")
Back to Journals » Drug Design, Development and Therapy » Volume 14
Computational and Pharmacological Investigation of (E)-2-(4-Methoxybenzylidene)Cyclopentanone for Therapeutic Potential in Neurological Disorders
Authors Farooq S, Khan A, Iqbal MS
Received 11 October 2019
Accepted for publication 24 July 2020
Published 7 September 2020 Volume 2020:14 Pages 3601—3614
DOI https://doi.org/10.2147/DDDT.S234345
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Anastasios Lymperopoulos
Sabah Farooq,1 Arif-ullah Khan,1 Muhammad Shahid Iqbal2
1Riphah Institute of Pharmaceutical Sciences, Riphah International University, Islamabad, Pakistan; 2Department of Clinical Pharmacy, College of Pharmacy, Prince Sattam Bin Abdulaziz University, Alkharj, Saudi Arabia
Correspondence: Arif-ullah Khan
Riphah Institute of Pharmaceutical Sciences, Riphah International University, 7th Avenue, Sector G-7/4, Islamabad, Pakistan
Tel +92-51-289-1835-38
Fax +92-51-2891471, 2890690
Email [email protected]
Purpose: This study involved the computational and pharmacological evaluation of (E)-2-(4-methoxybenzylidene)cyclopentan-1-one (A2K10).
Methods: In silico studies were conducted through virtual screening. Morris water and Y-maze tests were conducted to evaluate Alzheimer’s disease. Acute epilepsy haloperidol,and hyperalgesia were used to calculate the epilepsy model, with Parkinson’s disease and mechanical allodynia at a dose of 1– 10 mg/kg in the mouse model.
Results: A2K10 exhibited the highest binding affinity against α7 nicotinic acetylcholine receptors (− 256.02 kcal/mol). A2K10 decreased escape latency in the Morris water test during different trials. In the Y-maze test, A2K10 dose-dependently increased spontaneous alteration behavior, with maximum effect of 75.5%± 0.86%. Furthermore, A2K10 delayed onset of pentylenetetrazole-induced myoclonic jerks and tonic–clonic seizures and decreased duration of tonic–clonic convulsions in mice, with maximum effect of 93.8± 5.30, 77.8± 2.91, and 12.9± 1.99 seconds, respectively. In the haloperidol-induced Parkinson’s disease model, A2K10 significantly prolonged hanging time and reduced tardive dyskinesia. Moreover, A2K10 extended latency in hot-plate hyperalgesia and increased the paw-withdrawal threshold in mechanical allodynia. In toxicity studies, no mortality was observed.
Conclusion: Overall, the results indicated that A2K10 has potential as an anti-Alzheimer’s, antiepileptic, antiparkinsonian, and analgesic therapeutic compound.
Keywords: computational pharmacology, anti-Alzheimer, antiepileptic, antiparkinsonism , analgesic
Introduction
In developing countries, neurological disorders contribute to more than 6% of the global burden of diseases, a major cause of morbidity and mortality worldwide.1 The human body can be protected from the harmful consequences of reactive oxygen species by natural antioxidants, thus preventing the incidence of many chronic disorders. One of the most common neurodegenerative disorders in elderly people is Alzheimer’s disease (AD). It primarily involves loss of memory, and in the nucleus basalis of Meynert, cholinergic neurons are almost completely destroyed.2 In the US, it is considered the sixth-leading cause of death and disability.
Epilepsy is characterized by periodic and involuntary seizures that may or may not be followed by uncontrolled body movements called convulsions. More than 50 million people in the world are suffering from epilepsy, about 80% of these from low- and middle-income countries.3 Parkinson’s disease (PD) is a movement disorder characterized by loss of dopaminergic neurons in the substantia nigra followed by muscular rigidity, bradycardia, and resting tremors; however its cause still remains unknown. Recently, PD has become less prevalent in Eastern countries due to genetic and environmental factors. Nevertheless, PD incidence is high among Africans, Americans, and Japanese.4
Neuropathic pain is one of most serious complications associated with chemotherapy. About 80%–90% patients receiving chemotherapeutic drugs experience peripheral neuropathy.5 The (E)-2-(4-methoxybenzylidene)cyclopentan-1-one investigated in present research project is an analogue of curcumin. The chemical name of curcumin is 1,7-bis(4-hydroxy-3-methoxyphenyl)-1,6-heptadiene-3,5-dione, and it is obtained from the rhizome of Curcuma longa through different methods. Antitumor, antiviral, anti-inflammatory, antibacterial, anticoagulant, antifungal, and antioxidant effects of curcumin have been reported.6 (E)-2-(4-methoxybenzylidene)cyclopentan-1-one (A2K10) has already been synthesized and chemically characterized. An ionic liquid dimethylammonium dimethylcarbamate (DIMCARB)-–catalyzed reaction was developed for the synthesis of monoarylidene cyclopentanone, which was used as an intermediate for the synthesis of A2K10. Reaction was performed by using a catalytic amount of DIMCARB in green solvent (H2O + EtOH). DIMCARB is an adduct of CO2 and dimethylamine. The A2K10 obtained by this reaction was approximately 98% pure.7 In this research, we investigated A2K10 for its antioxidant, anti-AD, antiepileptic, anti-PD, and analgesic activities by employing in silico, in vitro, and in vivo techniques to explore its therapeutic potential in various neurological disorders. The chemical structure of A2K10 is shown in Figure 1.
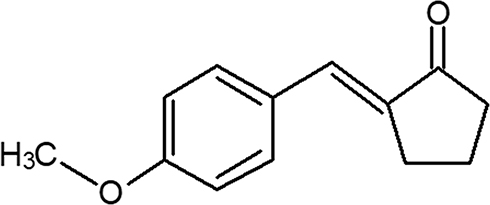 |
Figure 1 Chemical structure of (E)-2-(4-methoxybenzylidene)cyclopentan-1-one (A2K10). |
Methods
Chemicals
Diazepam (Valium, 10 mg/2 mL injection) was obtained from Roche Pharmaceuticals. Cisplatin, levodopa, haloperidol, scopolamine, ascorbic acid, methanol, normal saline, ethanol, pentylenetetrazole (Ptz), and dimethyl sulfoxide (DMSO) were purchased from Sigma-Aldrich. All chemicals used were of analytic grade.
Animals
Adult BALB/c mice of either sex weighing 25–30 g were housed in a controlled temperature (22°C–25°C). Animals were kept in clean plastic cages and maintained on a standard diet with water ad libitum. Animals were divided into various groups. Experiments were carried out in accordance with the guidelines of the Institute of Laboratory Animal Resorces, Commission on Life Sciences University, National Research Council (1996), and approved by the Ethical Committee of Riphah Institute of Pharmaceutical Sciences (REC/RIPS/2017/012).8
Computational Study
Molecular docking of test compounds was carried out to study therapeutic effects, focusing on good binding affinity at target sites. The three-dimensional structure of the test compound (A2K10) was drawn with Discovery Studio Visualizer (DSV). After selection of possible targets,3-D structures of targets/receptors/macromolecules were taken from the RCSB Protein Data Bank (PDB; https://www.rcsb.org/pdb) and modified with DSV software. The PubChem database was used to obtain the structures of different standard drugs (https://pubchem.ncbi.nlm.nih.gov/search), which were then modified in DSV to get 3-D structures. PatchDock was used to study the binding affinity of A2K10 and standard drugs with targets based on their atomic contact energy (ACE; kcal/mol). A2K10 were docked against targets involved in AD: acetylcholinesterase (AChE; PDB ID 2WHP), butyrylcholinesterase (BuChE; PDB ID 1P0Q), N-methyl-D-aspartate (NMDA; PDB ID 5B3J), catechol-O-methyltransferase (COMT; PDB ID 5LSA), and β-secretase (BACE; PDB ID 6EJ2).Reference drugs against these targets were tacrine (PubChem CID 1935), tolcapone (PubChem CID 4659569), and donepezil (PubChem CID 3152). Targets involved in epilepsy include dual-specificity protein phosphatase 13 (DUSP13; PDB ID 2PQ5), γ-aminobutyric acid type A (GABAA; PDB ID 6A96), calcium release–activated calcium channel (Orai; PDB ID 4HKS). Reference drugs used against these targets were carbamazepine (PubChem CID 2554), diazepam (PubChem CID 3016), and phenytoin (PubChem CID 1775). A2K10 was also docked against multiple targets involved in PD, including dopamine receptor D2 (PDB ID 6CM4), GABAB, (PDB ID 4MS3), histamine receptor H1 (PDB ID 3RZE) and muscarinic receptor M1 (PDB ID 5CXV). Reference drugs used against these targets were levodopa (PubChem CID 6047), valproic acid (PubChem CID 4659569), promethazine (PubChem CID 4927), and orphenadrine (PubChem CID 4601). Targets modulating pain taken into account during this study included phospholipase A2 (PLA2; PDB ID 3U8B), α7 nicotinic receptors (α7nAChRs; PDB, ID 3SQ9),TLR4, PDB ID 3FXI), TNFα (PDB ID 2TNF), PPARγ (PDB ID 6ENQ), cyclooxygenase 2 (COX2, PDB ID 5KIR), and α4β2 receptors (PDB ID 5KXI). 4-Methoxy benzoic acid (PubChem CID 7478), acetylcholine (PubChem CID 101,536,190), ibudilast (PubChem CID 3671), aspirin (PubChem CID 2244), and pioglitazone (PubChem CID 4829) were used as reference drugs. XML files of structures of standard drugs were converted to PDB format using Open Babel JUI software.
Antioxidant Activity
A test-sample solution was prepared in DMSO using ascorbic acid as a standard. DPPH solution was prepared in methanol and added in dilutions to A2K10. Then, the mixture of DMSO solution and dilutions were placed in the dark for 30 minutes. Ultraviolet spectrophotometry was used to measure absorbance at 517 nm.9 Free-radical scavenging was calculated:
% DPPH radical scavenging = [(A–B)/A] × 100
where, A is absorbance of control and B absorbance of test sample.
Anti-Alzheimer’s Models
Morris Water Maze (MWM) Test
Mice were divided into five groups (n=5). Group I was injected with normal saline (10 mL/kg) intraperitoneally (IP) and used as negative control. Groups II, III, and IV were given various doses of test compound (1, 3, and 10 mg/kg respectively). Group V was given donepezil (3 mg/kg IP) once/day for 5 days and used as positive control. On the first day, swimming training was given to all mice for 60 seconds without a platform. With the platform in place, the mouse was given trial sessions for 4 days. The mouse was allowed to remain on the platform for 10 seconds after it had located it. If the mouse failed to locate the platform within 120 seconds, it was placed on it for 10 seconds and then removed. On day 5, mice were individually subjected to a probe-trial session without a platform. Each mouse was allowed to swim for 120 seconds to search for it, and escape latencywas determined.10 Anti-AD activity was indicated when there is a decrease in escape latency.
Y-Maze Test
A Y-maze apparatus with three arms was used. Grouping of mice was the same as in the MWM. At 30 minutes after receiving saline, A2K10, or donepezil, each mouse was placed at the center of the apparatus, then allowed to move freely for three sessions (8 minutes each). The successive entry of the mice into three arms in overlapping triplet sets was termed spontaneous alteration behavior. Scopolamine (2 mg/kg) was also used in a group to check results in the diseased group. Alteration behavior percentage was calculated as: (successive triplet sets [entries into three arms consecutively]/total number of entries –2] × 100.11 Anti-AD activity was indicated by an increase in alteration behavior percentage.
Antiepileptic Assays
Mice were divided into five groups (n=5). Group I was injected with normal saline (10 mL/kg IP) and used as negative control. Groups II, III, and IV were given various doses of A2K10 (1, 3, and 10 mg/kg respectively). Group V received diazepam (1 mg/kg IP) and was used as positive control. At 30 minutes after injection of saline, A2K10, or diazepam, Ptz) was injected IP at a dose of 90 mg/kg and each mouse observed for 30 minutes for the onset of myoclonic and tonic–clonic seizures and time span of tonic–clonic seizures. Drugs that showed anticonvulsant-effect delays in the onset of these seizures and reduced the duration of tonic–clonic convulsions.12 The mortality of mice was also observed: mortality = number of mice dead after seizures/total number of mice used × 100).
Anti-Parkinsonism Techniques
Hanging Test
Mice were divided into six groups (n=6). Group I was injected with normal saline (10 mL/kg IP) and was used as negative control. Group II was given haloperidol (1 mg/kg IP). Groups III, IV, and V were administered (1, 3 and 10 mg/kg IP, respectively) for 1 week. Group VI was given IPthe positive control — levodopa (30 mg/kg IP). Saline, test compound, and levodopa were given 30 minutes before the administration of haloperidol. Tests were performed on day 8. To check neuromuscular strength, mice were lifted by their tails, placed on a horizontal grid, and supported until they grabbed the grid with both their fore- and hind-paws. The grid was inverted to allow mice to hang upside down for 30 seconds, Five chances were given to animals with 1-minute intervals, and maximum hanging time was recorded.13
Tardive Dyskinesia
Tardive dyskinesia means rapid vacuous chewing movements (VCMs). Grouping of mice was the same as in the hanging test.Mice were placed individually in small cages for observation of oral dyskinesia. Ten minutes was given to animals to get used to the experimental cage. VCMs were measured by the number of mouth openings in the vertical plane. Under the floor and behind the back wall, mirrors were positioned and the animal faced away from observer for measurement of oral dyskinesia for 5 minutes.13
Cisplatin-Induced Neuropathic Pain
Thermal Hyperalgesia
Animals were divided into six groups (n=6). Group I was administered normal saline (10 mL/kg IP) as negative control. Group II was injected with cisplatin (5 mg/kg IP) once a week for 25 days (days 0, 8, 15, and 22). Groups III, IV, and V were given A2K10 (1, 3, and 10 mg/kg IP, respectively). Group VI received tramadol hydrochloride (30 mg/kg IP) as positive control. A2K10 was injected from day 8 to day 25. Cisplatin was injected 30 minutes before A2K10. Tests were performed on day 0 and from day 8 to day 25, with intervals of 4 days. A hot plate maintained at 54°C±0.5°C was used to measure latency A 30-seconds cutoff was used to avoid paw-tissue damage.
Mechanical Allodynia
In response to mechanical stimulation, paw-withdrawal threshold (PWTs) were measured using the up-and-down method. Grouping of mice was the same as in thermal hyperalgesia. Mice were placed in individual cages on an elevated wire-mesh platform and allowed to get used to the apparatus for at least 30 minutes. Force was be applied using a von Frey hair kit to the mid-plantar region of each hindpaw. Baseline readings were obtained before experimental testing.14
Acute Toxicity
A group of five mice was administered 500 mg/kg A2K10 IP. Mice were observed for 24 hours for any mortality.15
Statistical Analysis
Data were analyzed and are expressed as mean ± SEM. One-way ANOVAs were utilized to analyze results, followed by Tukey’s post hoctest. p<0.05 was taken as significantly significant. Bar charts were assessed using GraphPad Prism 6.
Results
Docking Evaluation
Computational analysis of A2K10 was carried out against various targets involved in different neurological diseases, including AD, epilepsy, PD, and neuropathic pain. It showed different values of ACE with these targets, which showed its binding affinity. The best interaction was against α7nAChR, with good interactions against PPARγ, TNFα, and COX2. The ACE value against α7nAChR was −256.02 kcal/mol. Interactions of A2K10 and acetylcholine against α7nAChRis are presented in Figure 2. ACE values, H-bonds, and residues forming H-bonds are also presented in Tables 1–3. Targets, reference drugs, and interactions are presented in Figures S1–S21.
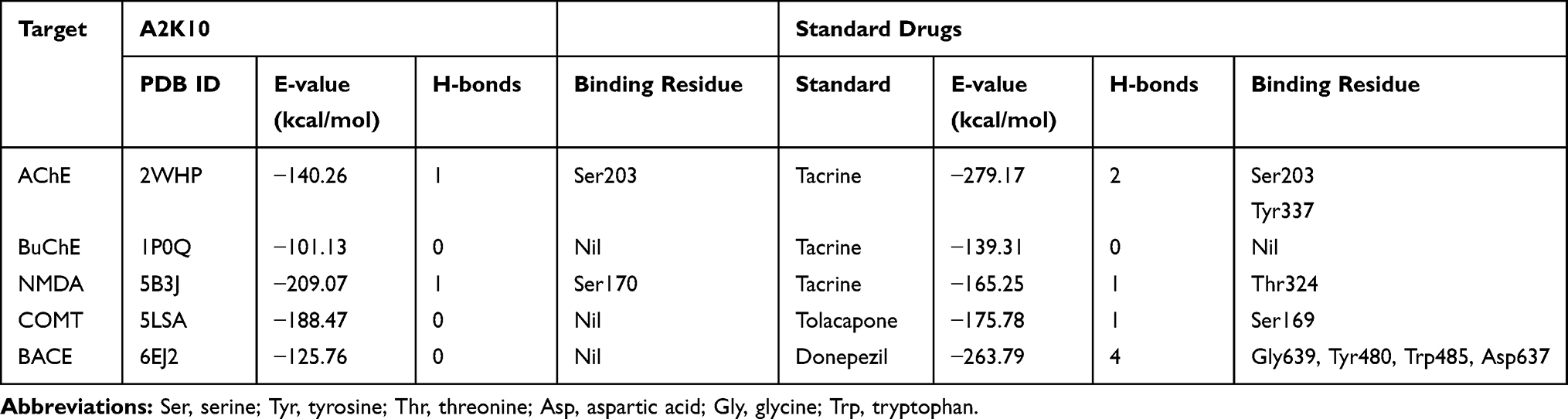 |
Table 1 ACE Values (kcal/mol), H-bonds, and Residues Forming H-bonds of Best-Docked Poses of (E)-2-(4-methoxybenzylidene)cyclopentan-1-one (A2K10) and Standard Drugs with AcetylcholinestErase (AChE), Butyrylcholinesterase (BuChE), N-methyl-D-aspartate (NMDA), catechol-O-methyltransferase (COMT), and β-secretase (BACE) |
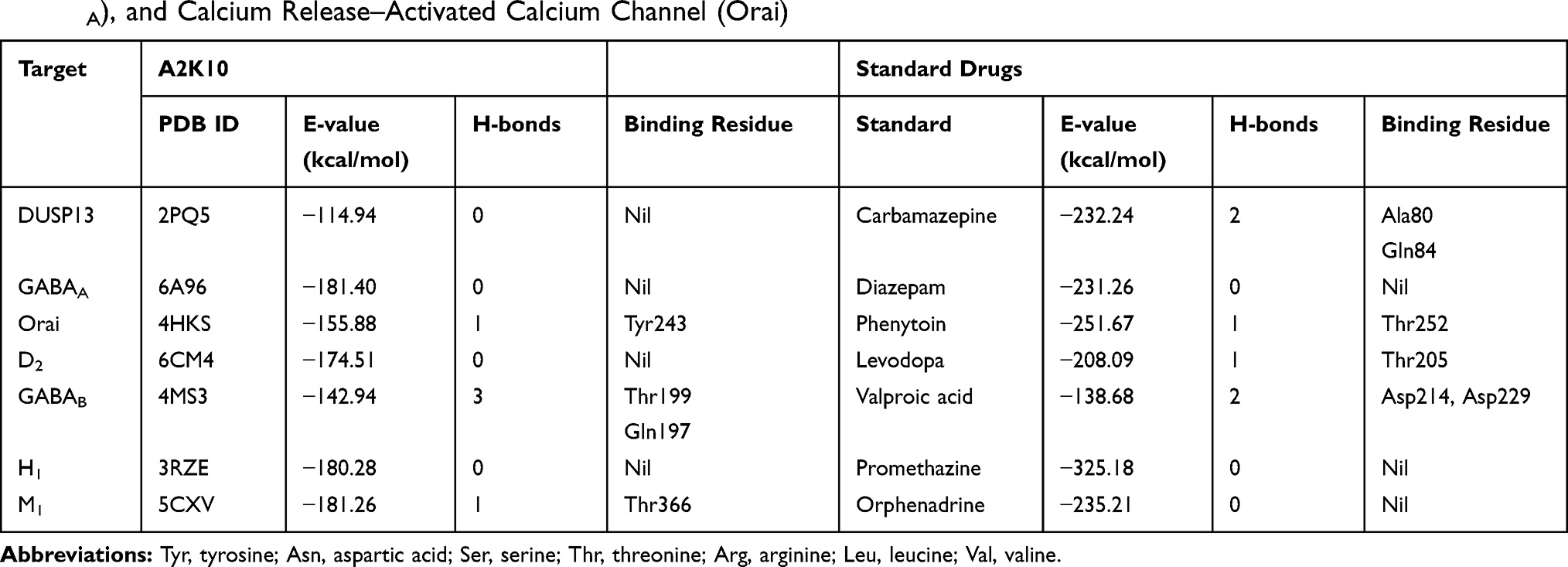 |
Table 2 ACE Values (kcal/mol), H-bonds, and Residues Forming H-bonds of Best-Docked Poses of (E)-2-(4-methoxybenzylidene)cyclopentan-1-one (A2K10) and standard drugs with dual-specificity protein phosphatase 13 (DUSP13), γ-aminobutyric acid A (GABAA), and Calcium Release–Activated Calcium Channel (Orai) |
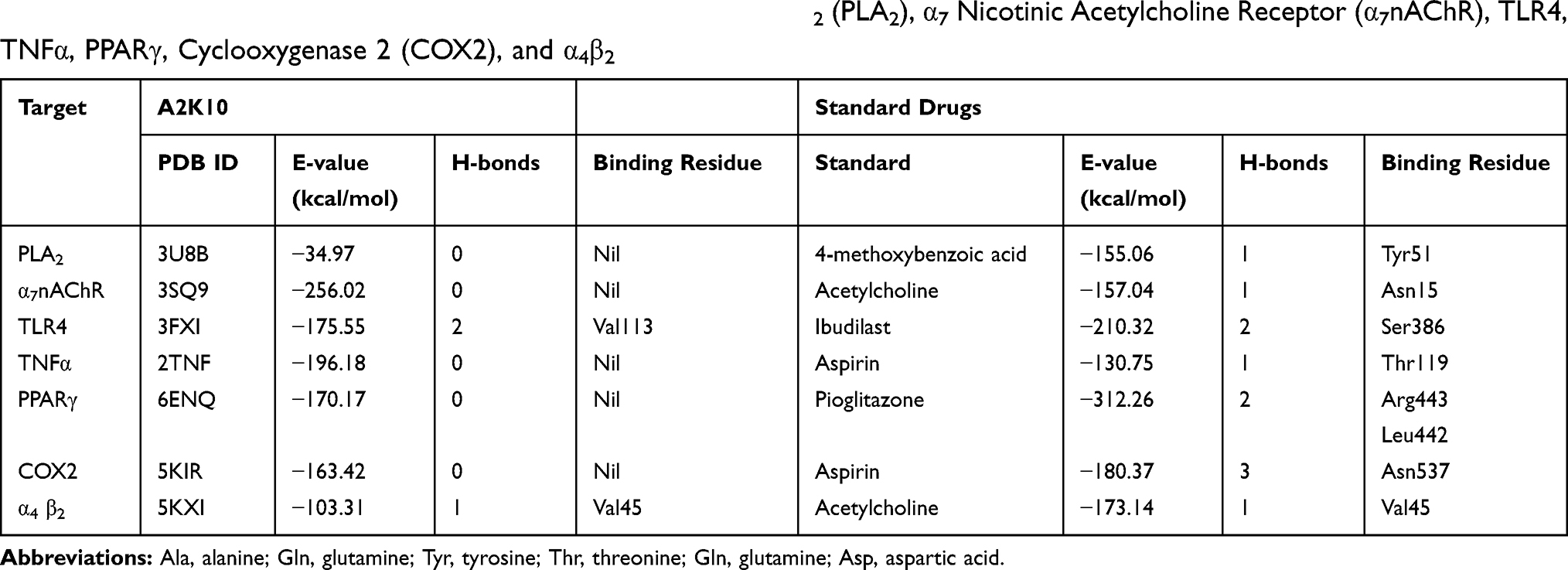 |
Table 3 ACE Values (kcal/mol), H-bonds, and Residues Forming H-bonds of Best-Docked Poses of (E)-2-(4-methoxybenzylidene)cyclopentan-1-one (A2K10) and standard drugs with Phospholipase A2 (PLA2), α7 Nicotinic Acetylcholine Receptor (α7nAChR), TLR4, TNFα, PPARγ, Cyclooxygenase 2 (COX2), and α4β2 |
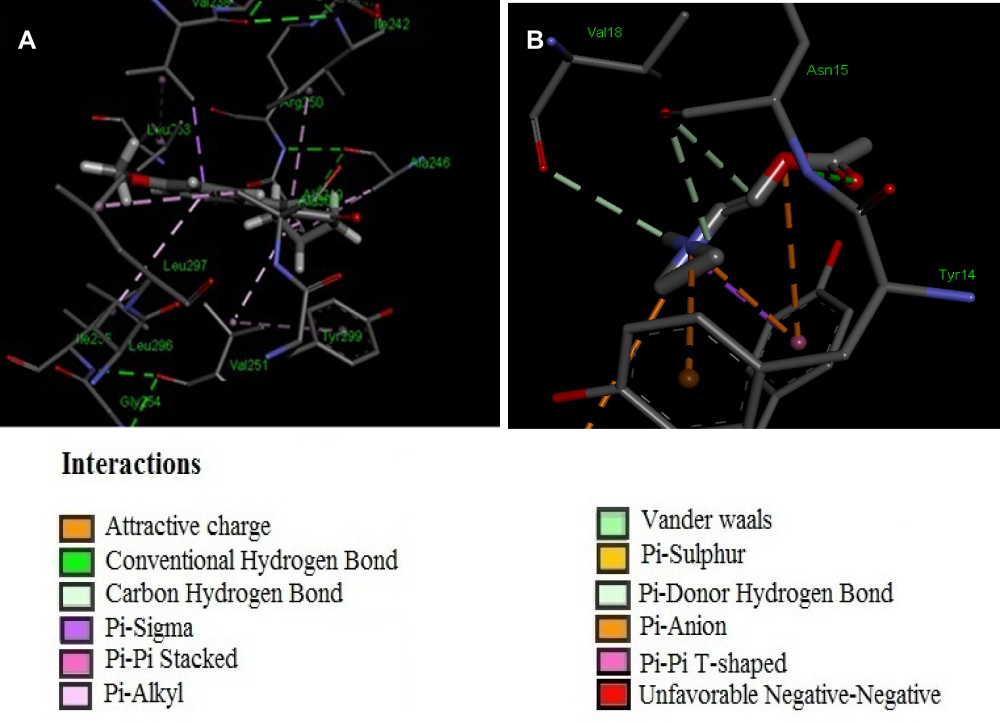 |
Figure 2 (A, B) Interactions of (E)-2-(4-methoxybenzylidene)cyclopentan-1-one (A2K10) and acetylcholine with α7 nicotinic acetylcholine receptors (α7nAChRs), respectively, drawn with Discovery Studio Visualizer 2016. |
DPPH Free Radical–Scavenging Effect
At concentrations of 1, 3, 10, 100, 300, 700, and 1,000,µg/mL, A2K10 exhibited free radical–scavenging effects of 0.82%±0.14%, 1.96%±0.44%, 4.05%±0.01%, 7.20%±0.44%, 8.68%±2.33%, 10.95%±2.56%, and 18.48%±0.006%, respectively. Ascorbic acid at concentrations of 1, 3, 10, 100, 300, 700, and 1,000 µg/mL caused 2.16%±0.18%, 4.24%±0.33%, 4.31%±0.45%, 15.95%±0.72%, 64.18%±0.14%, 92.22%±0.02%, and 93.29%±0.09% free radical–scavenging effects, respectively (Figure 3).
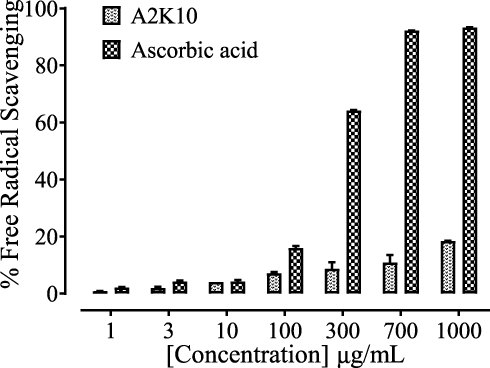 |
Figure 3 Bar chart showing inhibitory effect of (E)-2-(4-methoxybenzylidene)cyclopentan-1-one (A2K10) and ascorbic acid against DPPH free radical–scavenging activity. Values shown as means ± SEM, n=3–4. |
Effect on Escape Latency
A2K10 dose-dependently (1–10 mg/kg) decreased escape latency in the MWM. In the saline (10 mL/kg)-treatment group, escape latency in trials 1–3 was 28.4±0.74, 23.6±0.60, and 19.9±0.72 seconds, respectively. Treatment with A2K10 (1 mg/kg) reduced escape latency in trials 1–3 to 18.6±0.61 (p<0.001 vs saline group), 16.4±0.88 (p<0.001 vs saline group) and 13.7±0.41 (p<0.001 vs saline group) seconds, respectively. Treatment with A2K10 (3 mg/kg) decreased escape latency in trials 1–3 to 15.9±0.46 (p<0.001 vs saline group), 13.5±0.48 (p<0.001 vs saline group) and 11.4±0.66 (p<0.001 vs saline group) seconds, respectively. Treatment with A2K10 (10 mg/kg) decreased escape latency in trials 1–3 to 11.2±0.78 (p<0.001 vs saline group), 8.4±0.51 (p<0.001 vs saline group), and 7.6±0.42 (p<0.001 vs saline group) seconds, respectively. For donepezil (3 mg/kg), escape latency in trials 1–3 was recorded as 22.0±0.95 (p<0.001 vs saline group), 18.4±0.95 (p<0.001 vs saline group), and 14.9±0.95 (p<0.001 vs saline group) seconds, respectively (Figure 4).
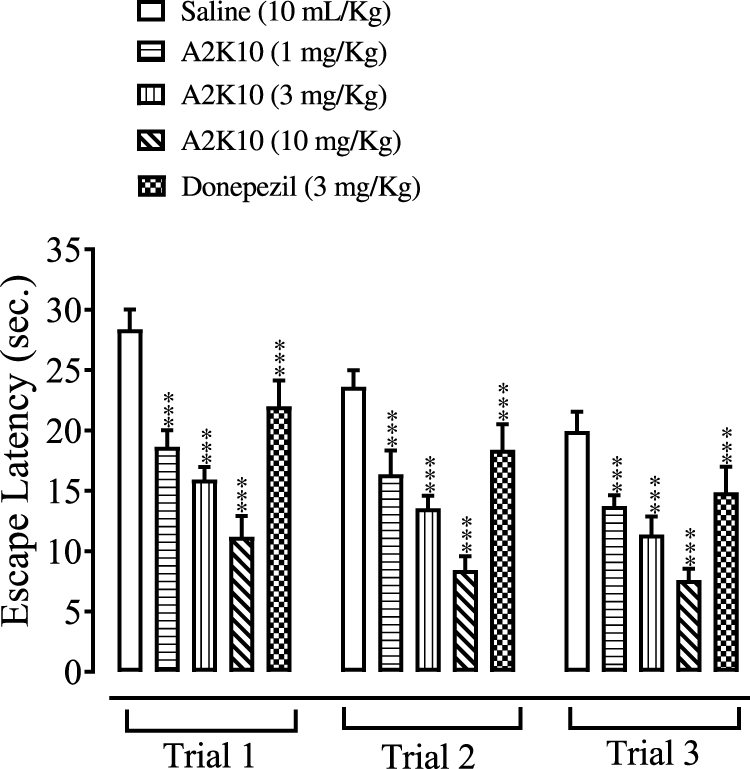 |
Figure 4 Bar chart showing effect of (E)-2-(4-methoxybenzylidene)cyclopentan-1-one (A2K10) and donepezil on escape latency of mice in trials 1, 2, and 3 in the Morris water maze (MWM) test. Data presented as means± SEM, n=5. ***p<0.001 vs saline group on one-way ANOVA with Tukey’s post hoc test |
Effect on Alteration Behavior
A2K10 dose-dependently (1–10 mg/kg) increased spontaneous alteration behavior of mice in the Y-maze. The saline (10 mL/kg)-treatment group showed alteration behavior of 49%±1.26%. Treatment with A2K10 (1 mg/kg) showed increased alteration behavior of 56.1%±0.93% (p<0.001 vs saline group). Treatment with A2K10 (3 mg/kg) showed increased alteration behavior of 63.2%±1.26% (p<0.001 vs saline group), while treatment with A2K10 (10 mg/kg) showed maximum alteration behavior of 75.5%±0.86% (p<0.001 vs saline group). The donepezil (3 mg/kg)-injected group showed alteration behavior of 64.5%±0.81% (p<0.001 vs saline group), as presented in Figure 5. A2K10 (10 mg/kg) increased spontaneous alteration behavior of mice in the Y-maze. The saline treatment group showed alteration behavior of 48.8%±2.62%, scopolamine (2 mg/kg) showed 25.9%±0.96%, A2K10 (10 mg/kg) showed 65.9%±2.32%, and donepezil showed alteration behavior of 74.02%±1.80%, as presented in Figure 6.
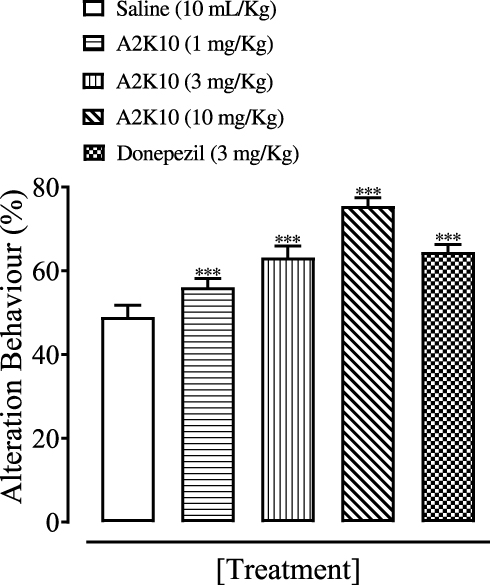 |
Figure 5 Bar chart showing effect of (E)-2-(4-methoxybenzylidene)cyclopentan-1-one (A2K10) and donepezil on alterations in behavior of mice in the Y-maze test. Data presented as means ± SEM, n=5.***p<0.001 vs saline group on one-way ANOVA with Tukey’s post hoc test. |
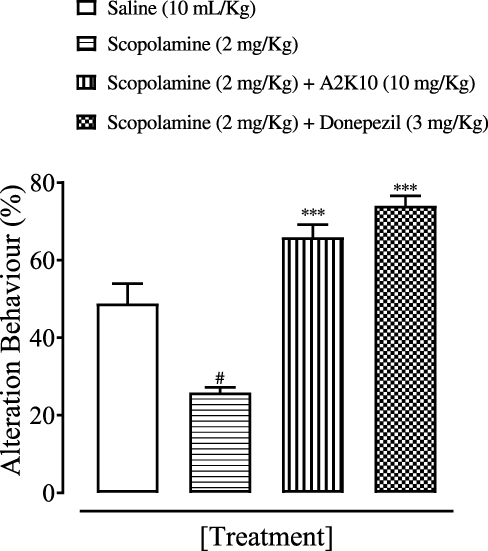 |
Figure 6 Bar chart showing effect of (E)-2-(4-methoxybenzylidene)cyclopentan-1-one (A2K10) and donepezil on alterations in behavior of mice with scopolamine-induced amnesiain the Y-maze test. Data presented as means ± SEM. #p<0.01 vs saline group and ***p<0.001 vs scopolamine group on one-way ANOVA with Tukey’s post hoc test. |
Effect on Number of Entries
A2K10 dose-dependently (1–10 mg/kg) increased the number of entries in the Y-maze test. In group receiving saline (10 mL/kg), the number of entries on days 1– 3 was 22.0±0.41, 18.3±0.58 and 16.8±0.40, respectively. In the A2K10 (1 mg/kg) group, the number of entries on days 1–3 increased to 33.5±1.05 (p<0.001 vs saline group), 21.1±0.74 (p<0.001 vs saline group), and 19.2±0.38 (p<0.001 vs saline group), respectively. In the A2K10 (3 mg/kg)-injected group, the number of entries on days 1–3 increased to 34.3±0.51 (p<0.001 vs saline group), 26.5±0.55 (p<0.001 vs saline group), and 23.2±0.58 (p<0.001 vs saline group), respectively. In the A2K10 (10 mg/kg)-injected group, the number of entries on days 1–3 increased to 45.8±0.68 (p<0.001 vs saline group), 37.9±0.77 (p<0.001 vs saline group), and 26.5±0.67 (p<0.001 vs saline group), respectively. In the donepezil (3 mg/kg)-treated group, the number of entries on days 1–3 recorded was 26.1±0.49 (p<0.001 vs saline group), 24.4±0.99 (p<0.001 vs saline group), and 21.6±0.52 (p<0.001 vs saline group), respectively (Figure 7).
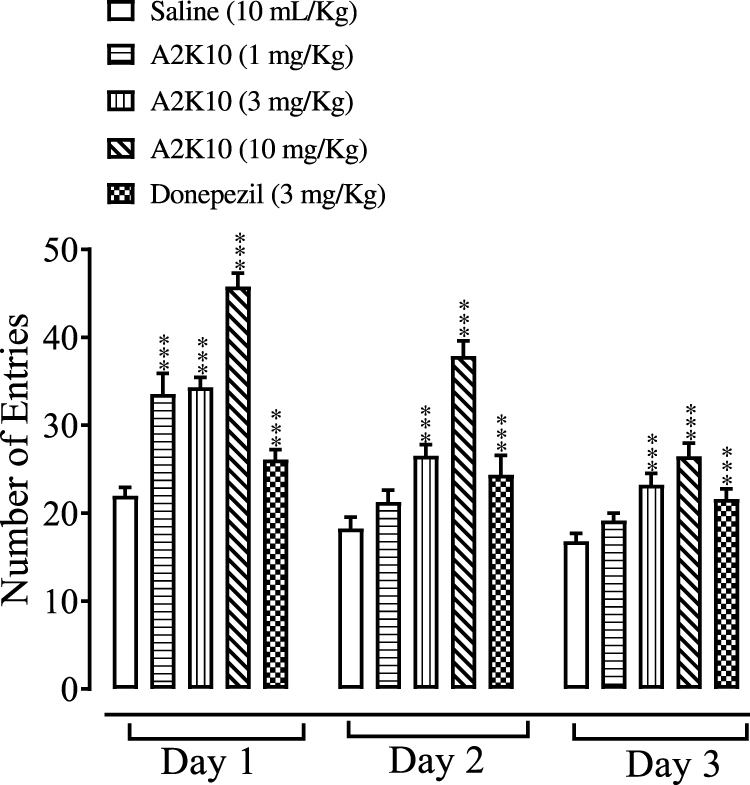 |
Figure 7 Bar chart showing effect of (E)-2-(4-methoxybenzylidene)cyclopentan-1-one (A2K10) and donepezil on number of mouse entries in the Y-maze test on days 1, 2, and 3. Data presented as means ± SEM, n=5. ***p<0.001 vs saline group on one-way ANOVA with Tukey’s post hoc test. |
Effect on Ptz-Induced Seizures
A2K10 dose-dependently (1–10 mg/kg) delayed onset of Ptz-induced myoclonic and tonic–clonic convulsions, decreased duration of tonic–clonic convulsions, and improved mortality. In the saline (10 mL/kg)-treatment group, onset of myoclonic, tonic–clonic convulsions, and duration of tonic–clonic convulsions were 16.8±5.28, 19.8±6.07, and 43.5±5.21 seconds, respectively. In the A2K10 (1 mg/kg) group, onset of myoclonic and tonic–clonic convulsions increased to 55.1±5.06 and 64.4±1.76 seconds (p<0.001 vs saline group), respectively, while duration of tonic–clonic convulsions decreased to 25.4±1.43 seconds. In the A2K10 (3 mg/kg) group, onset of myoclonic and tonic–clonic convulsions extended to 75.0±3.07 and 77.8±2.91 seconds (p<0.001 vs saline group), respectively, while duration of tonic–clonic convulsions decreased to 20.9±1.94 seconds (p<0.05 vs saline group). In the A2K10 (10 mg/kg)-treated group, onset of myoclonic and tonic–clonic convulsions extended to 93.8±5.29 and 77.8±2.91 seconds (p<0.001 vs saline group), respectively, while duration of tonic–clonic convulsions decreased to 12.9±1.99 seconds (p<0.001 vs saline group). In the group treated with diazepam (1 mg/kg), onset of myoclonic and tonic–clonic convulsions extended to 90.9±6.59 and 117.4±5.45 seconds (p<0.001 vs saline group) respectively, while duration of tonic–clonic convulsions decreased to 12.5±2.28 seconds (p<0.001 vs saline group), as shown in Figure 8. A2K10 at 1, 3, and 10 mg/kg decreased mortality rates to 80% (p<0.01 vs saline group), 60% (p<0.001 vs saline group), and 20% (p<0.001 vs saline group), respectively. In the group treated with diazepam (1 mg/kg), no mortality (p<0.001 vs saline group) was observed, as presented in Table 4.
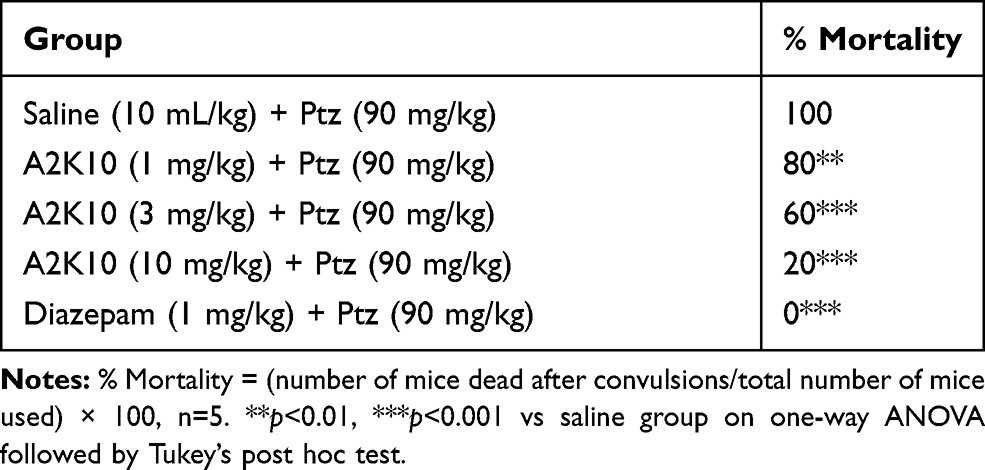 |
Table 4 Effect of (E)-2-(4-methoxybenzylidene)cyclopentan-1-one (A2K10) and Diazepam on Pentylenetetrazole (Ptz)-Induced Seizure Mortality in Mice |
 |
Figure 8 Bar chart showing effects of (E)-2-(4-methoxybenzylidene)cyclopentan-1-one (A2K10) and diazepam on onset of pentylenetetrazole (Ptz)-induced myoclonic jerks, tonic–clonic seizures, and duration of tonic–clonic seizures in mice. Data presented as means ± SEM (n=5).*p<0.05, ***p<0.001 vs saline group on one-way ANOVA with Tukey’s post hoc test. |
Effect on Hanging Time
On the first day, there was no significant change seen in hanging time of mice, but A2K10 had caused a dose-dependent (1–10 mg/kg) increase in hanging time by the eighth day. In the saline (10 mL/kg)-treatment group, hanging duration on the first and eighth days was 16.3±0.24 and 17.3±1.02 seconds, respectively. In the group treated with haloperidol (1 mg/kg), hanging time on the first and eighth days had decreased to 8.8±0.34 and 3.5±0.30 seconds (p<0.001 vs saline group), respectively. In the A2K10 (1 mg/kg)-injected group, hanging time on the first and eighth days had increased to 7.5±0.47 and 14.3±0.81 seconds (p<0.001 vs haloperidol group), respectively. In the A2K10 (3 mg/kg)-injected group, hanging time on the first and eighth days had increased to 9.4±0.45 and 17.3±0.70 seconds (p<0.001 vs haloperidol group), respectively. In the A2K10 (10 mg/kg)-injected group, hanging time on the first and eighth days had increased to 11.1±0.93 and 20.7±0.66 seconds (p<0.001 vs haloperidol group), respectively. In the group treated with levodopa (30 mg/kg), hanging time on the first and eighth day had increased to 18.8±1.69 and 21.0±0.55 seconds (p<0.001 vs haloperidol group), respectively (Figure 9).
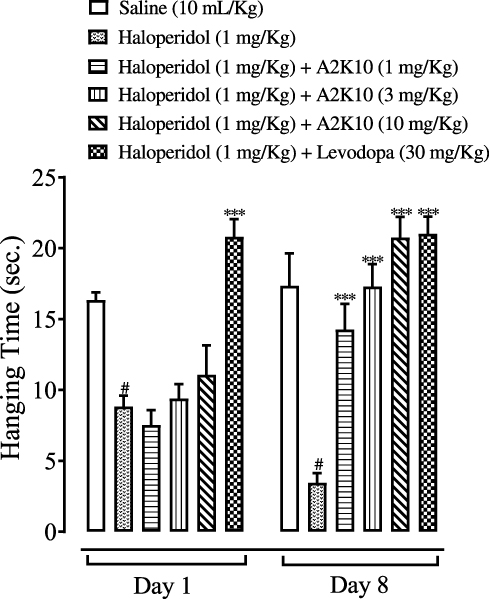 |
Figure 9 Bar chart showing effects of (E)-2-(4-methoxybenzylidene)cyclopentan-1-one (A2K10) and levodopa on hanging time of mice in haloperidol-induced Parkinson’s disease (PD). Data presented as means ± SEM (n=5).#p<0.001 vs saline group and ***p<0.001 vs haloperidol group on one-way ANOVA with Tukey’s post hoc test. |
Effect on VCMs
A2K10 had dose-dependently (1–10 mg/kg) decreased VCMs in mice by the eighth day of experiment. The group treated with saline (10 mL/kg) showed7.2±0.37 and 6.4±0.51 VCMs on the first and eighth days, respectively. The group treated with haloperidol (1 mg/kg) showed 46.2±1.20 and 70.6±0.93 (p<0.001 vs saline group) VCMs on the first and eighth days, respectively. In the A2K10 (1 mg/kg) group, VCMs on the first and eighth days had reduced to 45.6±1.21 and 36.6±0.93 (p<0.001 vs haloperidol group), respectively. In the A2K10 (3 mg/kg) group, VCMs on the first and eighth days had reduced to 44.2±1.28 and 22.4±0.93 (p<0.001 vs haloperidol group), respectively. In the A2K10 (10 mg/kg)-injected group, VCMs on the first and eighth days had reduced to 43.8±1.16 and 13.4±0.93 (p<0.001 vs haloperidol group), respectively. In the levodopa (30 mg/kg)-injected group, VCMs on the first and eighth days had reduced to 14.8±0.97 and 11.2±0.66 (p<0.001 vs haloperidol group), respectively (Figure 10).
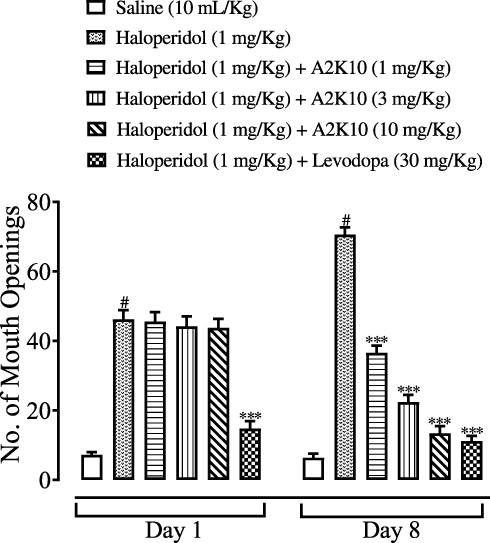 |
Figure 10 Bar chart showing effects of (E)-2-(4-methoxybenzylidene)cyclopentan-1-one (A2K10) and levodopa on number of mouth openings in haloperidol-induced Parkinson’s disease (PD) model in mice. Data presented as means ± SEM (n=5). #p<0.001 vs saline group and ***p<0.001 vs haloperidol group on one-way ANOVA with Tukey’s post hoc test. |
Effect on Hot-Plate Nociception
A2K10 dose-dependently (1–10 mg/kg) increased the escape latency of mice on hot-plate assays. The group treated with saline (10 mL/kg) showed latency to thermal sensitivity at days 0, 8, 12, 16, 20, and 24 of 17.5±1.07, 21.2±0.32, 16.4±0.60, 18.4±0.53, 20.6±0.45, and 19.4±0.51 seconds, respectively. In the cisplatin (5 mg/kg)-injected group, latency had reduced to 16.7±1.16 (p<0.001 vs saline group), 6.2±0.76 (p<0.001 vs saline group), 5.4±0.58 (p<0.001 vs saline group), 4±0.34 (p<0.001 vs saline group), 3.3±0.34 (p<0.001 vs saline group), and 2.8±0.25 seconds (p<0.001 vs saline group), respectively. In the group treated with A2K10 (1 mg/kg), latency had increased to 17.0±1.18, 6.7±0.75 (p<0.001 vs cisplatin group), 5.9±0.78, 6.1±0.56, 5.7±0.27, and 5.4±0.24 seconds, respectively. In the A2K10 (3 mg/kg) group, latency had increased to 16.8±0.72, 7.0±0.73, 8.7±0.72, 11.0±0.58 (p<0.001 vs cisplatin group), 11.8±0.61 (p<0.001 vs cisplatin group), and 12.4±0.64 seconds (p<0.001 vs cisplatin group), respectively. In tthe A2K10 (10 mg/kg) group, latency had increased to 18.0±0.72, 8.3±0.26, 9.3±0.64 (p<0.001 vs cisplatin group), 11.7±0.65 (p<0.001 vs cisplatin group), 12.9±0.57 (p<0.001 vs cisplatin group), and 15.3±0.68 seconds (p<0.001 vs cisplatin group), respectively. In the tramadol (30 mg/kg)-injected group, latency had increased to 18.1±0.47, 10.6±0.70 (p<0.001 vs cisplatin group), 11.1±0.74 (p<0.001 vs cisplatin group), 12.1±0.65 (p<0.001 vs cisplatin group), 14.3±0.70 (p<0.001 vs cisplatin group), and 17.1±0.45 seconds (p<0.001 vs cisplatin group), respectively (Figure 11).
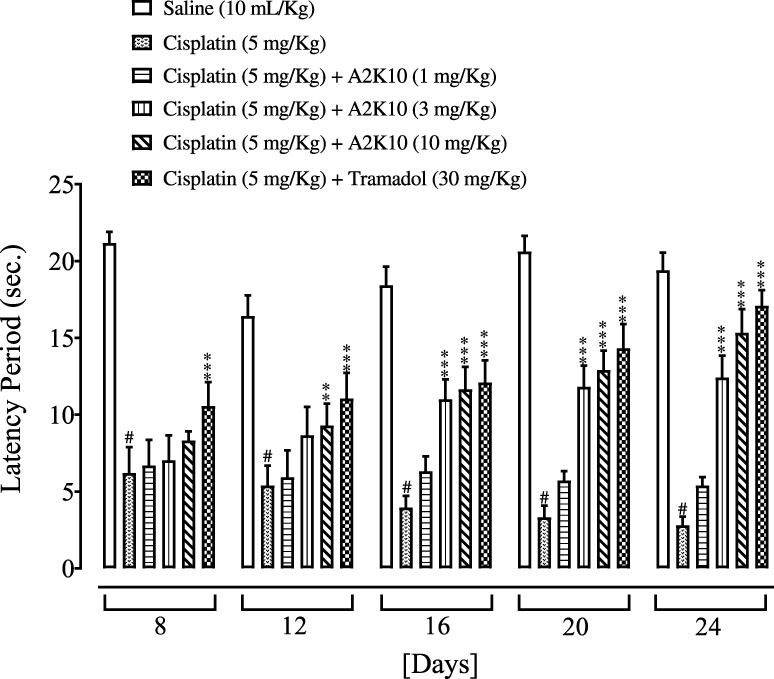 |
Figure 11 Bar chart showing effects of (E)-2-(4-methoxybenzylidene)cyclopentan-1-one (A2K10) and tramadol on mouse escape latency in cisplatin-induced neuropathic pain. Data presented as means ± SEM (n=5). #p<0.001 vs saline group and **p<0.01 and ***p<0.001 vs cisplatin group on one-way ANOVA with Tukey’s post hoc test. |
Effect on Paw-Withdrawal Threshold
A2K10 dose-dependently (1–10 mg/kg) suppressed cisplatin-induced mechanical allodynia. The group treated with saline (10 mL/kg) showed PWT at days 0, 8, 12, 16, 20, and 24 of 2±0, 2±0, 2±0, 2±0, 2±0, and 2±0 g, respectively. In the cisplatin (5 mg/kg)-injected group, mechanical PWT had reduced to 2±0 (p<0.001 vs saline group), 1.4±0.15 (p<0.001 vs saline group), 0.9±0.08 (p<0.001 vs saline group), 0.6±0.06 (p<0.001 vs saline group), 0.6±0.06 (p<0.001 vs saline group), and 0.3±0.05 g (p<0.001 vs saline group) g, respectively. In the A2K10 (1 mg/kg) group, mechanical PWT had increased to 2±0, 1.2±0.08, 0.9±0.11, 0.9±0.11, 0.9±0.08, and 0.6±0.08 g, respectively. In the A2K10 (3 mg/kg) group, mechanical PWT had increased to 2±0, 1.1±0.1, 0.9±0.14, 1±0.1 (p<0.01 vs cisplatin group), 1.1±0.1, and 0.7±0.08 g, respectively. In the A2K10 (10 mg/kg) group, mechanical PWT had further increased to 2±0, 1.2±0.08, 1.2±0, 1.2±0.11, 1.3±0.13 (p<0.001 vs cisplatin group), and 1.5±0.06 g (p<0.001 vs cisplatin group), respectively. In the tramadol (30 mg/kg)-injected group, mechanical PWT was 2±0, 1.3±0.1 (p<0.001 vs cisplatin group), 1.9±0.06 (p<0.001 vs cisplatin group), 1.9±0.06 (p<0.001 vs cisplatin group), 1.9±0.06 (p<0.001 vs cisplatin group), and 1.9±0.05 (p<0.001 vs cisplatin group) g, respectively (Figure 12).
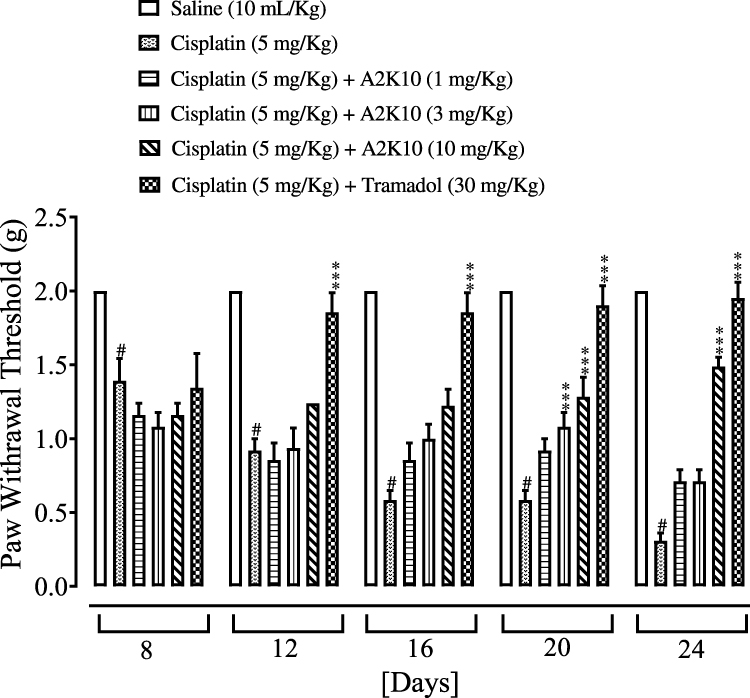 |
Figure 12 Bar chart showing effects of (E)-2-(4-methoxybenzylidene)cyclopentan-1-one (A2K10) and tramadol on mouse paw–withdrawal threshold in cisplatin-induced neuropathic pain. Data presented as means ± SEM (n=5). #p<0.001 vs saline group and ***p<0.001 vs cisplatin group on one-way ANOVA with Tukey’s post hoc test. |
Acute Toxicity
A2K10 caused no mortality at a dose of 500 mg/kg.
Discussion
This study was performed to investigate docking, anti-AD, antiepileptic, anti-PD, and analgesic effects of A2K10 in mice. An important component of drug-discovery programs is computational analysis. The n silico analytical tool PatchDock was used to assess the binding affinity of A2K10, taking into consideration ACE values, number of hydrogen bonds, and amino-acid residues forming hydrogen bonds against protein targets. Depending upon the ACE values and number of hydrogen bonds, postdocking analysis revealed a binding-affinity order of A2K10 against target proteins involved in AD of NMDA > COMT > AChE > BACE > BuChE. Among target proteins interceding epilepsy, the ligand receptor–affinity order was GABAA > Orai > DUSP13. GABAA has good binding affinity. The ligand’s order of binding affinity against PD-associated receptors was M1 > H1 > D2 > GABAB. Computational analysis of A2K10 was also carried out against receptors involved in pain. The order of binding affinity was α7nAChR > TNFα > TLR4 > PPARγ > COX2 > α4β2 > PLA2. α7nAChR had the maximum ACE value with no hydrogen bond.
DPPH assays were used to check antioxidant potential. Discoloration of the solution indicated the DPPH free radical–scavenging effect. Studies have shown that the free radical–scavenging mechanism depends upon the hydroxyl groups present on the antioxidant molecule. Increased DPPH free radical–scavenging activity showed antioxidant properties.16 A2K10 showed a slight increase in DPPH free radical–scavenging effect.
In order to study anti-AD activity, MWM and Y-maze tests were used.10,11 Memory impairment can be due to neurodegeneration in hippocampus and presence of amyloid plaques in the hippocampus of the brain. Spontaneous alteration behavior was studied to check memory. Enhanced cognitive performance is associated with increased of spontaneous alteration behavior.11 A2K10 dose-dependently increased the number of entries and spontaneous alteration behavior of mice in the Y-maze test. Anti-AD activity of A2K10 was revealed by its dose-dependent increase in escape latency in the MWM. Mostly neurofibrillary lesions, β-amyloid plaques, inflammatory processes, acetylcholine deficiency, and damaged cholinergic neurons are involved in AD.17 A2K10 possesses good binding affinity against NMDA receptors, and studies have shown that memory and learning can be enhanced by blockade of NMDA receptors.18
In accordance with the in silico studies, the antiepileptic action of A2K10 can be due to the mechanism of GABAA activation. Valproic acid is the main antiepileptic drug that mainly inhibits sodium channels and increases brain levels of GABA. GABAergic neurons in the thalamus are mainly involved in the control of seizures. Ptz is a GABAA (inhibitory neurotransmitter) antagonist that induces seizures by reducing GABAergic neurotransmission.19 A2K10 caused dose-dependent delays in the onset of myoclonic and tonic–clonic convulsions, along with a reduction in the duration of tonic–clonic convulsions and decreased mortality of mice. A2K10 showed lower antiepileptic activity compared to diazepam, suggesting that antiepileptic action of the test compound was possibly due to the activation of GABAA receptors.
In silico studies showed that M1 and H1 were binding sites that might be involved in the anti-PD effect of A2K10. PD is characterized mainly by the degeneration of neurons in the substantia nigra of brain accompanied by reduced dopamine release and movement disorders, including tremors and slow movement. Haloperidol is an antipsychotic drug that induces PD by blocking dopamine receptors, resulting in dopamine turnover followed by oxidative stress.20 The hanging test was used to check neuromuscular strength, and A2K10 dose-dependently increased the hanging timeand reduced VCMs in mice. Studies have shown that the cholinergic system is involved in maintaining functioning of basal ganglia, and muscarinic antagonists were used in PD treatment.21 A2K10 showed more effect compared to levodopa which may be due to binding affinity with NMDA and M1 receptors. α7nAChR, PPARγ, TNFα, and COX2 were the main sites with good binding affinity, so they might be involved in the analgesic effect of A2K10. Cisplatin is involved in most types of tumors. Among common chemotherapeutic drugs used in cancer chemotherapy, it has good therapeutic efficacy against many agents. Platinum-based drugs in particular have a dose-limiting side effect known as chemotherapy-induced neuropathic pain. Painful sensory neuropathy produced in the distal extremities are mainly produced by platinum-based drugs, ie cisplatin and oxaliplatin.22 Production of proinflammatory cytokines is modulated by the activation ofα7nAChR,23 and antinociception to acute mechanical and thermal stimuli can be produced by these receptors.24 PPARγ agonists are involved in the reduction of neuropathic pain by inhibiting expression of chemokines that are inflammatory mediators.25 Prostaglandins produced by COX2 play an important role in enhancing pain and inflammation. Therefore, pain and inflammation can be reduced by blocking COX2 receptors.26
Conclusion
The present study reveals that A2K10 possesses high affinity against α7nAChR, NMDA, M1, and GABAA targets. In vitro and in vivo investigation revealed its antioxidant, antiepileptic, and analgesic effects, with positive responses against memory and cognitive deficits as well. As such, the therapeutic potential of A2K10 in various neurological disorders should be explored.
Acknowledgments
We are grateful to Dr Zia Ud Din for the gift of A2K10. We are thankful to Dr Muzaffar Abbas for his valuable guidance and Komal Naeem for her editorial cooperation.
Disclosure
The authors report no conflicts of interest for this work.
References
1. Sarfo SS, Akassi J, Badu E, et al. Profile of neurological disorders in an adult neurology clinic in Kumasi, Ghana. J Neurol Sci. 2016;3:69–74.
2. Bales KR, Tzavara ET, Wu S, et al. Cholinergic dysfunction in a mouse model of Alzheimer disease is reversed by an anti-abantibody. J Clin Invest. 2006;116(3):825–832. doi:10.1172/JCI27120
3. Saxena S, Li S. Defeating epilepsy: a global public health commitment. Epilepsia Open. 2017;2(2):153–155. doi:10.1002/epi4.12010
4. Abbas MM, Xu Z, Tan LCS. Epidemiology of Parkinson’s disease-east versus west. Mov Disord Clin Pract. 2017;5(1):14–28. doi:10.1002/mdc3.12568
5. Lin H, Heo BH, Yoon MH. A new rat model of cisplatin induced neuropathic pain. Korean J Pain. 2015;28(4):236–243. doi:10.3344/kjp.2015.28.4.236
6. Yeung AWK, Horbańczuk M, Tzvetkov NT, et al. Curcumin: total-scale analysis of the scientific literature. Molecules. 2019;24(7):1393. doi:10.3390/molecules24071393
7. Din ZU, Filho ER. Optimized one-pot synthesis of monoarylidene and unsymmetrical diarylidene cycloalkanones. Arab J Chem. 2016.
8. National Research Council. Guide for the Care and Use of Laboratory Animals. Washington: National Academy Press; 1996. 1–7.
9. Kumar S, Kumar V, Chandrashekhar MS. In vitro anti-oxidant and alpha-amylase inhibitory activity of isolated fractions from methanolic extract of Asystasiadalzelliana leaves. Int J Pharmtech Res. 2011;3:889–894.
10. Morris R. Developments of a water-maze procedure for studying spatial learning in the rat. J Neurosci Methods. 1984;1984(11):47–60. doi:10.1016/0165-0270(84)90007-4
11. Ali T, Badshah H, Kim TH, et al. Melatonin attenuates D-galactose-induced memory impairment, neuroinflammation and neurodegeneration via RAGE/NF-KB/JNK signaling pathway in aging mouse model. J Pineal Res. 2015;58(1):71–85. doi:10.1111/jpi.12194
12. Bum EN, Ngoupaye GT, Talla E, et al. The anticonvulsant and sedative properties of stems of Cissusquadrangularis in mice. Afr J Pharm Pharmacol. 2008;2:42–47.
13. Bagewadi HG, Khan A. Investigation of antiparkinsonian effect of Aloe vera on haloperidol induced experimental animal model. Indian J Pharm Biol Res. 2015;3(01):108–113. doi:10.30750/ijpbr.3.1.15
14. Marcus DJ, Zee M, Hughes A, et al. Tolerance to the antinociceptive effects of chronic morphine requires c-Jun N-terminal kinase. Mol Pain. 2015;11:15–31. doi:10.1186/s12990-015-0031-4
15. Baghel SS, Dangi S, Soni P, et al. Acute toxicity study of aqueous extract of Cocciniaindica (roots). Asian J Res Pharm Sci. 2011;1:23–25.
16. Mensor LL, Menezes FS, Leitao GG, et al. Screening of Brazilian plant extracts for antioxidant activity by the use of DPPH free radical method. Phytother Res. 2001;15(2):127–130. doi:10.1002/ptr.687
17. Lee GY, Lee C, Park GH, et al. Amelioration of scopolamine-induced learning and memory impairment by pinene in C57BL/6 mice. Evid Based Complementary Altern Med. 2017;1:1–9.
18. Li F, Tsien JZ. Memory and the NMDA receptors. N Engl J Med. 2009;361(3):302–303. doi:10.1056/NEJMcibr0902052
19. Okada R, Negishi N, Nagaya H. The role of the nigrotegmental GABAergic pathway in the propagation of pentylenetetrazol-induced seizures. Brain Res. 1989;480(1–2):383–387. doi:10.1016/0006-8993(89)90212-6
20. Saeed A, Shakir L, Khan MA, et al. Haloperidol induced Parkinson’s disease mice model and motor-function modulation with pyridine-3-carboxylic acid. Biomed Res Ther. 2017;4(05):1305–1317. doi:10.15419/bmrat.v4i05.169
21. Xiang Z, Thompson AD, Jones CK, et al. Roles of the M1 muscarinic acetylcholine receptor subtype in the regulation of basal ganglia function and implications for the treatment of Parkinson’s disease. J Pharmacol Exp Ther. 2011;340(3):595–603. doi:10.1124/jpet.111.187856
22. Joseph EK, Levine JD. Comparison of oxaliplatin- and cisplatin-induced painful peripheral neuropathy in the rat. J Pain. 2009;10(5):534–541. doi:10.1016/j.jpain.2008.12.003
23. Jonge WD, Ulloa L. The alpha7 nicotinic acetylcholine receptor as a pharmacological target for inflammation. Br J Pharmacol. 2007;151(7):915–929. doi:10.1038/sj.bjp.0707264
24. Gurun MS, Parkar R, Eisenach JC, et al. The effect of peripherally administered CDP-choline in an acute inflammatory pain model: the role of α7 nicotinic acetylcholine receptor. Int Anaesth Res Soc. 2009;108:1680–1687.
25. Freitag CM, Miller RJ. Peroxisome proliferator-activated receptor agonists modulate neuropathic pain: a link to chemokines? Front Cell Neurosci. 2014;8:238. doi:10.3389/fncel.2014.00238
26. Zarghi A, Arfaei S. Selective COX-2 inhibitors: a review of their structure-activity relationships. Iran J Pharm Res. 2011;10(4):655–683.
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.