Back to Journals » OncoTargets and Therapy » Volume 13
Construction of a circRNA-Related ceRNA Prognostic Regulatory Network in Breast Cancer
Authors Song H, Sun J, Kong W, Ji Y ![]() , Xu D, Wang J
, Xu D, Wang J ![]()
Received 24 June 2020
Accepted for publication 4 August 2020
Published 20 August 2020 Volume 2020:13 Pages 8347—8358
DOI https://doi.org/10.2147/OTT.S266507
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Arseniy Yuzhalin
Huan Song,1,* Jian Sun,2,* Weimin Kong,2,* Ye Ji,1 Dian Xu,1 Jianming Wang1
1Department of Epidemiology, Center for Global Health, School of Public Health, Nanjing Medical University, Nanjing 211166, People’s Republic of China; 2Department of Thoracic Surgery, The First People’s Hospital of Yancheng City, Yancheng 224006, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Jianming Wang Department of Epidemiology
Center for Global Health, School of Public Health, Nanjing Medical University, Nanjing 211166, People’s Republic of China
Tel +86-25-86868438
Email [email protected]
Purpose: Accumulating evidence has indicated that circRNAs are closely involved in tumorigenesis and progression of human cancers. However, the molecular mechanism underlying function of circRNAs in breast cancer has not been thoroughly elucidated. Currently, we aimed to characterize the circRNA-related competing endogenous RNA (ceRNA) regulatory network in breast cancer and to construct prognostic model.
Materials and Methods: First, we constructed circRNA expression profiles for paired breast cancer in a Chinese population using a human circRNA microarray. Expression profiles of circRNAs, miRNAs, and mRNAs were retrieved from our circRNA dataset, the Gene Expression Omnibus (GEO) and The Cancer Genome Atlas (TCGA) databases. We applied the limma and edgeR packages to identify differentially expressed RNAs. Weighted gene correlation network analysis (WGCNA) was used to identify critical modules of mRNAs. Next, a ceRNA network was established based on circRNA–miRNA and miRNA–mRNA intersections. Both Cox regression analysis and ROC curve analysis were performed to generate prognostic model. Additionally, we performed Gene Set Enrichment Analysis (GSEA) on prognostic signatures.
Results: Total of 59 circRNAs, 98 miRNAs and 3966 mRNAs were identified as differentially expressed RNAs. We first identified 38 miRNA-mRNA pairs and 38 circRNA-miRNA pairs to construct the circRNA–miRNA-mRNA regulatory network and then generated a prognostic model based on 7 signatures (MMD, SLC29A4, CREB5, FOS, ANKRD29, MYOCD, and PIGR), and patients with high-risk scores presented poor prognosis. Several cancer-related pathways were enriched, including the TGF-β pathway, the focal adhesion pathway, and the JAK-STAT signaling pathway, and 20 prognostic ceRNA regulatory networks were subsequently identified.
Conclusion: In all, we screened a series of dysregulated circRNAs, miRNAs, and mRNAs, and constructed circRNA-related ceRNA network in breast cancer. Our findings may help to deepen the understanding of circRNA-related regulatory mechanisms. Moreover, we generated a prognostic model that provided new insight into postoperative management for breast cancer.
Keywords: ncRNA, ceRNA, circRNA, miRNA, mRNA, breast cancer, TCGA
Introduction
Breast cancer is the second most common cancer and is the leading cause of cancer-related death among females worldwide with over 2 million newly diagnosed cases and more than 60 thousand deaths every year.1 Despite advances in treatment, the mortality rate of breast cancer remains high, mainly due to its high frequency of metastasis, chemotherapy resistance, and recurrence. Some preventable causes of breast cancer have been identified, including aging, nulliparity, early menarche, late menopause, family history, and use of fertility drugs or oral contraceptives.2–4 Recently, genetic and epigenetic factors have garnered increasing attention and have exhibited excellent application prospects in the diagnosis and treatment of breast cancer.
Noncoding RNA (ncRNA) is a functional RNA molecule that is transcribed from DNA but is not translated into proteins. Epigenetic related ncRNAs include tRNAs (transfer RNAs), rRNAs (ribosomal RNAs), miRNAs (microRNAs), siRNAs (small interfering RNAs), lncRNAs (long ncRNAs), circular RNAs (circRNAs), and piRNAs (Piwi-interacting RNAs). LncRNAs are characterized as being longer than 200 bp, while siRNAs, snRNAs, miRNAs, and piRNAs are shorter than 200 bp.5 Recently, ncRNAs have been shown to be involved in the pathogenesis and progression of various diseases through the mechanism of ceRNA function.6–8 For instance, the lncRNA MYOSLID may function as a ceRNA to regulate MCL-1 expression by sponging miR-29c-3p in gastric cancer,9 and ciRS-7 could bind miR-7, serving as a therapeutic target by reducing EGFR-RAF1 activity in colorectal cancer.10
CircRNAs are a novel type of endogenous ncRNA that were considered an outcome of “splicing error” in the 1970s.11,12 Advances in high-throughput sequencing technology have facilitated the understanding of circRNA function. CircRNAs can be classified as exotic, intronic, exon-intron, or intergenic.11,13 Compared to corresponding linear RNAs, circRNAs are characterized by their covalently closed loop structure without a 5ʹ cap or a 3ʹ poly tail, which makes them resistant to RNase.14 Accumulating evidence has demonstrated specific expression patterns of circRNAs in cells, tissues, or different stages of diseases, presenting promising diagnostic and prognostic implications.15,16 In terms of molecular mechanisms, circRNAs may function as competitive endogenous RNAs, they may bind proteins, and they may encode proteins,17,18 participating in the occurrence and development of cardiovascular diseases, neurological disorders, and cancers.19–21 However, the circRNA-related ceRNA network motifs and the underlying prognostic values of these RNAs in breast cancer have not been thoroughly elucidated.
In the current study, we combined our previously constructed circRNA database with the Gene Expression Omnibus (GEO) database and The Cancer Genome Atlas (TCGA) database to explore the circRNA-miRNA-mRNA axis in breast cancer.22 The GEO database and TCGA databases provide a large number of publicly available RNA-seq data regarding various cancers, including breast cancer.23 Additionally, we attempted to construct a circRNA-related ceRNA regulatory network based on the dysregulated expression profiles in breast cancer, and we sought to further explore its pathogenesis and progression and to find the underlying prognostic and therapeutic biomarkers of breast cancer.
Materials and Methods
Patients and Data Collection
First, we recruited four patients with breast cancer who had undergone a mastectomy in the First People’s Hospital of Yixing City, China, in March 2016. We constructed circRNA expression profiles for breast cancer; the profiles were derived from four pairs of cancer tissues and adjacent normal-appearing tissues using Arraystar human circRNA microarray V2 (Catalog No: AS-CR-H-V2.0).24 The circRNA expression profiles were recruited from the GEO database (https://www.ncbi.nlm.nih.gov/geo/) with the filter criteria of 1) the expression dataset was acquired from tumor and normal tissues in patients with breast cancer; 2) the detection method was microarray analysis; and 3) the sample size was at least three. Finally, the dataset GSE101123, consisting of eight breast cancer and three normal tissues, was selected. The level-three RNA-seq raw count data of miRNA (containing 1090 breast cancer and 103 normal tissues) and mRNA (including 1096 breast cancer and 112 normal tissues) were extracted from the TCGA data portal (https://portal.gdc.cancer.gov/, access date: August 14, 2019), except for the expression data of males. Additionally, we downloaded the corresponding clinical characteristics from TCGA, including age, survival time, survival status, clinical stage, and T, N, and M stages. The flow chart of the analysis is illustrated in Figure 1.
 |
Figure 1 Flowchart of circRNA-related ceRNA regulatory network analysis in breast cancer. |
Identification of Differentially Expressed circRNAs, miRNAs and mRNAs
First, we extracted and sorted the expression matrixes by Perl (https://www.perl.org/get.html) and identified the differential expression using R3.6.0 (https://www.r-project.org/). For differentially expressed circRNA (DEcircRNA) analysis, we first integrated our previous circRNA dataset with GSE101123 by batch normalization using the “sva” package and then applied the “limma” package to screen DEcircRNAs with a filter criterion of |Fold Change| > 1 and FDR < 0.05. The filter criteria for differentially expressed miRNAs (DEmiRNAs) and mRNAs (DEmRNAs) were |log2(fold-change)| > 2 and FDR < 0.05 using the “edgeR” package. We applied the “pheatmap” package to construct the heatmap of DEcircRNAs, DEmiRNAs, and DEmRNAs. Afterward, we established the coexpression network for DEmRNAs in breast cancer using a weighted gene correlation network analysis (WGCNA) package. First, we used goodSampleGenes and meanFPKM =0.5 to remove the unqualified genes. Then, we picked the best soft-thresholding power by the pickSoftThreshold function. Adjacency was converted into a TOM, and genes with high correlation were grouped into different modules according to TOM-based dissimilarity with minModuleSize =30. Ultimately, the DEmRNAs involved in the highest correlated modules were candidates for further investigation, and we performed logarithmic transformation for homogenization.
Construction of the circRNA-miRNA-mRNA Regulatory Network
We employed the cancer-specific circRNA database (CSCD, http://gb.whu.edu.cn/CSCD/) and the circular RNA interactome (CircInteractome, https://circinteractome.nia.nih.gov/) database to predict circRNA-miRNA interactions. The target miRNAs were further identified by overlapping with DEmiRNAs obtained from the TCGA database. Pairs of miRNA-mRNA interactions were predicted by three bioinformatic databases, namely, miRDB (https://mirdb.org/download.html), miRTarBase (http://mirtarbase.mbc.nctu.edu.tw/php/index.php) and TargetScan (http://www.targetscan.org/vert_72/). Only the interactions that were consistently predicted by the three databases were selected for further analysis. Then, we screened the target mRNAs by intersecting with the DEmRNAs involved in the key modules based on WGCNA. Finally, based on miRNA-mRNA pairs and circRNA-miRNA interactions, we established the circRNA-miRNA-mRNA regulatory network and visualized it with Cytoscape 3.7.1 software (https://cytoscape.org/).
Identification of Prognostic Signatures in Breast Cancer
To investigate the prognostic value of targeted mRNAs involved in the circRNA-related ceRNA network of breast cancer, we constructed a prognostic model. Briefly, we identified the prognostic signatures based on Cox regression analysis using the “survival” package and selected the key signatures according to the best Akaike information criterion (AIC) using the “MASS” package. These signatures were subsequently applied to construct a prognostic model and generate a risk score based on the expression of each signature and its corresponding coefficients. We also performed a receiver operating characteristic curve (ROC) to evaluate the risk score. Patients with risk scores lower than the median values were classified into a low-risk group, and the rest were classified into a high-risk group.
Functional Analysis of Prognostic Signatures in the circRNA-Related Network
To predict the potential functions of prognostic signatures involved in the circRNA-related ceRNA network, we performed Gene Set Enrichment Analysis (GSEA, https://www.gsea-msigdb.org/gsea/index.jsp).25 For the phenotypic file, we defined patients with targeted mRNA expression greater than the median level as the “high-risk” subgroup and defined the rest as the “low-risk” subgroup. We applied the C2 (c2.cp.kegg.v7.1.symbols.gmt) subcollection as reference gene sets. Thresholds for significance were determined by 1000 permutation analyses, and FDR < 0.05 was considered to be significant.
Ethics Statement
This study was approved by the Ethics Committee of Nanjing Medical University. After informed consent was obtained from all participants, questionnaires were used to collect demographic data. This study was conducted in accordance with the Declaration of Helsinki. Data downloaded from the public database followed the data access policy.
Results
Differentially Expressed circRNAs, miRNAs, and mRNAs in Breast Cancer
Compared with normal breast tissues, a total of 59 DEcircRNAs were identified from circRNA microarray analyses of breast-cancer tissues, including 54 upregulated circRNAs and 5 downregulated circRNAs (Figure 2A). The basic characteristics of the top 4 DEcircRNAs (hsa_circ_0062682, hsa_circ_0007798, hsa_circ_0092342, and hsa_circ_0065173) are described in Table 1. A total of 98 DEmiRNAs were identified, including 78 upregulated miRNAs and 20 downregulated miRNAs (Figure 2B). Among the DEmiRNAs, miR-133b (log2(fold-change) = −6.96, FDR < 0.05) was the most downregulated, and miR-122-5p (log2(fold-change) = 7.79, FDR < 0.05) was the most upregulated. Meanwhile, 3966 DEmRNAs, consisting of 2906 upregulated mRNAs and 1060 downregulated mRNAs, were detected between breast-cancer tissues and normal tissues (Figure 2C), of which UCN3 (log2(fold-change) = 9.09, FDR < 0.05) and MYH2 (log2(fold-change) = −8.59, FDR < 0.05) were ranked as the most upregulated and downregulated mRNAs, respectively.
 |
Table 1 Characteristics of the Top Four DEcircRNAs in Breast Cancer |
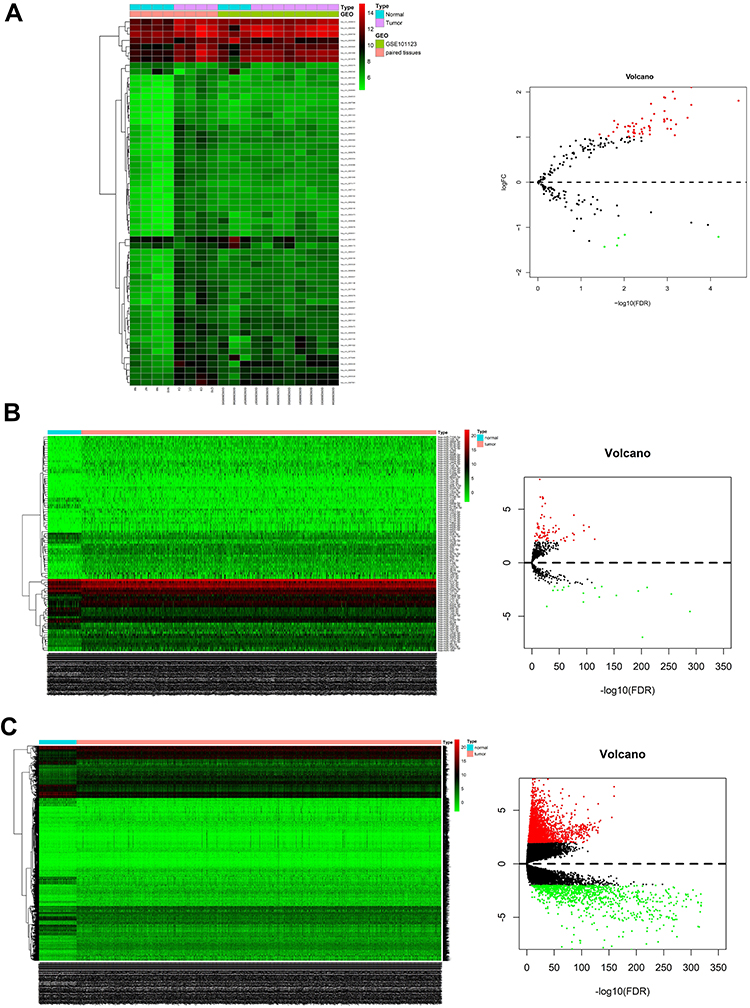 |
Figure 2 Differentially expressed genes in breast cancer. (A) Heatmap and volcano of DEcircRNAs identified from previous circRNA datasets and GEO databases using the limma package (|log2(fold-change) >1| and FDR < 0.05); (B) Heatmap and volcano of DEmiRNAs identified from TCGA database using the edgeR package (|log2(fold-change) >2| and FDR < 0.05); (C) Heatmap and volcano of DEmRNAs identified from TCGA database using the edgeR package (|log2(fold-change) > 2| and FDR < 0.05). |
Identification of the Coexpression Network for DEmRNAs in Breast Cancer
To identify the key modules of mRNAs in breast cancer, we performed WGCNA on the DEmRNAs. In brief, β =4 was selected as the best soft threshold with a scale-free R2 = 0.96. Subsequently, the highly correlated modules were merged with 75% relevance, and a total of 11 coexpression modules were generated, with the number of mRNAs contained in these modules ranging from 44 to 1400 (Figure 3A). As shown in Figure 3B, the most significant module associated with breast cancer and the normal trait was the turquoise module, which was composed of 1400 mRNAs that were candidates for further analysis (P = 2×10−262). The scatterplots of Gene Significance vs Module Membership in the turquoise module suggested the highest correlation (Figure 3C).
 |
Figure 3 Weighted gene correlation network analysis (WGCNA) of mRNAs in breast cancer. (A) Identification of a coexpression module of DEmRNAs in breast cancer. (B) The correlation between the DEmRNA module and clinical traits. (C) Scatter plot of module eigengenes in the turquoise modules. P < 0.05. Abbreviation: DEmRNAs, differentially expressed mRNAs. |
Construction of the ceRNA Regulatory Network in Breast Cancer
To clarify the circRNA-related ceRNA regulatory mechanism, a circRNA-miRNA-mRNA network was constructed. We used the CSCD database for the miRNA response element (MRE) prediction of DEcircRNAs, and the CircInteractome database was subsequently applied for those not included in the CSCD. After intersecting with the DEmiRNAs identified from the TCGA database, a total of 46 miRNAs were identified. Next, we acquired 1761 predicted mRNAs of these 46 miRNAs by overlapping the three databases (miRDB, miRTarBase, and TargetScan). A total of 30 predicted mRNAs remained after overlapping with DEmRNAs involved in key modules of DEmRNAs by WGCNA. Finally, a total of 38 circRNA-miRNA pairs, including 29 circRNAs and 18 miRNAs, and 38 miRNA-mRNA pairs, including 18 miRNAs and 30 mRNAs, remained for further ceRNA network construction. The ceRNA network based on 76 intersections is illustrated in Figure 4A.
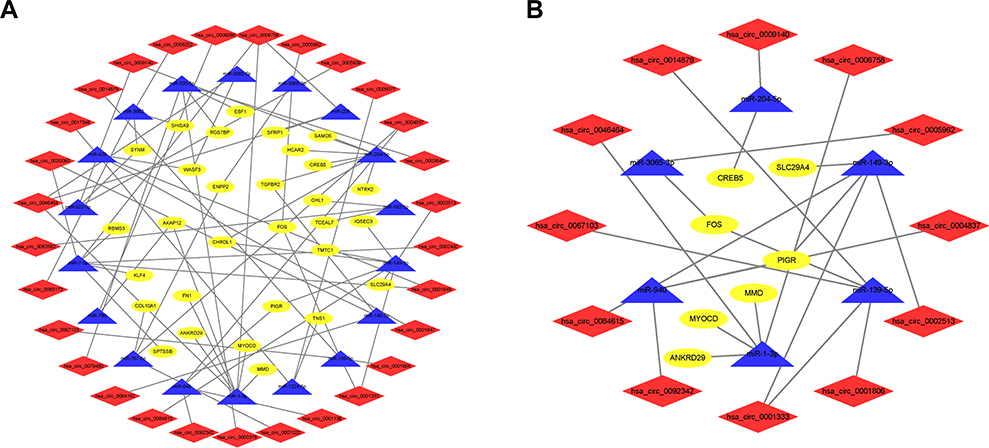 |
Figure 4 CircRNA-miRNA-mRNA ceRNA regulatory network in breast cancer. (A) The ceRNA regulatory network in breast cancer. (B) The prognostic ceRNA regulatory network in breast cancer. |
Exploration of the Prognostic Model and Corresponding ceRNA Network
To estimate the prognostic values of mRNAs involved in the circRNA-related ceRNA regulatory network, we attempted to construct a prognostic model. A total of 30 mRNAs involved in the circRNA-related ceRNA network were applied to establish the prognostic model. Then, 7 signatures were identified as optimally prognostic signatures based on stepAIC function, including MMD, SLC29A4, CREB5, FOS, ANKRD29, MYOCD and PIGR. Kaplan–Meier analysis of these 7 prognostic signatures for patients with breast cancer is shown in Supplemental Figure 1. Lower expression of ANKRD29 and PIGR was associated with poor prognosis in patients with breast cancer (P = 0.0371 and P = 0.00632, respectively).
After that step, a risk score for the prediction of prognosis was generated as follows: Risk score=ExpressionMMD*0.21041-ExpressionSLC29A4*0.11003-ExpressionCREB5*0.14338 -ExpressionFOS*0.07729-ExpressionANKRD29*0.12044+ExpressionMYOCD*0.16679-ExpressionPIGR*0.09200. We found that the risk score can accurately predict the prognosis of patients with an area under the curve (AUC) = 0.833 (Figure 5A). Next, we classified patients into high- and low-risk groups based on the median risk score. As shown in Figure 5B, the overall survival rate of patients in the high-risk group was lower than that of patients in the low-risk group (P = 1.356×10−6). Based on the univariate Cox regression analysis, the age (HR:1.036, 95% CI: 1.021–1.051), clinical-stage (HR:2.768, 95% CI: 1.932–3.964; stage III-IV vs stage I-II), T stage (HR:1.925, 95% CI: 1.291–2.869; T3-4 vs T1-2), M stage (HR:5.907, 95% CI: 3.244–10.757; M1 vs M0), N stage (HR:2.162, 95% CI: 1.486–3.147; N1-3 vs N0) and risk score (HR:2.469, 95% CI: 1.680–3.628) were related to the overall survival (Figure 5C). As shown in Figure 5D, the multivariate Cox regression analysis indicated that the risk score was an independent risk factor for overall survival (HR: 2.305, 95% CI: 1.533–3.464). In addition, we observed that 7 signatures were involved in 20 circRNA-related ceRNA networks (Figure 4B).
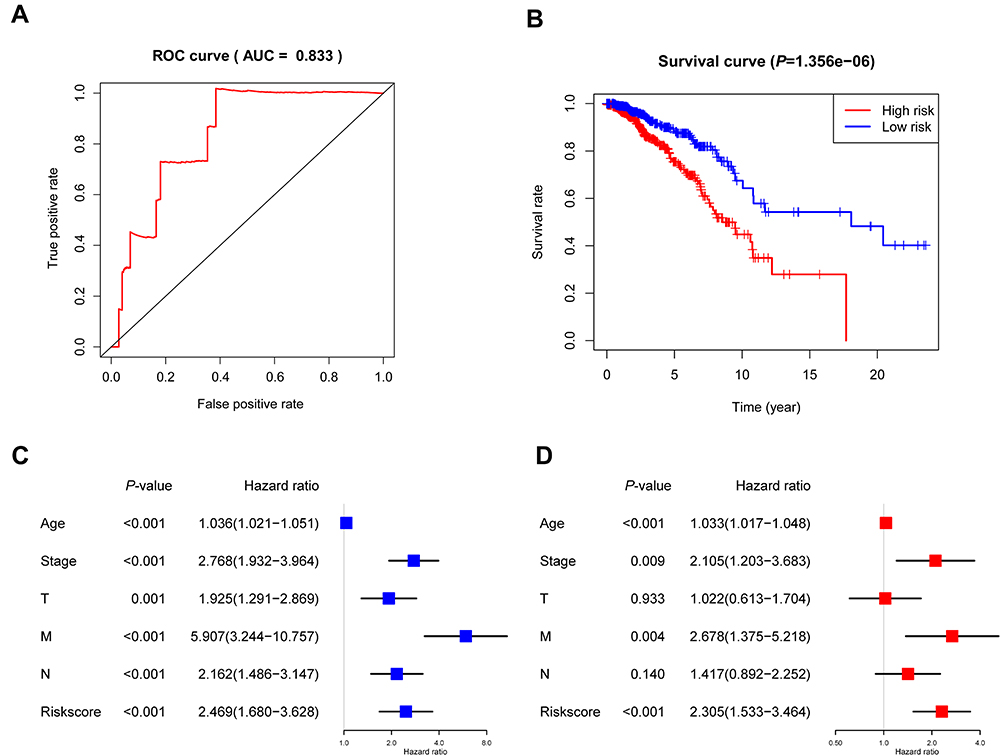 |
Figure 5 Construction of 7 signature-based prognostic models. (A) The ROC curve to evaluate the risk score. (B) Kaplan–Meier analysis for patients with high and low risk scores for breast cancer. (C) Univariate Cox regression analysis. (D) Multivariable Cox regression analysis. P < 0.05. Abbreviation: ROC, receiver operating characteristic curve. |
GSEA Analysis for Prognostic Signatures in the ceRNA Regulatory Network
We performed GSEA analysis on 1096 patients from high-risk and low-risk groups to identify the cancer-related pathways involved in the 7 prognostic signatures. A total of 27 pathways were enriched in patients with high expression of MMD, 1 pathway for SLC29A4, 4 pathways for CREB5, 21 pathways for FOS, 14 pathways for ANKRD29, 30 pathways for MYOCD and 9 pathways for PIGR. As shown in Figure 6, multiple cancer-related pathways were enriched, including the TGF-β pathway (NES = 2.35, FDR < 0.001) for MMD, ECM receptor interaction for CREB5 (NES = 2.37, FDR < 0.001), ECM receptor interaction for FOS (NES = 2.09, FDR<0.001), cell cycle for ANKRD29 (NES = −2.27, FDR<0.001), cell cycle for MYOCD (NES = −2.18, FDR < 0.001) and JAK STAT signaling pathway (NES = −1.90, FDR = 0.04) for PIGR.
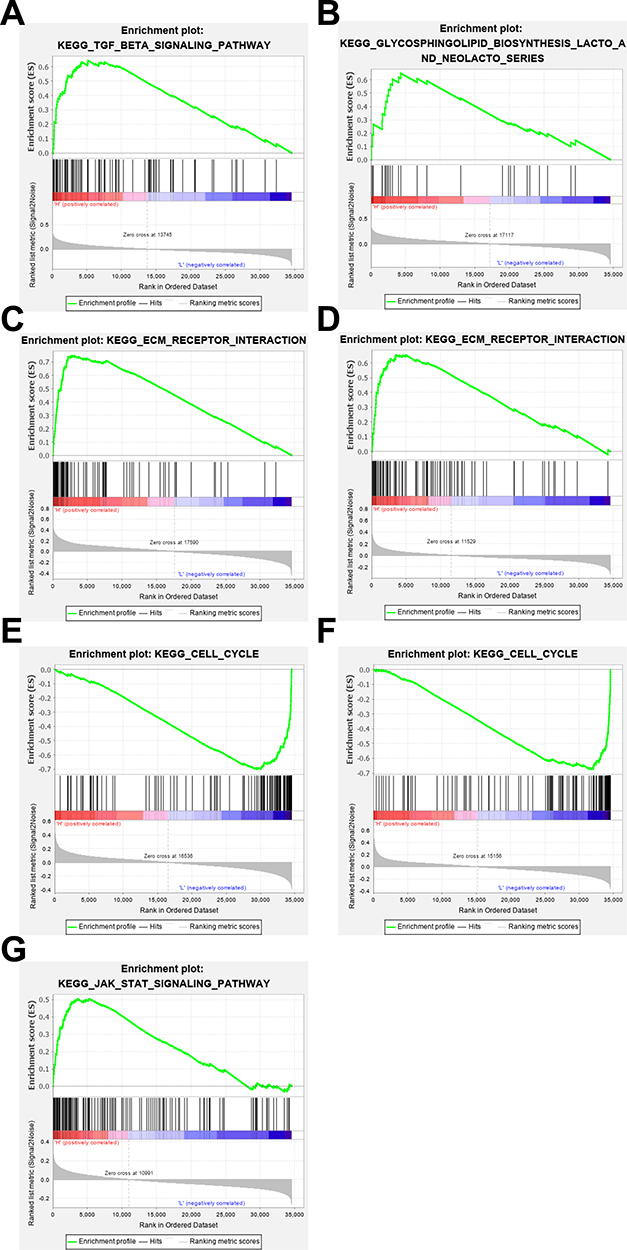 |
Figure 6 GSEA analysis of these 7 signatures in breast cancer. (A) GSEA analysis of MMD; (B) GSEA analysis of SLC29A4; (C) GSEA analysis of CREB5; (D) GSEA analysis of FOS; (E) GSEA analysis of ANKRD29; (F) GSEA analysis of MYOCD; (G) GSEA analysis of PIGR. (FDR < 0.05). |
Discussion
Breast cancer is one of the most common malignant tumors among females in the world. It is imperative to clarify the molecular mechanisms of carcinogenesis, explore prognostic factors, and search for therapeutic biomarkers of breast cancer. NcRNAs are a novel type of RNA that is primarily expressed in eukaryotes and plays an essential role in maintaining regular physiological activity.26–30 Although the ceRNA regulatory network has been widely verified, the circRNA-related ceRNA network has rarely been studied. In the current study, we established a circRNA-related ceRNA regulatory network and prognostic model in breast cancer by bioinformatic analysis. Expression datasets of DEcircRNAs, DEmiRNAs, and DEmRNAs between breast-cancer tissues and adjacent normal mammary glands were identified based on our previous circRNA dataset, the GEO database and TCGA database.31,32 Then, we constructed a circRNA-miRNA-mRNA regulatory network to investigate the underlying molecular mechanisms of DEcircRNAs. We constructed a prognostic model derived from 7 signatures in the ceRNA network, and functional analysis suggested that the 7 signatures were significantly enriched in cancer-related pathways, thereby demonstrating promise as prognostic and therapeutic biomarkers of breast cancer.
With the rapid development of high-throughput sequencing technology, the biogenesis and function of circRNAs have been widely explored. Accumulating evidence has confirmed the capacity of circRNAs to serve as a biomarker in the diagnosis or prognosis of cancer due to its covalently closed loop structure. For instance, a decrease in circMTO1 can predict poor survival among patients with hepatocellular carcinoma.33 Wang et al reported that hsa_circ_0077837 and hsa_circ_0001821 could serve as potential biomarkers for lung cancer, whereas hsa_circ_0001073 and hsa_circ_0001495 showed value as diagnostic molecules.34 Circ-KIAA1244, a gastric cancer-derived circRNA, was reported to be a novel circulating molecular biomarker of gastric cancer.35 Xu et al reported that hsa_circ_0005230 could mechanically sponge miR-1238 and miR-1299 to serve its oncogenic functions in cholangiocarcinoma.36 In the current study, we identified 59 DEcircRNAs in breast cancer, including hsa_circ_0062682, hsa_circ_0007798, hsa_circ_0092342, and hsa_circ_0065173. Among these DEcircRNAs, hsa_circ_0065173 has been previously found to be altered in breast cancer.37
Several molecular functions of circRNAs have been identified, including sponging microRNA, binding to RNA-binding proteins and serving protein translation functions.38,39 CircRNAs are enriched with miRNA binding sites, which causes them to act as miRNA sponges and then participate in the pathogenesis and development of various diseases. Zhang et al reported that the downregulation of circRNA_100269 in gastric cancer may suppress cell line proliferation (in AGS and MKN28 cells) by targeting miR-630.40 CircRNA ZFR could stimulate the progression of non-small-cell lung cancer by acting as a miR-101-3p sponge to enhance CUL4B expression.41 In this study, we successfully constructed a circRNA-miRNA-mRNA axis, including 29 DEcircRNAs, 18 DEmiRNAs, and 30 DEmRNAs, which might help to elucidate the circRNA-related regulatory mechanisms in breast cancer. In addition, we successfully constructed a molecular prognostic model originating from 7 signatures involved in the circRNA-related ceRNA regulatory network (MMD, SLC29A4, CREB5, FOS, ANKRD29, MYOCD and PIGR), providing new insight into postoperative management for breast cancer. Meanwhile, functional analysis indicated that these 7 signatures were significantly enriched in cancer-related pathways, including the TGF-β pathway, the focal adhesion pathway, ECM-receptor interaction, the cell cycle pathway and the JAK-STAT signaling pathway. We confirmed 7 signatures in 20 ceRNA networks, which might help elucidate the mechanisms of circRNA in breast cancer.
In this study, we combined the publicly available TCGA and GEO databases with our previous circRNA dataset for bioinformatic analysis, identified 59 DEcircRNAs, 98 DEmiRNAs, and 3966 DEmRNAs and constructed a breast cancer-related ceRNA regulatory network, including 38 circRNA-miRNA pairs and 38 miRNA-mRNA pair intersections. We also successfully generated a prognostic model based on 7 signatures and 20 prognostic ceRNA axes. Multiple cancer-related pathways were involved in the ceRNA network, providing new insight into the underlying pathogenesis of breast cancer and suggesting therapeutic targets for patients with this disease. The results of this study may help to deepen the understanding of circRNA-related regulatory mechanisms. Moreover, we generated a prognostic model that provided new insight into postoperative management for breast cancer.
However, it should be noted that our findings are based on a microarray analysis of a small sample followed by bioinformatic analysis. Further in-depth studies should be conducted to verify the underlying ceRNA mechanism in breast cancer.
Data Sharing Statement
All data generated or analyzed during this study are included in this published article.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agreed to be accountable for all aspects of the work.
Disclosure
The authors report no conflicts of interest for this work.
References
1. Bray F, Ferlay J, Soerjomataram I, et al. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68(6):394–424. doi:10.3322/caac.21492
2. Al Qadire M, Alkhalaileh M, Hina H. Risk factors for breast cancer among jordanian women: a case-control study. Iran J Public Health. 2018;47(1):49–56.
3. Majeed W, Aslam B, Javed I, et al. Breast cancer: major risk factors and recent developments in treatment. Asian Pacific j Cancer Prevention. 2014;15(8):3353–3358. doi:10.7314/apjcp.2014.15.8.3353
4. Sun YS, Zhao Z, Yang ZN, et al. Risk factors and preventions of breast cancer. Int J Biol Sci. 2017;13(11):1387–1397. doi:10.7150/ijbs.21635
5. Brosnan CA, Voinnet O. The long and the short of noncoding RNAs. Curr Opin Cell Biol. 2009;21(3):416–425. doi:10.1016/j.ceb.2009.04.001
6. Long J, Xiong J, Bai Y, et al. Construction and investigation of a lncRNA-Associated ceRNA regulatory network in cholangiocarcinoma. Front Oncol. 2019;9:649. doi:10.3389/fonc.2019.00649
7. Anastasiadou E, Jacob LS, Slack FJ. Non-coding RNA networks in cancer. Nat Rev Cancer. 2018;18(1):5–18. doi:10.1038/nrc.2017.99
8. Liu Q, Zhang W, Wu Z, et al. Construction of a circular RNA-microRNA-messengerRNA regulatory network in stomach adenocarcinoma. J Cell Biochem. 2019. doi:10.1002/jcb.29368
9. Han Y, Wu N, Jiang M, et al. Long non-coding RNA MYOSLID functions as a competing endogenous RNA to regulate MCL-1 expression by sponging miR-29c-3p in gastric cancer. Cell Prolif. 2019:e12678. doi:10.1111/cpr.12678.
10. Weng W, Wei Q, Toden S, et al. Circular RNA ciRS-7-A promising prognostic biomarker and a potential therapeutic target in colorectal cancer. Clin Cancer Res. 2017;23(14):3918–3928. doi:10.1158/1078-0432.CCR-16-2541
11. Hsu MT, Coca-Prados M. Electron microscopic evidence for the circular form of RNA in the cytoplasm of eukaryotic cells. Nature. 1979;280(5720):339–340. doi:10.1038/280339a0
12. Sanger HL, Klotz G, Riesner D, et al. Viroids are single-stranded covalently closed circular RNA molecules existing as highly base-paired rod-like structures. Proc Natl Acad Sci U S A. 1976;73(11):3852–3856. doi:10.1073/pnas.73.11.3852
13. Kristensen LS, Andersen MS, Stagsted LVW, et al. The biogenesis, biology and characterization of circular RNAs. Nat Rev Genet. 2019;20:675–691. doi:10.1038/s41576-019-0158-7
14. Suzuki H, Zuo Y, Wang J, et al. Characterization of RNase R-digested cellular RNA source that consists of lariat and circular RNAs from pre-mRNA splicing. Nucleic Acids Res. 2006;34(8):e63. doi:10.1093/nar/gkl151
15. Nicolet BP, Engels S, Aglialoro F, et al. Circular RNA expression in human hematopoietic cells is widespread and cell-type specific. Nucleic Acids Res. 2018;46(16):8168–8180. doi:10.1093/nar/gky721
16. Salzman J, Chen RE, Olsen MN, et al. Cell-type specific features of circular RNA expression. PLoS Genet. 2013;9(9):e1003777. doi:10.1371/journal.pgen.1003777
17. Rybak-Wolf A, Stottmeister C, Glazar P, et al. Circular RNAs in the mammalian brain are highly abundant, conserved, and dynamically expressed. Mol Cell. 2015;58(5):870–885. doi:10.1016/j.molcel.2015.03.027
18. He J, Xie Q, Xu H, et al. Circular RNAs and cancer. Cancer Lett. 2017;396:138–144. doi:10.1016/j.canlet.2017.03.027
19. Kristensen LS, Hansen TB, Veno MT, et al. Circular RNAs in cancer: opportunities and challenges in the field. Oncogene. 2018;37(5):555–565. doi:10.1038/onc.2017.361
20. Fan X, Weng X, Zhao Y, et al. Circular RNAs in cardiovascular disease: an overview. Biomed Res Int. 2017;2017:5135781. doi:10.1155/2017/5135781
21. Lu S, Yang X, Wang C, et al. Current status and potential role of circular RNAs in neurological disorders. J Neurochem. 2019;150(3):237–248. doi:10.1111/jnc.14724
22. Lü L, Sun J, Shi P, et al. Identification of circular RNAs as a promising new class of diagnostic biomarkers for human breast cancer. Oncotarget. 2017;8(27):44096–44107. doi:10.18632/oncotarget.17307
23. Tomczak K, Czerwinska P, Wiznerowicz M. The Cancer Genome Atlas (TCGA): an immeasurable source of knowledge. Contemporary Oncol. 2015;19(1A):A6877. doi:10.5114/wo.2014.47136
24. Shi P, Sun J, He B, et al. Profiles of differentially expressed circRNAs in esophageal and breast cancer. Cancer Manag Res. 2018;10:2207–2221. doi:10.2147/cmar.s167863
25. Subramanian A, Tamayo P, Mootha VK, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A. 2005;102(43):15545–15550. doi:10.1073/pnas.0506580102
26. Song H, Xu D, Shi P, et al. Upregulated circ RNA hsa_circ_0000337 promotes cell proliferation, migration, and invasion of esophageal squamous cell carcinoma. Cancer Manag Res. 2019;11:1997–2006. doi:10.2147/CMAR.S195546
27. Sun S, Wang W, Luo X, et al. Circular RNA circ-ADD3 inhibits hepatocellular carcinoma metastasis through facilitating EZH2 degradation via CDK1-mediated ubiquitination. Am J Cancer Res. 2019;9(8):1695–1707.
28. Ju HQ, Zhao Q, Wang F, et al. A circRNA signature predicts postoperative recurrence in stage II/III colon cancer. EMBO Mol Med. 2019:e10168. doi:10.15252/emmm.201810168.
29. Kolling M, Haddad G, Wegmann U, et al. Circular RNAs in urine of kidney transplant patients with acute T cell-mediated allograft rejection. Clin Chem. 2019. doi:10.1373/clinchem.2019.305854
30. Dong Z, Deng L, Peng Q, et al. CircRNA expression profiles and function prediction in peripheral blood mononuclear cells of patients with acute ischemic stroke. J Cell Physiol. 2019. doi:10.1002/jcp.29165
31. Yang L, Wang S, Zhang Q, et al. Clinical significance of the immune microenvironment in ovarian cancer patients. Mol Omics. 2018;14(5):341–351. doi:10.1039/c8mo00128f
32. Wang S, Zhang Q, Yu C, et al. Immune cell infiltration-based signature for prognosis and immunogenomic analysis in breast cancer. Brief Bioinform. 2020. doi:10.1093/bib/bbaa026
33. Han D, Li J, Wang H, et al. Circular RNA circMTO1 acts as the sponge of microRNA-9 to suppress hepatocellular carcinoma progression. Hepatology. 2017;66(4):1151–1164. doi:10.1002/hep.29270
34. Wu Y, Xie Z, Chen J, et al. Circular RNA circTADA2A promotes osteosarcoma progression and metastasis by sponging miR-203a-3p and regulating CREB3 expression. Mol Cancer. 2019;18(1):73. doi:10.1186/s12943-019-1007-1
35. Tang W, Fu K, Sun H, et al. CircRNA microarray profiling identifies a novel circulating biomarker for detection of gastric cancer. Mol Cancer. 2018;17(1):137. doi:10.1186/s12943-018-0888-8
36. Xu Y, Yao Y, Liu Y, et al. Elevation of circular RNA circ_0005230 facilitates cell growth and metastasis via sponging miR-1238 and miR-1299 in cholangiocarcinoma. Aging. 2019;11(7):1907–1917. doi:10.18632/aging.101872
37. Afzali F, Salimi M. Unearthing regulatory axes of breast cancer circRNAs networks to find novel targets and fathom pivotal mechanisms. Interdiscip Sci. 2019;11(4):711–722. doi:10.1007/s12539-019-00339-6
38. Qiu M, Xia W, Chen R, et al. The circular RNA circPRKCI promotes tumor growth in lung adenocarcinoma. Cancer Res. 2018;78(11):2839–2851. doi:10.1158/0008-5472.CAN-17-2808
39. Zhang R, Zhu Z, Shen W, et al. Golgi membrane protein 1 (GOLM1) promotes growth and metastasis of breast cancer cells via regulating matrix metalloproteinase-13 (MMP13). Med Sci Monitor. 2019;25:847–855. doi:10.12659/MSM.911667
40. Zhang Y, Liu H, Li W, et al. CircRNA_100269 is downregulated in gastric cancer and suppresses tumor cell growth by targeting miR-630. Aging. 2017;9(6):1585–1594. doi:10.18632/aging.101254
41. Zhang H, Wang X, Hu B, et al. Circular RNA ZFR accelerates non-small cell lung cancer progression by acting as a miR-101-3p sponge to enhance CUL4B expression. Artificial Cells, Nanomed Biotechnol. 2019;47(1):3410–3416. doi:10.1080/21691401.2019.1652623
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.