")
Back to Journals » Open Access Journal of Clinical Trials » Volume 7
Conducting clinical trials in emerging markets of sub-Saharan Africa: review of guidelines and resources for foreign sponsors
Authors Puppalwar G, Mourya M, Kadhe G, Mane A
Received 10 November 2014
Accepted for publication 7 January 2015
Published 24 March 2015 Volume 2015:7 Pages 23—34
DOI https://doi.org/10.2147/OAJCT.S77316
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Greg Martin
Gaurav Puppalwar, Meenakshi Mourya, Ganesh Kadhe, Amey Mane
Medical Affairs, Wockhardt Limited, Wockhardt Towers, Bandra Kurla Complex, Mumbai, India
Abstract: Clinical trials provide a foundation for new drug development processes, as well as for product license extensions for existing therapies. The reduction in the amount of time and cost to conduct a clinical trial becomes important, as competition to bring a new drug to the market is increasing, and so is the search for new markets. Kenya, Nigeria, Tanzania, Uganda, and Zambia offer a diverse patient population, as well as a comparatively research-friendly and ambitious government to develop these countries as pharmaceutical and health sectors of excellence. All these countries have their own guidelines to conduct clinical trials that feature some similarities and some subtle differences. Over the last decade, the guidelines have been evolving to provide a good ground to foreign sponsors, which carry out clinical trials while keeping the interest of patients as a priority. In the advent of these evolving guidelines, it becomes important for a foreign sponsor to understand and be aware of these guidelines before carrying out clinical trials. The present paper attempts to collect and compile all information available regarding the guidelines on the conduct of trials by a foreign sponsor in these five countries, which are available at government websites and search engines. The information gathered was organized into simplified flowcharts for easy understanding and usage. A clear understanding of the guidelines can effectively reduce the challenges faced for conducting clinical trials in these countries.
Keywords: informed consent, ethics, drug development, emerging markets
Introduction
Clinical trials are the mainstay in new drug development processes, as well as for product license extensions for existing therapies. Often, clinical trials are conducted on varied populations to ensure that the drug is safe and effective in varying ethnic groups.1
Over the past decade, there has been a dramatic increase in the number of registered clinical trials.2 As per the clinical trials registry, the number of registered clinical trials has increased by 3,006% from the year 2000 (number=5,635) to 2014 (as of September 19, 2014, n=175,013).3 While the cost and time to bring a new drug to market has intensified, the pressure to reduce the amount of time and cost to market, the fierce competition for patients due to increased regulatory requirements for licensure and recruiting across a broad range of patients has also increased.4,5 The escalating costs of the research and development of new drugs, as well as reduced access to large populations of treatment-naïve patients, has led the pharmaceutical industry to devise ways to reduce the time in getting the drugs licensed and approved in newer locations.1,5–7 This has also led to the globalization and migration of clinical trials to offshore locations or developing nations/emerging markets. Presently, about 40% of clinical trials are being conducted in emerging markets of Asia, Latin America, and Africa.1,3,8
Despite the fact that developing countries are usually under-represented in research due to a lack of commercial viability and trained researchers, Africa is emerging as an important destination.1 In the year 2013 alone, 4,060 clinical trials were conducted, and all major pharmaceutical companies have their presence in Africa.3 Moreover, conducting trials in low- and middle-income countries can be positive for the trial site as it raises research standards, and brings health improvements and badly needed investment.1,6,9,10 In Africa, research-led solutions and reductions in disease burden have also brought the greatest impact to high rates of early mortality.1,11 Sub-Saharan Africa (SSA), excluding South Africa, is notable in this regard; however; conducting clinical trials in SSA often has its own challenges, such as differing regulatory laws and evolving guidelines.12 Considering scientific factors, stringent regulations, and the International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use (ICH)-Good Clinical Practice (GCP) guidelines, it becomes crucial for stakeholders to be aware of the challenges faced when conducting clinical trials in SSA.10,11 The International Ethical Guidelines for Biomedical Research involving Human Subjects as the minimum requirement were adopted at the Health Research Ethics seminar in Africa held at Arusha, Tanzania, on January 15, 2001. However, different SSA countries follow different practices, and these guidelines have not yet received widespread utility.9
We attempted to present these differing approaches with regard to the differences in guidelines of conducting trials and taking care of patient’s interests in five emerging pharmaceutical markets of SSA (Kenya, Nigeria, Tanzania, Uganda, and Zambia), as they offer a diverse patient population and a comparatively research-friendly and ambitious government to develop these countries as pharmaceutical and health sectors of excellence. Further, these countries have a set of standard regulatory guidelines to follow for conducting a clinical trial.
Methods
We collected information about the conduct of trials by foreign sponsors in these countries (Kenya, Nigeria, Tanzania, Uganda, and Zambia) from various sources. We searched for guidelines on the conduct of clinical trials and guidelines for the ethical conduct of human research on the Websites owned by government organizations such as the Ministry of Health, National Drug Authority (NDA), Food and Drug Authority, and National Council for Science and Technology (NCST) of these countries. We also searched the clinical trial registry Websites of these countries for information. A literature search was also conducted via databases such as PubMed, MEDLINE, Google Scholar, and the Cochrane Library. Once collected, we compiled the information and organized it into simplified flowcharts for easy understanding and usage.
Results
Clinical trials in Kenya
In Kenya, the legal framework for science and technology came into existence in 1979 under the Science and Technology Act. It established the NCST and all the public research institutes. The NCST oversees and coordinates all research in Kenya and advises the government on all matters related to research. The Pharmacy and Poisons Board (PPB) is the national drug regulatory authority in Kenya, which was established under Cap 244 Laws of Kenya. In 2011, the Expert Committee on Clinical Trials of the PPB has developed guidelines to easily navigate the Kenyan clinical trial authorization process. The PPB has the mandate to ensure that clinical trials involving the use of new investigational drugs and older drugs for new conditions or diseases, or investigational devices in human subjects, are in compliance with national regulations (including procedures) to protect the safety of all participants. For drugs that are already registered, approval is not required from the Expert Committee on Clinical Trials, and the studies can be directly initiated after approval from the ethics review committees of the corresponding institutes. The steps in conducting clinical trials in Kenya are represented in Figure 1.
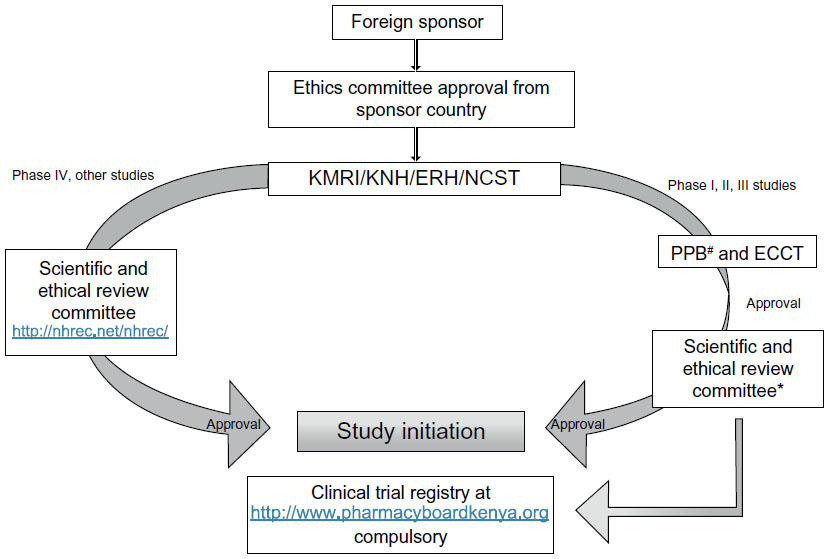 | Figure 1 Flow chart for conducting clinical trials in Kenya. |
The first step to conduct any study in Kenya is to file an application to conduct a clinical trial, followed by a review of the application by the PPB, and submitting a clinical protocol upon its approval.
Application requirements
An application to conduct a clinical trial has to be made by a sponsor or sponsor’s representative, known as the Applicant, along with an application fee of USD $1,000.00, which has to be submitted to the registrar of the PPB.13,14 For multisite trials, only one application with an addendum featuring specific site details (such as staff and capacity) has to be filed by the sponsor, but there should be a coordinating principal investigator (PI) who should be responsible for all the sites.
The application is required to conduct a clinical trial for any study that intends to use human subjects for the testing of unregistered medicines, vaccines, or medical devices; studies intended to generate data on a product that is registered in Kenya based on foreign-generated data; or any study that is going to use an investigational product/device on human beings. Clinical trials are mandatory for already registered medicines if there are changes in clinical indications, target population(s), routes of administration, and/or dosage.14
Procedures for the acceptance, review, and approval of applications
The applications are reviewed by the clinical trial unit of the Division of Medicines Information and Pharmacovigilance of the PPB. The decision on the status of application is communicated to the applicant within 30 days of filing the application.14
Qualifications and responsibilities of investigators, sponsors, and monitors
A qualified Kenyan resident with relevant experience can be a PI of the clinical trial. He or she should have been trained in GCP within the last 3 years. All the sponsors, investigators, and monitors should assume responsibilities, as provided in the ICH-GCP guidelines.15
Clinical trial protocol
A clinical trial protocol should be written as per the ICH-GCP guidelines (Chapter 6) and should describe the objectives, design, methodology, statistical considerations, and organization of the clinical trial.15
Requirements concerning informed consent
Ethical clearance must be obtained for biomedical research. The researchers or sponsors wishing to conduct a clinical trial are often advised to be affiliated with the Kenya Medical Research Institute (KMRI), Kenyatta National Hospital, Eldoret Referral Hospital, and/or Aga Khan Hospital, which have ethics clearance committees. The research guidelines are, however, only followed by the KMRI.16
For ethical clearance, the trial has to fulfill seven basic requirements that provide a systematic and coherent framework for determining the ethical aspects of the trial. As per these requirements, the trial should add value to the improvements in health, knowledge, or well-being of humans; and the trial should be scientifically valid with a clear objective and a feasible methodology. Further, the trial should employ fair subject selection with clearly defined inclusion and exclusion criteria, and it should have a favorable risk–benefit ratio. As a prerequisite, the trial must have undergone an independent review by individuals unaffiliated with the clinical research to help minimize the potential impact of conflicts of interest of the sponsors. In Kenya, evaluation of scientific research is done through scientific and ethical review committees of the KMRI, Kenyatta National Hospital, Eldoret Referral Hospital, and/or Aga Khan Hospital. The participants should have willingly given informed consent to participate in the trial. The language used in the informed consent provided to the participants should either be English or Kiswahili, and the local spoken language of the area, where applicable. Lastly, potential and enrolled subjects should be respected, even if they refuse to be enrolled.16
The ethics guidelines focus on the need to develop the research capacity in Kenya, as well as on the reasonable availability of the intervention to the people of Kenya. For sponsors, two major ethical considerations are highlighted:
- An external sponsoring agency should submit the research protocol for ethical and scientific review, according to the standards of the country of the sponsoring agency; and
- The ethical standards applied should be no less exacting than they would be in the case of research carried out in that country.
The investigator’s brochure
The investigator’s brochure (IB) should be written as per the ICH-GCP guidelines for unregistered products and must contain information regarding:15
- The physical, chemical, and pharmaceutical properties of the drug/s;
- The pharmacological aspects, including its metabolites in all animal species tested;
- The pharmacokinetics and metabolism including its biological transformation in all animal species tested;
- The toxicological effects in any animal species tested under a single-dose study, a repeated-dose study, or a special study;
- Results of clinical pharmacokinetic studies;
- Information regarding the safety, pharmacodynamics, efficacy, and dose responses that were obtained from previous clinical trials in humans; and
- More details provided in the ICH-GCP guidelines may be followed when compiling information on this part.
For registered products being investigated for new conditions, the latest periodic safety update report, a certificate of analysis, and a good manufacturing practices (GMP) inspection certificate should also be submitted.
Investigational new drug (IND) dossier
The PPB may inspect the manufacture of investigational medicinal product (IMP) in the same way as in the case of marketed drug products. For INDs not registered by the PPB, chemistry and manufacturing information should be presented in a concise manner and should include details about the active pharmaceutical ingredient, IMP, and alterations in IMP, if any. IMPs should be properly labeled and relabeled (the remaining IMP from previously manufactured batches) in accordance with GMP principles.17 The sponsor has to bear certain responsibilities for IMPs, as follows:
- Ensure the timely delivery of investigational product(s) to the investigator(s).
- Maintain records that document shipment, receipt, disposition, return, and destruction of the investigational product(s).
- Maintain a system for retrieving investigational products and documenting this retrieval (eg, for deficient product recall, reclaim after trial completion, expired product reclaim).
- Maintain a system for the disposition of unused investigational product(s) and for the documentation of this disposition.
- Take steps to ensure that the investigational product(s) are stable over the period of use.
- Maintain sufficient quantities of the investigational product(s) used in the trials to reconfirm specifications, should this become necessary, and maintain records of batch sample analyses and characteristics.
Safety reporting of suspected unexpected serious adverse reactions (SUSARs)
The sponsor is required to provide initial reports of SUSARs to the PPB as soon as possible, but within 7 calendar days of the notification of the SUSARs, with follow-up reports being provided within a further 8 calendar days. A summary of serious adverse events (SAEs) and SUSARs should be submitted every 6 months from the day of approval of the study.
For periodic safety update reports, the report should be submitted at least 6 months after authorization, until it has been placed in and been on the market for the first 2 years. After this period, it has to be submitted annually for the subsequent 2 years, and thereafter at an interval of 3 years. The sponsor should be able to provide the report immediately upon request.
Requirements concerning the Data and Safety Monitoring Board (DSMB)/Data Monitoring Committee (DMC)
The sponsor must submit the composition of the DSMB and reports of the DSMB to the PPB within 2 weeks of the deliberations.
Protocol amendments
Any protocol amendment that affects the conduct/management of the trial, the safety of the subjects, and the manufacture of the product necessitating changes to the protocol, consent form, and trial sites should be immediately notified to the PPB.
Information on ongoing trials
The sponsor and/or PI must submit progress reports containing the DSMB report, SAE/SUSAR log, and the number of subjects enrolled to the PPB on a 6-month basis from the date of initiation of the clinical trial. The applicant has to submit an updated copy of the IB and a favorable opinion from an independent ethics committee for annual renewal of the trial 6 weeks prior to the expiry of previous approval. An acknowledgment from the PPB is a must for continuing the study.
Post-trial information
The sponsor has to submit a summary of the clinical study report (CSR) within 1 year of the end of the study; the final CSR has to be submitted to the PPB along with interim reports.
Inspection of clinical trial sites
The PPB can conduct an announced or unannounced inspection of the clinical trial (investigator) sites, sponsor’s office, data management center, contract research organization, or any other establishment related to the trial.14
Termination of the clinical trial
For the premature termination of a trial, the PPB has to be informed within 15 days with the reason(s) for the termination and its impact on the proposed or ongoing clinical trials with respect to the IMP. For the trial that is completed, a closing report has to be submitted within 60 days, followed by a final CSR within 1 year after the trial closure unless otherwise justified.
Conditions for clinical trial import license
A copy of the endorsed clinical trial import license and/or evidence of delivery to the approved investigator(s)/trial center(s) on the importation and supply of each consignment of the product has to be submitted to the PPB.14,16
Clinical trials in Nigeria
In Nigeria, the National Agency for Food and Drug Administration and Control (NAFDAC) is the regulatory body that oversees research and clinical trials.18,19 Clinical trial registration can be done at the Nigeria Clinical Trials Registry;20 the registration is a prerequisite if the study is to be published in an International Committee of Medical Journal Editors member journal. Ethical research practices, in adherence to ethical guidelines and the protection of human research participants, are governed by the National Health Research Ethics Committee (NHREC). In addition, the institutes conducting the research can have their own internal health research ethics committee (HREC) that has to be registered with the NHREC. The registration is valid for 2 years, after which a reregistration with NHREC has to be done. A foreign sponsor has to present the protocol to HREC, which has a maximum of 3 months from the date of receipt of a valid application to give its decision to the applicant. For a multisite trial, the HREC of each site has to be approached if sites are less than three. In the event that the trial sites are more than three, the sponsor can directly approach the NHREC.20 In the event that some animal samples have to be sent out of Nigeria, a material transfer agreement has to be signed by all the stakeholders and then reviewed by the HREC. For ethical clearance, the trial has to fulfill all the requirements, as specified by the guidelines in Kenya discussed earlier.
The guidelines for conducting a clinical trial in Nigeria are broadly similar to those in Kenya. A clinical trial is mandatory for new or relatively new chemical entities, drugs for new indications, drugs for new population groups, new combinations, new dosages, and new drug delivery systems.21 For drugs already registered in other countries, the approval has to be sought from an institutional HREC, that will have to revert its decision within 3 months. The application to carry out the clinical trial has to be submitted to NAFDAC with a submission fee of Nigerian Naira (NGN) 5,000 + 5% value-added tax. In addition, an approval/authorization fee of NGN 750,000 for imported drugs and NGN 250,000 for locally developed drugs is charged for the industry-sponsored trial.21 The clinical trial application form should be accompanied with the protocol, informed consent form, IB, evidence of agreement between the sponsor and the investigator, evidence of registration of the institutional review board (IRB) with the NHREC, a list of IRB members, the minutes of meeting held to approve the protocol and informed consent form, evidence to show that the investigator(s) have undergone basic GCP trainings in the last 2 years, the curriculum vitae of the investigators, a sample of all case report forms or electronic case report forms for the study, evidence of insurance coverage for the trial participants, the name and qualification of the trial monitor, and a list and charter of the DSMB.22 The study is to be conducted as per the approved protocol, NAFDAC-GCP guidelines, and the Declaration of Helsinki.21,23 The GCP inspection is done by the NAFDAC-GCP inspectors. The SAEs have to be submitted to NAFDAC within 7 working days of notification. The final study report and any related information also needs to be submitted to NAFDAC.21 The steps followed when conducting clinical trials in Nigeria are represented in Figure 2.
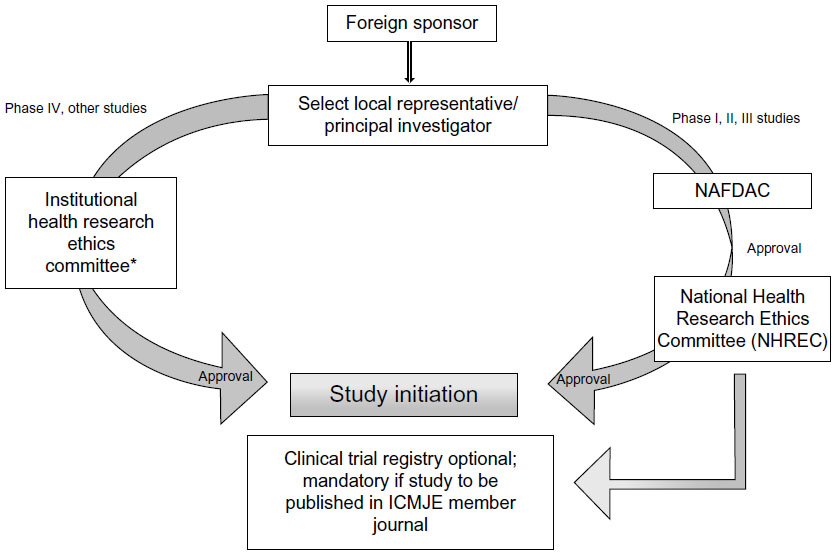 | Figure 2 Flow chart for conducting clinical trials in Nigeria. |
Clinical trials in Tanzania
The Tanzania Food and Drugs Authority (TFDA) oversees the conduct of clinical trials in Tanzania.24 The guidelines for the conduct of clinical trials in Tanzania have been made under the provisions of Section 63(1) of the Tanzania Food, Drugs and Cosmetics Act, 2003.24 A clinical trial is required for all unregistered and registered drugs in case of change in dosage, indication, target population, or route of administration. Approval is not required for the postmarketing study of an already registered drug. A foreign sponsor wishing to conduct a clinical trial in Tanzania should submit an application with the prescribed fee, an ethical clearance certificate issued by any approved institute for medical research, and other relevant information to the TFDA. The Medical Research Coordinating Committee coordinates and ensures that the research in Tanzania follows the country’s ethics requirements. The National Health Research Ethics Review Sub-Committee (NatHREC) of the Medical Research Coordinating Committee registers the trial, reviews the ethics, and approves and monitors the research in Tanzania.25 The NatHREC supports research institutions in Tanzania to establish their own independent ethics committees/IRBs.
For ethical clearance, four copies of the research proposal with a fee of USD $300.00 per proposal are sent to the Director General of the National Institute for Medical Research. The application, along with the filled, signed, and stamped application form, should include a covering letter; general investigation plan; capacity-building plans including training and updating of staff involved in the trial; clinical protocol; IB; declarations by the sponsor, investigators, and monitor(s) in the prescribed format; financial declaration by the sponsor and/or PI in prescribed format; a certified copy of the insurance of study participants; and an IMP dossier. The proposal is reviewed for its scientific validity, ethical content, capability of the investigators, data ownership and material transfer issues, budget issues and justification, and local contact institution. The entire process of ethical clearance usually takes around 4–6 weeks. For Phase I, II, and III trials, ethical clearance or a copy of the acknowledgment of submission of the study protocol from the NatHREC is required during the application to TFDA. The study can be initiated only after obtaining the ethical clearance certificate. All information regarding trial updates, the end of the trial, SAEs, safety reports, and CSRs has to be submitted to the TFDA.9,26,27 The steps involved in conducting clinical trials in Tanzania are represented in Figure 3.
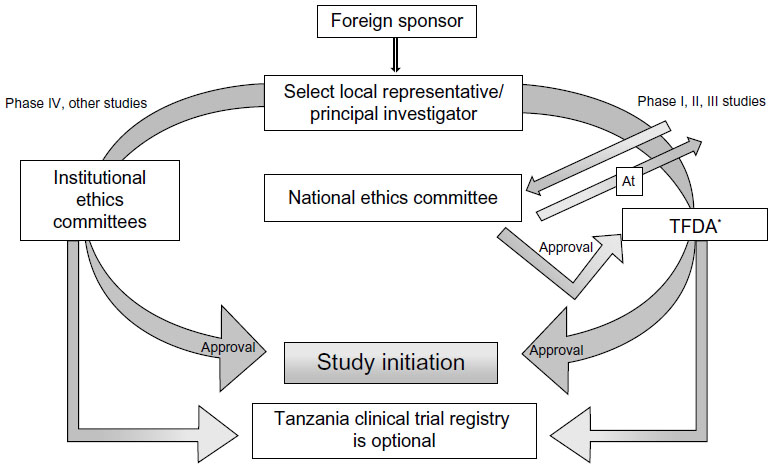 | Figure 3 Flow chart for conducting clinical trials in Tanzania. |
Clinical trials in Uganda
In Uganda, the national guidelines for research were formulated in 2007 by the Uganda National Council for Science and Technology (UNCST), a semiautonomous government agency.28 The participation of humans in clinical trials in Uganda is overseen at the institutional level by the Institutional Review Committees (IRCs) (synonymous to IRB), and at the national level by UNCST. The UNCST liaises with the Research Secretariat in the Office of the President, for national security reasons, to clear researchers to undertake their projects in Uganda.28 For clinical trials, an additional requirement is to obtain the NDA’s authorization to import and/or use the trial drug/device in Uganda. The NDA regulates the conduct of clinical trials as per section 40 of the National Drug Policy and Authority Act, 2000 edition. The steps involved in conducting clinical trials in Uganda are represented in Figure 4.
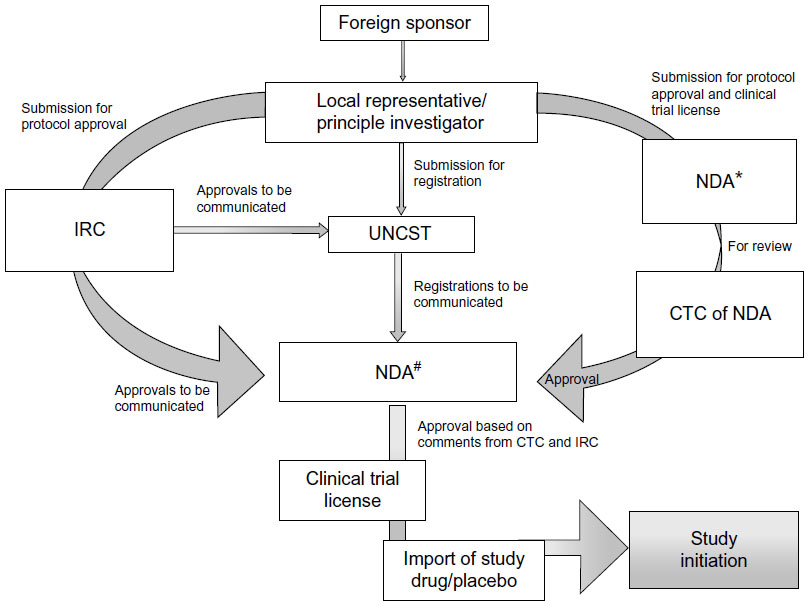 | Figure 4 Flow chart for conducting clinical trials in Uganda. |
The application to conduct a clinical trial in Uganda has to be submitted in a clinical trial application format (available at http://www.nda.or.ug) to the Executive Secretary/Registrar of the NDA. The application should be accompanied by complete, legible copies of key (peer-reviewed) publications supporting the information. The NDA requires approval from the IRC and registration with UNCST before arriving at a decision to conduct any clinical trial. Some institutions have scientific committees (SCs) within institutions as an internal review mechanism for research proposals. In that case, the SC exists in the institute that the sponsor has collaborated with, and approval has to be granted from the SC before the IRC. A DSMB should be established before the commencement of a clinical trial, and its composition should be submitted to the IRC and the UNCST for record purposes.
In the case of research on potentially hazardous substances of a physical, chemical, biological, or any other nature, approval also has to be granted from the Institutional Bio-safety Committees. The IRC should be an independent body of medical and nonmedical members to verify the safety, integrity, and human rights of the subjects participating in a particular trial. The applications for conducting a trial are judged on four ethical research principles that include respect for persons, beneficence to research participants, nonmaleficence, and justice.28 The application should be accompanied by the other relevant documents such as the IB, drug description, pack size, labeling, etc, as previously discussed for the other countries. The IRC has to respond with a decision within 60 days of submission of the application for approval. The NDA is authorized to conduct an announced or unannounced audit of the clinical trial site. All the SAEs, expected and unexpected, are also reported to the IRC and NDA as soon as possible, but no later than 7 calendar days upon receiving notice of such an event. Additional follow-up information should be made available to the NDA as soon as possible, but in any case, not later than 15 calendar days. An interim report for trials lasting for more than 6 months should be submitted at 6-month intervals, or as requested by NDA. The interim report should include the number of patients treated so far, number and type of SAEs reported, the number of discontinued patients, and the reason for discontinuation. The End of Study Summary Report and Product Accountability/Disposal Report should be submitted within 3 months from the Last Patient Out/Last Patient Last Visit date to the NDA. A final report on the trial findings should be submitted no later than 3 months after the completion of the whole trial.28,29
Clinical trials in Zambia
The Pharmaceutical Regulatory Authority (PRA) has formulated guidelines for ethical research in Zambia that are aligned with the World Health Organization (WHO) and GCP, as per the requirement of the Pharmaceutical Act (number 14) of 2004 and the National Health Research Act, 2013.30,31 The guidelines are broadly similar to the guidelines for research in Kenya. The steps involved in conducting clinical trials in Zambia are represented in Figure 5.
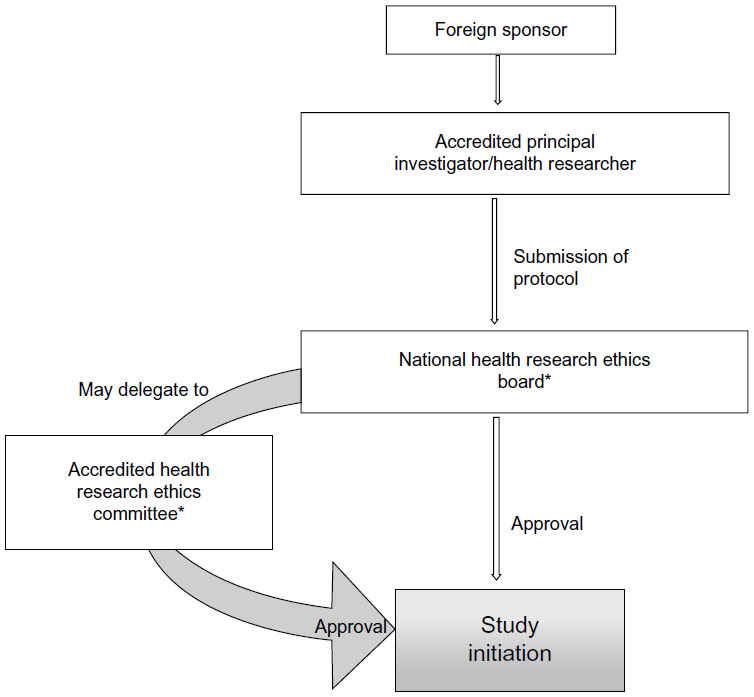 | Figure 5 Flow chart for conducting clinical trials in Zambia. |
The application for conducting a trial along with the relevant documents has to be submitted to the PRA. Approval from the ethics committee is a must to initiate any trial, and the NHREC regulates ethics on human subjects. HRECs that are registered with the NHREC oversee the ethical conduct of the trial at an institutional level. The PRA has the authority to monitor, evaluate, and conduct audits at the trial site of the clinical trial being conducted in Zambia. It is mandatory to include a Zambian in the research team as a principal or coprincipal researcher to carry out the trial. The dissemination of health research information for the trial conducted in Zambia has to be first disseminated locally before being disseminated outside Zambia; the PRA should be notified in writing, citing the research title and the ethical approval obtained from the board before publishing the results. All the SAEs, final study report, and other documents are to be submitted to the PRA.30,31
Discussion
To market a new pharmaceutical drug with reduced costs and in less time is the need of the hour. Clinical trials/research are gaining popularity in the emerging markets, so as to combat with the issues of cost and time.7 As growth opportunities are gradually reducing from traditional pharmaceutical markets, SSA is emerging as an important destination for many pharmaceutical companies to carry out clinical trials and research.1,9,10 Pharmaceutical growth reflects economic strength, accompanied by increasing health care spending. Africa is the second most favored destination in terms of pharmaceutical spending, with a 10.6% compound annual growth rate (CAGR) through 2016.33 According to the Economist Intelligence Unit, the economy of SSA countries is expanding very quickly when compared to the other countries in the world.32
The countries of Kenya, Nigeria, Tanzania, Uganda, and Zambia have the reputation of being emerging pharmaceutical markets in SSA, as per the IMS Health data. As compared to other SSA countries (except South Africa), the nascent market access capabilities, the potential to fulfill pharmaceutical opportunities, friendly coordination with adequate government policies to foster research and drug development, comparatively better availability of skilled investigators and staff, better transport facilities, comparatively higher numbers of hospitals that can recruit patients, and registered clinical trial support services make Kenya, Nigeria, Tanzania, Uganda, and Zambia suitable for the study. Secondly, these are Anglophone countries where people speak and understand English, which makes it easy to communicate with them, whereas others are Francophone (French-speaking) or Lusophone (Portuguese-speaking) countries. This has led to an increased scope of conducting regulatory clinical trials in these regions.33
The conduct of clinical trials in these countries is supported by the government, nongovernment organizations, and private sector investments. This has also led to a decline in epidemic diseases such as HIV/AIDS, malaria, etc, in SSA.34–37 In 2002, out of 29.4 million HIV-infected people, only 50,000 had access to antiretroviral treatments. As SSA became an emerging market for clinical trials, many multinational pharmaceutical companies, in collaboration with the local health care department, have predominantly focused on and succeeded in reducing the infections by improving access to newer therapies to the people of SSA. Uganda, Tanzania, Zambia, and other countries have their success stories in fighting AIDS. The United Nations Program on HIV and AIDS, as well as governments working with nongovernment organizations and their own people have helped to raise awareness about HIV, even providing free treatment.33 Some of these free treatments are funded by a combination of government resources and donor contributions, showing partnerships at work. This has led to a 32% decline in HIV deaths in SSA from 2005 to 2011, whereas a 24% decline in children newly infected with HIV has also been observed.35–37
Although the strategy underpinning a successful market strategy in SSA countries (ie, the selection of location and portfolio, along with economic strength considerations) is similar to those elsewhere in the world, SSA is facing some hurdles to attract multinational corporations for clinical research.33 There is a diversity in the unforeseen and logistical challenges and complexities associated with the conduct of clinical trials in these countries. Political instability, inadequate training of staff, and language barriers, as well as complex and multifaceted sets of markets and cultural barriers, form the basis of the limitations to identifying appropriate study sites.11,38 Further, the noncompliance of regulatory bodies and ethics committees in following the timelines to respond to application hinders clinical research. As per the guidelines, the decision on the status of application from the date of receipt should be communicated to the applicant within 30 days, 28–42 days, 60 days, and 90 days of filing the application in Kenya, Tanzania, Uganda, and Nigeria, respectively. Streamlining the activities and strictly following the timelines for communication will further encourage clinical research activities in these countries.
Another challenge is in getting informed consent from the patients to participate in the trial. Informed consent of the patient participating in the trial is an essential requisite of ethical research needed by the approving authorities. It provides relevant information to the potential participants regarding the nature of the research procedure, the scientific purpose, and alternatives to study.39 A lack of education, and the inability to analyze and interpret the meaning of consent, leads to questions on the full and comprehensive understanding of the consent procedure.10,39 Some unfortunate incidents have led the governments of these countries to make stricter laws for ethical conduct of clinical trials and to protect the participants.10,40 The process of getting informed consent has become stringent, and it is mandatory to explain the procedure, conduct of the trial, effect of the drug, long-term implications, and compensation available to the participants before enrolling them in the trial. For sponsors, it is crucial to understand these guidelines and regulatory laws so as to not be involved in any issues that could hinder the conduct of the trial and malign the name of the sponsor.41
Many patients are skeptical about signing the informed consent document, and a need for a moderator is often felt. Thus, it is must to have local ground staff who can communicate with the patients and clear their doubts and anxieties about the study and its procedures in the local language.11,41
In the advent of these evolving guidelines, it becomes important for a sponsor to fully understand and be aware of these guidelines before carrying out clinical trials. A trial investigator/sponsor should also take responsibility to ensure that participants have a clear understanding of the rules and procedures they need to follow for their own health and for the validity of the trial.41 The participants should be made aware of the compensations they will get. Further, interministerial platforms to coordinate policy and action for conducting clinical trials are still under argument.42 There is a need to develop a permanent platform within a single ministry to undertake these policies and regulations.
Although Africa is expected to reach USD $30 billion in pharmaceutical spending by 2016, it is still lagging behind in terms of health care infrastructure development and the conduct of clinical research. Pharmaceutical companies are striving hard to accelerate clinical trials in SSA, keeping in view the scenario of other markets and the positive aspects of the SSA countries. The government of these countries is also taking steps to boost the pharmaceutical companies and bring SSA at par with the other markets for improving access to drugs for their population.32 As an example, in Nigeria, the government launched the growing National Health Insurance Scheme in 2008 as part of a commitment to ensure universal health coverage by 2015.33 Likewise, from 2007 to 2011, the Kenyan government has expended 4.7% of their entire yearly budget on health care and commits to spending 15% of its national yearly budget in the coming years.43 Similar steps are also being taken in other countries of SSA.33,43
There is a need to understand that an individual country has its own requirements that cannot be underestimated or overseen to achieve a “one size fit all” approach. Instead of one blanket approach, the promotion of intersectoral collaboration through flexibility and cooperation might prove beneficial in achieving a clear understanding of the guidelines coupled with appropriate planning, skilled communication with the trial participants, the development of a sound strategy, and transparency in conducting clinical trials in these countries.42
Conclusion
Conducting clinical trials in SSA is important both for the sponsor and for the participants of the trial who are in need of otherwise unavailable or unaffordable medical care. However, there is a need to follow the guidelines of the respective countries, which provide clearly articulated standards of GCP in research that are relevant to local realities and contexts, ensuring sound scientific and ethical standards. Compliance with these standards will provide the public with assurance that the rights, safety, and well-being of the trial participants are protected. At the same time, the sponsors can also reap the benefits of the study and serve patients in the best possible way.
The clinical study conducted in the emerging markets of SSA should act as a boon to the pharmaceutical industry, as well as to the natives of these countries, leaving no question or doubt unanswered, while maintaining proper harmonization and respecting humanity.
Disclosure
The authors report no conflicts of interest in this work.
References
Lang T, Siribaddana S. Clinical trials have gone global: is this a good thing? PLoS Med. 2012;9(6):e1001228. | |
Petryna A. Clinical trials offshored: on private sector science and public health. Biosocieties. 2007;2:21–40. | |
ClinicalTrials.gov [webpage on the Internet]. Trends, charts, and maps. Bethesda, MD: US National Institutes of Health; 2014. Available from: http://clinicaltrials.gov/ct2/resources/trends. Accessed September 19, 2014. | |
Rehnquist J. The Globalization of Clinical Trials: A Growing Challenge in Protecting Human Subjects. Washington, DC: Department of Health and Human Services, Office of the Inspector General; 2001. | |
AT Kearney, Inc. Make Your Move: Taking Clinical Trials to the Best Location. Chicago, IL: AT Kearney, Inc.; 2006. Available from: http://www.atkearney.com/documents/10192/312631/EA+vol+IX+no+1-Make+Your+Move.pdf/bb05c14b-2709-4ff1-828f-8ef851f303de. Accessed September 19, 2014. | |
Brouwer E [webpage on the Internet]. Clinical trials in developing countries. Global Medicine (5), the Netherlands. Available from: http://globalmedicine.nl/issues/issue-5/clinical-trials-in-developing-countries-2/. Accessed September 19, 2014. | |
Soni U, Singh M. Clinical trials outsourcing: good or bad? Drug Designing. 2013;2(1):1–3. | |
ClinicalTrials.gov [homepage on the Internet]. ClinicalTrials.gov. Bethesda, MD: US National Institutes of Health; 2014. Available from: http://clinicaltrials.gov/. Accessed September 19, 2014. | |
Kumaranayake L, Mujinja P, Hongoro C, Mpembeni R. How do countries regulate the health sector? Evidence from Tanzania and Zimbabwe. Health Policy Plan. 2000;15(4):357–367. | |
The Wemos Foundation. The Clinical Trials Industry in Kenya: Realities, Risks and Challenges. Amsterdam, the Netherlands: The Wemos Foundation; 2014. Available from: http://www.wemos.nl/files/Documenten%20Informatief/Bestanden%20voor%20’Medicijnen’/Boekje%20Kenia%20(1).pdf. Accessed September 19, 2014. | |
Shannon R. Supplying the Needs of Emerging Markets – Understanding Clinical Trial Logistics. Craigavon, UK: Clinical Trial Services; 2006. Available from: http://www.almacgroup.com/wp-content/uploads/CTS_Emerging_Markets_Apr_062.pdf; Accessed September 19, 2014. | |
Mwangoka G, Ogutu B, Msambichaka B, Mzee T, Salim N, Kafuruki S, Mpina M, et al. Experience and challenges from clinical trials with malaria vaccines in Africa. Malar J. 2013;12:86–94. | |
Pharmacy and Poisons Board: Downloads. Available from: http://pharmacyboardkenya.org/?page_id=401; Accessed on February 24, 2015. | |
Expert Committee on Clinical Trials. Guidelines for Applications to Conduct Clinical Trials in Kenya. Kenya: Republic of Kenya, Pharmacy and Poisons Board; 2011. Available from: http://apps.who.int/medicinedocs/documents/s21351en/s21351en.pdf. Accessed September 19, 2014. | |
International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use. ICH Harmonised Tripartite Guideline: Guideline for Good Clinical Practice E6(R1). Geneva, Switzerland: ICH; 1996. Available from: http://www.ich.org/fileadmin/Public_Web_Site/ICH_Products/Guidelines/Efficacy/E6/E6_R1_Guideline.pdf. Accessed September 19, 2014. | |
National Council for Science and Technology. Guidelines for Ethical Conduct of Biomedical Research Involving Human Subjects in Kenya. Nairobi, Kenya: Republic of Kenya; 2004. Available from: https://webapps.sph.harvard.edu/live/gremap/files/ke_NCST_guidelines.pdf. Accessed September 19, 2014. | |
World Health Organization. WHO Good Manufacturing Practices for Pharmaceutical Products: Main Principles. Geneva, Switzerland: World Health Organization; 2011. Available from: http://www.who.int/medicines/areas/quality_safety/quality_assurance/TRS986annex2.pdf?ua=1. Accessed September 19, 2014. | |
Wikipedia [webpage on the Internet]. National Agency for Food and Drug Administration and Control. San Francisco, CA: Wikipedia; 2014. Available from: http://en.wikipedia.org/wiki/National_Agency_for_Food_and_Drug_Administration_and_Control; Accessed on September 19, 2014. | |
National Agency for Food and Drug Administration and Control [homepage on the Internet]. NAFDAC. Abuja, Nigeria: National Agency for Food and Drug Administration and Control; 2014. Available from: http://www.nafdac.gov.ng/. Accessed September 19, 2014. | |
National Health Research Ethics Commitee, Nigeria. Available from: http://nhrec.net/nhrec/nigeria-clinical-trials-registry/; Accessed on February 24, 2015. | |
National Agency for Food and Drug Administration and Control. Regulatory and Registration Directorate: Documentation Guidelines for Clinical Trials in Nigeria. Abuja, Nigeria: National Agency for Food and Drug Administration and Control. Available from: http://www.nafdac.gov.ng/images/GUIDELINES/CLINICAL_TRIAL/GUIDELINES_FOR_CLINICAL_TRIALS_IN_NIGERIA.pdf. Accessed September 19, 2014. | |
Federal Ministry of Health. National Code of Health Research Ethics. Abuja, Nigeria: Federal Ministry of Health; 2007. Available from: http://www.nhrec.net/nhrec/NCHRE_Aug%2007.pdf. Accessed September 19, 2014. | |
World Medical Association, Inc. Declaration of Helsinki. Ferney-Voltaire, France: World Medical Association; 2014. Available from: http://www.wma.net/en/20activities/10ethics/10helsinki/index.html.pdf?print-media-type&footer-right=[page]/. Accessed September 19, 2014. | |
Tanzania Food and Drugs Authority [webpage on the Internet]. TFDA. Dar es Salaam, Tanzania: TFDA ICT department; 2014. Available from: http://tfda.or.tz/. Accessed September 19, 2014. | |
National Institute for Medical Research Tanzania [webpage on the Internet]. Health research ethics. Dar es Salaam, Tanzania: The National Institute for Medical Research (NIMR); 2014. Available from: http://www.nimr.or.tz/ethical-guidelines/. Accessed September 19, 2014. | |
Clerk of the National Assembly. The Tanzania Food, Drugs and Cosmetics Act, 2003. Dar es Salaam, Tanzania: Clerk of the National Assembly; 2003. Available from: http://ihi.eprints.org/667/1/ihi.eprints.pdf_(124).pdf. Accessed September 19, 2014. | |
Tanzania Food and Drugs Authority. Guidelines for Application to Conduct Clinical Trials in Tanzania. Dar es Salaam, Tanzania: Tanzania Food and Drugs Authority; 2009. Available from: http://apps.who.int/medicinedocs/documents/s21352en/s21352en.pdf. Accessed September 19, 2014. | |
Uganda National Council for Science and Technology. National Guidelines for Research Involving Humans as Research Participants. Kampala, Uganda: Uganda National Council for Science and Technology; 2007. Available from: http://www.uncst.go.ug/dmdocuments/Guideline,%20Human%20Subjects%20Guidelines%20Marc.pdf. Accessed September 19, 2014. | |
National Drug Authority. National Drug Authority Guidelines for the Conduct of Clinical Trials. Kampala, Uganda: The Republic of Uganda Ministry of Health; 2008. Available from: http://www.nda.or.ug/docs/CLINICAL.pdf. Accessed September 19, 2014. | |
Pharmaceutical Regulatory Authority. Guidelines on Regulating the Conduct of Clinical Trial in Human Participants. Geneva, Switzerland: World Health Organization; 2004. Available from: http://www.who.int/medicines/areas/coordination/zambia_clinical_trials.pdf; Accessed on September 19, 2014. | |
National Health Research. The National Health Research Act, 2013. Lusaka, Zambia: National Health Research; 2013. Available from: http://www.zambialii.org/files/zm/legislation/act/2013/2/Health%20%20Research%20%20Act%202013.pdf. Accessed September 19, 2014. | |
Economist Intelligence Unit. Into Africa: Emerging Opportunities for Business. A Special Report from the Economist Intelligence Unit. London, UK: Economist Intelligence Unit; 2013. Available from: http://www.i-conferences.org/wp-content/uploads/2013/07/2012-Rapport-EIU-on-Africa.pdf. Accessed December 22, 2014. | |
IMS Health Incorporated. Africa: A Ripe Opportunity. Understanding the Pharmaceutical Market Opportunity and Developing Sustainable Business Models in Africa. London, UK. IMS Health Incorporated, 2012. Available from: http://www.imshealth.com/ims/Global/Content/Insights/Featured%20Topics/Emerging%20Markets/IMS_Africa_Opportunity_Whitepaper.pdf. Accessed December 22, 2014. | |
O’Meara WP, Mangeni JN, Steketee R, Greenwood B. Changes in the burden of malaria in sub-Saharan Africa. Lancet Infect Dis. 2010;10(8):545–555. | |
Shah A [webpage on the Internet]. AIDS in Africa. GlobalIssues.org; 2009. Available from: http://www.globalissues.org/article/90/aids-in-africa. Accessed December 22, 2014. | |
UNAIDS. Regional Fact Sheet 2012: Sub-Saharan Africa. Geneva, Switzerland: UNAIDS; 2012. Available from: http://www.unaids.org/sites/default/files/en/media/unaids/contentassets/documents/epidemiology/2012/gr2012/2012_FS_regional_ssa_en.pdf. Accessed December 22, 2014. | |
Ogunbodede EO. HIV/AIDS situation in Africa. Int Dent J. 2004;54(6 Suppl 1):352–360. | |
Fisher Clinical Services. New challenges for global clinical trials: Managing supply logistics in an expanding clinical trial universe. Available from: http://www.fisherclinicalservices.com/wp-content/uploads/2014/03/Whitepaper_Global-Clinical-Trial-Challenges.pdf. Accessed September 19, 2014. | |
Department of Health. Guidelines for Good Practice in the Conduct of Clinical Trials with Human Participants in South Africa. 2nd ed. Pretoria, South Africa: Department of Health; 2006. Available from: http://www.kznhealth.gov.za/research/guideline2.pdf. Accessed September 19, 2014. | |
Okonta PI. Ethics of clinical trials in Nigeria. Niger Med J. 2014;55(3):188–194. | |
Angell M. The ethics of clinical research in the Third World. N Engl J Med. 1997;337(12):847–849. | |
Okello AL, Bardosh K, Smith J, Welburn SC. One health: past successes and future challenges in three African contexts. PLoS Negl Trop Dis. 2014;8(5):e2884. | |
African Economic Outlook. Kenya 2012. Abidjan, Ivory Coast: African Development Bank Group; 2012. Available from: http://www.africaneconomicoutlook.org/fileadmin/uploads/aeo/PDF/Kenya%20Full%20PDF%20Country%20Note.pdf. Accessed December 22, 2014. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.