Back to Journals » International Journal of General Medicine » Volume 14
Construction and Integrated Analysis of Competitive Endogenous Long Non-Coding RNA Network in Thoracic Aortic Dissection
Authors Shao Y ![]() , Luo J, Ye L, Ran HY, Shi HM, Zhang C, Wu QC
, Luo J, Ye L, Ran HY, Shi HM, Zhang C, Wu QC
Received 25 August 2021
Accepted for publication 29 September 2021
Published 16 October 2021 Volume 2021:14 Pages 6863—6873
DOI https://doi.org/10.2147/IJGM.S335082
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Scott Fraser
Yue Shao,1 Jun Luo,1 Liu Ye,2 Hao-Yu Ran,1 Hao-Ming Shi,1 Cheng Zhang,1 Qing-Chen Wu1
1Department of Cardiothoracic Surgery, The First Affiliated Hospital of Chongqing Medical University, Chongqing, People’s Republic of China; 2The First Branch, The First Affiliated Hospital of Chongqing Medical University, Chongqing, People’s Republic of China
Correspondence: Cheng Zhang; Qing-Chen Wu
Department of Cardiothoracic Surgery, The First Affiliated Hospital of Chongqing Medical University, Chongqing, People’s Republic of China
Email [email protected]; [email protected]
Background: Long non-coding RNAs (lncRNAs) can act as a competitive endogenous RNA (ceRNA) to regulate gene expression by sequestering the microRNA (miRNA). However, the lncRNA-miRNA-mRNA ceRNA network in thoracic aortic dissection (TAD) has been rarely documented.
Methods: Three Gene Expression Omnibus (GEO) datasets were used to detect differentially expressed mRNAs, miRNAs, and lncRNAs in TAD. Gene ontology and the Kyoto Encyclopedia of Genes and Genomes (KEGG) analyses were conducted for the differentially expressed mRNAs. A protein–protein interaction network for differentially expressed mRNAs was also constructed, and hub genes were identified. We established a ceRNA network of TAD based on the differentially expressed miRNAs, mRNAs and lncRNAs, and verified our results using an independent dataset and quantitative real-time PCR (qRT-PCR).
Results: In TAD, 267 lncRNAs, 81 miRNAs, and 346 mRNAs were identified as differentially expressed. The established ceRNA network consisted of seven lncRNA nodes, three mRNA nodes, and three miRNA nodes, and the expression of miRNAs in TAD was opposite to that of lncRNAs and mRNAs. Subsequently, an independent GEO dataset and qRT-PCR were used to validate the expression of three mRNAs. In addition, the expression differences in SLC7A5, associated miRNA and lncRNA were verified. According to gene set enrichment analysis of SLC7A5, the most significant KEGG pathway was considerably enriched in spliceosome and pentose phosphate pathway.
Conclusion: We established a novel ceRNA regulatory network in TAD, which provides valuable information for further research in the molecular mechanisms of TAD.
Keywords: bioinformatics analysis, ceRNA, expression profile, thoracic aortic dissection
Introduction
Thoracic aortic dissection (TAD), one of the most fatal vascular diseases, is characterized by the separation of thoracic aortic medial layers.1 According to a previous population survey, 17.6% of TAD patients were declared dead upon arrival at the hospital, and mortality rates for hospitalized patients can reach 21.4% within 24 hours.2 The most common risk factors for aortic dissection are hypertension, connective tissue disorders, trauma, and aging.3,4 However, the potential molecular mechanisms of TAD are unknown, making prevention and treatment difficult. Therefore, advances in sequencing technology could help researchers in identifying candidate pathogenic genes that promote expansion and facilitate dissection.5
LncRNAs are the largest class of noncoding RNAs, and most lncRNAs have not been confirmed in function so far. However, growing evidence suggests that lncRNAs may play a key role in transcriptional, post-transcriptional and epigenetic levels.6 Recently, aberrant expression of a large number of lncRNAs has been found to be associated with the occurrence and progression of multiple vascular diseases.7 However, little is known about the molecular mechanism of lncRNAs in TAD.
LncRNAs are thought to act via various mechanisms. In 2011, a complex hypothesis of competitive endogenous RNA (ceRNA) was proposed.8 Accumulating researches prove that lncRNA can inhibit the function of miRNA through sharing at least one miRNA response elements (MRE), thereby regulating target gene protein expression.9,10 The competition between lncRNAs and mRNAs with common miRNA target sites widely exists in vascular diseases.11,12 However, so far, few studies have demonstrated the lncRNA-miRNA-mRNA ceRNA network of TAD.
Elucidating the lncRNA-associated ceRNA network in TAD may help to develop new therapeutic targets for TAD. In addition, the analysis of TAD-associated ceRNA network is still rarely reported. In this study, a ceRNA network, consisting of seven lncRNA nodes, three mRNA nodes, and three miRNA nodes, was successfully constructed. The expression of mRNAs in the ceRNA network was verified using external datasets and qRT-PCR. The aim of this research was to provide further insights into the development and progression of TAD.
Methods
Gene Expression Datasets
Four datasets (GSE52093, GSE98770, GSE147026 and GSE153434) were downloaded from the Gene Express Omnibus (GEO) database (www.ncbi.nlm.nih.gov/geo/), which contain gene expression profiles of TAD and healthy controls.
Three datasets containing mRNA microarray data for 31 TAD patients and healthy controls (12 samples from GSE52093, 11 samples from GSE98770, and 8 samples from GSE147026) were downloaded. The GSE98770 dataset contained microRNA (miRNA) microarray data for 6 TAD patients and 5 healthy controls. The lncRNA microarray data of 4 TAD patients and 4 healthy controls were from GSE147026. Finally, the mRNA microarray data of 10 TAD patients and 10 healthy controls were downloaded from GSE153434 for independent external validation. The detail information of datasets is shown in Supplementary Table 1.
Clinical Samples
Aortic wall tissues were collected during surgical operations in 25 TAD patients who had been admitted at the First Affiliated Hospital of Chongqing Medical University between October 2020 and May 2021. Additionally, 15 healthy aortic tissues were collected from organ donors who were free of Marfan syndrome and other aortic diseases. The basic clinical characteristics of the study population are shown in Table 1. Ethical approval was obtained from the ethics committee of the First Affiliated Hospital of Chongqing Medical University, and informed consents were obtained from all participating patients (ethics approval number: 2018-022-2). The study complied with the Declaration of Helsinki guidelines.
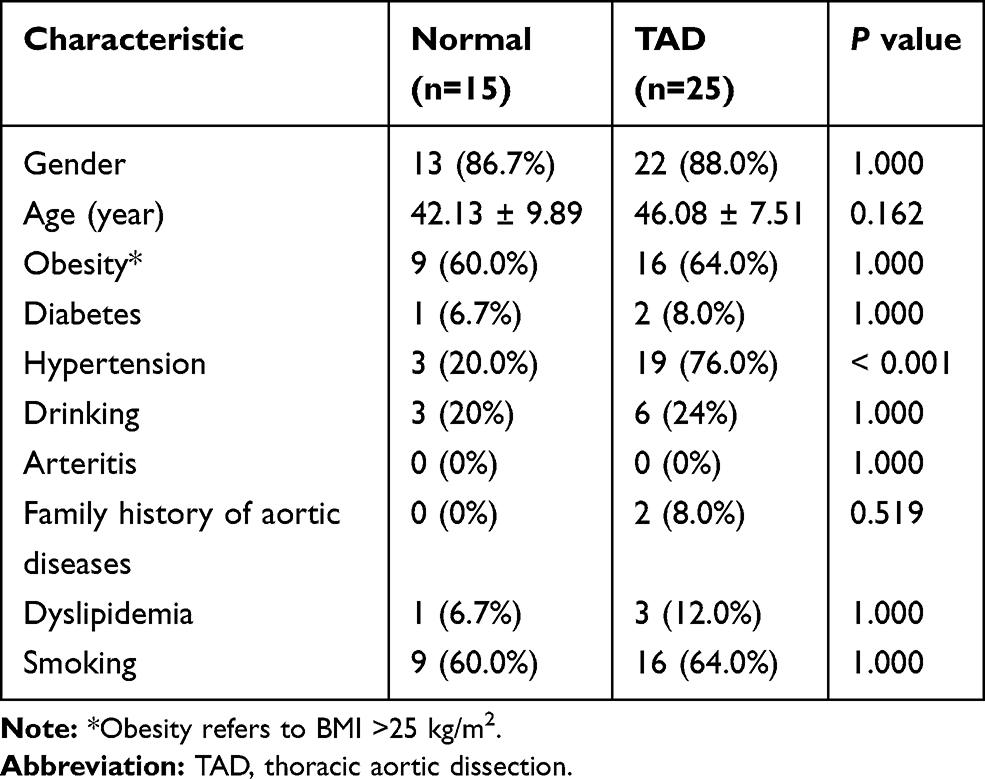 |
Table 1 Baseline Demographic Data |
Differentially Expressed Analysis
We merged TAD and normal sample data from the three datasets, and removed the batch effect using ComBat in “sva” R package. The mRNA expression data were normalized with the NormalizeBetweenArrays function of the “limma” package, and a PCA plot was constructed using “ggplot2” package. The differentially expressed mRNAs (DEmRNAs) were identified with thresholds of |log2foldChange(FC)| >1.0 and adjusted P value <0.05, differentially expressed miRNAs (DEmiRNAs) with |log2FC| >1 and P value <0.05, and differentially expressed lncRNAs (DElncRNAs) with |log2FC| >1 and P value <0.05. Volcano Plots were applied to identify DEmRNAs, DEmiRNAs, and DElncRNAs. Next, we used the pheatmap R package to scale and cluster the rows based on the z score of normalized expression between various conditions.
Construction of Protein-Protein Interaction (PPI) Network and Screening for Hub Genes
The identified DEmRNAs were input into Search Tool for the Retrieval of Interacting Genes (STRING; version 11.5; https://string-db.org/) to generate a potential PPI network and detect the possible relationships with a minimum required interaction score of 0.4, removing disconnected nodes. Hub gene plays a key role in biological processes and often affects other genes in related pathways. CytoHubba, a valid plug-in unit of the Cytoscape, was used to identify hub genes accurately by 12 topological analysis methods. Finally, the top 20 hub genes were screened using Maximal Clique Centrality (MCC).
Gene Ontology (GO), Kyoto Encyclopedia of Genes and Genomes (KEGG) Pathway Analyses and Gene Set Enrichment Analysis (GSEA)
We used “clusterprofiler” and “ggplot2” packages to do GO and the KEGG pathway enrichment analyses for upregulated and downregulated genes, respectively. The GO terms were divided into three types: Biological Process, Cellular Component, and Molecular Function, and the three descriptions with the lowest P value, respectively, were shown in the figure Furthermore, GSEA of SLC7A5 was performed using the GSEA software (Broad Institute, Cambridge, MA, USA).
Construction of a lncRNA-Related ceRNA Network
The ceRNA hypothesis states that lncRNA can regulate mRNA expression through chelating and binding miRNA. Therefore, we used the following steps to build a ceRNA regulated network: the intersection of three databases including Targetscan (http://www.targetscan.org/), miRTarBase (http://mirtarbase.mbc.nctu.edu.tw/), and miRDB (http://www.mirdb.org/) was used as a potential target mRNAs of DEmiRNAs; the lncRNA-miRNA interaction was predicted using DIANA-LncBase v2 (www.microrna.gr/LncBase) with thresholds of miTG-score >0.85;13 the intersecting miRNAs negatively regulated by lncRNAs and mRNAs were chosen to build the ceRNA network, whose graph was created using Cytoscape version 3.7.1 (http://www.cytoscape.org/). A detailed flow chart of the design is shown in Supplementary Figure 1.
Total RNA Extraction and Quantitative Real-Time Polymerase Chain Reaction
Total RNA was isolated from aortic wall tissue using TRIzol reagent (Takara, Dalian), and was reverse transcribed using the PrimeScript™ RT Reagent Kit (Takara, Japan). Then, diluted cDNA was used as a template in a qPCR reaction and amplified using mRNA primers labeled with a TB™ Green Premix Ex Taq™ (Takara, Japan). Relative expression levels were obtained by normalizing to 18S ribosomal RNA (18S). The experiment was performed in triplicate with three replicates each time. The details of the used primers are shown in Supplementary Table 2.
Statistical Analysis
Statistical analyses were performed by R software (R version 4.1.0; https://www.r-project.org/). The Wilcoxon rank sum test was used to compare differences in qRT-PCR. Expression data were presented using the median and interquartile range (IQR). Correlation coefficients (r) between mRNA and lncRNA were assessed using the Pearson correlation method. The baseline data of patients used Fisher’s exact test and Student’s t-test. Statistical significance was considered at P < 0.05.
Results
Detection of DEmRNAs, DEmiRNAs and DElncRNAs
The gene expression profiles of all the samples from different datasets were subjected to a PCA before and after normalization (Figures 1A and B). According to the PCA, there was a significant reduction in batch effects after normalization. The DEmRNAs were identified after batch correction and standardization of the microarray results from GSE52093, GSE98770 and GSE147026 datasets. In the merged dataset, a total of 346 DEmRNAs were screened out, where 161 were upregulated and 185 downregulated. The volcano plot (Figure 1C) and clustered heat map (Figure 1D) of DEmRNAs displayed distinct differences between normal and TAD tissues. Totally, 1347 miRNAs were detected in 11 samples from the GSE98770 miRNA array dataset, as well as 81 DEmiRNAs, 47 of which were upregulated and 34 of which were downregulated. The volcano plot (Figure 2A) and heat map (Figure 2B) displayed the aberrantly expressed miRNA molecules. In addition, 267 DElncRNAs (87 upregulated and 180 downregulated) were identified in the GSE147026 dataset, which were shown in Figures 2C and D.
 |
Figure 1 Principal component analysis (PCA) of the gene expression datasets and differently expressed mRNAs in TAD. The points of the scatter plots visualize the samples based on the top two principal components of gene expression profiles without (A) and with (B) the removal of batch effect. The colors represent samples from three different datasets, respectively. (C) The volcano graph shows the distribution of differently expressed mRNAs. (D) The heatmap of the mRNAs expression in TAD. In the volcano graph, the X-axis represents the fold changes of differently expressed mRNAs, and the Y-axis represents the adjusted p-value, red dots present up-regulated genes and blue dots present down-regulated genes (|log2foldChange| >1.0 and adjusted P value <0.05). |
 |
Figure 2 Differently expressed miRNAs and lncRNAs in TAD. (A) The volcano graph shows the distribution of differently expressed miRNAs. (B) The heatmap of the miRNAs expression in TAD. (C) The volcano graph shows the distribution of differently expressed lncRNAs. (D) The heatmap of the lncRNAs expression in TAD. In the volcano graph, the X-axis represents the fold changes of differently expressed miRNAs and lncRNAs, and the Y-axis represents the p-value, red dots present up-regulated genes and blue dots present down-regulated genes (|log2foldChange| >1.0 and P value <0.05). |
Functional Analyses
To explore the functional significance and potential regulators of DEmRNAs, GO annotation and KEGG pathway enrichment analyses were performed on upregulated and downregulated mRNAs, respectively. Genes without official gene symbols or transcript annotations in US National Center for Biotechnology Information or Ensembl were removed. The GO and KEGG analyses displayed the top 12 enriched terms of the upregulated and downregulated mRNAs (Figures 3A and B). With regard to biological process, the downregulated DEmRNAs were enriched in muscle cell development, muscle contraction, and muscle system process; the top three enriched terms of the upregulated DEmRNAs were nuclear division, mitotic nuclear division, and chromosome segregation. In the molecular functions category, downregulated DEmRNAs were enriched in protein serine/threonine phosphatase inhibitor activity, structural constituent of muscle, and ion channel binding; the upregulated DEmRNAs were enriched in endopeptidase inhibitor activity, peptidase regulator activity, and peptidase inhibitor activity. In cellular component, chromosomal region, condensed chromosome/centromeric region, and condensed chromosome were the top three enriched terms in upregulated DEmRNAs; contractile fiber, sarcomere, and contractile fiber part were the top three enriched terms in downregulated DEmRNAs. The three most significantly enriched KEGG pathways, when considering the downregulated DEmRNAs, were hypertrophic cardiomyopathy, calcium signaling pathway, and dilated cardiomyopathy. In the upregulated DEmRNAs, TNF signaling pathway, cell cycle, and mineral absorption were the top three enriched pathways. The specific genes involved in each term are listed in the Supplementary Tables 3 and 4.
 |
Figure 3 Gene ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analyses of differently expressed mRNA. (A) GO and KEGG pathway analyses of up-regulated genes. (B) GO and KEGG pathway analyses of down-regulated genes. (C) Protein–protein interaction network of differently expressed mRNA (red indicates up-regulation, and green indicates down-regulation). (D) Protein–protein interaction network of top 20 mRNAs. The color changes from dark red to light red indicate the MCC score changes from high to low. |
The protein-protein interaction networks among DEmRNAs was built using String in Cytoscape (Figure 3C), where the isolated nodes were excluded. The centrality degree of each node was evaluated using cytoHubba. Twenty genes ranked by MCC method including AURKA, NCAPGCENPE, CCNB2AURKB, UBE2C, RRM2, CENPF, ASPM, MKI67, CDC20, KIF20A, MCM10, ASF1B, BIRC5, FOXM1, ESPL1, HJURP, CDCA5 and TRIP13 were defined as hub genes (Figure 3D). The core network suggested that hub genes were connected to each other, forming a dense hub.
Construction of the ceRNA Network
Because CeRNA acts as a sponge for miRNA, the expression of miRNA was opposite that of mRNA and lncRNA in the TAD and health groups. The ceRNA network is shown in Figure 4. The lncRNA-miRNA-mRNA network was constructed according to the steps listed in the methods, including seven lncRNAs, three miRNAs, and three mRNAs. In this study, seven lncRNA‑miRNA pairs and three miRNA‑mRNA pairs were mined. In addition, seven pairs of lncRNA‑miRNA‑mRNA interactions were identified.
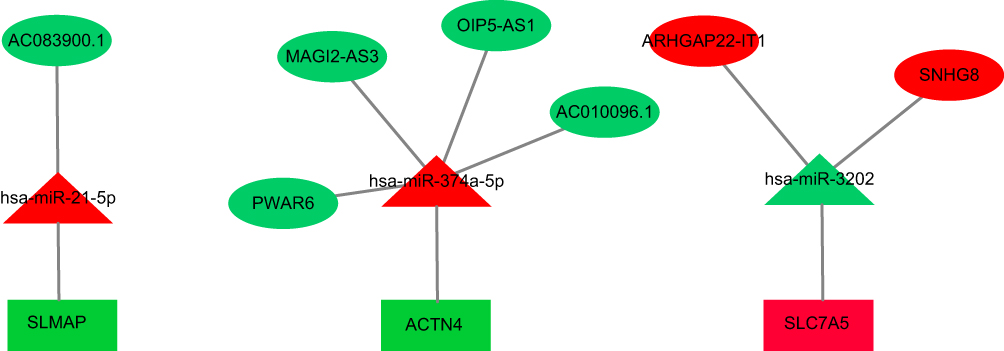 |
Figure 4 The miRNA-lncRNA-mRNA ceRNA network is composed of 7 lncRNAs, 3 miRNAs and 3 mRNAs. Rectangles, triangles and ellipses represent mRNAs, miRNA and lncRNAs, respectively, while red indicates up-regulation, and green indicates down-regulation. |
Patient Characteristics
A total of 25 patients with TAD and 15 controls met our inclusion criteria. Demographic and clinical information of all patients is summarized in Table 1. Except for the presence of more severe hypertension patients (76.0%) in the TAD group, there were no significant differences in age, gender, or other characteristics.
Validation of Differentially Expressed ceRNAs
The SLMAP, ACTN4, and SLC7A5 genes were selected for further tests using GSE153434 and qRT-PCR to validate our findings (Figure 5). In GSE153434, ACTN4 and SLC7A5 were remarkably downregulated in the TAD group, whereas SLMAP showed no statistically significant difference. Furthermore, qRT-PCR showed a substantial increase in SLC7A5 expression in TAD (P value< 0.05), while the expression of SLMAP and ACTN4 were not significantly different. Next, we used qRT-PCR to test for SLC7A5-linked miRNAs and lncRNAs. Interestingly, hsa-miR-3202 was found to have low expression in TAD, but ARHGAP22-IT1 and SNHG8−2 were highly expressed (Figure 6A–C). The correlation between expression levels of SLC7A5 and associated lncRNAs (ARHGAP22-IT1 and SNHG8-2) were examined using a Pearson correlation coefficient. The expression of two lncRNA-mRNA pairs was significantly positively correlated (r> 0.4, P value< 0.05, Figures 6D and E).
 |
Figure 5 Independent dataset (GSE153434) and quantitative real-time PCR validation of the three key mRNA between TAD and normal controls. (A–C) SLMAP, ACTN4 and SLC7A5 expression based on GSE153434 for TAD. (D–F) Relative mRNA expression of SLMAP, ACTN4 and SLC7A5 was detected by RT-PCR. All experiments were performed in triplicate and results were presented as Median ± interquartile range. |
 |
Figure 6 Validation of the expression of representative dysregulated lncRNAs and miRNAs by quantitative qRT-PCR in TAD, the correlation between expression levels of ceRNAs, and GESE analysis of SLC7A5. (A) Validation of the expression of hsa-miR-3202. (B) Validation of the expression of ARHGAP22-IT1. (C) Validation of the expression of SNHG8. (D) correlation between expression levels of SLC7A5 and ARHGAP22-IT1. (E) correlation between expression levels of SLC7A5 and SNHG8. (F) GSEA analysis of SLC7A5. |
Based on the GSEA standard recommendations, GSEA results with a false discovery rate (FDR) q-value <0.25, nominal FDR <0.05, and |Normalized Enrichment Score (NES)| >1 were considered significant. According to the GSEA of the SLC7A5, the most significant KEGG pathway was considerably enriched in spliceosome and pentose phosphate pathway (PPP), which may be associated with pathophysiological development of TAD (Figure 6F).
Discussion
Over the past few decades, numerous researchers studied the functions of non-coding RNA and enhanced our understanding of various biological processes. LncRNAs are a type of non-coding transcripts that have over 200 nucleotides.14 Increasing evidence demonstrate that lncRNAs use miRNA as a bridge to regulate the expression of mRNA. The ceRNA regulatory network extends the functional genetic information in the genome and is essential in vascular diseases.8,12,15,16 In this study, data from the GEO database were used to generate triple networks based on the ceRNA theory. The ceRNA network consisted of seven lncRNA nodes, three mRNA nodes, and three miRNA nodes. Subsequently, the expression of three mRNAs was validated using an independent GEO dataset and qRT-PCR. Furthermore, the difference in SLC7A5 expression was verified. Finally, the qRT-PCR results for miRNA and lncRNA associated to SLC7A5 were consistent with our expectations.
Previous studies have shown that the transformation of the phenotype of vascular smooth muscle cells (SMC) from contractile to synthetic, which plays a key role in aortic wall degeneration.17,18 This VSMCs switching is marked with the enhanced proliferation, migration, antiapoptosis, up-regulation of synthetic markers, and down-regulation of contractile markers.19 Wang et al demonstrated that VSMCs from dissecting aorta proliferated faster than VSMCs from normal aortic tissue.18 In addition, the expression level of genes involved in proliferation increased.20
In this study, the DEmRNAs were subjected to GO and KEGG enrichment analyses. A large number of significantly enriched GO terms, such as mitotic nuclear division, muscle contraction, contractile fiber, contractile fiber component, sarcomere, structural constituent of muscle, and ion channel binding, were consistent with previous studies on TAD.5,21 Key KEGG pathways were highly enriched in cell cycle, mineral absorption, dilated cardiomyopathy, hypertrophic cardiomyopathy, and TNF signaling pathway, which were also in line with previous reports.22–24
Three DEmRNAs were retained in the ceRNA network after screening. The expression of SLC7A5, which was consistent with the external dataset, was confirmed using qRT-PCR. To ensure high accuracy, we focused our discussion on SLC7A5, related miRNA and lncRNA. SLC7A5, also known as LAT1, interacts with the glycoprotein SLC3A2 to form a high-affinity transporter. The transporter mediates uptake of large neutral amino acids.25,26 Previous studies found that SLC7A5 is overexpressed in a variety of cancers and plays a role in proliferation and metastasis.27–29 Liu et al demonstrated that SLC7A5-mediated L amino acid transport system is essential for the proliferation and survival of vascular smooth muscle cell (VSMC).30 Notably, increased strain of the blood vessel wall and vascular injury can induce arterial remodeling by stimulating the proliferation of VSMC.31,32
The expression of hsa-miR-3202 was found to be lower in patients with early-onset post-stroke depression.33 Under high glucose conditions, Huang et al discovered that hsa-miR-3202 promotes H5V cell apoptosis by directly targeting Fas apoptosis inhibitor 2 (FAIM2).34 To explore the role of hsa-miR-3202, we used bioinformatics tools to predict its target gene SLC7A5. The results demonstrated that hsa-miR-3202 inhibits the proliferation of VSMC by suppressing the expression of SLC7A5.
Despite the numerous DElncRNAs that have been found in various human diseases, only a few studies have investigated the role of lncRNAs in TAD. In this study, we found two DElncRNA that were positively correlated with the expression of SLC7A5. A previous study reported that ARHGAP22 Intronic Transcript 1(ARHGAP22‐IT1) was upregulated in lung adenocarcinoma patients.35 Small Nucleolar RNA Host Gene 8 (SNHG8) can promote the proliferation and migration of multiple tumor cells through serving as sponge for different miRNAs.36–38 Our results revealed that the ARHGAP22-IT1 and SNHG8 were overexpressed in TAD patients. According to the ceRNA hypothesis, the ARHGAP22-IT1 and SNHG8 can competitively bind to hsa-miR-3202 and upregulate SLC7A5, thus promoting VSMC proliferation and TAD development.
The GSEA and KEGG pathway enrichment of the SLC7A5 implicated that the spliceosome and pentose phosphate pathway (PPP) were most significantly regulated. Because RNA splicing is involved in diverse aspects of cell proliferation, survival, and differentiation. The PPP, a basic glucose metabolism pathway, is crucial for cell growth. Stimulation of PPP can strengthen nucleotide production and DNA synthesis, accelerating cell proliferation.39 This suggests that SLC7A5 may promote the proliferation of VSMC through two pathways, which may affect the occurrence and development of TAD. However, this hypothesis needs further research to verify.
The present study contains several limitations. Firstly, the sample size was relatively small, which could have lowered the accuracy of results verification. Secondly, the ceRNA network was constructed by combining three different GSE datasets, and there could have been missing and overlapping data between the databases. Therefore, a complete lncRNA/miRNA/mRNA profile should be performed in future studies. Furthermore, all samples were obtained from a single center, introducing a regional bias, necessitating further multicenter research. Finally, the specific functions of the ceRNAs in TAD are unclear, and future research can investigate the potential molecular mechanisms using Western blotting and luciferase reporter assay.
Conclusion
We used bioinformatics tools to identify the differentially expressed lncRNAs, miRNAs, and mRNAs of TAD. Importantly, we effectively established and verified the lncRNA-associated ceRNA regulatory network, which may lay a foundation for further studies of lncRNAs in TAD. Our study emphasized the important role of ceRNAs in pathogenesis of TAD and provide novel lncRNAs as potential therapeutic targets, although more studies are needed to confirm our findings.
Data Sharing Statement
The data used to support the findings of this study are available from the corresponding author (Qing-chen Wu, Email: [email protected]) upon request.
Ethics Approval
The project was approved by the ethics committee of the First Affiliated Hospital of Chongqing Medical University, and informed consents were obtained from all participating patients (ethics approval number: 2018-022-2).
Acknowledgment
This project was supported by two grants from the Health Bureau of Chongqing (2020MSXM131 and 2021MSXM005).
Disclosure
The authors declare no conflicts of interest.
References
1. Shah A, Khoynezhad A. Thoracic endovascular repair for acute type A aortic dissection: operative technique. Ann Cardiothorac Surg. 2016;5(4):389–396. doi:10.21037/acs.2016.07.08
2. Melvinsdottir IH, Lund SH, Agnarsson BA, Sigvaldason K, Gudbjartsson T, Geirsson A. The incidence and mortality of acute thoracic aortic dissection: results from a whole nation study. Eur J Cardiothorac Surg. 2016;50(6):1111–1117. doi:10.1093/ejcts/ezw235
3. Gawinecka J, Schönrath F, von Eckardstein A. Acute aortic dissection: pathogenesis, risk factors and diagnosis. Swiss Med Wkly. 2017;147:w14489. doi:10.4414/smw.2017.14489
4. Nienaber CA, Eagle KA. Aortic dissection: new frontiers in diagnosis and management: part II: therapeutic management and follow-up. Circulation. 2003;108(6):772–778. doi:10.1161/01.CIR.0000087400.48663.19
5. Wang W, Liu Q, Wang Y, et al. Verification of hub genes in the expression profile of aortic dissection. PLoS One. 2019;14(11):e0224922. doi:10.1371/journal.pone.0224922
6. Xiong XD, Ren X, Cai MY, Yang JW, Liu X, Yang JM. Long non-coding RNAs: an emerging powerhouse in the battle between life and death of tumor cells. Drug Resist Updat. 2016;26:28–42. doi:10.1016/j.drup.2016.04.001
7. Lu D, Thum T. RNA-based diagnostic and therapeutic strategies for cardiovascular disease. Nat Rev Cardiol. 2019;16(11):661–674. doi:10.1038/s41569-019-0218-x
8. Salmena L, Poliseno L, Tay Y, Kats L, Pandolfi PP. A ceRNA hypothesis: the rosetta stone of a hidden RNA language? Cell. 2011;146(3):353–358. doi:10.1016/j.cell.2011.07.014
9. Yu F, Chen B, Dong P, Zheng J. HOTAIR epigenetically modulates PTEN expression via MicroRNA-29b: a novel mechanism in regulation of liver fibrosis. Mol Ther. 2020;28(12):2703. doi:10.1016/j.ymthe.2020.10.021
10. Ye Y, Li S-L, Wang S-Y, Passos GA. Construction and analysis of mRNA, miRNA, lncRNA, and TF regulatory networks reveal the key genes associated with prostate cancer. PLoS One. 2018;13(8):e0198055. doi:10.1371/journal.pone.0198055
11. Liang H, Su X, Wu Q, et al. LncRNA 2810403D21Rik/Mirf promotes ischemic myocardial injury by regulating autophagy through targeting Mir26a. Autophagy. 2020;16(6):1077–1091. doi:10.1080/15548627.2019.1659610
12. Yu XH, Deng WY, Chen JJ, et al. LncRNA kcnq1ot1 promotes lipid accumulation and accelerates atherosclerosis via functioning as a ceRNA through the miR-452-3p/HDAC3/ABCA1 axis. Cell Death Dis. 2020;11(12):1043. doi:10.1038/s41419-020-03263-6
13. Paraskevopoulou MD, Vlachos IS, Karagkouni D, et al. DIANA-LncBase v2: indexing microRNA targets on non-coding transcripts. Nucleic Acids Res. 2016;44(D1):D231–D238. doi:10.1093/nar/gkv1270
14. Roberts TC, Morris KV, Weinberg MS. Perspectives on the mechanism of transcriptional regulation by long non-coding RNAs. Epigenetics. 2014;9(1):13–20. doi:10.4161/epi.26700
15. Glenfield C, McLysaght A. Pseudogenes provide evolutionary evidence for the competitive endogenous RNA hypothesis. Mol Biol Evol. 2018;35(12):2886–2899. doi:10.1093/molbev/msy183
16. Liu K, Liu C, Zhang Z. lncRNA GAS5 acts as a ceRNA for miR-21 in suppressing PDGF-bb-induced proliferation and migration in vascular smooth muscle cells. J Cell Biochem. 2019;120(9):15233–15240. doi:10.1002/jcb.28789
17. Majesky MW. Developmental basis of vascular smooth muscle diversity. Arterioscler Thromb Vasc Biol. 2007;27(6):1248–1258. doi:10.1161/ATVBAHA.107.141069
18. Wang L, Zhang J, Fu W, Guo D, Jiang J, Wang Y. Association of smooth muscle cell phenotypes with extracellular matrix disorders in thoracic aortic dissection. J Vasc Surg. 2012;56(6):1698–1709.e1. doi:10.1016/j.jvs.2012.05.084
19. Zhu SB, Zhu J, Zhou ZZ, Xi EP, Wang RP, Zhang Y. TGF-β1 induces human aortic vascular smooth muscle cell phenotype switch through PI3K/AKT/ID2 signaling. Am J Transl Res. 2015;7(12):2764–2774. doi:1943-8141/AJTR0014193.
20. Müller BT, Modlich O, Prisack HB, et al. Gene expression profiles in the acutely dissected human aorta. Eur J Vasc Endovasc Surg. 2002;24(4):356–364. doi:10.1053/ejvs.2002.1731
21. Wang W, Wang Y, Piao H, et al. Bioinformatics analysis reveals MicroRNA-193a-3p regulates ACTG2 to control phenotype switch in human vascular smooth muscle cells. Front Genet. 2021;11:572707. doi:10.3389/fgene.2020.572707
22. Su Y, Li Q, Zheng Z, Wei X, Hou P. Integrative bioinformatics analysis of miRNA and mRNA expression profiles and identification of associated miRNA-mRNA network in aortic dissection. Medicine. 2019;98(24):e16013. doi:10.1097/MD.0000000000016013
23. Fang J, Pan Z, Yu H, et al. Regulatory master genes identification and drug repositioning by integrative mRNA-miRNA network analysis for acute type A aortic dissection. Front Pharmacol. 2021;11:575765. doi:10.3389/fphar.2020.575765
24. Gao H, Sun X, Liu Y, et al. Analysis of hub genes and the mechanism of immune infiltration in stanford type a aortic dissection. Front Cardiovasc Med. 2021;8:680065. doi:10.3389/fcvm.2021.680065
25. Scalise M, Galluccio M, Console L, Pochini L, Indiveri C. The human SLC7A5 (LAT1): the intriguing histidine/large neutral amino acid transporter and its relevance to human health. Front Chem. 2018;6:243. doi:10.3389/fchem.2018.00243
26. Mastroberardino L, Spindler B, Pfeiffer R, et al. Amino-acid transport by heterodimers of 4F2hc/CD98 and members of a permease family. Nature. 1998;395(6699):288–291. doi:10.1038/26246
27. Xu M, Sakamoto S, Matsushima J, et al. Up-regulation of LAT1 during antiandrogen therapy contributes to progression in prostate cancer cells. J Urol. 2016;195(5):1588–1597. doi:10.1016/j.juro.2015.11.071
28. Ding K, Tan S, Huang X, et al. GSE1 predicts poor survival outcome in gastric cancer patients by SLC7A5 enhancement of tumor growth and metastasis. J Biol Chem. 2018;293(11):3949–3964. doi:10.1074/jbc.RA117.001103
29. Li Y, Wang W, Wu X, Ling S, Ma Y, Huang P. SLC7A5 serves as a prognostic factor of breast cancer and promotes cell proliferation through activating AKT/mTORC1 signaling pathway. Ann Transl Med. 2021;9(10):892. doi:10.21037/atm-21-2247
30. Liu XM, Reyna SV, Ensenat D, et al. Platelet-derived growth factor stimulates LAT1 gene expression in vascular smooth muscle: role in cell growth. FASEB J. 2004;18(6):768–770. doi:10.1096/fj.03-0886fje
31. Reyna SV, Ensenat D, Johnson FK, Wang H, Schafer AI, Durante W. Cyclic strain stimulates L-proline transport in vascular smooth muscle cells. Am J Hypertens. 2004;17(8):712–717. doi:10.1016/j.amjhyper.2004.03.673
32. Chappell J, Harman JL, Narasimhan VM, et al. Extensive proliferation of a subset of differentiated, yet plastic, medial vascular smooth muscle cells contributes to neointimal formation in mouse injury and atherosclerosis models. Circ Res. 2016;119(12):1313–1323. doi:10.1161/CIRCRESAHA.116.309799
33. Zhang Y, Cheng L, Chen Y, Yang GY, Liu J, Zeng L. Clinical predictor and circulating microRNA profile expression in patients with early onset post-stroke depression. J Affect Disord. 2016;193:51–58. doi:10.1016/j.jad.2015.12.061
34. Huang X, Xie H, Xue G, Ye M, Zhang L. MiR-3202 - promoted H5V cell apoptosis by directly targeting Fas Apoptotic Inhibitory Molecule 2 (FAIM2) in high glucose condition. Med Sci Monit. 2017;23:975–983. doi:10.12659/msm.899443
35. Yang Z, Li H, Wang Z, et al. Microarray expression profile of long non-coding RNAs in human lung adenocarcinoma. Thorac Cancer. 2018;9(10):1312–1322. doi:10.1111/1759-7714.12845
36. Xuan L, Sun Z, Wang J, Gao S. lncRNA SNHG8 promotes ovarian cancer progression through serving as sponge for miR-1270 to regulate S100A11 expression. J Gene Med. 2021;e3315. doi:10.1002/jgm.3315
37. Zhen Y, Ye Y, Wang H, et al. Knockdown of SNHG8 repressed the growth, migration, and invasion of colorectal cancer cells by directly sponging with miR-663. Biomed Pharmacother. 2019;116:109000. doi:10.1016/j.biopha.2019.109000
38. Shi Z, Zhang H, Jie S, et al. Long non-coding RNA SNHG8 promotes prostate cancer progression through repressing miR-384 and up-regulating HOXB7. J Gene Med. 2021;23(3):e3309. doi:10.1002/jgm.3309
39. Wu S, Wang H, Li Y, et al. Transcription factor YY1 promotes cell proliferation by directly activating the pentose phosphate pathway. Cancer Res. 2018;78(16):4549–4562. doi:10.1158/0008-5472.CAN-17-4047
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.