Back to Journals » International Journal of General Medicine » Volume 15
Construction of a circRNA-Mediated ceRNA Network Reveals Novel Biomarkers for Aortic Dissection
Authors Liu DB ![]() , He YF
, He YF ![]() , Chen GJ, Huang H, Xie XL, Lin WJ, Peng ZJ
, Chen GJ, Huang H, Xie XL, Lin WJ, Peng ZJ
Received 26 December 2021
Accepted for publication 18 March 2022
Published 12 April 2022 Volume 2022:15 Pages 3951—3964
DOI https://doi.org/10.2147/IJGM.S355906
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Scott Fraser
De-Bin Liu,1,* You-Fu He,2– 4,* Gui-Jian Chen,1 Hua Huang,1 Xu-Ling Xie,1 Wan-Jun Lin,1 Zhi-Jian Peng1
1Department of Cardiology, The Second People’s Hospital of Shantou, Shantou, Guangdong Province, People’s Republic of China; 2Department of Cardiology, Guizhou Provincial People’s Hospital, Guiyang, Guizhou Province, People’s Republic of China; 3Guizhou Provincial Cardiovascular Disease Clinical Medicine Research Center, Guiyang, Guizhou Province, People’s Republic of China; 4Medical College, Guizhou University, Guiyang, Guizhou Province, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Zhi-Jian Peng, Department of Cardiology, The Second People’s Hospital of Shantou, Shantou, 515000, Guangdong Province, People’s Republic of China, Tel +86 18316056382, Fax +86-754 88983534, Email [email protected]
Background: Aortic dissection (AD) is a rare and lethal disorder with its genetic basis remains largely unknown. Many studies have confirmed that circRNAs play important roles in various physiological and pathological processes. However, the roles of circRNAs in AD are still unclear and need further investigation. The present study aimed to elucidate the underlying molecular mechanisms of circRNAs regulation in AD based on the circRNA-associated competing endogenous RNA (ceRNA) network.
Methods: Expression profiles of circRNAs (GSE97745), miRNAs (GSE92427), and mRNAs (GSE52093) were downloaded from Gene Expression Omnibus (GEO) databases, and the differentially expressed RNAs (DERNAs) were subsequently identified by bioinformatics analysis. CircRNA–miRNA–mRNA ceRNA network, Gene Ontology (GO), and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analyses were used to predict the potential functions of circRNA-associated ceRNA network. RNA was isolated from human arterial blood samples after which qRT-PCR was performed to confirm the DERNAs.
Results: We identified 14 (5 up-regulated and 9 down-regulated) differentially expressed circRNAs (DEcircRNAs), 17 (8 up-regulated and 9 down-regulated) differentially expressed miRNAs (DEmiRNAs) and 527 (297 up-regulated and 230 down-regulated) differentially expressed mRNAs (DEmRNAs) (adjusted P-value < 0.05 and | log2FC | > 1.0). KEGG pathway analysis indicated that DEmRNAs were related to focal adhesion and extracellular matrix receptor interaction signaling pathways. Simultaneously, the present study constructed a ceRNA network based on 1 circRNAs (hsa_circRNA_082317), 1 miRNAs (hsa-miR-149-3p) and 10 mRNAs (MLEC, ENTPD7, SLC16A3, SLC7A8, TBC1D16, PAQR4, MAPK13, PIK3R2, ITGA5, SERPINA1). qRT-PCR demonstrated that hsa_circRNA_082317 and ITGA5 were significantly up-regulated, and hsa-miR-149-3p was dramatically down-regulated in AD (n = 3).
Conclusion: This is the first study to demonstrate the circRNA-associated ceRNA network is altered in AD, implying that circRNAs may play important roles in regulating the onset and progression and thus may serve as potential biomarkers for the diagnosis and treatment of AD.
Keywords: circRNA, miRNA, mRNA, ceRNA, aortic dissection, bioinformatics analyses
Graphical Abstract:

Introduction
Aortic dissection (AD) is a rare but often fatal condition.1 The separation of the aortic intima causes blood to leak into the space between the intima and the media, forming intramural hematoma and the true and false lumen along the artery’s long axis. To investigate patients with suspected aortic dissection, computed tomography angiography (CTA) is currently the international and widely accepted gold standard.2 Increased C-reactive protein (CRP) and D-dimer levels, which are nonspecific markers of systemic inflammation, have been linked to acute aortic dissection (AAD).3,4 There is currently no effective serum diagnostic or therapeutic marker for AD. As a result, new potential biomarkers for the diagnosis and prognosis of AD must be discovered as soon as possible.
At the RNA level, previous studies have revealed molecular changes in various aortic diseases.5 A new mechanism for the interaction of RNAs, known as the competing endogenous RNA (ceRNA) network, has received much attention in recent years. The hypothesis holds that miRNA is the core element in the ceRNA network, while transcripts such as circRNAs, lncRNAs, mRNAs, and other RNAs can act as miRNA sponges to competitively combine with the same miRNA response elements (MREs), and then represses the inhibition of miRNA on their target genes, whether in pathological or physiological situations.6 Previously, several miRNAs have been confirmed to be closely involved in cardiovascular diseases and may even be a new strategy for the diagnosis, therapy, or prediction of cardiovascular diseases. Certain miRNAs, such as miRNA-21,7 miRNA-134-5p,8 miRNA-145,9 miRNA-146b,10 have been reported to play definite roles in the development of AD.
Unlike miRNAs, circRNAs have high stability and tissue specificity, which form a continuous cycle of covalent closures without 5’ or 3’ polyadenylated tail, and are resistant to RNase R degradation or RNA exonuclease digestion. Recent evidence indicates that circRNAs are involved in various human diseases, such as cancer,11 Alzheimer’s disease,12 and cardiovascular diseases.13,14 However, no research has shown that circRNAs and related ceRNA networks can be used as diagnostic or prognostic markers in AD.
In this study, we first identified the DEmRNAs, DEmiRNAs, and DEcircRNAs and constructed a reliable circRNA-associated ceRNA regulatory network in AD based on GEO datasets using bioinformatics analysis. Finally, we identified novel circRNA-associated ceRNAs modules of AD and provided new evidence for the early diagnostics and prognostics for AD patients.
Materials and Methods
Data Collection
Gene Expression Omnibus (GEO) databases of the National Center for Biotechnology Information (NCBI) (https://www.ncbi.nlm.nih.gov/geo/)15 is an international public repository for high throughput microarray and sequence-based data. Three datasets (GSE52093, GSE92427, GSE97745) for AD were retrieved and downloaded from GEO databases. The mRNAs datasets GSE52093 contained Stanford type A AD (n = 7) and normal aortic tissue (n = 5), and the samples were tested by the GPL10558 Illumina HumanHT-12 V4.0 expression bead chip platform. GSE92427 miRNAs profiles, including AAD (n = 8), healthy (n = 8) and aortic aneurysm (n = 8) subjects, was analyzed with the GPL16770 platform [Agilent-031181 Unrestricted_Human_miRNA_V16.0_Microarray (miRBase release 16.0 miRNA ID version)]. The circRNAs microarray GSE97745 included 3 human TAD tissues and 3 age-matched normal donor tissues; the platform used for the microarray was GPL21825 074301 Arraystar Human CircRNA microarray V2.
Identification of DEcircRNAs, DEmiRNAs, and DEmRNAs
Limma,16 a Bioconductor package in R software, was applied to screen DEmRNAs, DEmiRNAs, and DEcircRNAs with thresholds of adjusted P-value <0.05 and | log2FC | > 1.0. Then, volcano plot and heat map of the DERNAs were constructed using the R software.
Gene Ontology (GO) and Pathway Enrichment Analysis of DEGs
The GO,17 including biological processes (BP), cellular components (CC), and molecular functions (MF), and KEGG18 enrichment analysis and visualization of DEmRNAs were performed by the R-packages “clusterProfiler”,19 “org.Hs.eg.db”, “enrichplot”, “ggplot2” with thresholds of P-value < 0.05 and q-value < 0.05.
Construction of miRNA–mRNA Pairs
In our study, we used miRNA IDs from miRBase (http://www.mirbase.org/)20 as the unified gene IDs. Target genes of DEmiRNAs were predicted by the Predictive Target Module of the miRWalk3.0 (http://mirwalk.umm.uni-heidelberg.de/),21 including miRDB,22 miRTarBase,23 and TargetScan databases. Any genes with at least one database were counted as target genes of DEmiRNAs. Then, the intersection of DEmiRNAs predicted target genes and DEmRNAs were identified by the online Venn diagram tool (Evenn,http://www.ehbio.com/test/venn/#/)24 and the overlapped genes were used to construct the miRNA–mRNA regulatory network using the Cytoscape software (Version 3.7.2,https://cytoscape.org/).25
Construction of circRNA–miRNA Pairs
circBase (http://www.circbase.org)26 is a database that merged and unified datasets of circRNAs. We unified the circRNAs IDs based on circBase and collected the detailed information of the circRNA ID, gene symbol, and genome location. To analyze the functional effects of DEcircRNAs and predict which circRNA acts as a miRNA sponge, we used a cancer-specific circRNA database (CSCD, https://gb.whu.edu.cn/CSCD)27 to predict the target relationship, by predicting the RNA binding protein (RBP) sites and MREs.
Reconstruction of the circRNA–miRNA–mRNA Network
Based on the identified DEmiRNA-DEmRNA and DEcircRNA-DEmiRNA relationship pairs, we screened the DEcircRNA–miRNA–mRNA interaction networks that are linked by the shared miRNAs predicted as downstream targets of DEcircRNAs and upstream regulators of DEmRNAs. Then, the DEcircRNA–miRNA–mRNA interaction networks were visualized by using Cytoscape software (Version 3.7.2, https://cytoscape.org/).
Patients and Tissue Specimens
The study was conducted following the Declaration of Helsinki, and the protocol was approved by the Second People’s Hospital of Shantou (protocol number: EC20210912(3)-P01). Informed, written consent was obtained from all participants. Arterial blood specimens were collected from three type B aortic dissection patients undergoing stent graft at the hospital in 2021. Arterial blood samples were obtained from three age- and gender-matched healthy control undergoing coronary angiography. After collection, plasma specimens were frozen by immersion in liquid nitrogen and stored in an ultralow temperature freezer at −80°C until use (DW-86L388A, Haier).
RNA Extraction and qRT-PCR
Total RNA was extracted from human arterial blood samples (three samples from AD and healthy, respectively) using Trizol total RNA isolation reagent (Invitrogen, 93289), according to the manufacturer’s instructions. For circRNA qRT-PCR analysis, DEcircRNAs, targeted DEmiRNAs, were selected for further investigation. Briefly, total RNA from each sample was digested with RNase R (Epicentre, Inc.) to remove linear RNAs and enrich the circRNAs. The differential expression of DEcircRNAs was confirmed through qRT-PCR (in triplicate) using the SYBR green PCR mix (Takara, RR820B) according to the manufacturer’s instructions. The sequence of circRNA results was obtained from the database “circBase” (http://www.circbase.org). For miRNA and mRNA analysis, total RNA was reverse-transcribed using random hexamer primers. GAPDH was employed as the loading control for mRNA and circRNA, and U6 RNA was amplified as the loading control for miRNAs. Finally, the relative expression of circRNAs, miRNAs, and mRNAs were analyzed and quantified by the comparative quantitative cycle (Cq) (2-ΔΔCq) method. All the primers were designed and synthesized by Sangon Biotech (Sangon Biotech, Shanghai, China), and the primer sequences are listed Table 1.
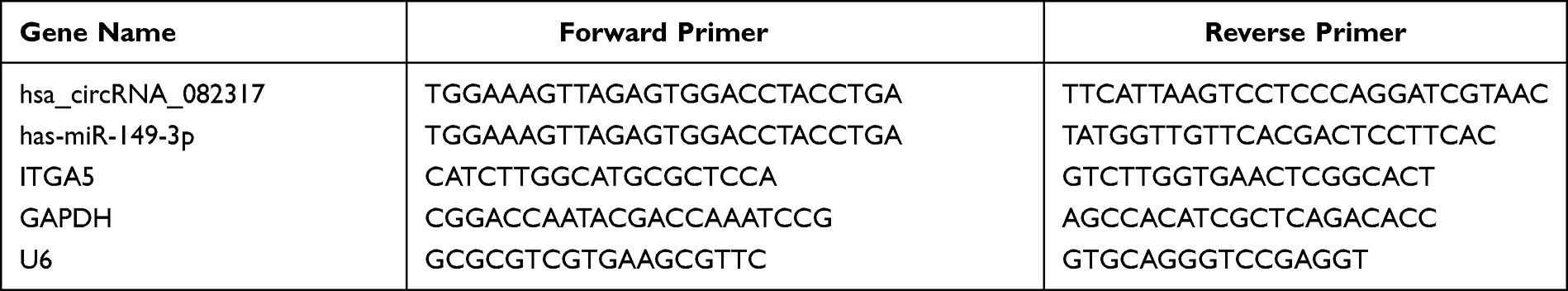 |
Table 1 Primer Sequences |
Statistics
All the data were analyzed using SPSS17.0 statistics software. The significance was assessed by paired Student’s t-test as appropriate (for the comparison of the two groups) and P-value <0.05 was considered statistically significant.
Results
Identification of Differential Expressed circRNAs, miRNAs and mRNAs
The flowchart of the study is shown below (Figure 1). In this study, we obtained three microarray datasets (GSE97745 circRNAs microarray, GSE 92427 miRNA profiles, GSE52093 mRNA datasets) for AD from the online databases GEO, and the fundamental information of the datasets were summarized below (Table 2). Differential expression analysis was performed using the Limma R package, with the criteria of adjusted P-value <0.05 and | log2FC | > 1.0. Based on the cut-off criteria, a total of 527 DEmRNAs (297 up-regulated and 230 down-regulated), 17 DEmiRNAs (8 up-regulated and 9 down-regulated), and 14 DEcircRNAs (5 up-regulated and 9 down-regulated) were identified when AD samples were compared with normal ascending aorta samples (Figure 2A–F).
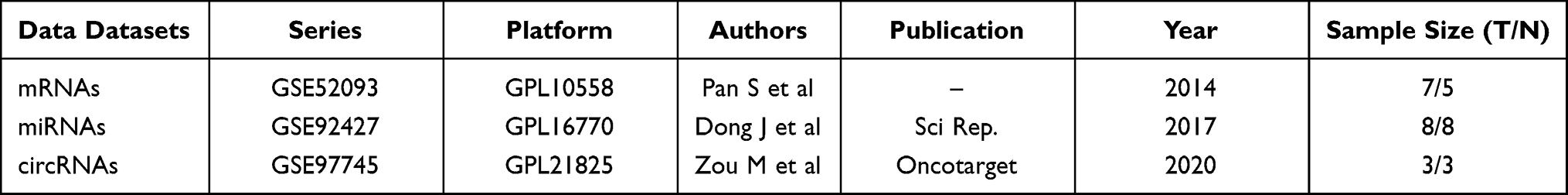 |
Table 2 The Fundamental Information of the Three Microarray Datasets from GEO |
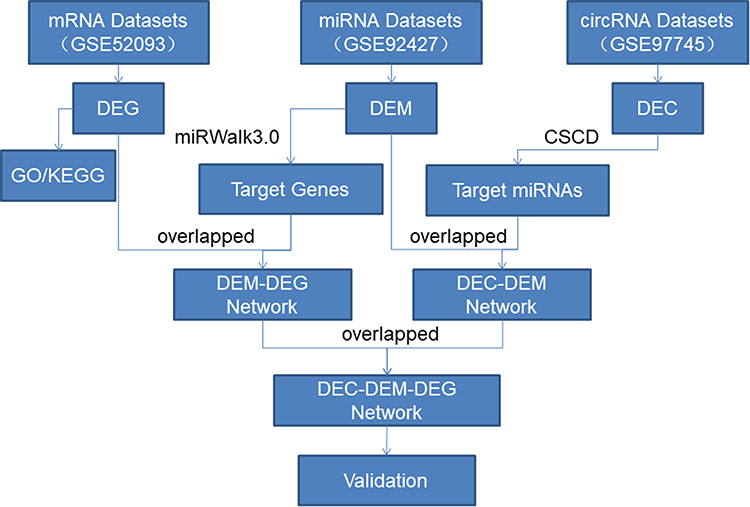 |
Figure 1 Flowchart of the approach utilized in this study. |
 |
Figure 2 Identification of differentially expressed RNAs (DERNAs). (A–C) Heatmap showing the expression of DEmRNAs, DEmiRNAs and DEcircRNAs, respectively; Each row in the heatmap represents a gene and each column represents a sample. The color scale at the right of the heatmap represents the raw Z-score ranging from blue (low expression) to red (high expression). (D–F) Volcano plot of DEmRNAs, DEmiRNAs and DEcircRNAs, respectively; The red nodes represent up-regulated; the green nodes represent down-regulated. |
GO and KEGG Pathway Annotation of DEmRNAs
To further investigate the corresponding biological functions of the DEmRNAs, GO and KEGG enrichment analyses were carried out using R packages “clusterProfiler” and “org.Hs.eg.db”. The GO enrichment analysis for DEmRNAs was divided into three main categories: BP, CC, and MF. GO enrichment analysis of DEmRNAs revealed that a total of 195, 43, and 6 GO terms were enriched in the BP, CC, and MF, respectively (p < 0.05) (Figure 3A, Table 3). For biological processes, GO terms were mainly associated with chromosome segregation (GO:0007059), nuclear chromosome segregation (GO:0098813), mitotic sister chromatid segregation (GO:0000070), muscle system process (GO:0003012), and regulation of mitotic sister chromatid separation (GO:0010965). For cellular components, GO terms were mainly included I band (GO:0031674), Z disc (GO:0030018), sarcomere (GO:0030017), myofibril (GO:0030016), and contractile fiber (contractile fiber). For molecular function, GO terms mainly involved calmodulin binding (GO:0005516), actin-binding (GO:0003779), structural constituent of muscle (GO:0008307), frizzled binding (GO:0005109), and histone kinase activity (GO:0035173). KEGG pathway analysis of the DEmRNAs revealed that 9 pathways were significantly enriched, of which the top 5 were Cell cycle (KEGG Pathway hsa04110), oocyte meiosis (KEGG Pathway hsa04114), DNA replication (KEGG Pathway hsa03030), focal adhesion (KEGG Pathway hsa04510) and hypertrophic cardiomyopathy (KEGG Pathway hsa05410) (Figure 3B, Table 4).
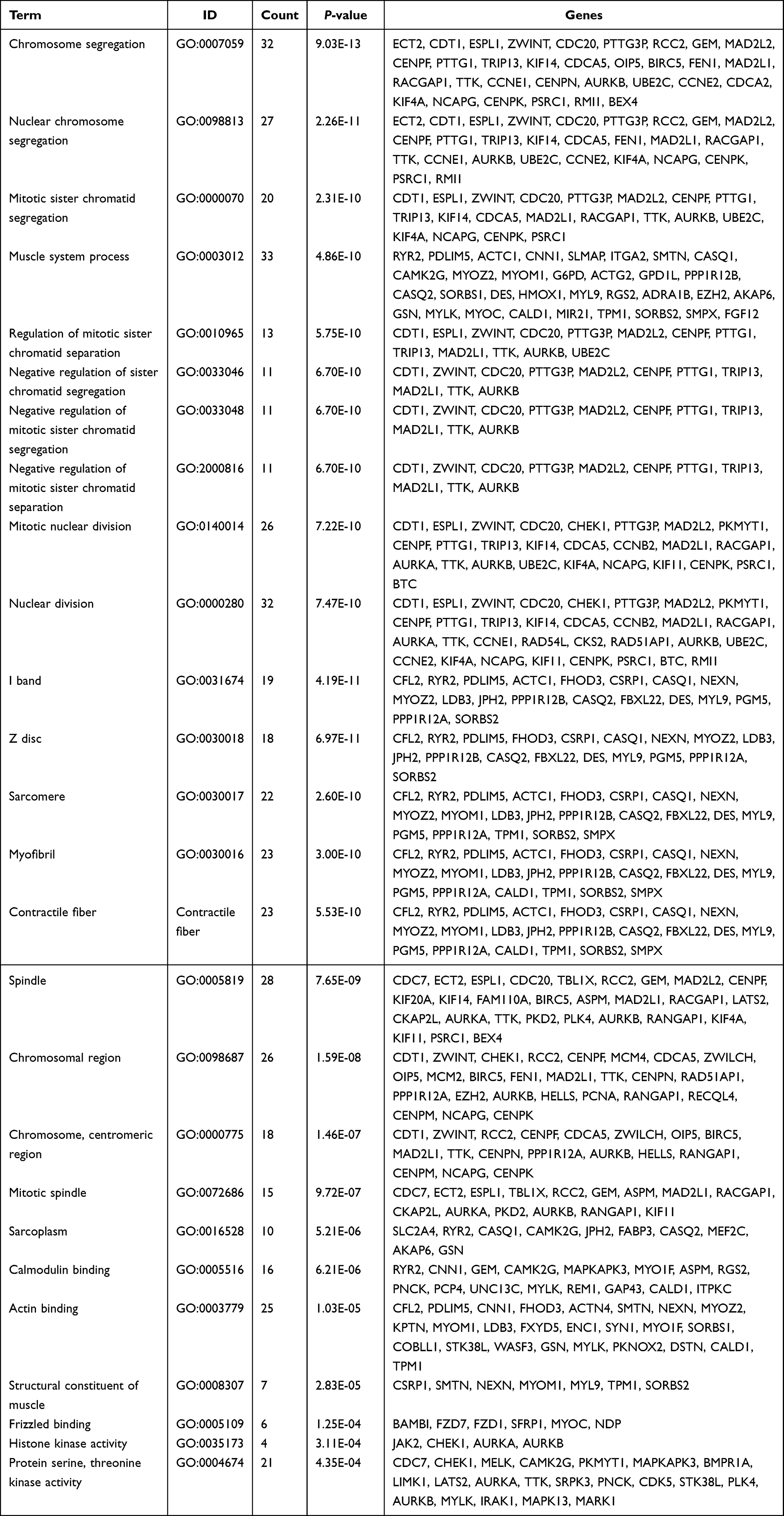 |
Table 3 GO Analysis for DEmRNAs of GSE52093 |
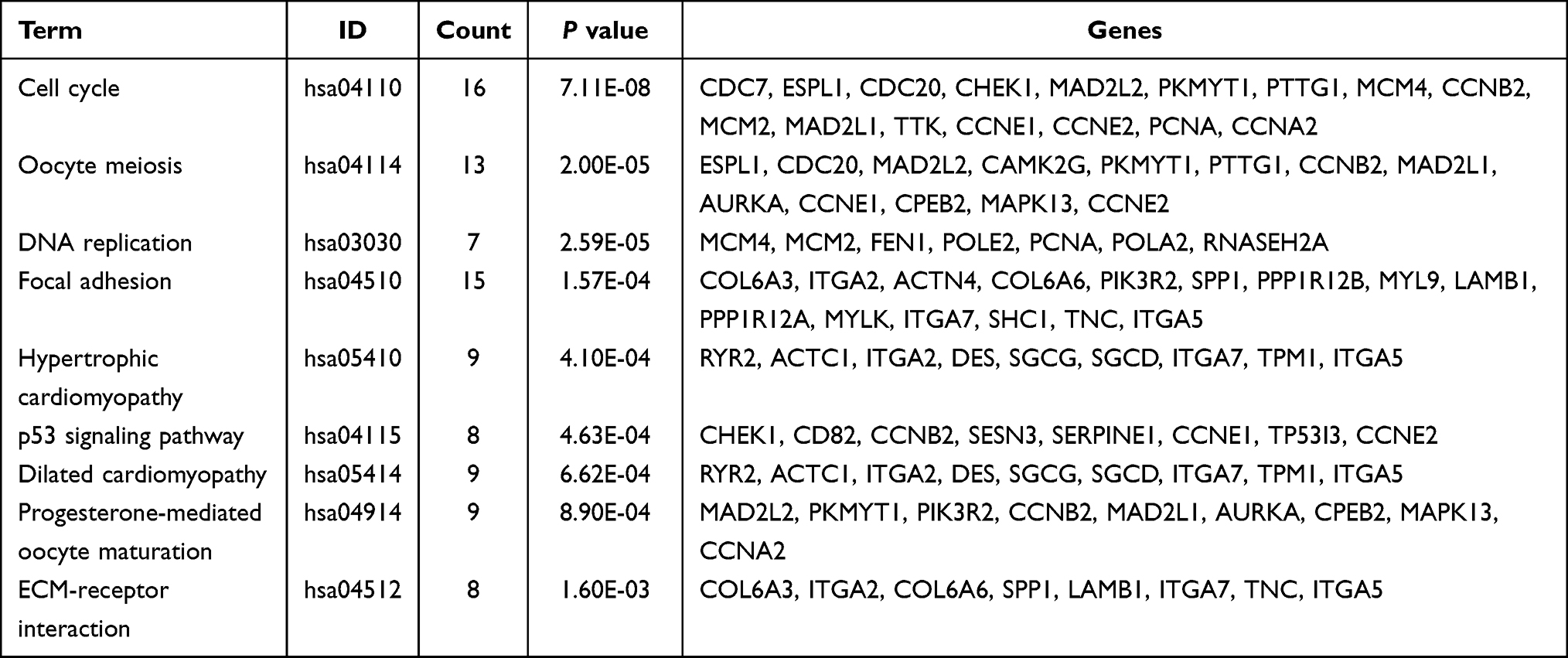 |
Table 4 KEGG Pathways Analysis for DEmRNAs of GSE52093 |
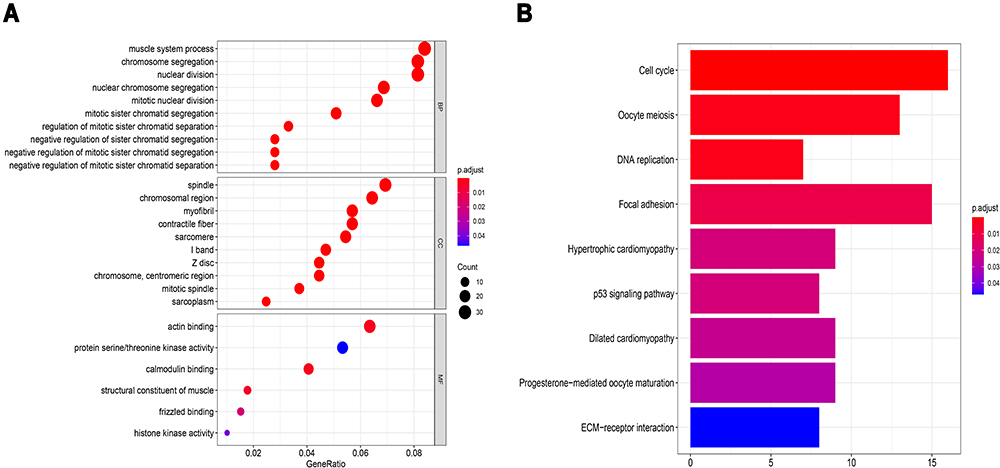 |
Figure 3 Bioinformatics enrichment analysis including GO analysis and KEGG pathway analysis for differentially expressed mRNAs of GSE52093 datasets. (A) Bubble diagram of Gene Ontology (GO) analysis of differentially expressed mRNAs revealed the enriched biological processes, cell components, and molecular functions. (B) Bar chart of Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analysis of targeted genes revealed the enriched signaling pathways. |
Construction of miRNA–mRNA Pairs
To further identify the potentially relevant miRNA–mRNA target interactions in AD progression, the miRWalk3.0 online database (http://mirwalk.umm.uni-heidelberg.de/) was utilized to predict the target genes of DEmiRNAs. 2933 genes were obtained and predicted as target genes of DEmiRNAs. These target genes and the DEmRNAs were compared, and 82 differentially expressed target genes were finally obtained (Figure 4A). Only the inversely correlated miRNA–mRNA pairs, including up-regulated miRNA-down-regulated mRNA pairs and down-regulated miRNA-up-regulated mRNA pairs, were considered as a potential miRNA–mRNA interaction. After filtering, there were 62 mRNA has an opposite interaction with DEmiRNAs. Cytoscape 3.7.2. the software was used to construct and analyze miRNA–mRNA regulatory network based on the identified 93 pairs of DEmiRNAs-DEmRNAs comprising 16 miRNAs (7 up-regulated and 9 down-regulated) and 62 DEmRNAs (43 up-regulated and 19 down-regulated) (Figure 4B, Table 5).
 |
Table 5 The Basic Characteristics of DEmiRNAs of GSE92427 |
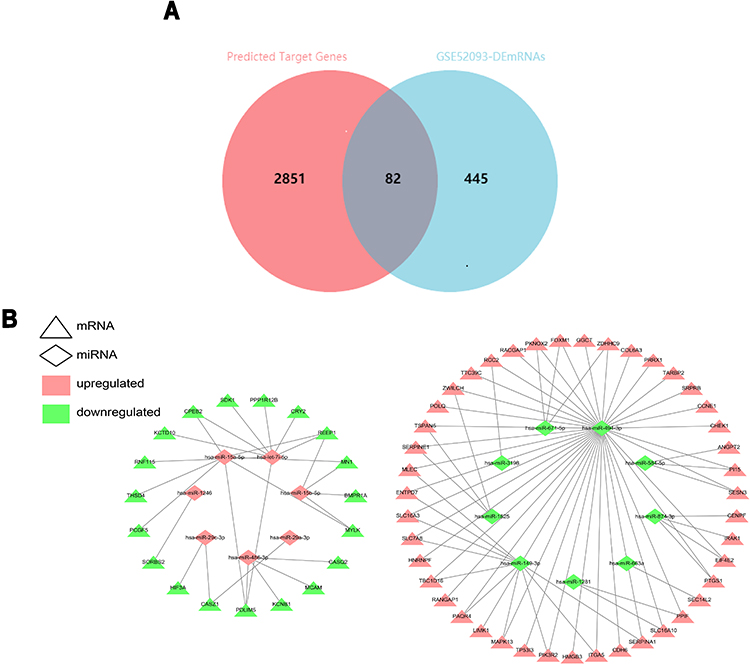 |
Figure 4 The Venn Diagrams and DEmiRNA-DEmRNA regulatory network. (A) Venn diagram showing overlap genes both in the differential expression miRNAs (DEmiRNAs) predicted target genes and DEmRNAs. The overlap areas represent differentially expressed target genes. (B) Visualization of the miRNA–mRNA network, which is composed of 16 miRNA nodes, 62 mRNA nodes, and 93 edges. The triangle nodes represent the mRNAs, and the diamond nodes represent the miRNAs. Red represents up-regulated expression, whereas green represents down-regulated expression. |
Construction of circRNA–miRNA Pairs
A total of 14 DEcircRNAs were annotated using the circBase database, of which 8 were known (3 up-regulated and 5 down-regulated) and 6 were novel (2 up-regulated and 4 down-regulated). Only the annotated DEcircRNAs in the circBase were used for further analysis (Table 6). Possible miRNA binding sites in DEcircRNAs were predicted using web tools Cancer-specific circRNA database (CSCD, https://gb.whu.edu.cn/CSCD) (Figure 5). After searching the CSCD database and comparing the DEmiRNAs-DEmRNAs pairs, we identified 1 circRNA–miRNA pair, resulting in, finally, the up-regulated hsa_circRNA_082317 adjusting its MREs: hsa-miR-149-3p (Figure 6A). Furthermore, we found that hsa_circRNA_082317 is generated by back splicing of ubiquitin-conjugating enzyme E2 (UBE2H) gene, which is located on the 7q32 chromosome, encodes a member of the E2 ubiquitin-conjugating enzyme family.
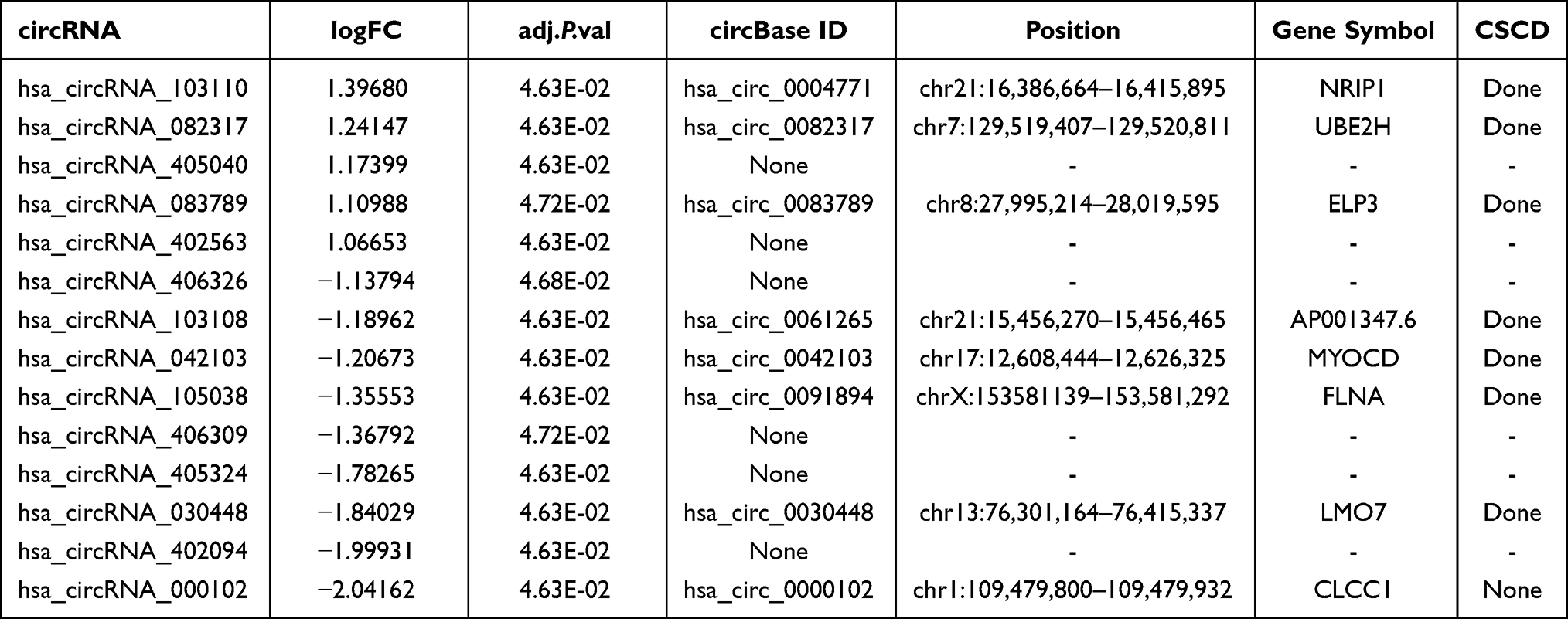 |
Table 6 The Primary Characteristics of the DEcircRNAs of GSE97745 |
 |
Figure 5 The fundamental structure modes of the candidate circRNAs predicted by CSCD. (A) Up-regulated circRNAs: hsa_circRNA_103110, hsa_circRNA_082317, hsa_circRNA_083789.(B) Down-regulated circRNAs: hsa_circRNA_103108, hsa_circRNA_105038, hsa_circRNA_030448. |
 |
Figure 6 CircRNA–miRNA–mRNA interaction network. (A) Visualization of the circRNA–miRNA network, which is composed of 1 circRNA node, 1 miRNA node and 1 edge. (B) Visualization of the circRNA–miRNA–mRNA network, which is composed of 1 circRNA node, 1 miRNA node, 10mRNA nodes, and 11 edges. Hexagon, Diamond, and Triangle nodes represent circRNAs, miRNAs, and mRNAs, respectively. Red represents up-regulated expression, whereas green represents down-regulated expression. |
Reconstruction of the circRNA–miRNA–mRNA Network
Based on the above intersections, we constructed a circRNA–miRNA–mRNA ceRNA regulatory network and visualized it via the Cytoscape software (version 3.7.2). The network featured a total of 12 nodes (one circRNA, one miRNA, ten mRNAs), 1 circRNA–miRNA relationship pair, and 10 miRNA–mRNA relationship pairs (Figure 6B). Ultimately, we have identified one up-regulated DEcircRNA (hsa_circRNA_082317), 1 down-regulated DEmiRNA (hsa-miR-149-3p) and 10 up-regulated DEmRNAs (MLEC, ENTPD7, SLC16A3, SLC7A8, TBC1D16, PAQR4, MAPK13, PIK3R2, ITGA5, SERPINA1). Based on the results of KEGG pathway analyses, our data showed that ITGA5 was significantly associated with the ECM–receptor interaction pathway and the focal adhesion pathway.
Validation of Candidates
To verify the authenticity of the ceRNA regulatory pathway in AD, we collected blood plasma samples from 3 AD patients and 3 healthy donors for qRT-PCR. The results have shown significant differences (P < 0.05) in the relative expression of hsa_circRNA_082317, hsa-miR-149-3p, and ITGA5 between AD patients and healthy control subjects (Figure 7).
 |
Figure 7 Validation of DERNAs expression. (A) Expression of hsa_circRNA_082317 between healthy donors aorta and aortic dissection; (B) Expression of hsa-miR-149-3p between healthy donors aorta and aortic dissection; (C) Expression of ITGA5 mRNA between healthy donors aorta and aortic dissection. Black represented healthy donors samples, and grey represented aortic dissection samples (n = 3 samples/group, * p ≤ 0.05, ** p ≤ 0.01, data are represented as mean ± SD). |
Discussion
AD is an aortic disease associated with rapid progression, high morbidity, and mortality. The annual incidence of aortic disease is about 0.03%. In AAD Stanford type A, mortality rates of up to 40–50% have been reported within the first 48 hours.28 With the advancement of diagnosis and treatment, AAD remains a significant challenge for physicians today. CTA is a common method for diagnosing AD.29 However, CTA has several disadvantages, including long detection time, inherent technical difficulties, and high costs.
Traditional plasma biomarkers like D-Dimer or CRP have certain value in the diagnosis of aortic disease,3,4 but there is evidence of misdiagnoses and missed diagnoses. As a result, patients with AAD who are not diagnosed and treated promptly may have poor outcomes. Due to the severity of AD, finding novel and specific biomarkers for early diagnosis and treatment is critical.
Studying the molecular mechanisms of the occurrence and development of AD is a major focus of current research. In the present study, we performed an analysis of dysregulated circRNAs, miRNAs and mRNAs between patients with AD and healthy controls. To our knowledge, this present study is the first to identify the interaction networks between circRNAs-miRNAs-mRNAs in aortic tissues of AD patients. The microarray datasets revealed that 527 DEmRNAs (297 up-regulated and 230 down-regulated), 17 DEmiRNAs (8 up-regulated and 9 down-regulated), and 14 DEcircRNAs (5 up-regulated and 9 down-regulated) were identified when aortic dissection samples were compared with normal ascending aorta samples (adjusted P-value <0.05 and | log2FC | > 1.0).
circRNA, as a novel endogenous RNA, has recently drawn extensive attention. Accumulating evidence has confirmed that circRNAs can act as “miRNA sponges” to repress the inhibition of miRNA on their target genes.30,31 Notably, to the best of our knowledge, the mechanism of the effect of circRNA on AD has not been investigated, even though previous studies have found that the differential expression of circRNAs between human AD tissues and normal control tissues is universal. Hence, we hypothesized that the dysregulation of ceRNA expression can affect AD pathogenicity and progression, and circRNA was selected as the entry point for studying the underlying mechanism of ceRNA.
In the present study, bioinformatics analyses have shown that up-regulated DEcircRNA (hsa_circRNA_082317) might compete with one of the key DEmiRNAs (hsa-miR-149-3p) to mediate target DEmRNA expression in AD. Based on the circBase database, we discovered that hsa_circRNA_082317 is formed from the ubiquitin conjugating enzyme E2 H (UBE2H) gene. In yeast and human placenta, UBE2H is a both structurally and functionally highly conserved, ubiquitin-dependent system protein.32,33 The UBE2H gene, which is located on the 7q32 chromosome, encodes a member of the E2 ubiquitin-conjugating enzyme family.34 Furthermore, UBE2H expression is increased following the activation of tumor necrosis factor-alpha/nuclear factor kappa B (TNF-α/NF-κB) signaling pathway in both human cardiac and skeletal muscles.35 Although the expression of hsa_circRNA_082317 is greatly increased, it is not yet clear whether hsa_circRNA_082317 contributes to the ubiquitin-dependent system in AD. This hypothesis needs to be further explored.
Further systemic bioinformatics analyses including the GO and KEGG pathway analysis were used to predict the functions of DEmRNAs, suggesting they may play an important role in regulating ECM–receptor interaction and focal adhesion. The ECM–receptor interaction, a micro-environmental pathway, that maintains cell and tissue homeostasis. ECM is an extremely complex and dynamic structure that provides mechanical support to cells and structural integrity to tissues. ECM and ECM receptors might play crucial roles in the aortic. As a kind of cell surface adhesion molecule, integrins interact with several signaling molecules and activate intracellular signaling pathways, such as the PI3K/AKT pathway. By connecting the actin cytoskeleton of the cells to the extracellular matrix, integrins can maintain cell morphology and regulate cell behaviors, such as in cell adhesion, proliferation, differentiation, and migration.36 Media degradation is a major histopathological feature of AD, which includes the degradation of the ECM (particularly elastic fibers and collagen) that is associated with the depletion of the smooth muscle cells (SMCs), consequently causing artery wall weakness and injury and, eventually, leading to the formation of AD.37,38 As a member of the integrin α-chain family, ITGA5 might be involved in the pathogenesis of AD by regulating cell–ECM adhesion and modulating adhesion-initiated signal transduction pathways.
Conclusion
Taken together, the present study successfully constructed a circRNA–miRNA–mRNA ceRNA network based on 1 circRNAs (hsa_circRNA_082317), 1 miRNAs (hsa-miR-149-3p) and 10 mRNAs (MLEC, ENTPD7, SLC16A3, SLC7A8, TBC1D16, PAQR4, MAPK13, PIK3R2, ITGA5, SERPINA1) in AD. Furthermore, significant up-regulation of hsa_circRNA_082317, ITGA5 and down-regulation of hsa-miR-149-3p were verified using qRT-PCR analysis. It was revealed that hsa_circRNA_082317 might compete with hsa-miR-149-3p to mediate target ITGA5, associated with the ECM–receptor interaction pathway and the focal adhesion pathway, in regulating the occurrence and development of AD. However, the sample size is too small, hsa_circRNA_082317, hsa-miR-149-3p and the mRNAs (MLEC, ENTPD7, SLC16A3, SLC7A8, TBC1D16, PAQR4, MAPK13, PIK3R2, ITGA5, SERPINA1) need further investigation to explore its potential to serve as diagnostic, prognostic, and therapeutic in AD.
Funding
This work supported by Guizhou Provincial Health Commission project (gzwkj 2021-102), Guizhou Provincial Traditional Chinese Medicine Project Fund (QZYY-2021-001), Guizhou Provincial High-level innovative talent training plan (Qian ke He Talent (2016) 4023), Guizhou Provincial Technology platform and talent team planning project (Qian Ke He Plantform Talent (2017) 5405), Guizhou Provincial Science and Technology Agency Project (Qian Ke He Basic-ZK (2022) General 268), and Guizhou Provincial Science and Technology Agency Project (Qian Ke He Basic-ZK (2021) General 358).
Disclosure
The authors report no conflicts of interest in this work.
References
1. LeMaire SA, Russell L. Epidemiology of thoracic aortic dissection. Nat Rev Cardiol. 2011;8(2):103–113. doi:10.1038/nrcardio.2010.187
2. Yoo SM, Lee HY, White CS. MDCT evaluation of acute aortic syndrome. Thorac Surg Clin. 2010;20(1):149–165. doi:10.1016/j.thorsurg.2009.12.011
3. Wen D, Du X, Dong JZ, Zhou XL, Ma CS. Value of D-dimer and C reactive protein in predicting inhospital death in acute aortic dissection. Heart. 2013;99(16):1192–1197. doi:10.1136/heartjnl-2013-304158
4. Weber T, Hogler S, Auer J, et al. D-dimer in acute aortic dissection. Chest. 2003;123(5):1375–1378. doi:10.1378/chest.123.5.1375
5. Cheng M, Yang Y, Xin H, et al. Non-coding RNAs in aortic dissection: from biomarkers to therapeutic targets. J Cell Mol Med. 2020;24(20):11622–11637. doi:10.1111/jcmm.15802
6. Salmena L, Poliseno L, Tay Y, Kats L, Pandolfi PP. A ceRNA hypothesis: the Rosetta Stone of a hidden RNA language?. Cell. 2011;146(3):353–358.
7. Huang X, Yue Z, Wu J, et al. MicroRNA-21 knockout exacerbates angiotensin II-induced thoracic aortic aneurysm and dissection in mice with abnormal transforming growth factor-beta-SMAD3 signaling. Arterioscler Thromb Vasc Biol. 2018;38(5):1086–1101. doi:10.1161/ATVBAHA.117.310694
8. Wang Y, Dong C-Q, Peng G-Y, et al. MicroRNA-134-5p regulates media degeneration through inhibiting VSMC phenotypic switch and migration in thoracic aortic dissection. Molecular Therapy - Nucleic Acids. 2019;16:284–294. doi:10.1016/j.omtn.2019.02.021
9. Li T, Liu C, Liu L, et al. Regulatory mechanism of microRNA-145 in the pathogenesis of acute aortic dissection. Yonsei Med J. 2019;60(4):352–359. doi:10.3349/ymj.2019.60.4.352
10. Li J, Zhou Q, He X, Cheng Y, Wang D. [Expression of miR-146b in peripheral blood serum and aortic tissues in patients with acute type Stanford A aortic dissection and its clinical significance]. Zhong Nan Da Xue Xue Bao Yi Xue Ban. 2017;42(10):1136–1142. Chinese. doi:10.11817/j.issn.1672-7347.2017.10.002
11. Li Y, Zheng Q, Bao C, et al. Circular RNA is enriched and stable in exosomes: a promising biomarker for cancer diagnosis. Cell Res. 2015;25(8):981–984. doi:10.1038/cr.2015.82
12. Lukiw WJ. Circular RNA (circRNA) in Alzheimer’s disease (AD). Front Genet. 2013;4:307. doi:10.3389/fgene.2013.00307
13. Jiang S, Guo C, Zhang W, et al. The Integrative Regulatory Network of circRNA, microRNA, and mRNA in Atrial Fibrillation. Front Genet. 2019;10:526. doi:10.3389/fgene.2019.00526
14. Burd CE, Jeck WR, Liu Y, Sanoff HK, Wang Z, Sharpless NE. Expression of linear and novel circular forms of an INK4/ARF-associated non-coding RNA correlates with atherosclerosis risk. PLoS Genet. 2010;6(12):e1001233. doi:10.1371/journal.pgen.1001233
15. Barrett T, Wilhite SE, Ledoux P, et al. NCBI GEO: archive for functional genomics data sets–update. Nucleic Acids Res. 2013;41(Database issue):D991–995. doi:10.1093/nar/gks1193
16. Ritchie ME, Phipson B, Wu D, et al. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015;43(7):e47. doi:10.1093/nar/gkv007
17. Ashburner M, Ball CA, Blake JA, et al. Gene ontology: tool for the unification of biology. The Gene ontology consortium. Nat Genet. 2000;25(1):25–29. doi:10.1038/75556
18. Kanehisa M, Goto S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000;28(1):27–30. doi:10.1093/nar/28.1.27
19. Yu G, Wang LG, Han Y, He QY. clusterProfiler: an R package for comparing biological themes among gene clusters. Omics. 2012;16(5):284–287. doi:10.1089/omi.2011.0118
20. Kozomara A, Birgaoanu M, Griffiths-Jones S. miRBase: from microRNA sequences to function. Nucleic Acids Res. 2019;47(D1):D155–D162. doi:10.1093/nar/gky1141
21. Dweep H, Sticht C, Pandey P, Gretz N. miRWalk–database: prediction of possible miRNA binding sites by “walking” the genes of three genomes. J Biomed Inform. 2011;44(5):839–847. doi:10.1016/j.jbi.2011.05.002
22. Chen Y, Wang X. miRDB: an online database for prediction of functional microRNA targets. Nucleic Acids Res. 2020;48(D1):D127–D131. doi:10.1093/nar/gkz757
23. Huang HY, Lin YC, Li J, et al. miRTarBase 2020: updates to the experimentally validated microRNA-target interaction database. Nucleic Acids Res. 2020;48(D1):D148–D154. doi:10.1093/nar/gkz896
24. Chen T, Zhang H, Liu Y, Liu YX, Huang L. EVenn: easy to create repeatable and editable Venn diagrams and Venn networks online. J Genet Genomics. 2021;48(9):863–866. doi:10.1016/j.jgg.2021.07.007
25. Shannon P, Markiel A, Ozier O, et al. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 2003;13(11):2498–2504. doi:10.1101/gr.1239303
26. Glazar P, Papavasileiou P, Rajewsky N. circBase: a database for circular RNAs. Rna. 2014;20(11):1666–1670. doi:10.1261/rna.043687.113
27. Xia S, Feng J, Chen K, et al. CSCD: a database for cancer-specific circular RNAs. Nucleic Acids Res. 2018;46(D1):D925–D929. doi:10.1093/nar/gkx863
28. Levy D, Goyal A, Grigorova Y, Farci F, Le JK. Aortic Dissection. In: StatPearls. Treasure Island (FL); 2021.
29. Weber TF, Ganten MK, Bockler D, Geisbusch P, Kauczor HU, von Tengg-kobligk H. Heartbeat-related displacement of the thoracic aorta in patients with chronic aortic dissection type B: quantification by dynamic CTA. Eur J Radiol. 2009;72(3):483–488. doi:10.1016/j.ejrad.2008.07.045
30. Hansen TB, Jensen TI, Clausen BH, et al. Natural RNA circles function as efficient microRNA sponges. Nature. 2013;495(7441):384–388. doi:10.1038/nature11993
31. Militello G, Weirick T, John D, Doring C, Dimmeler S, Uchida S. Screening and validation of lncRNAs and circRNAs as miRNA sponges. Brief Bioinform. 2017;18(5):780–788. doi:10.1093/bib/bbw053
32. Qin S, Nakajima B, Nomura M, Arfin SM. Cloning and characterization of a Saccharomyces cerevisiae gene encoding a new member of the ubiquitin-conjugating protein family. J Biol Chem. 1991;266(23):15549–15554. doi:10.1016/S0021-9258(18)98652-7
33. Kaiser P, Mandl S, Schweiger M, Schneider R. Characterization of functionally independent domains in the human ubiquitin conjugating enzyme UbcH2. FEBS Lett. 1995;377(2):193–196. doi:10.1016/0014-5793(95)01323-7
34. Vourc’h P, Martin I, Bonnet-Brilhault F, et al. Mutation screening and association study of the UBE2H gene on chromosome 7q32 in autistic disorder. Psychiatr Genet. 2003;13(4):221–225. doi:10.1097/00041444-200312000-00005
35. Li YP, Lecker SH, Chen Y, Waddell ID, Goldberg AL, Reid MB. TNF-alpha increases ubiquitin-conjugating activity in skeletal muscle by up-regulating UbcH2/E220k. FASEB J. 2003;17(9):1048–1057. doi:10.1096/fj.02-0759com
36. Hynes RO. Integrins: bidirectional, allosteric signaling machines. Cell. 2002;110(6):673–687. doi:10.1016/S0092-8674(02)00971-6
37. Didangelos A, Yin X, Mandal K, et al. Extracellular matrix composition and remodeling in human abdominal aortic aneurysms: a proteomics approach. Mol Cell Proteom. 2011;10(8):
38. Wang L, Zhang J, Fu W, Guo D, Jiang J, Wang Y. Association of smooth muscle cell phenotypes with extracellular matrix disorders in thoracic aortic dissection. J Vasc Surg. 2012;56(6):1698–1709, 1709 e1691. doi:10.1016/j.jvs.2012.05.084
 © 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.