")
Back to Journals » Cancer Management and Research » Volume 11
Connexin 32 downregulation is critical for chemoresistance in oxaliplatin-resistant HCC cells associated with EMT
Authors Yang Y , Yao JH , Du QY, Zhou YC , Yao TJ, Wu Q, Liu J , Ou YR
Received 31 January 2019
Accepted for publication 16 April 2019
Published 31 May 2019 Volume 2019:11 Pages 5133—5146
DOI https://doi.org/10.2147/CMAR.S203656
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Chien-Feng Li
Yan Yang,1 Jing-Hao Yao,1 Qian-Yu Du,1 Yong-Chun Zhou,2 Ting-Jing Yao,3 Qiong Wu,4 Jing Liu,1 Yu-Rong Ou4
1Department of Medical Oncology, The First Affiliated Hospital of Bengbu Medical College, Bengbu 233004, People’s Republic of China; 2Department of Radiation Oncology, The First Affiliated Hospital of Bengbu Medical College, Bengbu 233004, People’s Republic of China; 3Department of Surgical Oncology, The First Affiliated Hospital of Bengbu Medical College, Bengbu 233004, People’s Republic of China; 4Department of Pathology, The First Affiliated Hospital of Bengbu Medical College, Bengbu 233004, People’s Republic of China
Background: Oxaliplatin (OXA)-based chemotherapy is critical in the management of advanced hepatocellular carcinoma (HCC); however, acquired drug resistance has largely restricted its clinical efficacy. This study aims to explore the key mechanisms and regulatory factors determining chemosensitivity in HCC.
Methods: We developed OXA-resistant (OR) HCC cells and used multiple methods, including real-time RT-PCR, Western blot, immunofluorescence, transwell invasion assay, wound-healing assay, MTT assay, gene transfection, and immunohistochemistry to achieve our goals.
Results: We found that OR HCC cells showed a typical epithelial–mesenchymal transition (EMT) phenotype. Meanwhile, the expression of Cx32, a major member of the liver connexin (Cx) family, was lowly expressed in OR HCC cells. Downregulation of Cx32 in parental HCC cells led to EMT induction and thereby reduced OXA cytotoxicity, while Cx32 upregulation in OR HCC cells could reverse the EMT phenotype and partially restore chemosensitivity to OXA. Finally, in human HCC tissue samples, Cx32 was positively correlated with the expression of the EMT marker E-cadherin and negatively correlated with the expression of Vimentin.
Conclusion: Our findings demonstrated that downregulation of Cx32 may be an important determinant for HCC cells to acquire EMT-related acquired drug resistance to OXA, and targeting Cx32 could be a novel strategy to overcome OXA resistance in HCC.
Keywords: hepatocellular carcinoma, oxaliplatin chemoresistance, connexin32, epithelial-mesenchymal transition
Introduction
Hepatocellular carcinoma (HCC) is the fourth common malignancy worldwide,1 with characteristics of high aggressivity and poor prognosis. Hepatocarcinogenesis is concealed, and early diagnosis of HCC is difficult. Most HCC patients cannot undergo curative surgery due to locally extensive invasion or distant metastasis at the time of diagnosis. In recent years, targeted therapy and immunotherapy have improved the prognosis of patients with advanced HCC.2 However, their clinical benefits remain moderate, especially with the lack of effective predictive indicators, resulting in unsatisfactory efficacy in clinical practice. With the application of third-generation cytotoxic drugs, systemic chemotherapy is making a breakthrough in the treatment of advanced HCC. Specifically, oxaliplatin (OXA)-based systemic chemotherapy has become one of the most important options for advanced HCC.3,4 However, patients using platinum drugs often suffer from drug resistance during the clinical application,5 and the mechanisms underlying drug resistance remain largely unknown.
Epithelial–mesenchymal transition (EMT) refers to the process by which epithelial cells switch to mesenchymal cells through specific processes and is associated with chemoresistance, including to platinum drugs.6 Studies have revealed that cancer cells acquire the mesenchymal phenotype in the process of generating acquired drug resistance, and tumor cells in the mesenchymal differentiation state often exhibit the characteristics of primary drug resistance.7 Our previous study confirmed phenotypic changes consistent with the EMT phenotype in gemcitabine (GEM)-resistant HCC cells.8 It is generally recognized that during EMT, epithelial cells lose epithelial properties and acquire the characteristics of mesenchymal cells, resulting in decreased intercellular adhesion and increased cell motility and invasion capacities. At the molecular level, cells lowly express or lose epithelial markers such as E-cadherin and highly express mesenchymal molecular markers, including Vimentin and N-cadherin, with upregulation of transcription factors such as Snail, Slug, and Twist.9
Connexin (Cx) is the basic unit of gap junction (GJ), which communicates the cytoplasm of two adjacent cells by directly mediating the transmission of electrical, chemical, and metabolic substances, thereby playing important roles in a series of life events, including cellular activity synchronization, tissue homeostasis maintenance, and cell proliferation and apoptosis.10 Abnormal Cx and GJ levels are closely related to the occurrence and development of various tumors, including HCC.11 Cx32, forming 90% of hepatic GJs, is the major Cx isoform expressed in the liver.12 This gene was originally considered a tumor-suppressor gene13,14 and has been demonstrated to be implicated in multiple hepatocellular processes such as carcinogenesis, cell proliferation, apoptosis, invasion, and metastasis.15–18 More importantly, Cx32 can affect sensitivity to many chemotherapeutic drugs, including platinum drugs.19,20
In the current study, we successfully established three OXA-resistant (OR) HCC cell lines and found that they all acquired EMT characteristics. Meanwhile, among the three Cxs (Cx26, Cx32, and Cx43) expressed in the liver, Cx32 was the most remarkably downregulated protein during the process of drug resistance. Downregulation of Cx32 in parental HCC cells induced EMT and decreased OXA cytotoxicity, while overexpression of Cx32 in OR HCC cells reversed the mesenchymal phenotype and partially restored sensitivity to OXA. Finally, we further confirmed the associations of Cx32 with the EMT markers E-cadherin and Vimentin in human HCC tissue samples. These results indicated that downregulation of Cx32 is involved in OXA-resistance and EMT features of HCC cells, demonstrating that targeting Cx32 may provide a new strategy for overcoming HCC resistance to OXA.
Material and methods
Cell lines and culture
The human HCC HepG2, Huh7, and SMMC-7721 cell lines were purchased from the cell bank of the Shanghai Institutes for Biological Sciences (Shanghai, China) and cultured at 37°C in 5% CO2 in Dulbecco’s modified Eagle’s medium (DMEM, Invitrogen, Carlsbad, CA, USA) supplemented with 10% FBS (HyClone Laboratories, Inc., Logan, UT, USA), 100 U/mL penicillin (Gibco, Waltham, MA, USA), and 100 μg/mL streptomycin (Gibco).
Establishment of OR HCC cell lines
To develop HCC cell lines chronically resistant to OXA, the human HCC cell lines HepG2, Huh7, and SMMC-7721 were continuously exposed to increasing doses of OXA for at least 6 months. A small dose of OXA was used as the starting point, to ensure that surviving cells can exceed 70% confluency. After the resultant cells grew stably in the medium containing OXA, they were passaged by trypsinization and exposed to a higher concentration of OXA. The process was repeated until the resultant cells could grow stably in the medium with the indicated OXA concentration. By using this concentration increment method,21 three OR HCC cell lines displaying resistance to the growth inhibitory properties of OXA at the indicated concentrations were established.
Cell growth assessment by the MTT assay
Parental and OR HCC cells [(8–10)×103/well] were seeded at equal densities into 96-well culture plates for overnight incubation. OXA at different concentrations was then added for 24 hrs. MTT assay was performed as described previously.22
Western blot and qRT-PCR
Western blot, RNA isolation, and qRT-PCR assay were described in our previous studies.18,22 Primary antibodies and dilutions used for Western blot were: P-gp (1:500; Proteintech Group, Inc., Rosemont, IL, USA); breast cancer resistance protein (BCRP) (1:1,000; CST, Danvers, MA, USA); E-cadherin (1:1,000; Abcam, Cambridge, MA, USA); Vimentin (1:1,000; Abcam); Snail (1:1,000; Abcam); Slug (1:2,000; Abcam); Cx26 (1:500; Invitrogen); Cx32 (1:500; Sigma-Aldrich, St. Louis, MO, USA); Cx43 (1:2,000; Sigma-Aldrich); and β-actin (1:500; Santa Cruz Biotechnology, Santa Cruz, CA, USA). The primers used for qRT-PCR are listed in Table 1. Gene and protein expression levels were normalized to those of the internal controls (GAPDH or β-actin).
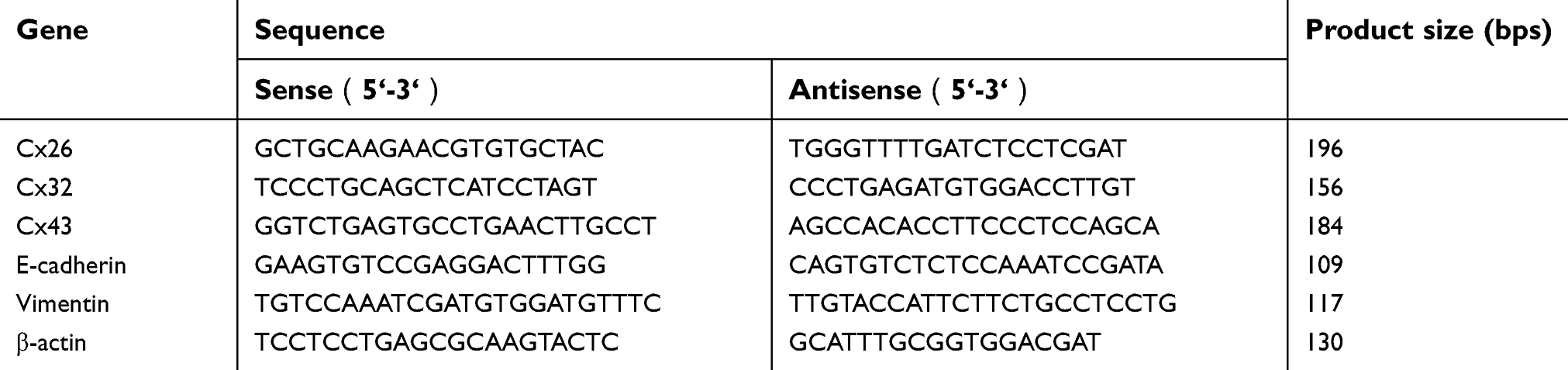 | Table 1 The primers used for qRT-PCR analysis |
Immunofluorescence
Immunofluorescence was performed as described previously.18,22 Primary antibodies targeting Cx32, E-cadherin, and Vimentin were used at 1:200 in 2% BSA. Alexa 488- or 568-conjugated secondary antibodies were added for 1 hr in the dark at room temperature. The cells were examined and photographed under a fluorescence microscope (Olympus, Tokyo, Japan).
Expression plasmids and gene silencing
The pEX-2 plasmid containing the full-length cDNA of human Cx32 (Gene ID: 2705) was purchased from Suzhou GenePharma Co., Ltd. (Suzhou, China). The empty plasmid pEX-2 was used as a negative control (NC). Cx32 siRNA fragments were also synthesized and supplied by GenePharma. The specific siRNA sequences for Cx32 and NC were described in our previous report.18 The transfection reagent was Lipofectamine™ 2000 (Invitrogen), and experiments were conducted strictly according to the manufacturer’s instructions.
Transwell invasion and wound healing assays
Transwell invasion and wound-healing assays to determine the invasion and migration capabilities of HCC cells were performed as described previously.8,18
Immunohistochemistry
A total of 34 paired samples of human HCC and matched adjacent non-cancerous tissues were collected as previously described.18 Anti-Cx32 (1:100, Sigma-Aldrich), anti-E-cadherin (1:50, Abcam), and anti-Vimentin (1:100, Abcam) primary antibodies were used for immunohistochemistry according to a two-step protocol. The intensity of positive signals was scored as: 0, negative (no staining); 1, weak (light yellow); 2, moderate (yellowish brown); 3, strong (brown). The extent of positivity was scored based on the percentage of positive cells: 0, <5%; 1, 5~25%; 2, 26~50%; 3, >50%. The staining index (SI) was determined as the final score by multiplying the above subscores and classified as negative (score 0–1), 1+ (score 2–3), 2+ (score 4–5), or 3+ (score ≥6). The study methodologies conformed to the standards set by the Declaration of Helsinki and were approved by the Ethics Committee of our institution. Written informed consent was obtained from the patients or their immediate family members.
Statistical analysis
All statistical analyses were performed with the SPSS version 19.0 statistical software (Chicago, IL, USA). Numerical data are mean ± SD and were compared by unpaired Student’s t-test. Differences between two groups in immunohistochemistry were evaluated by the Chi-square test or Wilcoxon rank test as appropriate. Spearman′s correlation was used to analyze the association of expressions of Cx32 and EMT markers. P<0.05 was considered statistically significant.
Results
Identification of OR HCC cell lines
In order to better simulate the clinical occurrence of acquired drug resistance to OXA, HepG2, Huh7, and SMMC-7721 cells displaying resistance to the growth inhibitory properties of OXA at concentrations of 5 μg/mL, 4 μg/mL, and 8 μg/mL, respectively, were eventually developed by concentration increment method as described in the “Material and methods” section. The resulting cells were designated as OR HCC cells, including HepG2/OXA, Huh7/OXA, and SMMC-7721/OXA. The drug-resistance features of these OR HCC lines were subsequently identified. MTT results showed that compared with the parental HCC cell lines, the growth inhibitory effect of OXA on OR HCC cells was significantly decreased (Figure 1A–C), and the expression levels of drug-related proteins P-gp and BCRP were significantly upregulated (Figure 1D–F). These results indicated that OR HCC cell lines were successfully established and could be used for subsequent drug-resistance studies.
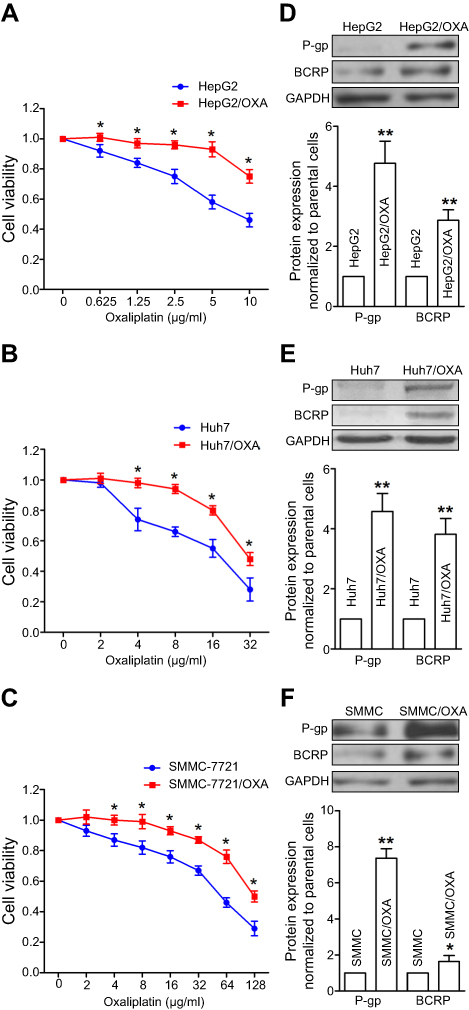 | Figure 1 Establishment and identification of oxaliplatin-resistant (OR) HCC cell lines. (A–C) Parental HepG2, Huh7, SMMC-7721 cells, and respective OR cells, including HepG2/OXA, Huh7/OXA, and SMMC-7721/OXA, were exposed to different concentrations of OXA for 24 hrs. Then, cell viability was detected by the MTT assay. The value of controls, treated with no oxaliplatin, was set at 1.0. (D–F) Induction of cell resistance markers in OR HCC cells, assessed by Western blot for P-gp and BCRP. Data obtained by densitometry represent the mean ± SD of target protein/GAPDH band densities of three determinations for each treatment condition. OR HCC cells were cultured in cell growth medium containing 5 μg/mL, 4 μg/mL, and 8 μg/mL OXA for HepG2, Huh7, and SMMC-7721, respectively. *P<0.05, and **P<0.01 vs parental control. Abbreviations: BCRP, breast cancer resistance protein; OXA, oxaliplatin; P-gp, p-glycoprotein; SMMC, SMMC-7721. |
EMT changes in OR HCC cells
Compared with the parental HCC cell lines, OR HCC cells showed typical EMT characteristics, with elongated spindle shape, loss of cell polarity, and increased formation of pseudopodia (Figure 2A–C). While the epithelial marker E-cadherin was downregulated at the mRNA level, the mesenchymal marker Vimentin was upregulated (Figure 2D). At the protein level, similar changes were observed (Figure 2E–G). Using the Huh7 cell line as an example, immunofluorescence further confirmed a decrease in the fluorescence intensity of E-cadherin but an increase in that of Vimentin in Huh7/OXA cells (Figure 2H). Thus, OR HCC cells acquired an EMT phenotype.
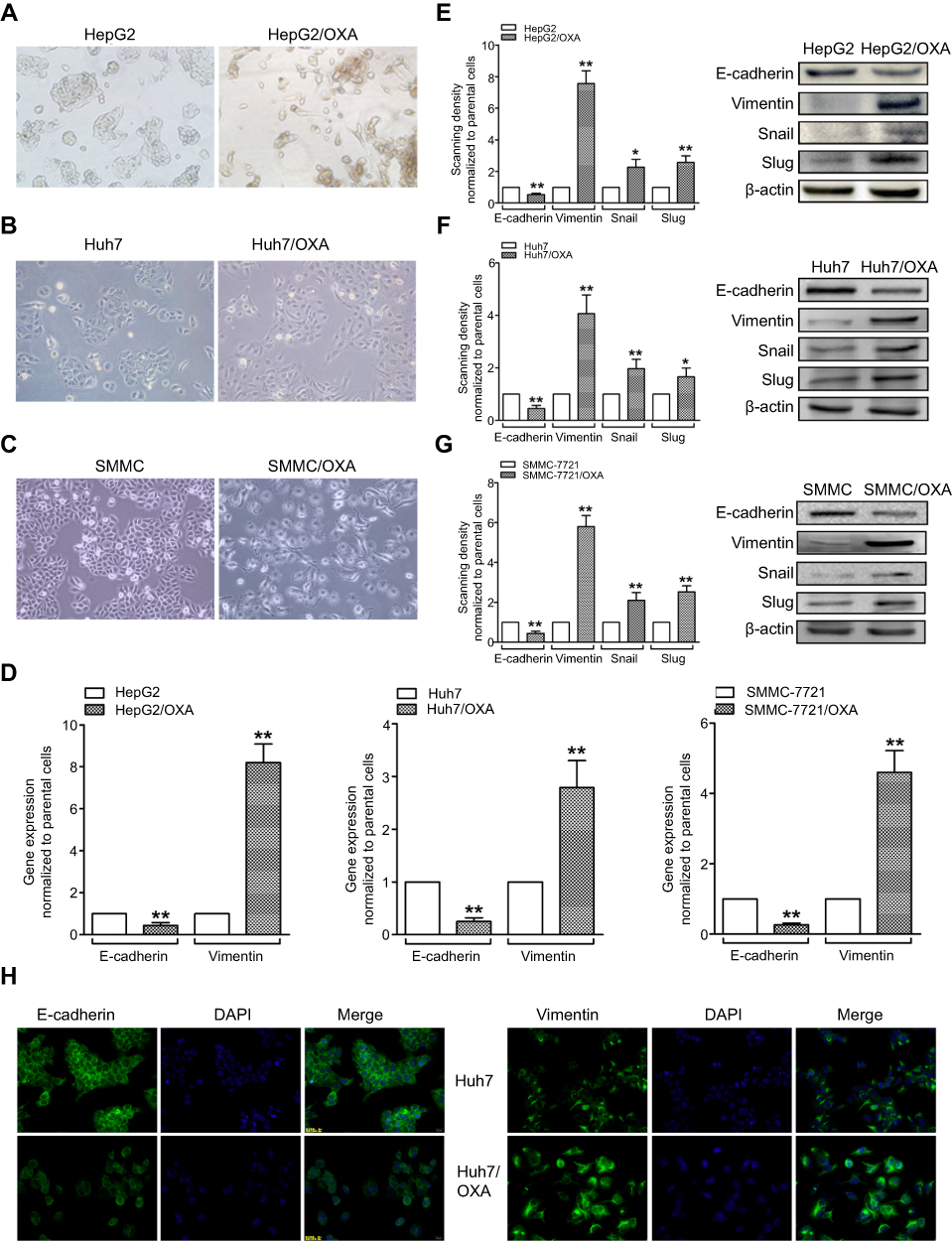 | Figure 2 OR HCC cells acquire the EMT phenotype. (A–C) Morphological changes of HepG2/OXA, Huh7/OXA, and SMMC-7721/OXA cells compared with parental counterparts, as assessed by microscopy. (D) qRT-PCR was performed to assess the mRNA levels of E-cadherin and Vimentin in parental and OR HCC cells. (E–G) Left panel, Western blot was conducted to measure the protein expression levels of E-cadherin, Vimentin, Snail, and Slug in parental and OR HCC cells. Right panel, quantitative results for the left panel. (H) Huh7/OXA cells expressed low level of E-cadherin but high level of Vimentin compared with parental Huh7 cells as evidenced by immunofluorescent staining (original magnification, ×200). OR HCC cells were cultured in cell growth medium containing 5 μg/mL, 4 μg/mL, and 8 μg/mL OXA for HepG2, Huh7, and SMMC-7721, respectively. *P<0.05, and **P<0.01 vs parental control. Abbreviations: EMT, epithelial–mesenchymal transition; HCC, hepatocellular carcinoma; OR, OXA-resistant; OXA, oxaliplatin. |
Increased invasion and migration activity in OR HCC cells
High invasion and migration abilities are important characteristics of tumor cells that undergo EMT. To further confirm the EMT progress in OR HCC cells, we subsequently performed the transwell invasion assay. The results showed significantly more OR HCC cells invading into the lower chamber compared with the parental HCC cells (Figure 3A–C). Consistently, OR HCC cells also had significantly enhanced migratory activity in vitro as evidenced by the wound-healing assay (Figure 3D–F).
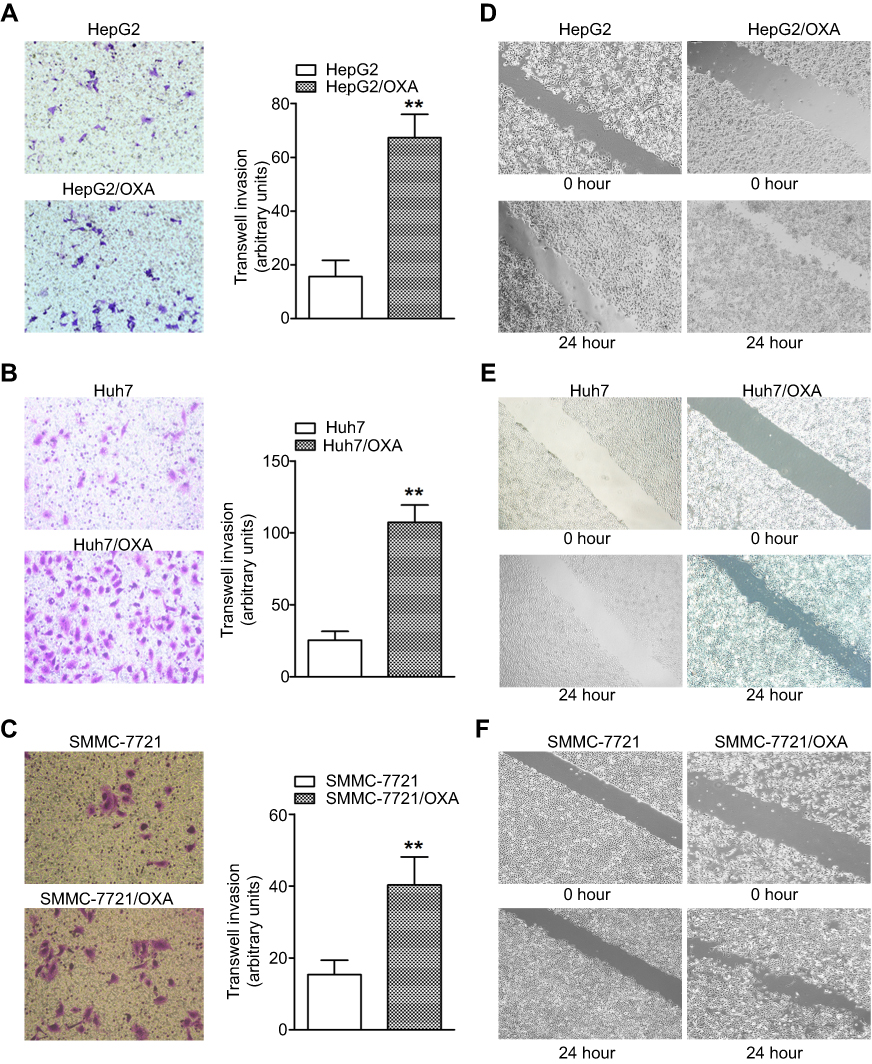 | Figure 3 OR HCC cells show increased invasion and migratory abilities. (A–C) The transwell assay was performed to measure the invasive abilities of HepG2/OXA, Huh7/OXA, and SMMC-7721/OXA cells compared with parental counterparts. (D–F) The wound-healing assay was performed to compare the migratory potential of HepG2 and HepG2/OXA cells, Huh7 and Huh7/OXA cells, SMMC-7721 and SMMC-7721/OXA cells, and the distance migrated was measured. OR HCC cells were cultured in cell growth medium containing 5 μg/mL, 4 μg/mL, and 8 μg/mL OXA for HepG2, Huh7, and SMMC-7721, respectively. **P<0.01 vs parental control. Abbreviations: HCC, hepatocellular carcinoma; OR, OXA-resistant; OXA, oxaliplatin. |
Downregulation of Cx32 in OR HCC cells
To assess the role of Cxs in the drug resistance of HCC cells to OXA, we detected the expression and distribution of Cx26, Cx32, and Cx43 in parental and resistant HCC cells. The results of qRT-PCR and Western blot showed that Cx43 expression was not significantly altered, while Cx26 and Cx32 levels were reduced in OR HCC cells compared with the respective parental cells. Most importantly, downregulation of Cx32 was the most obvious (Figure 4A–C). Subsequently, immunofluorescence revealed that Cx32 fluorescence was significantly decreased in SMMC-7721/OXA cells compared with parental SMMC7721 cells, and Cx32 signals were mainly located in the cytoplasm (Figure 4D). These results indicated that Cx32 may be associated with OXA-induced EMT in HCC cells.
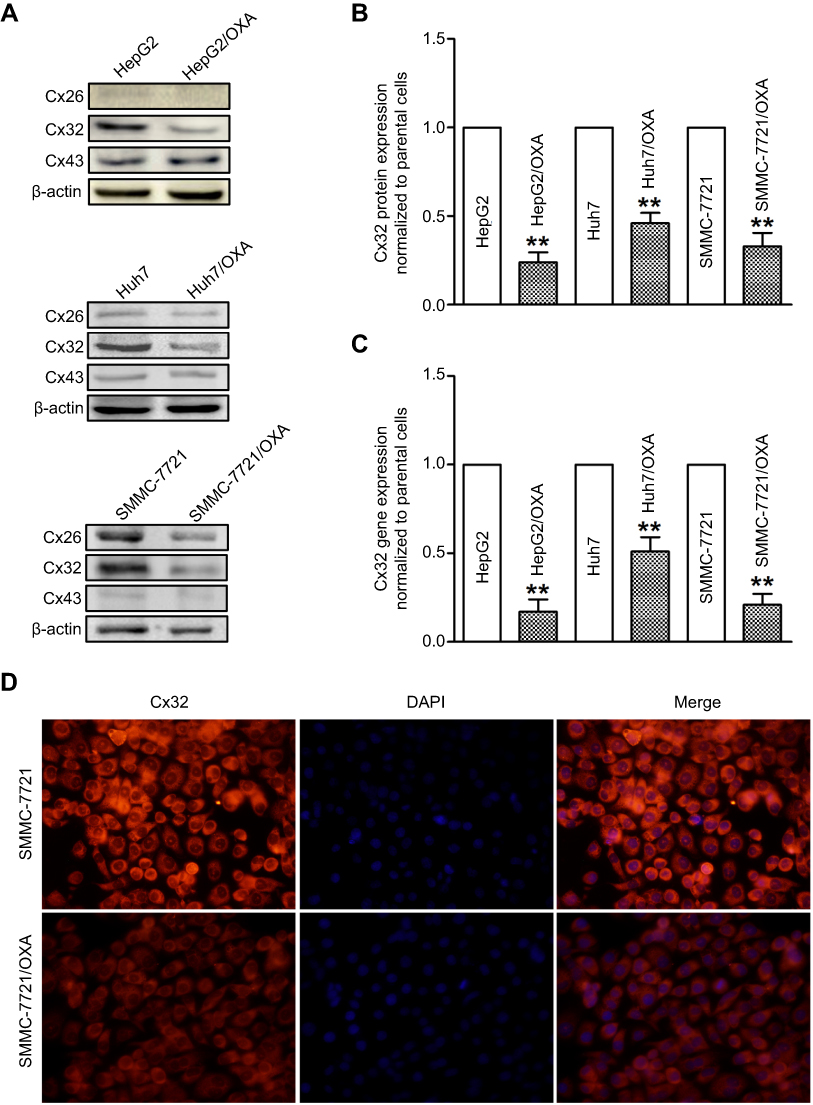 | Figure 4 OR HCC cells exhibit low expression of Cx32. (A) Western blot was performed to assess protein expression levels of Cx26, Cx32, and Cx43 in parental HepG2, Huh7, SMMC-7721 cells, and OR counterpart cells. (B) Quantitative results for Cx32 protein expression. Bar graphs were derived from densitometric scanning of blots for Cx32. (C) Decreased mRNA level of Cx32 was detected in OR HCC cells compared with parental HCC cells, as shown by qRT-PCR. (D) Immunofluorescent staining revealed decreased expression of Cx32, by showing that weaker fluorescence intensities for positive signals were observed, in SMMC-7721/OXA cells compared with parental SMMC-7721 cells (original magnification, ×200). OR HCC cells were cultured in cell growth medium containing 5 μg/mL, 4 μg/mL, and 8 μg/mL OXA for HepG2, Huh7, and SMMC-7721, respectively. **P<0.01 vs parental control. Abbreviations: Cx, connexin; HCC, hepatocellular carcinoma; OR, OXA-resistant; OXA, oxaliplatin. |
Downregulation of Cx32 induces EMT in HCC parental cells
To further investigate the role of Cx32 in OR HCC cells with EMT features, we first explored whether Cx32 silencing by its specific siRNA could induce EMT in parental cells. Since the effect of Cx32 on EMT procedure in HepG2 and SMMC-7721 parental cells was described in our previous report,18 we continued to use the Huh7 cell line as a tool to confirm whether Cx32 universally exerts this effect in the current study. Western blot analysis confirmed a decreased Cx32 expression in Huh7 cells transfected with specific Cx32 siRNAs, with siRNA3 exhibiting the most inhibition (Figure 5A); thus, Cx32 siRNA3 was used in the following gene silencing experiments. As shown in Figure 5B, irregular fibroblastoid morphologic changes were observed in Huh7 cells upon Cx32 downregulation. Meanwhile, the expression level of the epithelial marker E-cadherin was decreased, while those of the mesenchymal markers Vimentin and Snail were increased (Figure 5C). The invasive ability of Huh7 cells was markedly enhanced by Cx32 downregulation as assessed by transwell invasion assay (Figure 5D). In addition, downregulation of Cx32 by siRNA significantly reduced the inhibitory effect of OXA in Huh7 cells (Figure 5E). Taken together, these results suggested that parental HCC cells with depleted Cx32 generally exhibited the phenotypic characteristics of EMT and were less sensitive to chemotherapy with OXA.
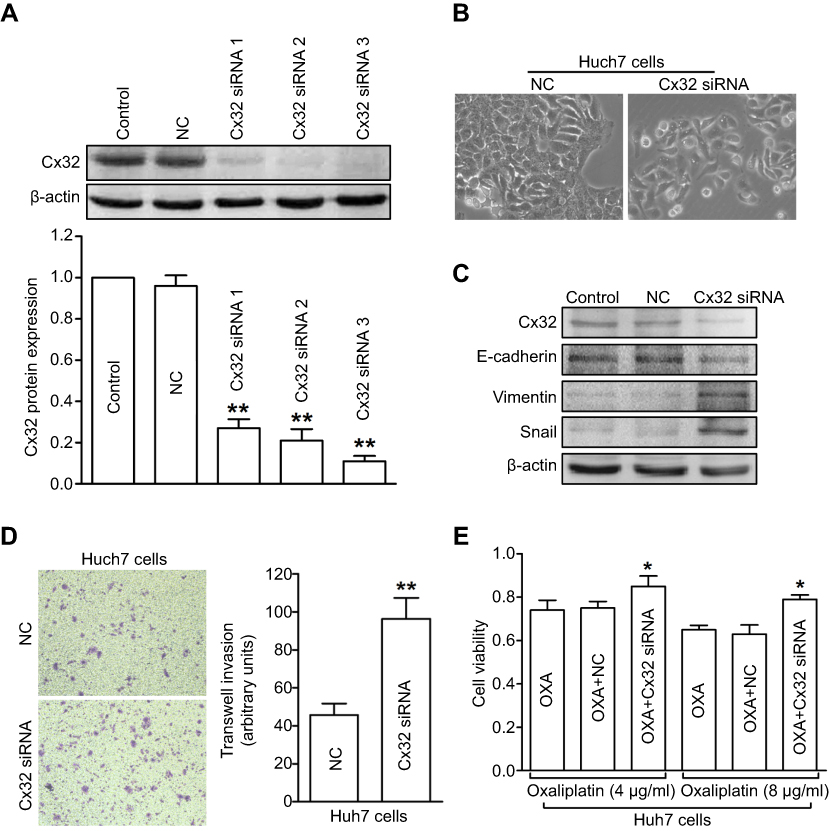 | Figure 5 Depletion of Cx32 in parental Huh7 cells induces EMT and reduces chemosensitivity to OXA. (A) Western blot was conducted to detect the inhibitory effect of Cx32 siRNA in Huh7 cells. (B) Cell morphological changes of Cx32 downregulated Huh7 cells as assessed by microscopy. (C) Decreased expression of E-cadherin but increased expression levels of Vimentin and Snail were found upon Cx32 silencing in Huh7 cells, as shown by Western blot. (D) Invasive ability was enhanced by Cx32 knockdown in Huh7 cells as assessed by the transwell invasion assay. (E) The MTT assay was performed to measure cell growth in Huh7 cells transfected with Cx32 siRNA 3. **P<0.01 vs NC (D), and *P<0.05 vs either OXA treatment in Huh7 cells or OXA treatment in Huh7 cells transfected with control siRNA (E). Abbreviations: Cx, connexin; EMT, epithelial–mesenchymal transition; NC, negative control; OXA, oxaliplatin. |
Upregulation of Cx32 reverses EMT in OR HCC cells
We next assessed whether specific upregulation of Cx32 by cDNA gene transfection could reverse EMT in OR HCC cells. We found that Huh7/OXA and SMMC-7721/OXA cells overexpressing Cx32 displayed a round cell-like morphology (Figure 6A). Moreover, E-cadherin expression was significantly increased, while Vimentin and Snail were significantly downregulated (Figure 6B). As expected, we found that upregulation of Cx32 markedly reduced invasive ability (Figure 6C) and partially restored OXA chemosensitivity (Figure 6D) in both Huh7/OXA and SMMC-7721/OXA cells. Altogether, these findings indicated that decreased Cx32 is involved in OXA resistance associated with EMT in HCC cells.
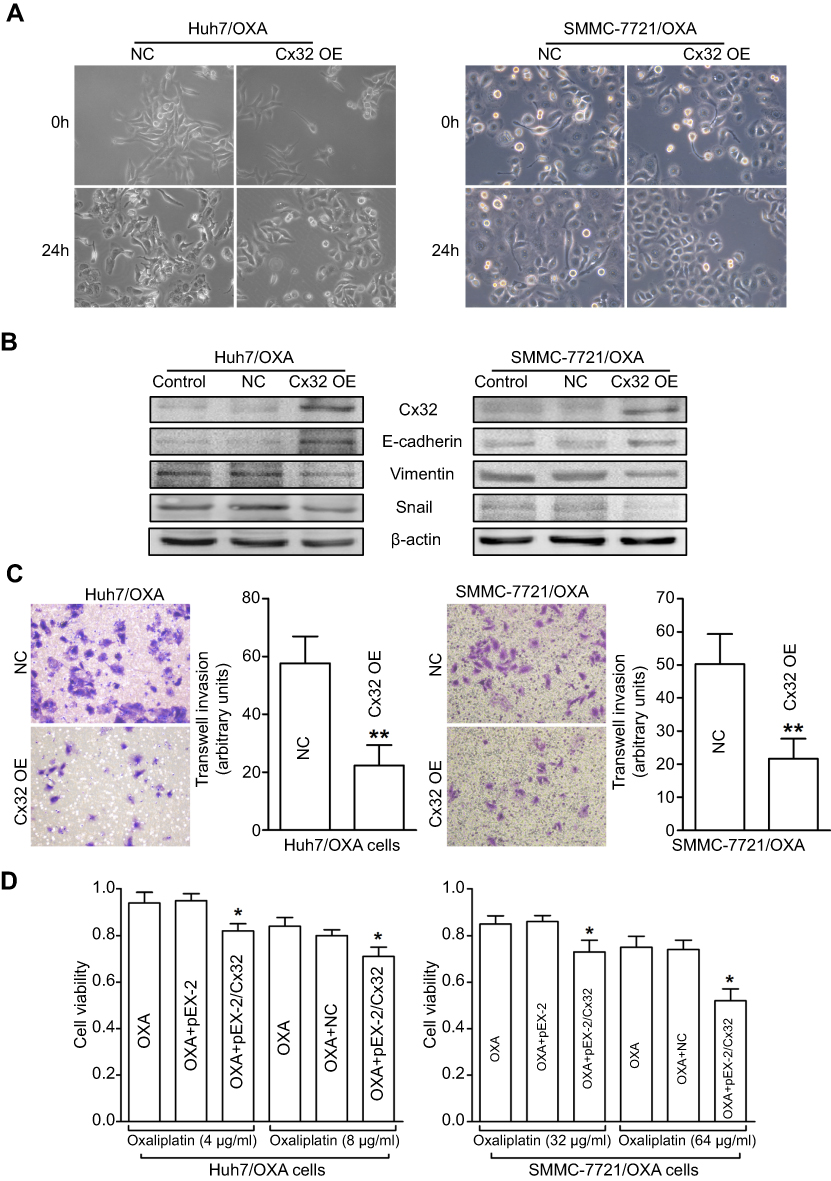 | Figure 6 Overexpression of Cx32 reverses the EMT phenotype and enhances chemosensitivity to OXA in OR HCC cells. (A) Cell morphological changes of Huh7/OXA and SMMC-7721/OXA cells transfected with pEX-2/hCx32. (B) Increased expression of E-cadherin and decreased Vimentin and Snail levels were detected in Huh7/OXA and SMMC-7721/OXA cells following Cx32 overexpression, as shown by Western blot. (C) Upregulation of Cx32 suppressed cell invasion in OR HCC cells Huh7/OXA and SMMC-7721/OXA, as assessed by the transwell invasion assay. (D) Huh7/OXA and SMMC-7721/OXA showed increased chemosensitivity to OXA after Cx32 overexpression by gene transfection, as determined by the MTT assay. Huh7/OXA and SMMC-7721/OXA cells were cultured in cell growth medium containing 4 μg/mL and 8 μg/mL OXA. **P<0.01 vs NC (C), and *P<0.05 vs either OXA treatment or OXA treatment in OR cells transfected with control plasmid (D). Abbreviations: Cx, connexin; EMT, epithelial–mesenchymal transition; NC, negative control; OE, overexpression; OR, OXA-resistant; OXA, oxaliplatin. |
Expression of Cx32, E-cadherin, and Vimentin in HCC tissues and adjacent nontumor tissues
Cx32 was abundantly expressed in the cell membrane in adjacent nontumor (ANT) tissues, but showed low and ectopic expression in the cytoplasm in HCC tissues (χ2=14.233, P=0.000) (Figure 7A). E-cadherin membranous expression rate in ANT tissues was as high as 94.1% (32/34), while in HCC its expression decreased to 70.6% (24/34) with overt membrane loss (χ2=6.476, P=0.023) (Figure 7A). Vimentin was almost not expressed in ANT tissues, but its positive expression rate in HCC tissues was 61.8% (21/34) (χ2=18.281, P=0.000) (Figure 7A). Differences in expression intensities of the above three indicators between HCC and ANT tissues were significant (Table 2). As for the quantitative SI, Cx32 and E-cadherin were concomitantly lower while Vimentin was higher in HCC tissues compared with ANT tissues (Figure 7B–D). Cx32 expression was demonstrated to be positively correlated with E-cadherin in HCC tissues by our previous study.18 In the present study, Cx32 expression was negatively associated with Vimentin expression as assessed by Spearman correlation analysis (Table 3). Taken together, these findings suggested tight regulatory relationships among Cx32, E-cadherin, and Vimentin during hepatocarcinogenesis.
 | Table 2 Expression of Cx32, E-cadherin, and Vimentin in HCC and matched adjacent nontumor tissues |
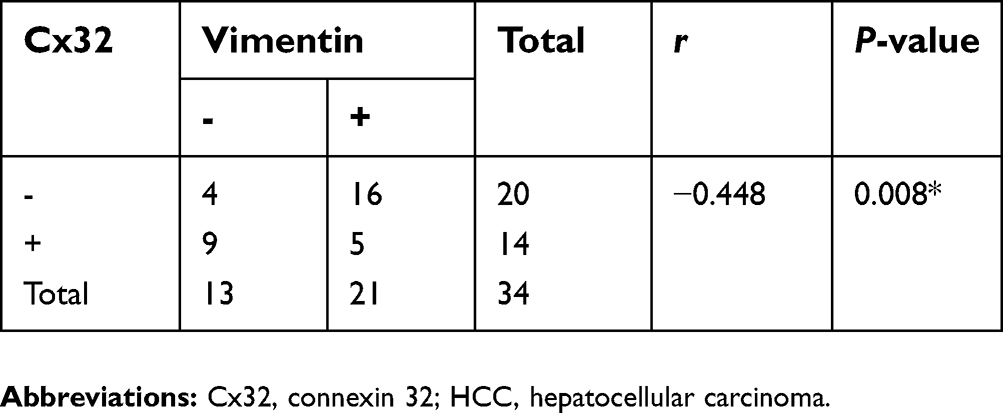 | Table 3 Correlation of Cx32 and Vimentin expression in 34 HCCs |
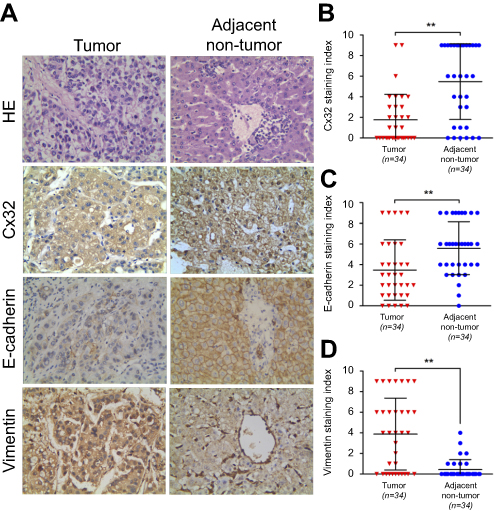 | Figure 7 Expression levels of Cx32, E-cadherin and Vimentin in HCC and matched adjacent nontumor (ANT) tissues. (A) Staining of Cx32, E-cadherin, and Vimentin was performed by immunohistochemistry, and representative images are shown. While Cx32 was weakly stained and mainly located in the cytoplasm in HCC tissues, Cx32 signals were distributed abundantly and linearly on the membrane in ANT tissues. Remarkably reduced membrane staining of E-cadherin was observed in HCC tissues compared with adjacent noncancerous normal liver tissues. HCC tissues showed strong staining of Vimentin in the cytoplasm, while ANT tissues had weak or no Vimentin expression (original magnification, ×400). (B–D) The staining indexes (SI) of Cx32 (B) and E-cadherin (C) were significantly lower, but that of Vimentin (D) was markedly higher in HCC tissues compared with normal paired samples. **P<0.01. Abbreviations: ANT, adjacent nontumor; Cx, connexin; HCC, hepatocellular carcinoma. |
Discussion
The treatment of unresectable locally advanced and metastatic HCC represents a challenge in the clinical practice. The prognosis of this patient population is extremely poor, and median overall survival remains only few months.23 Palliative chemotherapy, especially OXA-based systemic chemotherapy, is critical in the management of advanced HCC.3,4 However, OXA still inevitably encounters drug resistance in clinical application.5,24 Therefore, it is urgent to explore the mechanism underlying drug resistance to OXA in HCC, providing a theoretical basis for clinical rational use and overcoming drug resistance.
Increasing evidence has demonstrated that EMT is closely related to drug resistance in human tumors (including HCC). Yang et al developed OR colorectal cancer cells using the concentration increment method and observed a mesenchymal invasive phenotype consistent with EMT.21 Adriamycin induces EMT in breast cancer cells in vitro; however, only cells with EMT features exhibit high invasion ability and multidrug resistance.25 Subsequently, typical EMT phenotypic changes were observed in non-small cell lung cancer (NSCLC) cells resistant to cisplatin26 and docetaxel,27 HCC cells resistant to doxorubicin28 and GEM,8 glioma cells resistant to temozolomide,29 and ovarian cancer cells resistant to cisplatin.30 Management strategies, including direct overexpression of E-cadherin,31 inhibition of the EMT-related transcription factor Zeb1,27 and targeted knockout of Slug,32 could reverse the mesenchymal state of drug-resistant cells and restore sensitivity to antitumor therapy. Therefore, EMT is considered an important target for reversing tumor resistance.33,34 Therefore, we investigated the relationship between OXA resistance and EMT in HCC cells. Consistent with the above findings, we successfully obtained various OR HCC cell lines by chronic long-term induction of OXA, which also underwent EMT changes. Combined with our previous findings on GEM resistance in HCC cells,8 we consider that EMT may be a common molecular event after HCC cells acquire resistance to various chemotherapeutic drugs.
Cx proteins are implicated in a variety of biological events through GJ-dependent or non-GJ-dependent pathways.10 It was reported that overexpression of Cx26 or Cx43 inhibits the proliferation of breast cancer cells in vitro as well as tumorigenicity in vivo and promotes the cellular transition from mesenchymal to epithelial.35 Co-downregulation of Cx and E-cadherin was detected in NSCLC tissues, and overexpression of Cx43 in lung cancer cells recruits E-cadherin to the cell membrane.36 Cx43 also reverses the malignant phenotypes of glioma stem cells, in association with the upregulation of E-cadherin and its various downstream transcriptional target molecules.37 Since E-cadherin downregulation or deletion is an important promoter of the EMT process in tumor cells,38 it was not difficult to speculate that Cx proteins may have the potential to regulate EMT. A series of recent studies have confirmed this hypothesis. First, in cisplatin-resistant lung adenocarcinoma A549/CDDP cells, Cx43 could modulate EMT.26 Subsequent findings regarding doxorubicin resistance in HCC cells revealed that Cx32 is an important regulator of doxorubicin-induced EMT.28 In addition, we have demonstrated that Cx32 is downregulated in HCC cells during hepatocarcinogenesis, and decreased Cx32 induces EMT by activating the Wnt/β-catenin/Snail pathway and confers high invasion and metastasis characteristics in HCC cells.18 In view of the importance of OXA in systemic chemotherapy for advanced HCC and the close relationship between EMT and chemoresistance, we aimed to assess the molecular mechanisms underlying EMT-related OXA resistance in HCC cells and explore whether this process is associated with Cx regulation. We first observed different patterns of various Cx proteins accompanied by EMT acquisition in OR HCC cells, with no significant difference in Cx43 expression, while Cx26 and Cx32 were downregulated; the difference in Cx32 expression was the most obvious. These results were confirmed in different HCC parental cell lines and the corresponding OR cells. We hypothesized that such discrepancy may be related to the different roles of Cx proteins in HCC. Three major Cxs, including Cx26, Cx32, and Cx43, were reported to be expressed in human and rat livers. Since Cx32 is the major subtype protein constituting the functional GJ in the liver,12 studies assessing Cxs in HCC have been more focused on Cx32 in recent years. Our findings also supported the notion that Cx32 may be a very important player in the process of acquired drug resistance to OXA in HCC. Induction of EMT by Cx32 inhibition in HCC parental cell lines, including HepG2 and SMMC-7721 cells, was shown in our pioneer study.18 In the current work, we further assessed the role of Cx32 and found a Cx32 downregulation or loss in OR HCC cells. On the contrary, upon overexpression of Cx32 by gene transfection in OR HCC cells, EMT was reverted and chemosensitivity to OXA was partially restored. We finally confirmed the associations of Cx32 with EMT markers in human HCC tissues. These results together suggested that downregulation of Cx32 may be an important molecular event in HCC cells acquiring drug resistance to OXA and the related EMT.
The chemosensitization effect of Cx on various cytotoxic drugs (eg, OXA) has been confirmed by several studies.20,22,39 Although its exact mechanism of action remains not fully elucidated, research has focused on the following aspects: 1) The role of GJ-dependent communication functions. The GJ-mediated “bystander effect” has been proposed as a mechanism of cell death caused by distinct damage factors.40 That is, while damage factors such as cytotoxic drugs induce cell death or apoptosis, it may promote a certain death or injury signal. When this signal is small enough to be transmitted to adjacent cells through GJs, it causes adjacent cells to die, thereby expanding the cytotoxicity of the drug. 2) non-GJ-dependent hemi-channel effects. Six Cx proteins surround the central channel on the cell membrane to form a “hemi-channel.” A pair of hemi-channels on adjacent cells is docked by the carbon ends of their extracellular loops to form a whole GJ channel. The hemi-channel is also one of the signaling pathways by which small molecules are released outside the cell, leading to changes in the concentrations of intracellular ions (such as Ca2+) or small molecules (such as ATP), which are involved in the transduction of various intracellular signals.41 3) Exerting biological effects as an independent protein. Cx itself plays an important role in physiological and pathological processes such as cell growth, apoptosis, oncogenesis, tumor invasion, and metastasis by regulating intracellular signal transduction and possible downstream gene transcription.42,43 This study provided evidence, for the first time, that Cx32 could partly restore chemosensitivity in OR HCC cells by reversing EMT. In this process, we speculate that the effect of Cx32 may be related to its own independent function, because immunofluorescence revealed that Cx32 was mostly localized in the cytoplasm and did not fulfill the conditions for assembly into GJs; only Cx localized on the cell membrane can form functional hemi-channels and GJ full channels. Studies have demonstrated an inhibitory effect of tumor growth by Cx26 or Cx43 overexpression in vivo; however, overexpressed Cx proteins accumulate in the cytoplasm and function through non-GJ-dependent mechanisms, including regulation of EMT and antiangiogenesis.35 The latter study, together with the present results, suggested that Cx proteins accumulated in the cytoplasm may play a unique and complex role in regulating a variety of biological effects.
Conclusion
In conclusion, the present study established the role of Cx32 in EMT occurrence in HCC cells resistant to OXA. We found downregulated Cx32 accompanied with mesenchymal phenotypic features in OR HCC cells and Cx32 upregulation in OR HCC cells could partly restore chemosensitivity to OXA by reversing EMT. Therefore, targeting Cx32 by recruiting or stimulating its expression could constitute a promising approach for overcoming chemoresistance toward effective treatment of advanced HCC.
Consent for publication
The publication of this study was approved by the ethics committee of the First Affiliated Hospital of Bengbu Medical College. Written informed consent was obtained from the patients or their immediate family members.
Ethics approval
The usage of patients’ tissue samples in this study was approved by the ethics committee of the First Affiliated Hospital of Bengbu Medical College.
Data sharing statement
The data supporting the results in the manuscript can be obtained from the corresponding author based on reasonable request.
Acknowledgments
We thank Biochemical & Medical Engineering Research Center of Anhui Province and Scientific Research Platform of Bengbu Medical College for instruments support. This work was supported by the National Natural Science Foundation of China (No. 81402514), and internal grant from the First Affiliated Hospital of Bengbu Medical College (No. Byyfykj201801).
Disclosure
The authors report no conflicts of interest in this work.
References
1. Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68(6):394–424. doi:10.3322/caac.21492
2. Contratto M, Wu J. Targeted therapy or immunotherapy? Optimal treatment in hepatocellular carcinoma. World J Gastrointest Oncol. 2018;10(5):108–114. doi:10.4251/wjgo.v10.i5.108
3. Qin S, Bai Y, Lim HY, et al. Randomized, multicenter, open-label study of oxaliplatin plus fluorouracil/leucovorin versus doxorubicin as palliative chemotherapy in patients with advanced hepatocellular carcinoma from Asia. J Clin Oncol. 2013;31(28):3501–3508. doi:10.1200/JCO.2012.44.5643
4. Petrelli F, Coinu A, Borgonovo K, et al. Oxaliplatin-based chemotherapy: a new option in advanced hepatocellular carcinoma. a systematic review and pooled analysis. Clin Oncol (R Coll Radiol). 2014;26(8):488–496. doi:10.1016/j.clon.2014.04.031
5. Martin LP, Hamilton TC, Schilder RJ. Platinum resistance: the role of DNA repair pathways. Clin Cancer Res. 2008;14(5):1291–1295. doi:10.1158/1078-0432.CCR-07-2238
6. Brozovic A. The relationship between platinum drug resistance and epithelial-mesenchymal transition. Arch Toxicol. 2017;91(2):605–619. doi:10.1007/s00204-016-1912-7
7. Singh A, Settleman J. EMT, cancer stem cells and drug resistance: an emerging axis of evil in the war on cancer. Oncogene. 2010;29(34):4741–4751. doi:10.1038/onc.2010.215
8. Wang R, Li Y, Hou Y, et al. The PDGF-D/miR-106a/Twist1 pathway orchestrates epithelial-mesenchymal transition in gemcitabine resistance hepatoma cells. Oncotarget. 2015;6(9):7000–7010. doi:10.18632/oncotarget.3193
9. Sleeman JP, Thiery JP. SnapShot: the epithelial-mesenchymal transition. Cell. 2011;145(1):162 e161. doi:10.1016/j.cell.2011.03.029
10. Aasen T. Connexins: junctional and non-junctional modulators of proliferation. Cell Tissue Res. 2015;360(3):685–699. doi:10.1007/s00441-014-2078-3
11. Graham SV, Jiang JX, Mesnil M. Connexins and pannexins: important players in tumorigenesis, metastasis and potential therapeutics. Int J Mol Sci. 2018;19(6). doi:10.3390/ijms19061645
12. Maes M, Decrock E, Cogliati B, et al. Connexin and pannexin (hemi)channels in the liver. Front Physiol. 2014;4:405. doi:10.3389/fphys.2013.00405
13. King TJ, Lampe PD. The gap junction protein connexin32 is a mouse lung tumor suppressor. Cancer Res. 2004;64(20):7191–7196. doi:10.1158/0008-5472.CAN-04-0624
14. Fujimoto E, Sato H, Shirai S, et al. Connexin32 as a tumor suppressor gene in a metastatic renal cell carcinoma cell line. Oncogene. 2005;24(22):3684–3690. doi:10.1038/sj.onc.1208430
15. Nakashima Y, Ono T, Yamanoi A, El-Assal ON, Kohno H, Nagasue N. Expression of gap junction protein connexin32 in chronic hepatitis, liver cirrhosis, and hepatocellular carcinoma. J Gastroenterol. 2004;39(8):763–768. doi:10.1007/s00535-003-1386-2
16. Edwards GO, Jondhale S, Chen T, Chipman JK. A quantitative inverse relationship between connexin32 expression and cell proliferation in a rat hepatoma cell line. Toxicology. 2008;253(1–3):46–52. doi:10.1016/j.tox.2008.08.010
17. Zhao B, Zhao W, Wang Y, et al. Connexin32 regulates hepatoma cell metastasis and proliferation via the p53 and Akt pathways. Oncotarget. 2015;6(12):10116–10133. doi:10.18632/oncotarget.2687
18. Yang Y, Zhang N, Zhu J, et al. Downregulated connexin32 promotes EMT through the Wnt/beta-catenin pathway by targeting Snail expression in hepatocellular carcinoma. Int J Oncol. 2017;50(6):1977–1988. doi:10.3892/ijo.2017.3985
19. Wang Q, You T, Yuan D, et al. Cisplatin and oxaliplatin inhibit gap junctional communication by direct action and by reduction of connexin expression, thereby counteracting cytotoxic efficacy. J Pharmacol Exp Ther. 2010;333(3):903–911. doi:10.1124/jpet.109.165274
20. Sato A, Sekine M, Kobayashi M, Virgona N, Ota M, Yano T. Induction of the connexin 32 gene by epigallocatechin-3-gallate potentiates vinblastine-induced cytotoxicity in human renal carcinoma cells. Chemotherapy. 2013;59(3):192–199. doi:10.1159/000354715
21. Yang AD, Fan F, Camp ER, et al. Chronic oxaliplatin resistance induces epithelial-to-mesenchymal transition in colorectal cancer cell lines. Clin Cancer Res. 2006;12(14 Pt 1):4147–4153. doi:10.1158/1078-0432.CCR-06-0038
22. Yang Y, Zhu J, Zhang N, et al. Impaired gap junctions in human hepatocellular carcinoma limit intrinsic oxaliplatin chemosensitivity: a key role of connexin 26. Int J Oncol. 2016;48(2):703–713. doi:10.3892/ijo.2015.3266
23. Cauchy F, Soubrane O, Belghiti J. Liver resection for HCC: patient‘s selection and controversial scenarios. Best Pract Res Clin Gastroenterol. 2014;28(5):881–896. doi:10.1016/j.bpg.2014.08.013
24. Virag P, Fischer-Fodor E, Perde-Schrepler M, et al. Oxaliplatin induces different cellular and molecular chemoresistance patterns in colorectal cancer cell lines of identical origins. BMC Genomics. 2013;14:480. doi:10.1186/1471-2164-14-181
25. Li QQ, Xu JD, Wang WJ, et al. Twist1-mediated adriamycin-induced epithelial-mesenchymal transition relates to multidrug resistance and invasive potential in breast cancer cells. Clin Cancer Res. 2009;15(8):2657–2665. doi:10.1158/1078-0432.CCR-08-2372
26. Yu M, Zhang C, Li L, Dong S, Zhang N, Tong X. Cx43 reverses the resistance of A549 lung adenocarcinoma cells to cisplatin by inhibiting EMT. Oncol Rep. 2014;31(6):2751–2758. doi:10.3892/or.2014.3163
27. Ren J, Chen Y, Song H, Chen L, Wang R. Inhibition of ZEB1 reverses EMT and chemoresistance in docetaxel-resistant human lung adenocarcinoma cell line. J Cell Biochem. 2013;114(6):1395–1403. doi:10.1002/jcb.24481
28. Yu M, Han G, Qi B, Wu X. Cx32 reverses epithelial-mesenchymal transition in doxorubicin-resistant hepatocellular carcinoma. Oncol Rep. 2017;37(4):2121–2128. doi:10.3892/or.2017.5462
29. Wang J, Zhou F, Li Y, et al. Cdc20 overexpression is involved in temozolomide-resistant glioma cells with epithelial-mesenchymal transition. Cell Cycle. 2017;16(24):2355–2365. doi:10.1080/15384101.2017.1388972
30. Cao L, Wan Q, Li F, Tang CE. MiR-363 inhibits cisplatin chemoresistance of epithelial ovarian cancer by regulating snail-induced epithelial-mesenchymal transition. BMB Rep. 2018;51(9):456–461.
31. Witta SE, Gemmill RM, Hirsch FR, et al. Restoring E-cadherin expression increases sensitivity to epidermal growth factor receptor inhibitors in lung cancer cell lines. Cancer Res. 2006;66(2):944–950. doi:10.1158/0008-5472.CAN-05-1988
32. Chang TH, Tsai MF, Su KY, et al. Slug confers resistance to the epidermal growth factor receptor tyrosine kinase inhibitor. Am J Respir Crit Care Med. 2011;183(8):1071–1079. doi:10.1164/rccm.201009-1440OC
33. van Staalduinen J, Baker D, Ten Dijke P, van Dam H. Epithelial-mesenchymal-transition-inducing transcription factors: new targets for tackling chemoresistance in cancer? Oncogene. 2018. doi:10.1038/s41388-018-0378-x
34. Du B, Shim JS. Targeting Epithelial-Mesenchymal Transition (EMT) to overcome drug resistance in cancer. Molecules. 2016;21(7):965. doi:10.3390/molecules21070965
35. McLachlan E, Shao Q, Wang HL, Langlois S, Laird DW. Connexins act as tumor suppressors in three-dimensional mammary cell organoids by regulating differentiation and angiogenesis. Cancer Res. 2006;66(20):9886–9894. doi:10.1158/0008-5472.CAN-05-4302
36. Xu HT, Li QC, Zhang YX, et al. Connexin 43 recruits E-cadherin expression and inhibits the malignant behaviour of lung cancer cells. Folia Histochem Cytobiol. 2008;46(3):315–321. doi:10.2478/v10042-008-0057-9
37. Yu SC, Xiao HL, Jiang XF, et al. Connexin 43 reverses malignant phenotypes of glioma stem cells by modulating E-cadherin. Stem Cells. 2012;30(2):108–120. doi:10.1002/stem.1685
38. Gravdal K, Halvorsen OJ, Haukaas SA, Akslen LA. A switch from E-cadherin to N-cadherin expression indicates epithelial to mesenchymal transition and is of strong and independent importance for the progress of prostate cancer. Clin Cancer Res. 2007;13(23):7003–7011. doi:10.1158/1078-0432.CCR-07-1263
39. Wang SQ, Zhang SW, Zhang CZ, Zhao ZY, Wang YJ. Connexin 43 enhances oxaliplatin cytotoxicity in colorectal cancer cell lines. Cell Mol Biol (Noisy-le-grand). 2017;63(4):53–58. doi:10.14715/cmb/2017.63.4.9
40. Arora S, Heyza JR, Chalfin EC, Ruch RJ, Patrick SM. Gap junction intercellular communication positively regulates cisplatin toxicity by inducing DNA damage through bystander signaling. Cancers (Basel). 2018;10(10):368. doi:10.3390/cancers10100368
41. Quist AP, Rhee SK, Lin H, Lal R. Physiological role of gap-junctional hemichannels. Extracellular calcium-depen-dent isosmotic volume regulation. J Cell Biol. 2000;148(5):1063–1074.
42. Zhou JZ, Jiang JX. Gap junction and hemichannel-independent actions of connexins on cell and tissue functions–an update. FEBS Lett. 2014;588(8):1186–1192. doi:10.1016/j.febslet.2014.01.001
43. Yu M, Zou Q, Wu X, Han G, Tong X. Connexin 32 affects doxorubicin resistance in hepatocellular carcinoma cells mediated by Src/FAK signaling pathway. Biomed Pharmacother. 2017;95:1844–1852. doi:10.1016/j.biopha.2017.09.065
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.