we selected three microarray data (GSE94591, GSE142700-GPL26945, GSE142700-GPL19956) from GEO database, identified 10 DEcircRNA: hsa_circ_0017446, hsa_circ_0012152, hsa_circ_0005273, hsa_circ_0001947, hsa_circ_0071375, hsa_circ_0009581, hsa_circ_0077765, hsa_circ_0000497, hsa_circ_0058058, hsa_circ_0004277. Among the 10 identified DEcircRNAs, hsacirccirc0001947, Circcirc0058058 and hsacirc0004277 have been found to be correlated with AML.15–18 Existing research has suggested that hsacirc0001947 inhibits AML cell proliferation via the hsa-miR-329-5p/CREBRF axis.15 Circ0058058 knockdown accelerated leukocytes apoptosis and inhibited AML progression by targeting miR-4319 to down-regulated EIF5A2 expression, and circ0058058 was considered a possible therapeutic target for AML.16 Li et al12 has suggested that the level of hsacirc0004277 is down-regulated in newly diagnosed AML patients, while the expression of hsacirc0004277 is up-regulated in complete remission cases, and in relapsed/refractory cases, the level was again down-regulated. The above results suggested that hsacirc0004277 can be a diagnostic marker and therapeutic target in AML. Liu et al17 further suggested that circ0004277 hinders the development of AML by targeting miR-134-5p and up-regulating SSBP2. Of the remaining seven circRNAs. hsacirc0000497 can promote peritoneal metastasis of ovarian cancer by regulating epithelial-mesenchymal transition (EMT) in vitro.18 The remaining six circRNAs have not been found to be correlated with tumors.
To further identify and validate key circRNAs, a network of the correlations between circRNAs, miRNAs, and hub genes was established through Cytoscape. The network contains four circRNAs that have been mentioned above, four miRNAs and fourteen hub genes. Wang et al19 found that circSPI1 contributes to proliferation and apoptosis by interacting with hsa-miR-382-5p in AML. hsa-miR-150-5p20 has been found to be associated with the proliferation and growth of a variety of solid tumors, such as non-small cell lung cancer,21 gastric cancer,22 and cervical cancer.23 hsa-miR-454-3p24 was associated with metastatic invasion of solid tumors such as breast and esophageal cancers.
We further performed RT-qPCR to confirm that some circRNA and miRNA are expressed differently in AML and healthy controls. The results showed that hsa_circ_0009581 and hsa_circ_0005273 were significantly upregulated in AML patients; hsa_circ_0000497 and hsa_circ_0001947 were significantly downregulated in AML patients. Hsa-miR-150-5p was significantly downregulated and hsa-miR-454-3p was upregulated in AML patients. hsa_circ_0009581 and hsa-miR-150-5p, hsa_circ_0001947 and hsa-miR-454-3p expression were significantly negatively correlated. According to the results of bioinformatics analysis and experiments, we speculated that hsa_circ_0009581 through hsa-miR-150-5p and hsa_circ_0001947 through hsa-miR-454-3p promoted leukemia development. hsa_circ_0001947 and hsa_circ_0009581 may provide new directions in the pathogenesis of AML.
Data from the TCGA database and the GTEx project were compared using GEPIA 2, and validation of the above hub genes (MYB, HSP90B1, PIK3R1, SMAD2, PKMYT1, RAD51 and TGFBR2) revealed differential expression in both AML patients and controls. Survival analysis revealed that high RAD51 expression was associated with poor prognosis. RAD51 whose core function is DNA repair is an essential gene for cell survival it can protect cells from genetic instability. In normal cells, it is regulated tightly and its expression is low while in most tumor cells, its expression level is high. RAD51 expression has been found to be significantly increased in breast and pancreatic cancers and is strongly associated with poor prognosis.25,26 Seedhouse et al27 investigated the role of RAD51 polymorphisms in modulating susceptibility to the development of AML. Park et al28 suggested that hypermethylation of RAD51, MLH1, and ATM shows better overall survival than the non-hypermethylation group in a univariate analysis in AML patients. Inhibition of RAD51 expression enhances cytarabine and daunorubicin sensitivity in AML cells.29 In our study, survival analysis of RAD51 revealed that high expression was associated with a poor prognosis, consistent with existing literature findings.
Four of the above hub genes were reported to be correlated with tumorigenesis in leukemia. Proto-oncogene MYB can be an important transcription factor to promote the development of AML. When its inhibitor monensin was used to effectively reduce MYB expression, it resulted in induction of cell differentiation and promotion of apoptosis.30 Hyperactivation of the PI3K/AKT pathway is correlated with drug resistance and relapse in AML cells.31 Disease-free survival and overall survival were significantly reduced in AML patients with up-regulated PI3K/AKT signaling pathway.32 Glioma-related oncogene 1 (GLI1) can directly bind PI3KR1 and activate the PI3K/AKT/GSK3β/CDK signaling pathway, resulting in AML cell growth and reduced chemosensitivity.33 Hypoxia can activate the TGF-β/Smad2 signaling pathway and induce multidrug resistance in leukemia cells.34 Chen et al35 have suggested that miR-425 is a prognostic factor in AML, and SMAD2 is one of the predicted targets of miR-425. Dou et al36 also suggested that DARS-AS1 up-regulates TGFB1 expression by binding to miR-425 to regulate AML progression through the Smad2/3 pathway. Yu et al37 have identified significant differences in miRNA data for ILCregs (regulatory innate lymphoid cells)-related genes (TGFBR2 included) in AML patients. These previous studies confirmed that our constructed ceRNAs are involved in the development of AML and may be therapeutic targets.
This study suggests that DEcircRNAs take on a critical significance in AML development and may be the effective therapeutic targets. The data from this study provide a novel perspective for pathogenesis of AML. Furthermore, our further research will examine the exact role of the above circRNAs in AML development and verify the hub genes identified in this study.
Conclusions
In this study, differentially expressed circRNAs, miRNAs and mRNAs were selected from the published microarray data of AML by bioinformatics analysis, and CircRNA/miRNA/hub gene networks were built, which confirmed by experiment and provided novel ideas for genetic research of AML. Despite rigorous bioinformatics analysis of this study, due to the limitations of sample number, Further clinical experiments are required to confirm the key circRNAs and seven hub genes including RAD51.
Data Sharing Statement
The data used to support the findings of this study are available from the corresponding author upon request.
Funding
Authors received no specific funding for this work.
Disclosure
Authors declare no conflicts of interest.
References
1. Döhner H, Weisdorf DJ, Bloomfield CD. Acute myeloid leukemia. N Engl J Med. 2015;373(12):1136–1152. doi:10.1056/NEJMra1406184
2. Pollyea DA, Bixby D, Perl A, et al. NCCN guidelines insights: acute myeloid leukemia, version 2.2021. J Natl Compr Canc Netw. 2021;19(1):16–27. doi:10.6004/jnccn.2021.0002
3. Webster JA, Luznik L, Gojo I. Treatment of AML relapse after Allo-HCT. Front Oncol. 2021;11:812207. doi:10.3389/fonc.2021.812207
4. Kristensen LS, Andersen MS, Stagsted LVW, Ebbesen KK, Hansen TB, Kjems J. The biogenesis, biology and characterization of circular RNAs. Nat Rev Genet. 2019;20(11):675–691. doi:10.1038/s41576-019-0158-7
5. Su M, Xiao Y, Ma J, et al. Circular RNAs in cancer: emerging functions in hallmarks, stemness, resistance and roles as potential biomarkers. Mol Cancer. 2019;18(1):90. doi:10.1186/s12943-019-1002-6
6. Kun-Peng Z, Xiao-Long M, Chun-Lin Z. Overexpressed circPVT1, a potential new circular RNA biomarker, contributes to doxorubicin and cisplatin resistance of osteosarcoma cells by regulating ABCB1. Int J Biol Sci. 2018;14(3):321–330. doi:10.7150/ijbs.24360
7. Liu Y, Dong Y, Zhao L, Su L, Luo J. Circular RNA‑MTO1 suppresses breast cancer cell viability and reverses monastrol resistance through regulating the TRAF4/Eg5 axis. Int J Oncol. 2018;53(4):1752–1762. doi:10.3892/ijo.2018.4485
8. Chandran RK, Geetha N, Sakthivel KM, et al. Genomic amplification of BCR-ABL1 fusion gene and its impact on the disease progression mechanism in patients with chronic myelogenous leukemia. Gene. 2019;686:85–91. doi:10.1016/j.gene.2018.11.005
9. Gong Y, Mao J, Wu D, et al. Circ-ZEB1.33 promotes the proliferation of human HCC by sponging miR-200a-3p and upregulating CDK6. Cancer Cell Int. 2018;18:116. doi:10.1186/s12935-018-0602-3
10. Zhang L, Bu Z, Shen J, Shang L, Chen Y, Wang Y. A novel circular RNA (hsa_circ_0000370) increases cell viability and inhibits apoptosis of FLT3-ITD-positive acute myeloid leukemia cells by regulating miR-1299 and S100A7A. Biomed Pharmacother. 2020;122:109619. doi:10.1016/j.biopha.2019.109619
11. Fleischmann M, Schnetzke U, Hochhaus A, Scholl S. Management of acute myeloid leukemia: current treatment options and future perspectives. Cancers. 2021;13(22):5722. doi:10.3390/cancers13225722
12. Li W, Zhong C, Jiao J, et al. Characterization of hsa_circ_0004277 as a new biomarker for acute myeloid leukemia via circular RNA profile and bioinformatics analysis. Int J Mol Sci. 2017;18(3):597. doi:10.3390/ijms18030597
13. Chen H, Liu T, Liu J, et al. Circ-ANAPC7 is upregulated in acute myeloid leukemia and appears to target the MiR-181 family. Cell Physiol Biochem. 2018;47(5):1998–2007. doi:10.1159/000491468
14. Zhang S. The characteristics of circRNA as competing endogenous RNA in pathogenesis of acute myeloid leukemia. BMC Cancer. 2021;21(1):277. doi:10.1186/s12885-021-08029-7
15. Han F, Zhong C, Li W, et al. Hsa_circ_0001947 suppresses acute myeloid leukemia progression via targeting hsa-miR-329-5p/CREBRF axis. Epigenomics. 2020;12(11):935–953. doi:10.2217/epi-2019-0352
16. Zhang T, Zhou Y, Guan J, Cheng H. Circ_0058058 knockdown inhibits acute myeloid leukemia progression by sponging miR-4319 to regulate EIF5A2 expression. Cancer Biother Radiopharm. 2021;2021:1.
17. Liu Y, Chen X, Liu J, Jin Y, Wang W. Circular RNA circ_0004277 inhibits acute myeloid leukemia progression through MicroRNA-134-5p / single stranded DNA binding protein 2. Bioengineered. 2022;13(4):9662–9673. doi:10.1080/21655979.2022.2059609
18. Luo N, Sulaiman Z, Wang C, et al. Hsa_circ_0000497 and hsa_circ_0000918 contributed to peritoneal metastasis of ovarian cancer via ascites. J Transl Med. 2022;20(1):201. doi:10.1186/s12967-022-03404-9
19. Wang X, Jin P, Zhang Y, Wang K. CircSPI1 acts as an oncogene in acute myeloid leukemia through antagonizing SPI1 and interacting with microRNAs. Cell Death Dis. 2021;12(4):297. doi:10.1038/s41419-021-03566-2
20. Jin M, Shi C, Yang C, Liu J, Huang G. Upregulated circRNA ARHGAP10 predicts an unfavorable prognosis in NSCLC through regulation of the miR-150-5p/GLUT-1 axis. Mol Ther Nucleic Acids. 2019;18:219–231. doi:10.1016/j.omtn.2019.08.016
21. Wang S, Tang D, Wang W, et al. circLMTK2 acts as a sponge of miR-150-5p and promotes proliferation and metastasis in gastric cancer. Mol Cancer. 2019;18(1):162. doi:10.1186/s12943-019-1081-4
22. Cai H, Zhang P, Xu M, Yan L, Liu N, Wu X. Circular RNA hsa_circ_0000263 participates in cervical cancer development by regulating target gene of miR-150-5p. J Cell Physiol. 2019;234(7):11391–11400. doi:10.1002/jcp.27796
23. Ren L, Chen H, Song J, et al. MiR-454-3p-mediated Wnt/β-catenin signaling antagonists suppression promotes breast cancer metastasis. Theranostics. 2019;9(2):449–465. doi:10.7150/thno.29055
24. Yan A, Wang C, Zheng L, Zhou J, Zhang Y. MicroRNA-454-3p inhibits cell proliferation and invasion in esophageal cancer by targeting insulin-like growth factor 2 mRNA-binding protein 1. Oncol Lett. 2020;20(6):359. doi:10.3892/ol.2020.12223
25. Zhang X, Ma N, Yao W, Li S, Ren Z. RAD51 is a potential marker for prognosis and regulates cell proliferation in pancreatic cancer. Cancer Cell Int. 2019;19:356. doi:10.1186/s12935-019-1077-6
26. Wiegmans AP, Al-Ejeh F, Chee N, et al. Rad51 supports triple negative breast cancer metastasis. Oncotarget. 2014;5(10):3261–3272. doi:10.18632/oncotarget.1923
27. Seedhouse C, Faulkner R, Ashraf N, Das-Gupta E, Russell N. Polymorphisms in genes involved in homologous recombination repair interact to increase the risk of developing acute myeloid leukemia. Clin Cancer Res. 2004;10(8):2675–2680. doi:10.1158/1078-0432.CCR-03-0372
28. Park S, So MK, Huh J. Methylation of DNA repair genes as a prognostic biomarker in AML of a TCGA-LAML cohort. Clin Lab. 2022;68(7). doi:10.7754/Clin.Lab.2021.211025
29. Xie C, Drenberg C, Edwards H, et al. Panobinostat enhances cytarabine and daunorubicin sensitivities in AML cells through suppressing the expression of BRCA1, CHK1, and Rad51. PLoS One. 2013;8(11):e79106. doi:10.1371/journal.pone.0079106
30. Yusenko MV, Trentmann A, Andersson MK, et al. a novel potent MYB inhibitor, suppresses proliferation of acute myeloid leukemia and adenoid cystic carcinoma cells. Cancer Lett. 2020;479:61–70. doi:10.1016/j.canlet.2020.01.039
31. Park S, Chapuis N, Tamburini J, et al. Role of the PI3K/AKT and mTOR signaling pathways in acute myeloid leukemia. Haematologica. 2010;95(5):819–828. doi:10.3324/haematol.2009.013797
32. Kornblau SM, Womble M, Qiu YH, et al. Simultaneous activation of multiple signal transduction pathways confers poor prognosis in acute myelogenous leukemia. Blood. 2006;108(7):2358–2365. doi:10.1182/blood-2006-02-003475
33. Zhou C, Du J, Zhao L, et al. GLI1 reduces drug sensitivity by regulating cell cycle through PI3K/AKT/GSK3/CDK pathway in acute myeloid leukemia. Cell Death Dis. 2021;12(3):231. doi:10.1038/s41419-021-03504-2
34. Cui XY, Skretting G, Jing Y, Sun H, Sandset PM, Sun L. Hypoxia influences stem cell-like properties in multidrug resistant K562 leukemic cells. Blood Cells Mol Dis. 2013;51(3):177–184. doi:10.1016/j.bcmd.2013.05.003
35. Yang C, Shao T, Zhang H, et al. MiR-425 expression profiling in acute myeloid leukemia might guide the treatment choice between allogeneic transplantation and chemotherapy. J Transl Med. 2018;16(1):267. doi:10.1186/s12967-018-1647-8
36. Dou B, Jiang Z, Chen X, et al. Oncogenic long noncoding RNA DARS-AS1 in childhood acute myeloid leukemia by binding to microRNA-425. Technol Cancer Res Treat. 2020;19:1533033820965580. doi:10.1177/1533033820965580
37. Yu J, Li Y, Pan Y, et al. Deficient regulatory innate lymphoid cells and differential expression of miRNAs in acute myeloid leukemia quantified by next generation sequence. Cancer Manag Res. 2019;11:10969–10982. doi:10.2147/CMAR.S234327
 © 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.