")
Back to Journals » International Journal of Nanomedicine » Volume 10 » Issue 1
Construction and characterization of an anti-CD20 mAb nanocomb with exceptionally excellent lymphoma-suppressing activity
Authors Li H, Wu C, Chen T, Zhang G, Zhao H, Ke C, Xu Z
Received 31 December 2014
Accepted for publication 15 May 2015
Published 30 July 2015 Volume 2015:10(1) Pages 4783—4796
DOI https://doi.org/10.2147/IJN.S80129
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Lei Yang
Hua-Fei Li,1–3,* Cong Wu,4,* Ting Chen,5,* Ge Zhang,1 He Zhao,1 Chang-Hong Ke,1 Zheng Xu2
1International Joint Cancer Institute, Translation Medicine Institute, 2Planning Division, Scientific Research Department, 3Tumor Immunology and Gene Therapy Center, Eastern Hepatobiliary Surgery Hospital, 4Department of Laboratory Diagnosis, Changhai Hospital, 5Department of Cardiology, Changhai Hospital, the Second Military Medical University, Shanghai, People’s Republic of China
*These authors contributed equally to this work
Abstract: The CD20-directed monoclonal antibody rituximab (RTX) established a new era in the treatment of non-Hodgkin lymphoma (NHL); however, suboptimal response and/or resistance to RTX still limit its clinical merits. Although four effector mechanisms are validated to participate in CD20-based immunotherapy, including complement-dependent cytotoxicity, antibody-dependent cell-mediated cytotoxicity, caspase-dependent apoptosis, and lysosome-mediated programmed cell death (PCD), they could hardly be synchronously activated by any anti-CD20 mAb or mAb derivative until now. Herein, a novel mAb nanocomb (polyethylenimine polymer–RTX–tositumomab [PPRT nanocomb]) was firstly constructed through mass arming two different anti-CD20 mAbs (RTX and tositumomab) to one polymer by nanotechnology. Comparing with free mAbs, PPRT nanocomb possesses a comparable binding ability and reduced “off-rate” to surface CD20 of NHL cells. When treated by PPRT nanocomb, the caspase-dependent apoptosis was remarkably enhanced except for concurrently eliciting complement-dependent cytotoxicity, antibody-dependent cell-mediated cytotoxicity, and lysosome-mediated PCD. Besides, “cross-cell link”-assisted homotypic adhesion by PPRT nanocomb further enhanced the susceptibility to PCD of lymphoma cells. Pharmacokinetic assays revealed that PPRT nanocomb experienced a relatively reduced clearance from peripheral blood compared with free antibodies. With the cooperation of all the abovementioned superiorities, PPRT nanocomb exhibits exceptionally excellent in vivo antitumor activities in both disseminated and localized human NHL xenotransplant models.
Keywords: non-Hodgkin lymphoma, CD20, nanotechnology, rituximab, programmed cell death
Introduction
Non-Hodgkin lymphoma (NHL) is predominantly a cancer of B lymphocytes.1,2 Depending on the subtype of B-cell NHL, the disease can pursue an indolent course spanning years, such as in follicular lymphoma, or it can present aggressively during several weeks or months, such as in diffuse large B-cell lymphoma.3 Although the “watch and wait” approach is acceptable in asymptomatic patients with indolent NHL, patients with more aggressive subtypes require immediate and effective treatments.3,4 Traditional therapies for NHL, including chemotherapy and radiotherapy, are limited because of serious harmful side effects.2,3 In the past decades, a variety of basic and groundbreaking discoveries paved the way for the development of mAbs, the theoretical “magic bullets”, which could specifically identify and kill malignant cells, thereby reducing the detrimental side effects to normal tissues.5,6
CD20 is a B-cell differentiating antigen, which selectively expressed on the surface of mature and malignant B-cells, but not immature B-cells or plasma cells.7–9 Therefore, although anti-CD20 monoclonal antibodies (mAbs) target mature and malignant B lymphocytes, they spare B-cell progenitors, allowing normal B-cell regeneration, which makes CD20 one of the most ideal therapeutic targets.7,10–12 Despite the success of US Food and Drug Administration (FDA) approved rituximab (RTX) in patients with B-cell NHLs, considerable room for improved therapeutic index remains. Only about 15% of patients with follicular lymphoma respond to the initial treatment with RTX monotherapy, and the majority of responders become refractory to RTX.13 Mantle cell lymphoma and chronic lymphocytic leukemia/small lymphocytic lymphoma remain incurable, while up to 40% of those with diffuse large B-cell lymphoma fail initial treatment and most of those die from their disease.14 Only 48% of all NHL patients respond to RTX treatment, with less than 10% showing a complete remission (CR).8,15 When patients relapsing after an initial response to RTX were retreated, an overall response rate of only 40% was reported.13,16 Recent studies demonstrate that the limitations of RTX-based immunotherapy may be attributed to the exhaustion and/or disability of effector cells (such as natural killer cells) and complements.17–19 In order to improve the disease prognosis, many novel anti-CD20 mAbs or mAb derivatives with enhanced ability in inducing programmed cell death (PCD) have been developed.15,20–25
Presently, anti-CD20 mAbs can be broadly divided into two types with distinct lymphoma-killing mechanisms. Type I mAbs (RTX-like) can efficiently promote complement-dependent cytotoxicity (CDC) but are relatively weak in evoking PCD, while type II mAbs (tositumomab [Tos]-like) are relatively poor at complement activation but can effectively elicit lysosome-mediated PCD. Besides, both types are comparable in carrying out antibody-dependent cell-mediated cytotoxicity (ADCC).17,26,27 It appears that all the current anti-CD20 mAbs effective in evoking PCD are relatively ineffective in mediating CDC, and vice versa. Also, it is well established that, with the cross-linking of secondary antibodies, RTX can induce caspase-dependent apoptosis in malignant B-cells, which is different from the lysosome-mediated cell death induced by type II mAbs.27–31 However, this cross-linking can hardly be realized in vivo. It seems, although we acknowledge that the activation of more effector mechanisms may lead to improved curative effects,7,22,25,32,33 the abovementioned immunotherapeutic mechanisms could hardly be synchronously activated by any reported anti-CD20 mAb or mAb derivative until now.
In this study, a novel mAb nanocomb (polyethylenimine [PEI] polymer–RTX–Tos [PPRT nanocomb]) was successfully prepared from two different anti-CD20 mAbs of distinct types by nanotechnology. It is worth mentioning that nanomedicine is an emerging form of anticancer therapy based on the assembling of biological molecules into nanosized particles.34 Previous publications have proven that high leakiness of microvessels and lack of lymphatic drainage give rise to enhanced accumulation and retention of high-molecular weight (MW) compounds in tumor tissues. This phenomenon is called the enhanced permeability and retention effect, which is one of the most important superiorities of nanosized (from tens to hundreds of nanometers) particles.35–38 Recently, several studies confirmed that nano-polymer cross-linked anti-CD20 mAbs can induce remarkable caspase-dependent apoptosis in NHL cells, with the apoptotic efficiency being proportional to both the polymer chain length and valence (amount of antibody molecules per polymer) to some extent.39–42 In this work, PEI with a MW of 70 kDa was employed as a long-chain polymer for the cross-linking of both antibodies. The well-modified PPRT nanocomb can eliminate malignant B-cells by the activation of all the abovementioned CD20-based immunotherapeutic mechanisms, including CDC, ADCC, and direct induction of PCD through both caspase-dependent and -independent manners. Also, PPRT nanocomb exhibits exceptionally potent in vivo lymphoma-suppressing ability in both disseminated and localized human NHL xenotransplant models. Besides, the specific mechanisms concerning its outstanding PCD-inducing ability have been carefully explored.
Materials and methods
Cell lines, materials, and animals
Three human NHL cell lines (Ramos, Raji, and JeKo-1) were obtained from the American Type Culture Collection ([ATCC] Manassas, VA, USA). Cells were propagated in RPMI 1640 supplemented with 10% (v/v) fetal bovine serum (Thermo Fisher Scientific, Waltham, MA, USA). RTX (C2B8) and Tos (B1) were purchased from Hoffman-La Roche Ltd. (Basel, Switzerland) and GlaxoSmithKline plc (London, UK), respectively. PEI (70 kDa) was purchased from Sigma-Aldrich Co. (St Louis, MO, USA). Maleimide–polyethylene glycol–Succinimide (MPEGS) was obtained from Greative PEGworks (Chapel Hill, Illinois, USA). Four-week-old female SCID and ICR mice were purchased from the Shanghai Experimental Animal Center of the Chinese Academic of Sciences (Shanghai, People’s Republic of China) and housed in specific-pathogen-free conditions. All the experiments on live mice were approved by the Committee on Animals of the Second Military Medical University (Shanghai, People’s Republic of China) and all the methods were carried out in accordance with the approved guidelines.
Preparation of PPRT nanocomb
After RTX and Tos were thiolated by dithiothreitol as previously described,38,43 the PEI polymer was dissolved in phosphate-buffered saline (PBS) (1 mg/mL). The MPEGS linker was added to the polymer solution under stirring and N2 bubbling at room temperature for 4 hours (PEI:MPEGS =1:15). Then, equal amounts of RTX–SH and Tos–SH were slowly dropped into the MPEGS–PEI suspension (mAb/PEI =1,000:3.44). The reaction was conducted in an N2 environment for 6–8 hours. Unconjugated antibodies were separated by dialysis. The PPRT nanocomb was thus obtained, as shown in Figure 1A. The control sample PEI polymer–BSA (PPB) was constructed in the same way. Purified PPRT nanocomb was quantified by NanoVue™ (General Electric Company Healthcare, Cleveland, OH, USA) and analyzed with 8% sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS–PAGE) followed by Coomassie brilliant blue staining.
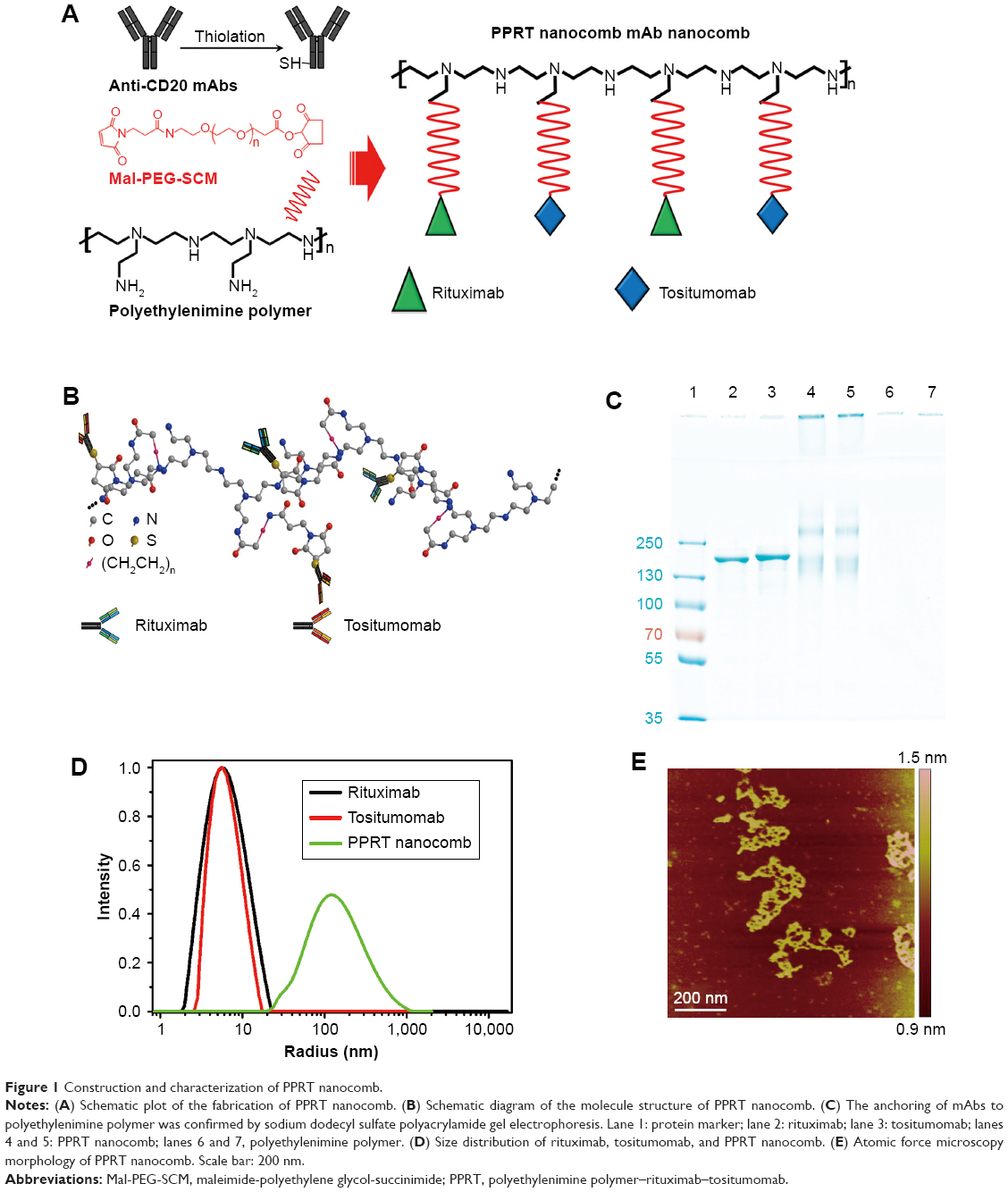 | Figure 1 Construction and characterization of PPRT nanocomb. |
Size and morphology characterization
RTX, Tos, and PPRT nanocomb were respectively diluted by Milli-Q water, and the size distribution was determined with a dynamic laser light scattering instrument (ALV/CGS3, ALV-Laser Vertriebsgesellschaft mbH, Langen, Germany) at a scattering angle of 30°.44 For morphology characterization, 10 μL PPRT nanocomb stocking suspension was spread onto freshly cleaved mica followed by gentle rinsing. The sample was air-dried and observed using atomic force microscopy (Veeco Instruments Inc, Woodbury, New York, US).
Weight-average molar mass analysis by static light scattering
The static light scattering measurements were carried out, varying the scattering angles (θ) from 40° to 140° with a 5° stepwise increase, with the dynamic laser light scattering instrument.45 The weight-average molar mass (Mm) was calculated by the following equation:46
| (1) |
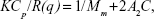
where 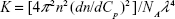 is optical contrast, with n being the refractive index of solvent, Cp being the PPRT nanocomb concentration, dn/dCp being the refractive index increment against Cp determined by a double-beam differential refraction meter (DMR-1021; Otsuka Electronics, Tokyo, Japan), λ being the incident wavelength, and NA being Avogadro’s number. R(q) is the Rayleigh ratio at a specific measurement angle. By measuring R(q) for various θ and Cp, values of Mm and A2 were estimated from typical Zimm plots.
is optical contrast, with n being the refractive index of solvent, Cp being the PPRT nanocomb concentration, dn/dCp being the refractive index increment against Cp determined by a double-beam differential refraction meter (DMR-1021; Otsuka Electronics, Tokyo, Japan), λ being the incident wavelength, and NA being Avogadro’s number. R(q) is the Rayleigh ratio at a specific measurement angle. By measuring R(q) for various θ and Cp, values of Mm and A2 were estimated from typical Zimm plots.
Labeling of free mAbs and PPRT nanocomb
The Alexa Fluor-488-labeled RTX (RTX-488) and Alexa Fluor-647-labeled Tos (Tos-647) were prepared using the Alexa Fluor-488/647 Protein Labeling Kit (Thermo Fisher Scientific) following the product information. Briefly, 50 μL of 1 M sodium bicarbonate (pH =9.0, provided with the labeling kit) was added to 0.5 mL of 2 mg/mL mAb solution. The resulting solution was then transferred to the vial of reactive dye (provided with the labeling kit) at room temperature with continuous stirring for 1 hour in the dark. The labeled mAbs were purified by the purification columns provided with the labeling kit. For the labeling of PPRT nanocomb, both RTX and Tos were respectively labeled with Alexa Fluor-488 and -647 before thiolation.
Confocal microscopy
For confocal microscopy, cells were placed onto poly-D-lysine (Sigma-Aldrich Co.)-coated microscope slides. Samples were fixed by 4% paraformaldehyde and permeabilized by 0.3% Triton X-100. After staining, samples were observed using a confocal microscope (Zeiss lsm 710; Carl Zeiss Meditec AG, Jena, Germany).
Binding activity
Raji cells were incubated with 10 μg/mL RTX-488, Tos-647, and Alexa Fluor-488/647-labeled PPRT nanocomb for 45 minutes at 4°C in the dark, respectively. After washing, cells were observed with a confocal microscope.
Off-rate assessment
Raji cells were incubated with 10 μg/mL RTX, Tos, or PPRT nanocomb for 1 hour. A humanized anti-human epidermal growth factor receptor-2 (Her-2) mAb, trastuzumab (Hoffman-La Roche Ltd.), was employed as an unspecific binding control. Then cells were washed and resuspended in mAb-free RPMI 1640 for different time intervals. After Alexa Fluor-488 labeled goat anti-human secondary antibody (GAH-488) staining, samples were analyzed by flow cytometry (FCM). The percentage of antibody remaining on cellular surface was calculated by the following equation:15,47
|
|
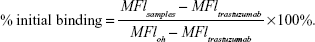
Where “MFIoh” is the mean fluorescence intensity (MFI) of cells after incubation with mAbs for 1 hour and labeled with GAH-488 without resuspension in antibody free culture medium.
Annexin V and propidium iodide staining
After a 16-hour treatment with 10 μg/mL RTX, Tos, or PPRT nanocomb, NHL cells were stained with Alexa Fluor-488 labeled Annexin V antibody, and propidium iodide (PI) and analyzed by two-channel FCM of FL-1 (annexin-V) and FL-2 (PI). Cells incubated with PPB and PPB + RTX + Tos were used as control treatments. For apoptosis inhibition assays, a cell-permeable pan-caspase inhibitor, Z-VAD-FMK (Promega Corporation, Fitchburg, WI, USA), was added 1 hour prior to the addition of therapeutic agents.
CDC and ADCC assays
NHL cells were incubated with 10 μg/mL anti-CD20 mAbs or PPRT nanocombs. For CDC assays, 5% (v/v) fresh human serum (donated by Dr Zheng Xu) was employed as a source of complement, while, for ADCC assays, human peripheral blood mononuclear cells (donated by Dr Cong Wu) were added as effector cells with an effector/target ratio of 25:1. After a 4-hour incubation, dead cells were assessed using CytoTox-Glo™ Cytotoxicity Assay kit (Promega Corporation) following the product information. Cells lysed by the lysis reagent (digitonin) offered in the assay kit were used as positive controls.
Lysosomal permeability
PPRT nanocomb- or mAb (10 μg/mL)-treated Raji cells were labeled with 200 nM Lyso-Tracker Red DND (Thermo Fisher Scientific) at 37°C for 10 minutes in the dark. Then FL-2 fluorescence (red) of labeled cells was assessed by FCM and confocal microscopy. Unlabeled cells were used as background controls.25,30
Mitochondrial membrane potentials and caspase activation assays
After a 16-hour treatment with 10 μg/mL RTX, Tos, and PPRT nanocomb, a JC-1 probe (Beyotime Biotechnology, Shanghai, People’s Republic of China) and the Vybrant® FAM Poly Caspases Assay Kit (Thermo Fisher Scientific) were employed to detect mitochondrial depolarization and caspase activation by FCM following the product information, respectively.
Homotypic adhesion determination
Raji cells were incubated with 2.5 μg/mL anti-CD20 mAbs or PPRT nanocombs for 8 hours and cell morphology was observed by light microscopy.
Pharmacokinetics
Three groups of three ICR mice were injected via the tail vein with 20 mg/kg RTX, Tos, and PPRT nanocomb, respectively, on day 0 and 1. After different time intervals, 40–60 μL venous blood was taken from the angular vein of the eyes. Serum concentrations of therapeutic antibodies were determined by enzyme-linked immunoassays.48,49 The pharmacokinetic parameters were analyzed by PKSolver software.50
Survival analysis
Five groups of ten eight-week-old female SCID mice were injected via the tail vein with 1×107 NHL (Ramos, Raji, and JeKo-1) cells. After a week, mice were randomly administered tail-vein injections of PBS, RTX, Tos, PPB + RTX + Tos, or PPRT nanocomb (with a total antibody amount of 15 mg/kg) every other day for five times. The mice were observed daily until natural death in a range of 120 days. All animals that survived were euthanatized at day 120.39,47
Treatment of established tumors in a localized human NHL xenotransplant model
Because neither Ramos nor Raji cells could form measurable subcutaneous tumors in SCID mice in our preliminary studies, only JeKo-1 cells were used in the localized model. Briefly, cells (1×107) were inoculated subcutaneously into the lateral flank of 8-week-old female SCID mice. When tumors reached about 0.8–1.0 cm in length, 20 mg/kg RTX, Tos, PPB + RTX + Tos, or PPRT nanocomb was respectively intravenously injected via tail vein weekly for three times.7,15,48 Tumor size was measured in two perpendicular diameters by a vernier caliper and estimated by the following function:38
| (3) |
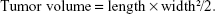
Statistical analysis
Statistical analysis was performed by Student’s t-test or one-way analysis of variance to identify significant differences unless otherwise indicated. Differences were considered significant at a P-value of <0.05.
Results
Characterization of PPRT nanocomb
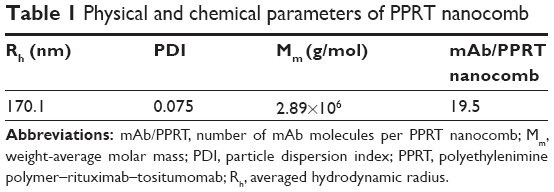
The characterization of PPRT nanocomb was firstly investigated after successful construction. The schematic diagram of PPRT nanocomb molecular structure is illustrated in Figure 1B. The anchoring of mAbs to PEI polymer was confirmed by SDS–PAGE. As illustrated in Figure 1C, recognizable stranded protein bands are observed near the sample wells of PPRT nanocomb (lanes 4 and 5) but not PEI polymer (lanes 6 and 7). By contrast, unmodified RTX (lane 2) and Tos (lane 3) both show clear protein bands at approximately 150 kDa. These results suggest that PPRT nanocomb was successfully prepared with a relatively high MW, which was further confirmed by dynamic light scattering (DLS) analysis (Figure 1D). As we can see, the mean radius of PPRT nanocomb was approximately 170 nm, compared with less than 7 nm for either free RTX or Tos. Also, the morphology of PPRT nanocomb was observed by atomic force microscopy, with the results displayed in Figure 1E.
More importantly, successful fabrication of PPRT nanocomb was validated by estimating antibody molecules per nanocomb according to the following equation:
|
|
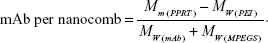
Here, Mm(PPRT) is the weight-average molar mass of PPRT nanocomb, which was estimated to be 2.89×106 g/mol by static light scattering analysis. The MW(PEI), MW(mAb), and MW(MPEGS) are, respectively, 70 kDa, 143.9 kDa, and 1 kDa according to their own product descriptions. Therefore, we can estimate that there are, on average, ~19.5 mAb molecules per nanocomb (Table 1).
 | Table 1 Physical and chemical parameters of PPRT nanocomb |
Cellular binding
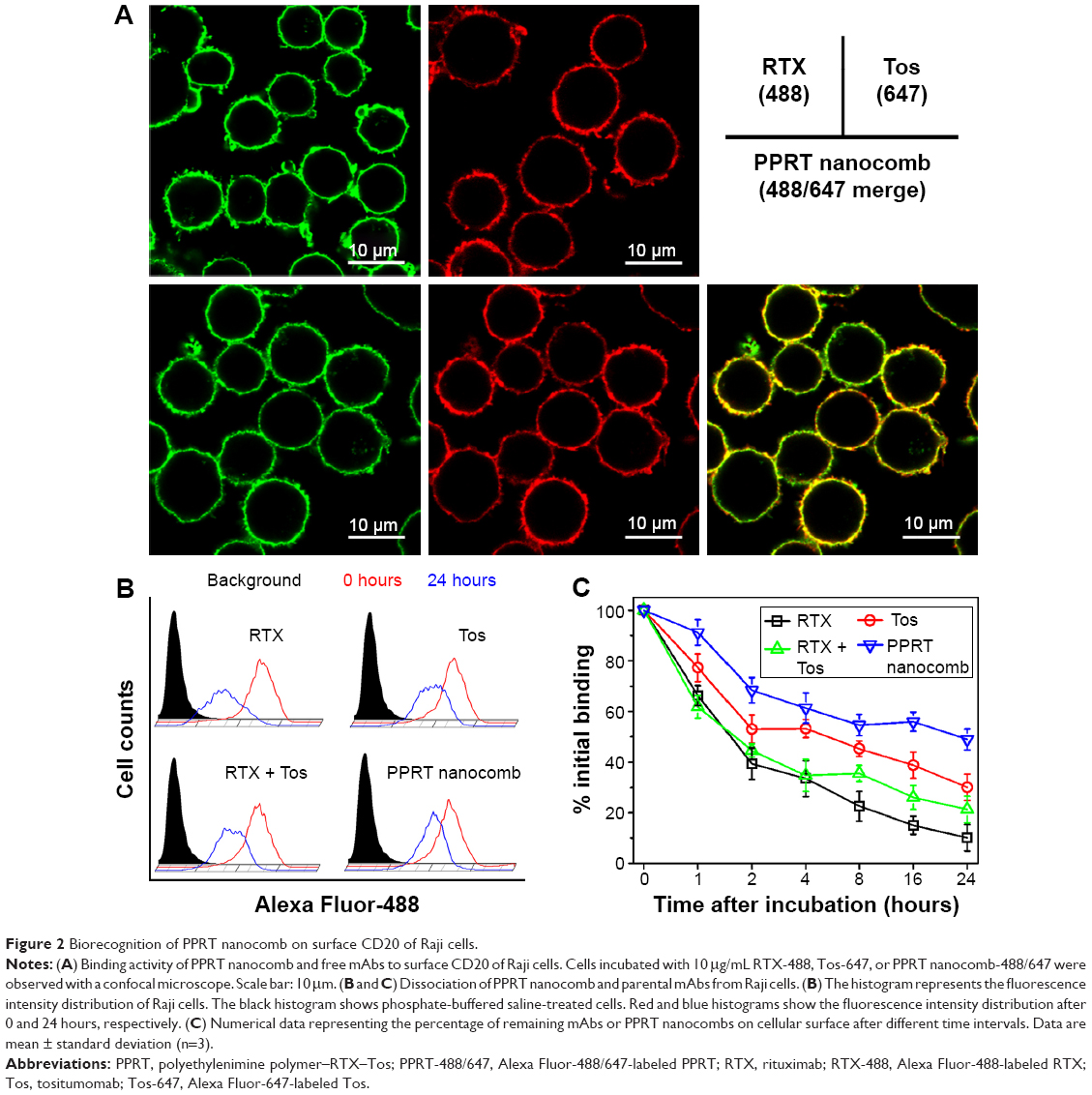
Figure 2A demonstrates that the exposure of Raji cells to RTX-488 or Tos-647 led to the decoration of cytomembranes with, respectively, green and red fluorescence, while the exposure to PPRT nanocomb led to both fluorescence decorations. The results suggest that the outstanding biorecognition between nanocomb and CD20 was not affected during the course of PPRT nanocomb preparation.
 | Figure 2 Biorecognition of PPRT nanocomb on surface CD20 of Raji cells. |
The binding “off-rate” experiments were performed by comparing the remaining ratio of PPRT nanocomb and free mAbs on cellular surface at different time intervals post-mAb incubation. As shown in Figure 2B and C, approximately 48.9%±4.1% of PPRT nanocomb remained on cellular surface after 24 hours, compared with 10.2%±2.2% of RTX (P<0.01) and 30.1%±5.2% of Tos (P<0.01), as well as 21.3%±5.2% of RTX + Tos (P<0.01). These results indicate that PPRT nanocomb possesses a reduced “off-rate” when compared with liberal mAbs.
In vitro tumor-killing ability
Figure 3A and B reveal that RTX (type I) but not Tos (type II) exhibits potent ability to mediate CDC. Just as per our expectation, the abilities of PPRT nanocomb to mediate both CDC and ADCC are not affected when compared with parental mAbs (RTX + Tos) or PPRT nanocomb components (PPB + RTX + Tos) (P>0.05). Figure 3C demonstrates that PPRT nanocomb can elicit a significantly higher level of PCD (characterized by annexin V+ subsets) than that induced by combination treatment with PPRT nanocomb components (PPB + RTX + Tos, P<0.01) in all the three NHL cell lines. Because PPB control treatment was unable to induce significant cell death, this excellent PCD-inducing ability has nothing to do with the PEI polymer.
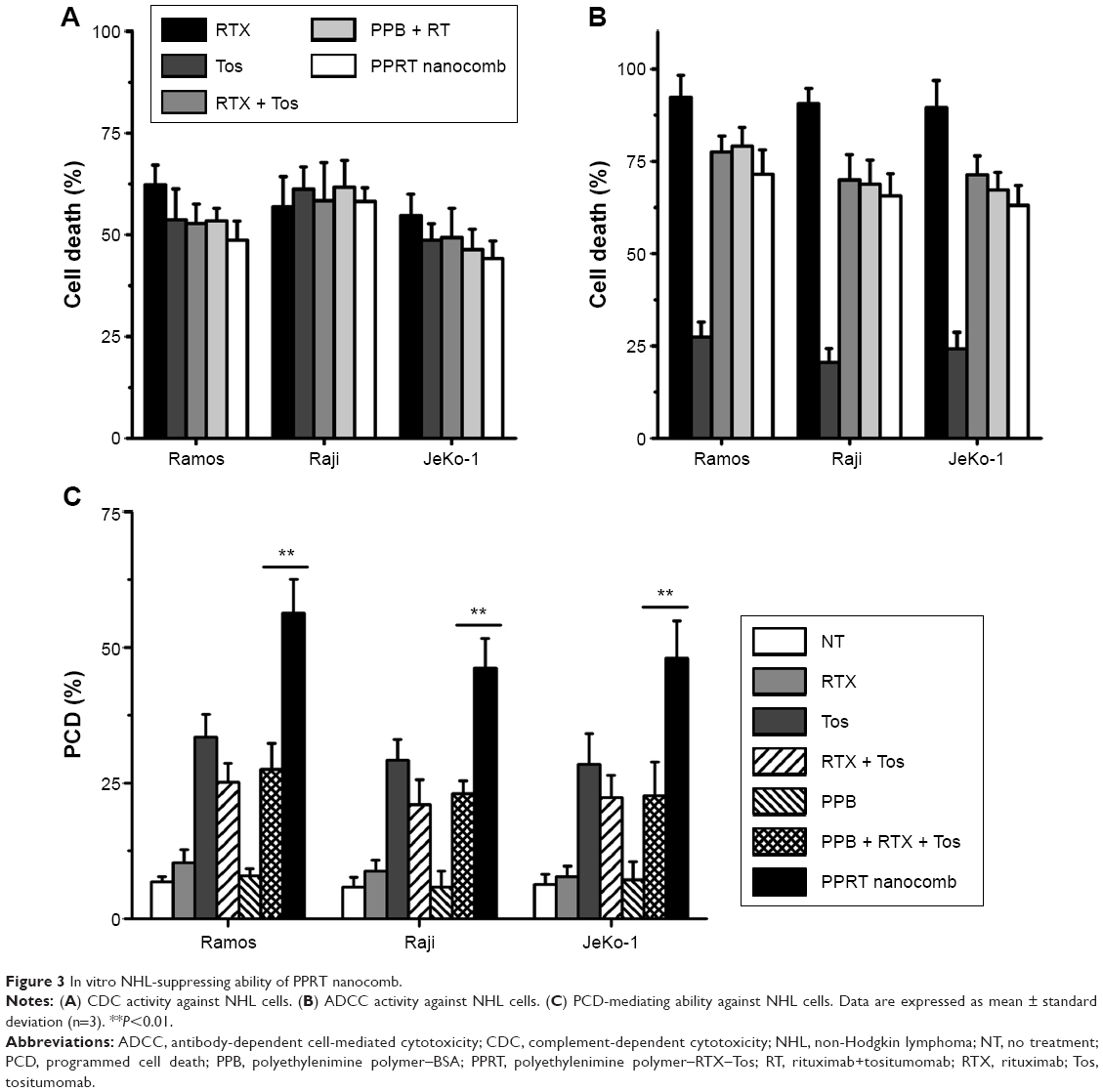 | Figure 3 In vitro NHL-suppressing ability of PPRT nanocomb. |
Involvement of lysosomes in PPRT nanocomb-induced PCD
Previous studies revealed that lysosomes play an important role in type II mAb (Tos)-induced PCD in leukemia and lymphoma cells.30,31 In order to characterize the lysosome involvement in PPRT nanocomb-evoked cell death, a Lyso-tracker was employed in our further experiments. Figure 4A reveals that the cellular fluorescence intentsity of FL-2 (lysosome tracker) subjected to visible alteration after the treatment with Tos (Figure 4Aa) or PPRT nanocombs (Figure 4Ab). Figure 4B indicates that the coefficient of variation of Lyso-tracker fluorescence (FL-2) increased from 28.1±2.5 to, respectively, 75.4±2.77 and 72.6±3.04 after the treatment with Tos and PPRT nanocomb, the results of which better describe the abovementioned fluorescence shift. Confocal microscopy images (Figure 4C) clearly explain this specific fluorescence shift. As we can see, in normal cells, cellular lysosomes were labeled as relatively small and confined organelles, while, after Tos treatment, an enlargement of red fluorescence-labeled compartments and diffusion of red fluorescence in the cytoplasm were clearly detected. Considering previous studies, we believe the Tos-treated cells were successively experiencing a swelling of lysosomes (enlargement of red compartments) and collapse of this compartment (diffusion of red fluorescence throughout the cytoplasm).25,30,31 Because Tos is one of the major components of PPRT nanocomb, similar lysosomal alteration was also observed in PPRT nanocomb-treated cells (Figure 4C). For further confirmation of lysosomal compartment collapse, we performed immunofluorescence staining for cathepsin B (a lysosomal component). In accordance, a substantial increase of green fluorescence (cathepsin B) was found throughout the cytoplasm of PPRT nanocomb-treated cells (Figure 4D).
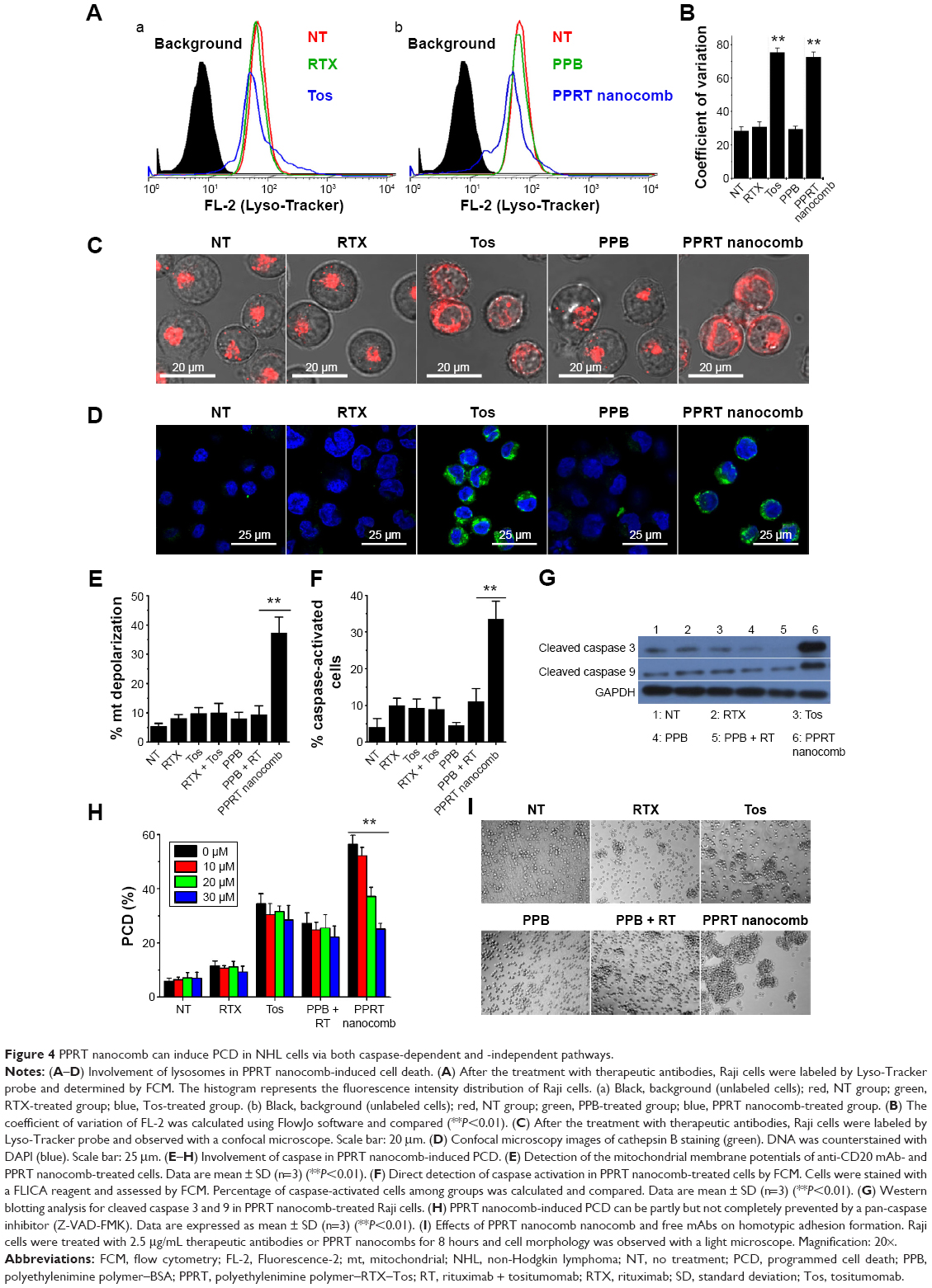 | Figure 4 PPRT nanocomb can induce PCD in NHL cells via both caspase-dependent and -independent pathways. |
Involvement of caspase in PPRT nanocomb-induced PCD
For determining the caspase involvement in PPRT nanocomb-induced PCD, we firstly detected the mitochondrial membrane potential of Raji cells by FCM post-JC-1 staining. Mitochondrial depolarization of cells was indicated by a decrease in FL-2 fluorescence intensity (JC-1 red). As shown in Figure 4E, cells treated with PPRT nanocomb experienced a significant mitochondrial depolarization compared with those treated with free mAbs or PPRT nanocomb components (PPB + RTX + Tos) (P<0.01). The caspase involvement was further confirmed by direct detection of caspase activation. As indicated in Figure 4F, cellular caspase can be efficiently activated by PPRT nanocomb but not the simple mixing of its components (PPB + RTX + Tos, P<0.01). Similar results were obtained by Western blotting analysis, with the results shown in Figure 4G. PCD inhibition results (Figure 4H) revealed that Z-VAD-FMK (a cell-permeable pan-caspase inhibitor) over a range of concentrations from 10 to 30 μM can hardly prevent free Tos- or RTX-induced PCD. However, PPRT nanocomb-evoked PCD can be significantly but not completely reduced in a dose-dependent manner (P<0.01).
PPRT nanocomb can evoke strong homotypic adhesion in targeting cells on account of cross-cell link
During our previous experiments, we accidentally found that PPRT nanocomb-treated cells experienced homotypic adhesion (HA) more obviously than free mAbs. For confirmation of this interesting phenomenon, Raji cells were incubated with 2.5 μg/mL anti-CD20 mAbs or PPRT nanocombs based on the results of our preliminary experiments, in which cells were incubated with different concentrations of therapeutic mAbs for various time intervals (data not shown). As displayed in Figure 4I, PPRT nanocomb exhibited the strongest HA-evoking ability among all the groups, which we consider as a potential contributor to the excellent PCD-inducing capability as discussed in the discussion section. Similar results were also observed in Ramos and JeKo-1 cells (Figure S1).
Pharmacokinetics
Table 2 demonstrates that the clearance (CL) of PPRT nanocomb from mouse peripheral blood was slower than that of free mAbs (CLPPRT: 13.31±3.12 L/h versus CLRTX: 12.3±0.9 L/h and CLTos: 7.71±0.99 L/h), with an elimination half-life (404.3±56.0 h) longer than that of unmodified RTX (257.3±28.7 h, P<0.01) and Tos (268.4±11.2 h, P<0.01). The reduced CL of PPRT nanocomb may contribute to increased circulation time in the blood vessels, resulting in long-lasting tumor-suppressing activity.
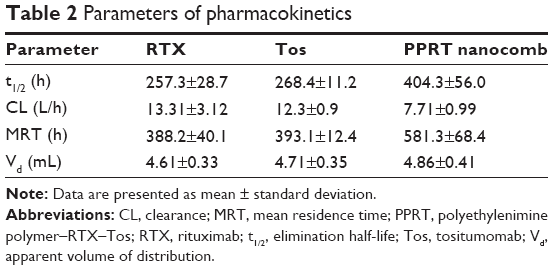 | Table 2 Parameters of pharmacokinetics |
PPRT nanocomb exhibits excellent NHL-suppressing ability in both disseminated and localized human NHL xenotransplant models
Before in vivo studies, the dose safety experiments were performed in healthy SCID mice. The results (shown in Figure S2) reveal that PPRT nanocomb demonstrated no toxicity to SCID with the therapeutic dose of 20 mg/kg in the following experiments. In the disseminated model, NHL-bearing mice were randomly administered tail-vein injections of PBS and therapeutic agents every other day for five times. The survival curves are shown in Figure 5A–C and the statistical results in Table S1. As we can see, RTX and Tos significantly prolonged the survival of NHL-bearing mice compared to PBS control (P<0.05). Similar results were seen with combination therapy of PPRT nanocomb components (PPB + RTX + Tos) (P<0.05) and were not statistically different compared to single injection of unmodified RTX and Tos. However, administration of PPRT nanocomb led to distinct prolongation of graft survival, with more than 50% of mice in complete remission (Ramos: 9/10, Raji: 7/10, JeKo-1: 6/10) indicated by long-term survival (>120 days post treatment).
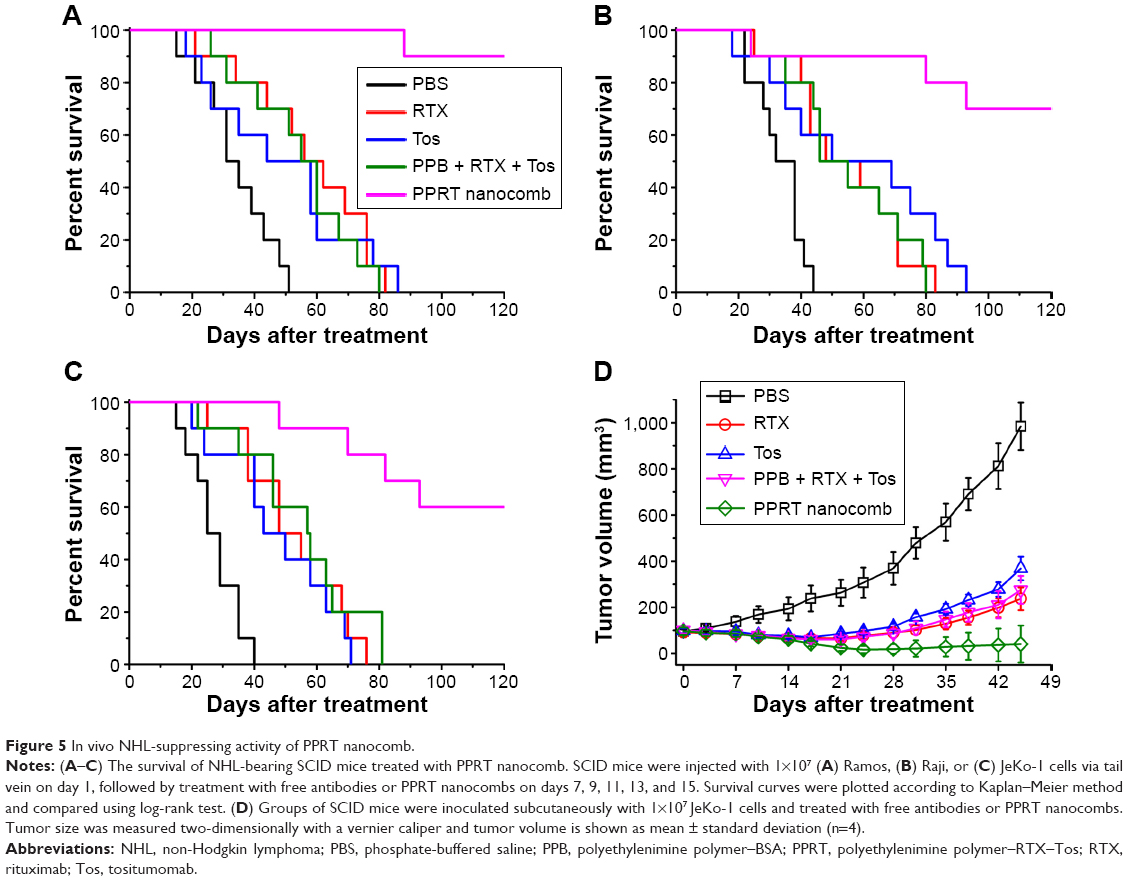 | Figure 5 In vivo NHL-suppressing activity of PPRT nanocomb. |
The outstanding NHL-suppressing activity was confirmed in a localized model. Figure 5D demonstrates that PPRT nanocomb-treated mice experienced a remarkable decrease in tumor burden compared with combination therapy of PPRT nanocomb components (PPB + RTX + Tos) as measured by tumor volume, with two out of four mice in complete remission characterized by no measurable mass, although immunotherapy by free antibodies can also induce a significant decrease of tumor burden compared with PBS control.
Discussion
The introduction of RTX for B-cell malignancies in 1997 inaugurated a new era of cancer therapy showcasing mAbs.1,3 Despite the uncontested success in the clinic, the application of RTX is still limited because of drug resistance, which is defined as a lack of response to an RTX-containing regimen, or progression within 6 months of treatment with an RTX-containing regimen.16 In the past decades, more and more novel anti-CD20 mAbs have been developed for improving the therapeutic efficacy against naïve and resistant NHLs.3,8,14 Presently, anti-CD20 mAbs can be grouped into two major categories (type I and type II) with different tumor-killing mechanisms, including CDC, ADCC, and direct induction of PCD.27 It is well established that cancer is usually multifactorial in nature, involving various redundant disease-mediating ligands and receptors, as well as crosstalk between signal cascades.51,52 Targeting only one or two pathways may not completely shut off a hallmark capability of cancer, allowing some malignant cells to survive until they or their progeny eventually adapt to the selective pressure imposed by the therapeutic agents.25,51–53 Therefore, activation of multiple tumor-suppression pathways may be an efficient way to improve therapeutic index and reduce drug resistance. However, all the current anti-CD20 mAbs effective in evoking PCD are relatively ineffective in activating complements, and vice versa. Although secondary antibody cross-linked anti-CD20 mAb can significantly induce caspase-dependent apoptosis in malignant B-cells, this cross-link can hardly be realized in vivo.28,29
In the present work, a novel mAb nanocomb (PPRT nanocomb) was successfully prepared by mass arming of two different anti-CD20 mAbs, RTX (type I) and Tos (type II), by nanotechnology. To our knowledge, this is the first study to report a mAb derivative consisting of two distinct antibodies. In vitro experimental results clearly prove that PPRT nanocomb can activate two distinct signal transduction pathways of PCD in targeting cells, including a lysosome-mediated pathway (induced by Tos) and a caspase-dependent pathway (induced by cross-link). Besides, because a great many mAbs tightly anchored to one nanocomb, PPRT nanocomb possesses the ability of binding to separate CD20 on neighboring cells, a phenomenon we termed as “cross-cell link”, which contributes to its outstanding HA-evoking ability. Previous studies proved that increased HA of cancer cells may cause increased susceptibility to apoptosis.54,55 Moreover, mAb-evoked HA of NHL cells can trigger specific intracellular alterations including lysosomal membrane permeabilization, mitochondrial depolarization, and phosphorylation or up/downregulation of proteins related to the apoptotic signal transduction pathways, culminating in PCD.11,15,24,26,30,31 Given previous publications and our findings, it would be reasonable to attribute the superior PCD-inducing ability of PPRT nanocomb partly to the “cross-cell link”-assisted HA. Further experimental results revealed that both CDC and ADCC can also be synchronously activated with the presence of complement and effector cells. As far as we know, no therapeutic agents except for our nanocomb could concurrently activate all the four known tumor-suppressing mechanisms of CD20-based immunotherapy (Figure 6) until now.
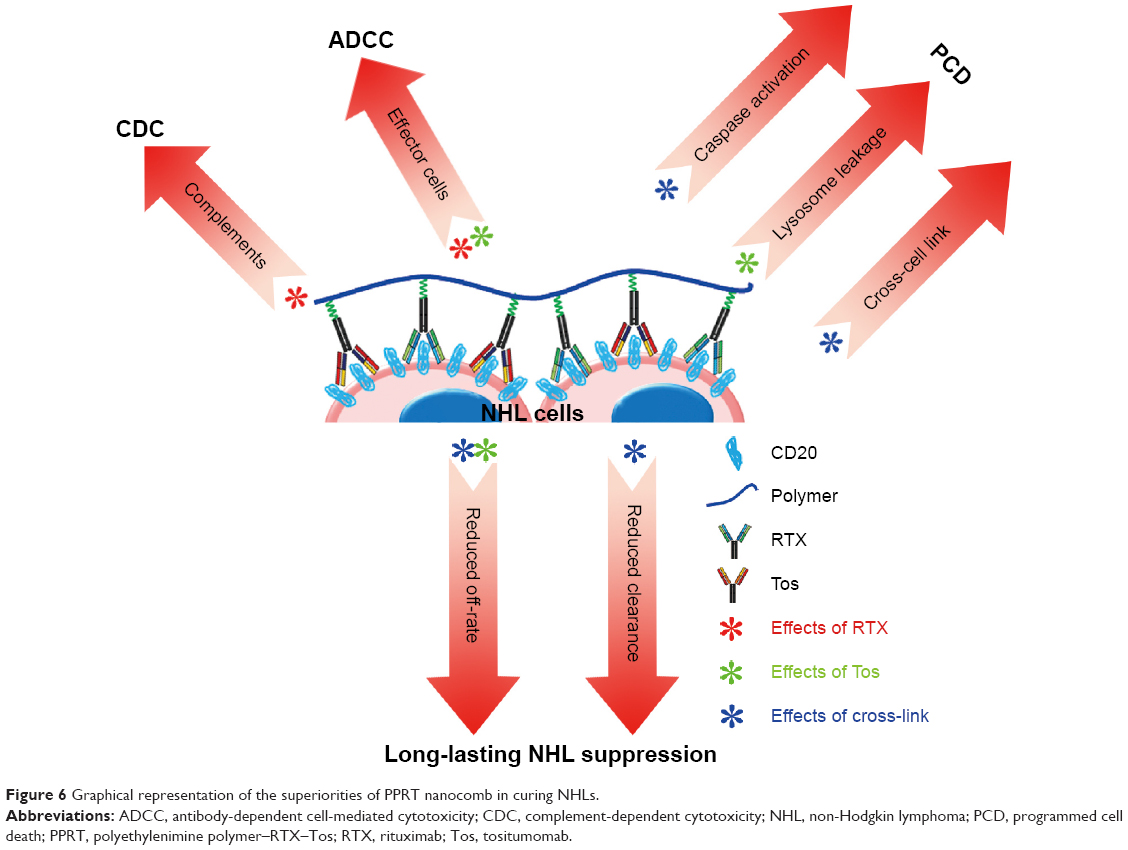 | Figure 6 Graphical representation of the superiorities of PPRT nanocomb in curing NHLs. |
Subsequent in vivo studies indicated that PPRT nanocomb was more effective than free parental antibodies for NHL inhibition in both disseminated and localized human NHL xenotransplant models. This exceptionally excellent NHL suppression ability may be due to the cooperation of the following mechanisms (Figure 6). Firstly, as discussed above, synchronous activation of all the known CD20-based immunotherapeutic mechanisms (including CDC, ADCC, caspase-dependent apoptosis, and lysosome-mediated cell death) make resistance to PPRT nanocomb therapy much more difficult. Secondly, “cross-cell link”-assisted HA further enhanced the susceptibility to PCD of lymphoma cells. Thirdly, reduced CL from peripheral blood, together with the decreased binding “off-rate” to surface CD20 on malignant B-cells, contributes to the durable and long-lasting anti-lymphoma ability of PPRT nanocomb. In conclusion, it should be appreciated that our PPRT nanocomb is more than just the sum of its parts and merits further evaluation as an NHL therapeutic in the clinic.
Acknowledgments
This work was financially supported by the National Natural Science Foundation of China (81171450) and the Youth Fund of the Second Military Medical University (2013QN14).
Disclosure
The authors report no conflicts of interest in this work.
References
Cheson BD, Leonard JP. Monoclonal antibody therapy for B-cell non-Hodgkin’s lymphoma. N Engl J Med. 2008;359(6):613–626. | ||
Shankland KR, Armitage JO, Hancock BW. Non-Hodgkin lymphoma. Lancet. 2012;380(9844):848–857. | ||
Lim SH, Levy R. Translational medicine in action: anti-CD20 therapy in lymphoma. J Immunol. 2014;193(4):1519–1524. | ||
Fisher RI, Gaynor ER, Dahlberg S, et al. Comparison of a standard regimen (CHOP) with three intensive chemotherapy regimens for advanced non-Hodgkin’s lymphoma. N Engl J Med. 1993;328(14): 1002–1006. | ||
Narayan VA, Mohwinckel M, Pisano G, Yang M, Manji HK. Beyond magic bullets: true innovation in health care. Nat Rev Drug Discov. 2013;12(2):85–86. | ||
Riether C, Schürch C, Ochsenbein AF. From “magic bullets” to specific cancer immunotherapy. Swiss Med Wkly. 2013;143:w13734. | ||
Chao MP, Alizadeh AA, Tang C, et al. Anti-CD47 antibody synergizes with rituximab to promote phagocytosis and eradicate non-Hodgkin lymphoma. Cell. 2010;142(5):699–713. | ||
Taylor RP, Lindorfer MA, Zent CS. Anti-CD20 antibody therapy for B-cell lymphomas. N Engl J Med. 2012;367(9):876–877; author reply 878. | ||
Tedder TF, Streuli M, Schlossman SF, Saito H. Isolation and structure of a cDNA encoding the B1 (CD20) cell-surface antigen of human B lymphocytes. Proc Natl Acad Sci U S A. 1988;85(1):208–212. | ||
Anderson KC, Bates MP, Slaughenhoupt BL, Pinkus GS, Schlossman SF, Nadler LM. Expression of human B cell-associated antigens on leukemias and lymphomas: a model of human B cell differentiation. Blood. 1984;63(6):1424–1433. | ||
Gupta P, Goldenberg DM, Rossi EA, et al. Dual-targeting immunotherapy of lymphoma: potent cytotoxicity of anti-CD20/CD74 bispecific antibodies in mantle cell and other lymphomas. Blood. 2012;119(16):3767–3778. | ||
Press OW, Appelbaum F, Ledbetter JA, et al. Monoclonal antibody 1F5 (anti-CD20) serotherapy of human B cell lymphomas. Blood. 1987;69(2):584–591. | ||
Davis TA, Grillo-López AJ, White CA, et al. Rituximab anti-CD20 monoclonal antibody therapy in non-Hodgkin’s lymphoma: safety and efficacy of re-treatment. J Clin Oncol. 2000;18(17):3135–3143. | ||
Ujjani C, Cheson BD. The current status and future impact of targeted therapies in non-Hodgkin lymphoma. Expert Rev Hematol. 2013;6(2):191–202; quiz 203. | ||
Zhao L, Tong Q, Qian W, et al. Eradication of non-Hodgkin lymphoma through the induction of tumor-specific T-cell immunity by CD20-Flex BiFP. Blood. 2013;122(26):4230–4236. | ||
Rezvani AR, Maloney DG. Rituximab resistance. Best Pract Res Clin Haematol. 2011;24(2):203–216. | ||
Taylor RP, Lindorfer MA. Immunotherapeutic mechanisms of anti-CD20 monoclonal antibodies. Curr Opin Immunol. 2008;20(4):444–449. | ||
Klepfish A, Schattner A, Ghoti H, Rachmilewitz EA. Addition of fresh frozen plasma as a source of complement to rituximab in advanced chronic lymphocytic leukaemia. Lancet Oncol. 2007;8(4):361–362. | ||
Bhat R, Watzl C. Serial killing of tumor cells by human natural killer cells – enhancement by therapeutic antibodies. PLoS One. 2007;2(3):e326. | ||
Alduaij W, Ivanov A, Honeychurch J, et al. Novel type II anti-CD20 monoclonal antibody (GA101) evokes homotypic adhesion and actin-dependent, lysosome-mediated cell death in B-cell malignancies. Blood. 2011;117(17):4519–4529. | ||
Herting F, Friess T, Bader S, et al. Enhanced anti-tumor activity of the glycoengineered type II CD20 antibody obinutuzumab (GA101) in combination with chemotherapy in xenograft models of human lymphoma. Leuk Lymphoma. 2014;55(9):2151–5160. | ||
Trinh KR, Vasuthasawat A, Steward KK, Yamada RE, Timmerman JM, Morrison SL. Anti-CD20-interferon-β fusion protein therapy of murine B-cell lymphomas. J Immunother. 2013;36(5):305–318. | ||
Beers SA, Chan CH, James S, et al. Type II (tositumomab) anti-CD20 monoclonal antibody out performs type I (rituximab-like) reagents in B-cell depletion regardless of complement activation. Blood. 2008;112(10):4170–4177. | ||
Mössner E, Brünker P, Moser S, et al. Increasing the efficacy of CD20 antibody therapy through the engineering of a new type II anti-CD20 antibody with enhanced direct and immune effector cell-mediated B-cell cytotoxicity. Blood. 2010;115(22):4393–4402. | ||
Zhao L, Xie F, Tong X, et al. Combating non-Hodgkin lymphoma by targeting both CD20 and HLA-DR through CD20-243 CrossMab. MAbs. 2014;6(3):740–748. | ||
Cragg MS, Walshe CA, Ivanov AO, Glennie MJ. The biology of CD20 and its potential as a target for mAb therapy. Curr Dir Autoimmun. 2005;8:140–174. | ||
Glennie MJ, French RR, Cragg MS, Taylor RP. Mechanisms of killing by anti-CD20 monoclonal antibodies. Mol Immunol. 2007;44(16):3823–3837. | ||
Riley JK, Sliwkowski MX. CD20: a gene in search of a function. Semin Oncol. 2000;27(6 Suppl 12):17–24. | ||
Hofmeister JK, Cooney D, Coggeshall KM. Clustered CD20 induced apoptosis: src-family kinase, the proximal regulator of tyrosine phosphorylation, calcium influx, and caspase 3-dependent apoptosis. Blood Cells Mol Dis. 2000;26(2):133–143. | ||
Ivanov A, Beers SA, Walshe CA, et al. Monoclonal antibodies directed to CD20 and HLA-DR can elicit homotypic adhesion followed by lysosome-mediated cell death in human lymphoma and leukemia cells. J Clin Invest. 2009;119(8):2143–2159. | ||
Grønbaek K, Jäättelä M. Engaging the lysosomal compartment to combat B cell malignancies. J Clin Invest. 2009;119(8):2133–2136. | ||
Jak M, van Bochove GG, van Lier RA, Eldering E, van Oers MH. CD40 stimulation sensitizes CLL cells to rituximab-induced cell death. Leukemia. 2011;25(6):968–978. | ||
Yang X, Zhang X, Fu ML, et al. Targeting the tumor microenvironment with interferon-β bridges innate and adaptive immune responses. Cancer Cell. 2014;25(1):37–48. | ||
Markman JL, Rekechenetskiy A, Holler E, Ljubimova JY. Nanomedicine therapeutic approaches to overcome cancer drug resistance. Adv Drug Deliv Rev. 2013;65(13–14):1866–1879. | ||
Seymour LW. Passive tumor targeting of soluble macromolecules and drug conjugates. Crit Rev Ther Drug Carrier Syst. 1992;9(2):135–187. | ||
Maeda H. Macromolecular therapeutics in cancer treatment: the EPR effect and beyond. J Control Release. 2012;164(2):138–144. | ||
Iyer AK, Khaled G, Fang J, Maeda H. Exploiting the enhanced permeability and retention effect for tumor targeting. Drug Discov Today. 2006;11(17–18):812–818. | ||
Li W, Zhao H, Qian W, et al. Chemotherapy for gastric cancer by finely tailoring anti-Her2 anchored dual targeting immunomicelles. Biomaterials. 2012;33(21):5349–5362. | ||
Wu K, Yang J, Liu J, Kopeček J. Coiled-coil based drug-free macromolecular therapeutics: in vivo efficacy. J Control Release. 2012;157(1):126–131. | ||
Wu K, Liu J, Johnson RN, Yang J, Kopecek J. Drug-free macromolecular therapeutics: induction of apoptosis by coiled-coil-mediated cross-linking of antigens on the cell surface. Angew Chem Int Ed Engl. 2010;49(8):1451–1455. | ||
Chu TW, Yang J, Kopeček J. Anti-CD20 multivalent HPMA copolymer-Fab’ conjugates for the direct induction of apoptosis. Biomaterials. 2012;33(29):7174–7181. | ||
Aluri SR, Shi P, Gustafson JA, et al. A hybrid protein-polymer nanoworm potentiates apoptosis better than a monoclonal antibody. ACS Nano. 2014;8(3):2064–2076. | ||
Huwyler J, Wu D, Pardridge WM. Brain drug delivery of small molecules using immunoliposomes. Proc Natl Acad Sci U S A. 1996;93(24):14164–14169. | ||
MacKay JA, Chen M, McDaniel JR, Liu W, Simnick AJ, Chilkoti A. Self-assembling chimeric polypeptide-doxorubicin conjugate nanoparticles that abolish tumours after a single injection. Nat Mater. 2009;8(12):993–999. | ||
Giacomelli C, Le Men L, Borsali R, et al. Phosphorylcholine-based pH-responsive diblock copolymer micelles as drug delivery vehicles: light scattering, electron microscopy, and fluorescence experiments. Biomacromolecules. 2006;7(3):817–828. | ||
Yoshimura T, Esumi K. Physicochemical properties of anionic triple-chain surfactants in alkaline solutions. J Colloid Interface Sci. 2004;276(2):450–455. | ||
Li B, Zhao L, Guo H, et al. Characterization of a rituximab variant with potent antitumor activity against rituximab-resistant B-cell lymphoma. Blood. 2009;114(24):5007–5015. | ||
Regazzi MB, Iacona I, Avanzini MA, et al. Pharmacokinetic behavior of rituximab: a study of different schedules of administration for heterogeneous clinical settings. Ther Drug Monit. 2005;27(6):785–792. | ||
Daydé D, Ternant D, Ohresser M, et al. Tumor burden influences exposure and response to rituximab: pharmacokinetic-pharmacodynamic modeling using a syngeneic bioluminescent murine model expressing human CD20. Blood. 2009;113(16):3765–3772. | ||
Zhang Y, Huo M, Zhou J, Xie S. PKSolver: an add-in program for pharmacokinetic and pharmacodynamic data analysis in Microsoft Excel. Comput Methods Programs Biomed. 2010;99(3):306–314. | ||
Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144(5):646–674. | ||
Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000;100(1):57–70. | ||
Wu C, Ying H, Grinnell C, et al. Simultaneous targeting of multiple disease mediators by a dual-variable-domain immunoglobulin. Nat Biotechnol. 2007;25(11):1290–1297. | ||
Fu Q, Cash SE, Andersen JJ, et al. CD43 in the nucleus and cytoplasm of lung cancer is a potential therapeutic target. Int J Cancer. 2013;132(8):1761–1770. | ||
Velentzas AD, Nezis IP, Stravopodis DJ, Papassideri IS, Margaritis LH. Mechanisms of programmed cell death during oogenesis in Drosophila virilis. Cell Tissue Res. 2007;327(2):399–414. |
Supplementary materials
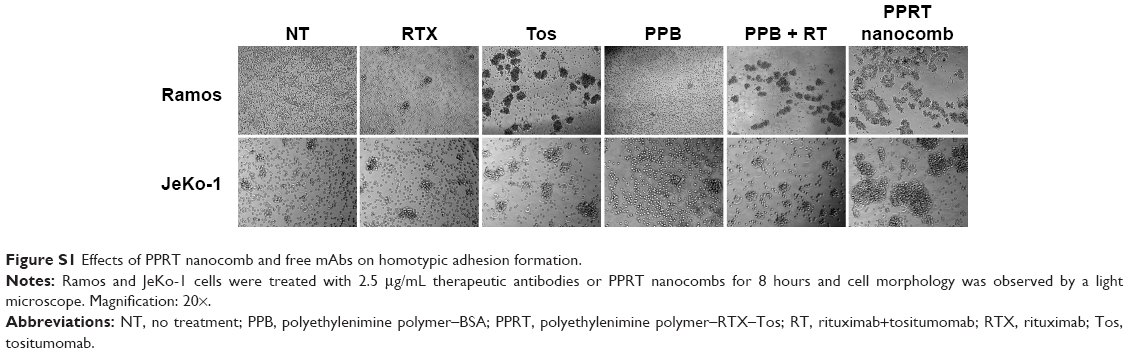 | Figure S1 Effects of PPRT nanocomb and free mAbs on homotypic adhesion formation. |
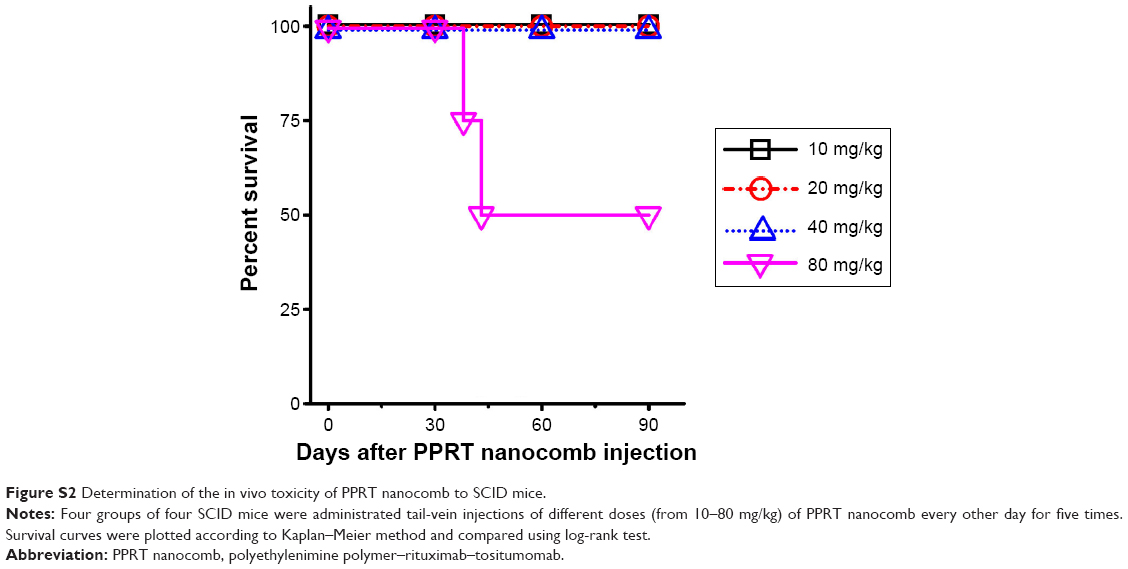 | Figure S2 Determination of the in vivo toxicity of PPRT nanocomb to SCID mice. |
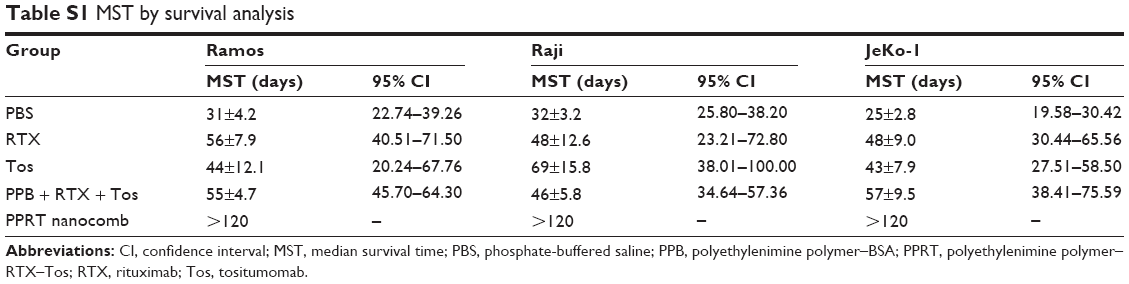 | Table S1 MST by survival analysis |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.