Back to Journals » OncoTargets and Therapy » Volume 13
Comprehensive Analysis of circRNA-miRNA-mRNA Network in Cervical Squamous Cell Carcinoma by Integrated Analysis
Authors Wu Q, Liu P, Lao G, Liu Y, Zhang W, Ma C
Received 17 March 2020
Accepted for publication 26 July 2020
Published 25 August 2020 Volume 2020:13 Pages 8641—8650
DOI https://doi.org/10.2147/OTT.S254323
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Sanjay Singh
Qiongwei Wu, Ping Liu, Guoying Lao, Yu Liu, Wenying Zhang, Chengbin Ma
Gynecology Department, Changning Maternity and Infant Health Hospital, Shanghai, People’s Republic of China
Correspondence: Ping Liu; Chengbin Ma
Gynecology Department, Changning Maternity and Infant Health Hospital, No. 786, Yuyuan Road, Changning District, Shanghai 200051, People’s Republic of China
Tel +86-13661900956; +86-13661900280
Email [email protected]; [email protected]
Purpose: Cervical squamous cell carcinoma (CSCC) seriously affects women’s health worldwide, and it is of great significance to illuminate the specific role of circRNAs in CSCC.
Materials and Methods: Three mRNA datasets, two miRNA datasets and one circRNA dataset of CSCC, downloaded from GEO, were utilized in this study. Differentially expressed mRNAs (DEmRNAs), miRNAs (DEmiRNAs) and circRNAs (DEcircRNAs) were identified, and a ceRNA (DEcircRNA-DEmiRNA-DEmRNA) regulatory network was constructed. GO and pathway analyses of DEcircRNAs and DEmRNAs in the ceRNA regulatory network were performed. Quantitative real-time polymerase chain reaction (qRT-PCR) validation of the expression of the selected DEmRNAs, DEmiRNAs and DEcircRNAs was performed.
Results: A total of 1356 DEmRNAs, 13 DEmiRNAs and 77 DEcircRNAs were obtained. The ceRNA network contained 3 circRNA-miRNA pairs and 158 miRNA-mRNA pairs, including 3 circRNAs, 3 miRNAs, and 138 mRNAs. Functional annotation of DEmRNAs in the ceRNA regulatory network revealed that these DEmRNAs were significantly enriched in cell cycle, p53 signalling pathway and DNA replication. The qRT-PCR results were generally consistent with those of our integrated analysis.
Conclusion: In conclusion, we speculate that the regulation of the hsa_circ_0000069/hsa-miR-125b-5p/CDKN2A and hsa_circ_0020594/hsa-let-7c-5p/CCNB2 axes may be involved in CSCC.
Keywords: cervical squamous cell carcinoma, CSCC, circRNA, ceRNA network, GEO
Introduction
Cervical cancer (CC) is one of the most widespread gynaecological cancers that threatens women’s health in developing countries.1 CC progresses through a long stage of precancerous lesions, called cervical intraepithelial neoplasia (CIN), which is classified into three grades: CIN 1, CIN 2 and CIN 3.2 Human papillomaviruses (HPVs) have been widely considered the main cause of CC.3 Cervical squamous cell carcinoma (CSCC) is the most frequent type, accounting for the majority of CCs.4 The molecular mechanisms of CSCC are complex and have not yet been fully elucidated. Hence, it is necessary to deeply understand the molecular mechanisms and pathways of CSCC progression.
In contrast to traditional linear RNAs, circRNAs are covalently closed circular molecules that have attracted wide attention from researchers.5 CircRNA, as one type of competing endogenous RNA (ceRNA), can bind to miRNAs, acting as a “miRNA sponge”, and regulate their target genes at the transcriptional or post-transcriptional level, thereby affecting the biological behaviour of tumours.6 The advent of microarray-based technology has facilitated studies on the molecular mechanisms of different tumours.7 However, investigations on CSCC are still limited, particularly with regard to circRNAs in CSCC. Wang et al suggested that hsa_circ_0101996 combined with hsa_circ_0101119 can serve as potential biomarkers for CSCC detection.8 Yi et al identified five circRNAs that may play critical roles in CC.1 Ma et al revealed that circRNA-000284 is involved in cell proliferation and invasion of CC by sponging miR-506.9 Therefore, more CSCC-related circRNAs need to be unearthed.
The current study performed an integrated analysis with datasets of mRNA, miRNA and circRNA expression profiles of CSCC collected from the GEO database. The identification of differentially expressed mRNAs (DEmRNAs), miRNAs (DEmiRNAs) and circRNAs (DEcircRNAs) and the construction of a ceRNA (DEcircRNA-DEmiRNA-DEmRNA) regulatory network were conducted. This present work sought to help elucidate the underlying mechanisms of CSCC pathogenesis and provide additional knowledge for CSCC.
Materials and Methods
Dataset Collection
To acquire the mRNA, miRNA and circRNA expression profiles of CSCC tumour tissues and normal cervix tissues, datasets in the GEO database with the following criteria were retrieved: selected datasets should be whole-genome mRNA/miRNA/circRNA expression profiles by array; these data were derived from CSCC tumour tissues and normal cervix tissues; (3) datasets were normalized or original. Three mRNA datasets, two miRNA datasets and one circRNA dataset were enrolled in this study (Table 1).
 |
Table 1 List of mRNA/miRNA/circRNA Study Samples from GEO |
Differential Expression Analysis
The raw data were visualized with the hist function, normalized with the log function, and centralized and standardized with the scale function. We calculated differential expression p-values and effect sizes from data either from classical or moderated t-tests (Limma, SMVar) for study and combined these p-values by the inverse normal method. Multiple comparison correction was performed by the Benjamini and Hochberg method to obtain the false discovery rate (FDR). By default, the FDR is controlled at 5%. MetaMA was utilized to acquire the DEmRNAs and DEmiRNAs with FDR < 0.05 and |Combined.ES| > 1. The Limma package in R was applied to acquire the DEcircRNAs with FDR < 0.05 and |log2 fold change| > 1. With the R package “pheatmap”, hierarchical clustering analysis of DEmRNAs, DEmiRNAs and DEcircRNAs was performed.
GO and Pathway Analysis of Host Genes of DEcircRNAs
GeneCodis3 (http://genecodis.cnb.csic.es/analysis) was applied to perform GO and KEGG pathway enrichment analysis of host genes of DEcircRNAs. The threshold was set as FDR < 0.05.
Construction of the ceRNA (DEcircRNA-DEmiRNA-DEmRNA) Regulatory Network
According to the results of the differential expression analysis, DEcircRNA–DEmiRNA interaction pairs were calculated with starBase v3.0 (http://starbase.sysu.edu.cn). The targeted DEmRNAs of DEmiRNAs were predicted with six bioinformatic algorithms (RNA22, miRanda, miRDB, miRWalk, PICTAR2 and TargetScan). Then, with miRWalk, the confirmed targeted mRNAs of miRNAs were obtained. Finally, the confirmed DEmiRNA-DEmRNA pairs derived from miRWalk and the DEmiRNA-DEmRNA pairs predicted by ≥4 algorithms were retained for network construction. The ceRNA (DEcircRNA-DEmiRNA-DEmRNA) regulatory network was constructed by combining circRNA-miRNA pairs with miRNA-mRNA pairs. Ultimately, Cytoscape 3.5.0 (http://cytoscape.org/) was used to visualize the regulatory network. With GeneCodis3, GO and KEGG pathway analyses of all DEmRNAs in the ceRNA regulatory network were performed. Statistical significance was defined as FDR < 0.05.
Validation in Quantitative Real-Time Polymerase Chain Reaction (qRT-PCR) and TCGA Database
Six paired CSCC tumour tissues and corresponding adjacent non-tumour tissues were obtained from 6 patients with CSCC. The clinical characteristics of the individuals included in this study are displayed in Table S1. We obtained written informed consent from every participant. This study was approved by the ethics committee of Changning Maternity and Infant Health Hospital and in accordance with the 1964 Helsinki declaration and its later amendments or comparable ethical standards.
Total RNA was isolated with the TRIzol reagent (Invitrogen, USA) following the manufacturer’s protocol. The qRT-PCRs were performed in an ABI 7300 Real-time PCR Detection System with SuperReal PreMix Plus (Invitrogen, USA). Relative gene expression was analysed by the 2-ΔΔCT method. Human GAPDH and ACTB were used as endogenous controls for mRNA expression in the analysis. Human U6 and GAPDH were used as endogenous controls for miRNA and circRNA expression in the analysis, respectively. The qRT-PCR was performed in triplicate. The p-value was assessed by one-way analysis of variance (ANOVA), and p < 0.05 was considered the criterion of statistical significance.
The mRNA and miRNA profiles of 252 CSCC cases and 2 adjacent non-tumour controls derived from the TCGA database were downloaded to validate the expression levels of selected DEmRNAs and DEmiRNAs. The difference in expression levels is displayed in box plots. In addition, to further investigate the prognostic value of selected DEmiRNAs and DEmRNAs, survival analysis was performed by using the survival package in R.
Results
DEmRNAs, DEmiRNAs and DEcircRNAs in CSCC Tumour Tissues Compared to Normal Cervix Tissues
A total of 1356 DEmRNAs (750 up- and 606 down-regulated mRNAs) and 13 DEmiRNAs (4 up- and 9 down-regulated miRNAs) were identified in CSCC compared to normal cervix tissues with FDR < 0.05 and |Combined.ES| > 1 (Figure 1A and B). Among them, CDKN2A and UPK1A were the most up-regulated and down-regulated mRNAs; hsa-miR-106b-5p and hsa-miR-125b-5p were the most up-regulated and down-regulated miRNAs, respectively (Table 2). A total of 77 DEcircRNAs (45 up- and 32 down-regulated circRNAs) were obtained in CSCC with FDR < 0.05, |log2 fold change| > 1 (Figure 1C). Among them, hsa_circRNA_104052 and hsa_circRNA_103384 were the most up-regulated and down-regulated circRNAs, respectively (Table 3).
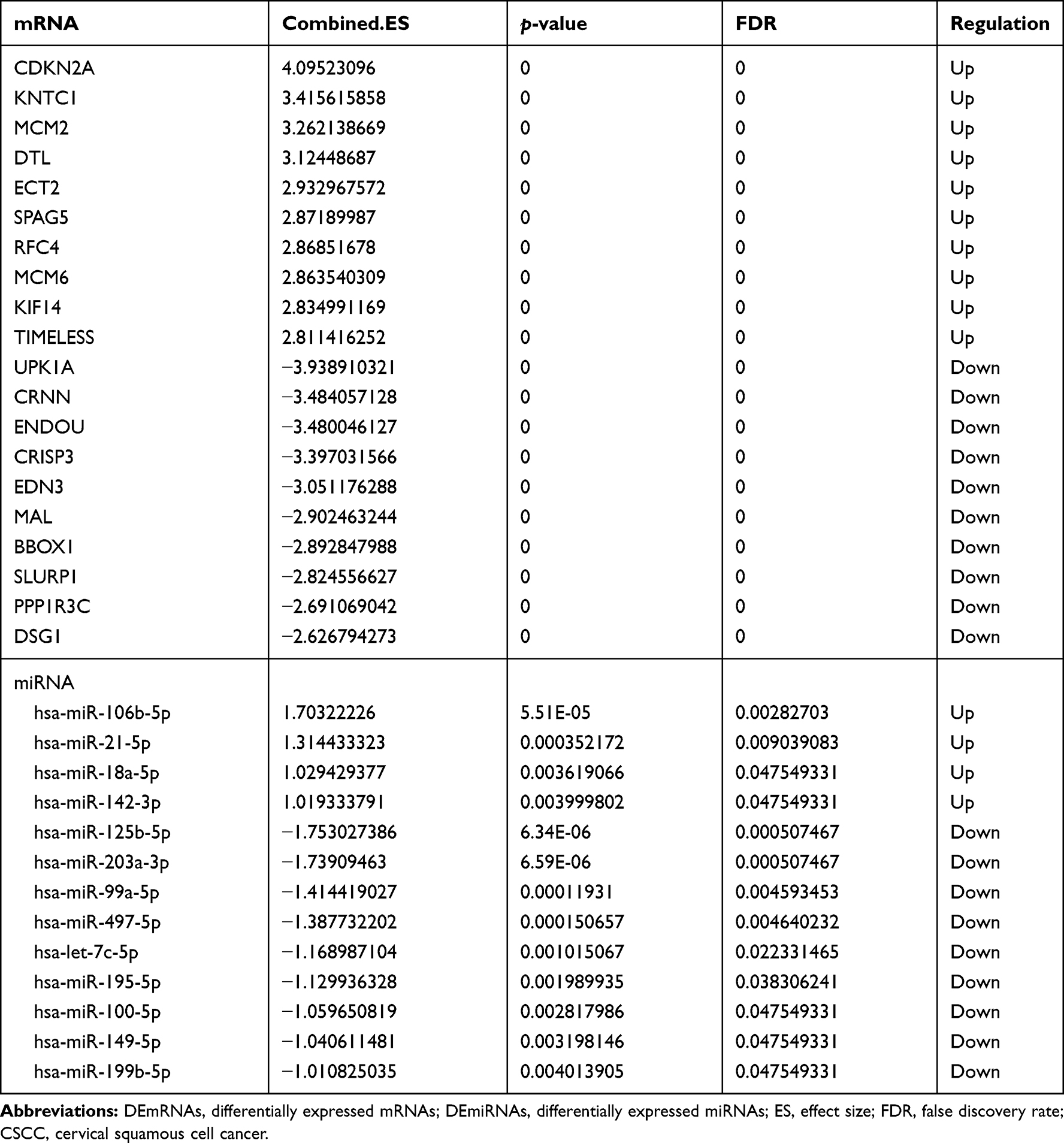 |
Table 2 Top 10 Up- and Down-Regulated DEmRNAs and DEmiRNAs in CSCC |
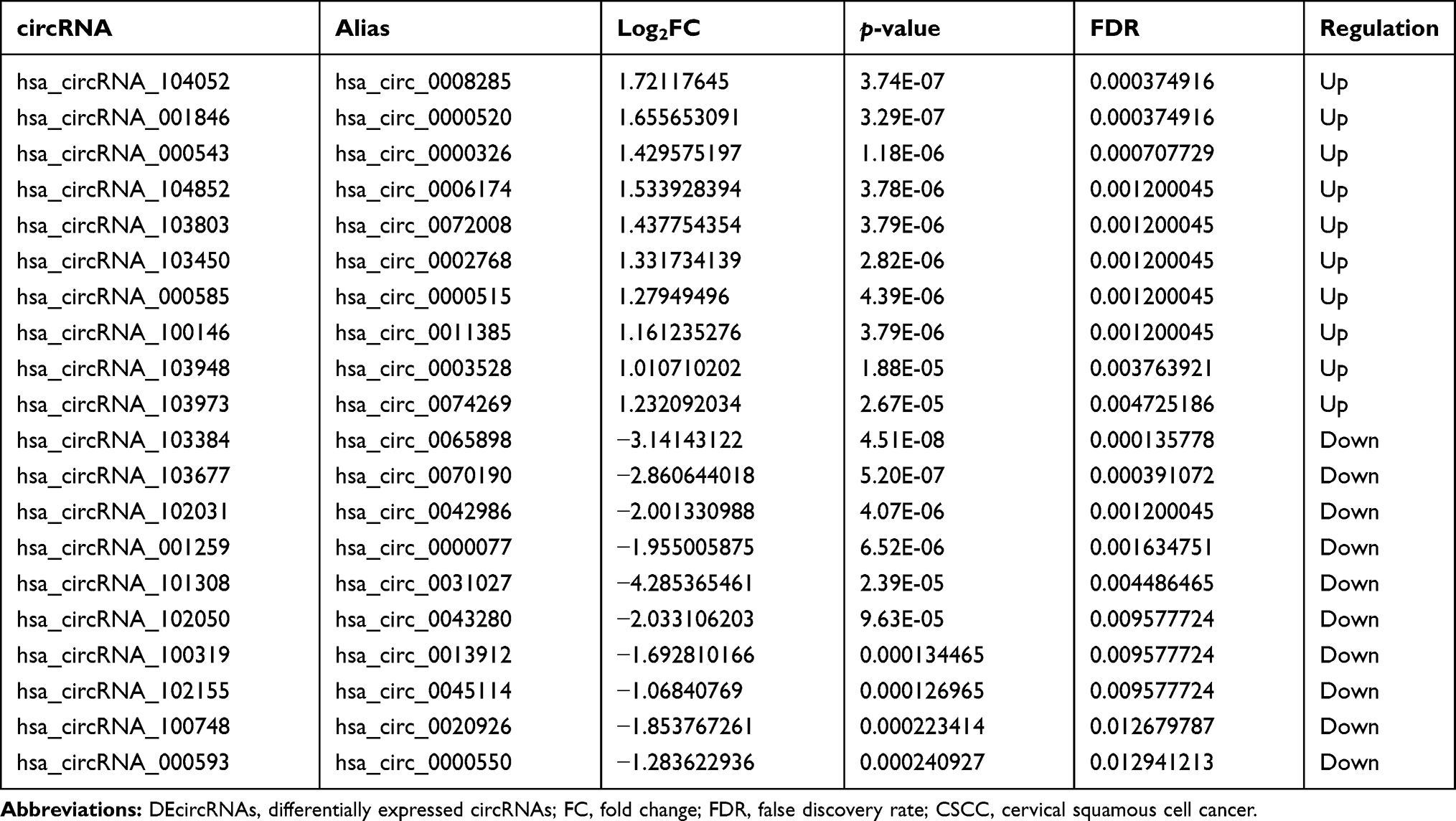 |
Table 3 Top 10 Up- and Down-Regulated DEcircRNAs in CSCC |
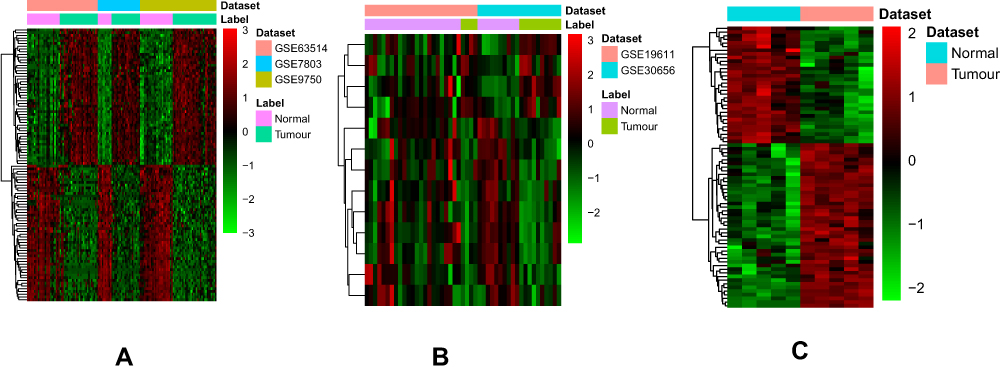 |
Figure 1 The heatmap of top 100 up- and down-regulated DEmRNAs (A), DEmiRNAs (B) and DEcircRNAs (C) between CSCC and normal cervix tissues. |
Functional Annotation of Host Genes of DEcircRNAs
A total of 73 host genes of DEcircRNAs were obtained. GO analysis indicated that these host genes were significantly enriched in platelet activation (FDR = 1.32E-02), protein phosphorylation (FDR = 1.35E-02), nucleus (FDR = 6.49E-06) and protein binding (FDR = 4.48E-04) (Figure 2A–C). According to the KEGG pathway enrichment analysis, several pathways were significantly enriched, including Protein processing in endoplasmic reticulum (FDR = 7.74E-04), RNA polymerase (FDR = 3.15E-02) and RNA transport (FDR = 3.42E-02) (Figure 2D).
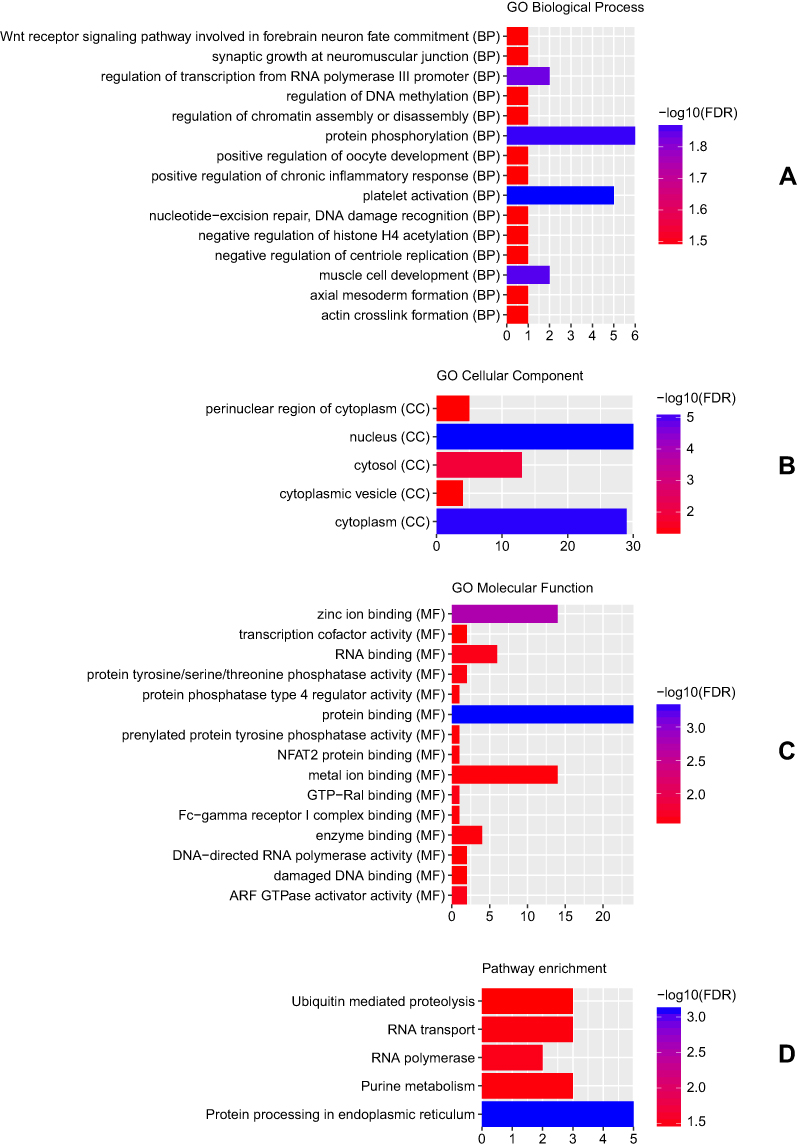 |
Figure 2 Significantly enriched GO terms and KEGG pathways of host genes of DEcircRNAs between CSCC tumour tissues and normal cervix tissues. (A) BP, biological process; (B) CC, cellular component; (C) MF, molecular function; (D) KEGG pathways. The x-axis shows counts of host genes enriched in GO terms or KEGG pathways and the y-axis shows GO terms or KEGG pathways. The colour scale represented -lg FDR. |
CeRNA (DEcircRNA-DEmiRNA-DEmRNA) Regulatory Network
The ceRNA network contained 3 circRNA-miRNA pairs and 158 miRNA-mRNA pairs, including 3 circRNAs, 3 miRNAs, and 138 mRNAs (Figure 3). GO enrichment analysis revealed that these DEmRNAs in ceRNA regulatory network were significantly enriched in DNA replication (FDR = 2.84E-14), mitotic cell cycle (FDR = 4.05E-10), nucleus (FDR = 1.50E-25) and protein binding (FDR = 4.52E-14) (Figure 4A–C). According to the KEGG pathway enrichment analysis, several pathways were significantly enriched, including Cell cycle (FDR = 1.23E-07), p53 signaling pathway (FDR = 2.71E-04) and DNA replication (FDR = 4.60E-03) (Figure 4D).
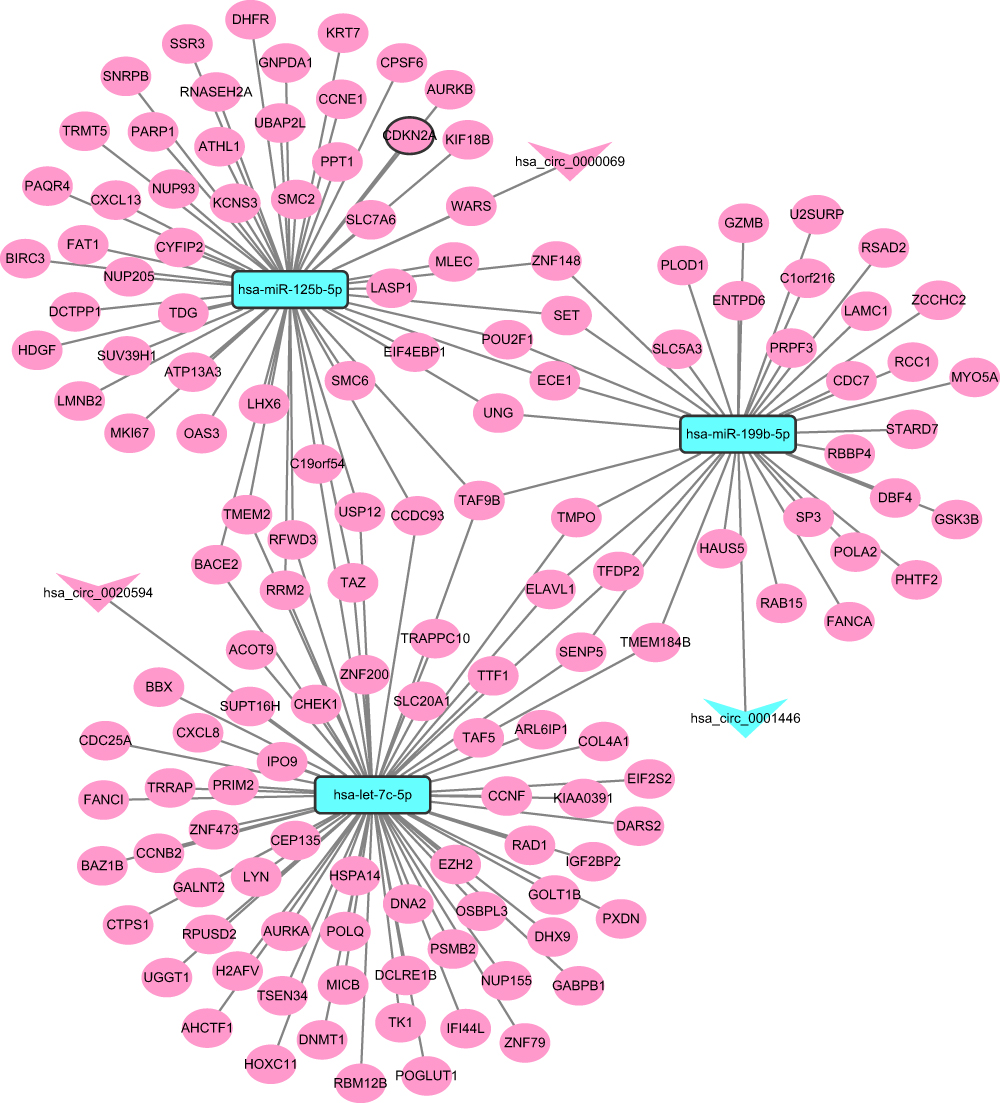 |
Figure 3 CeRNA (DEcircRNA-DEmiRNA-DEmRNA) regulatory network. The inverted triangles, rectangle and elliptical nodes indicate DEcircRNAs, DEmiRNAs and DEmRNAs, respectively. Red and blue colour represent up-regulation and down-regulation, respectively. Nodes with black border were DEcircRNA/DEmiRNA/DEmRNA derived from top 10 up- and down-regulated DEcircRNA/DEmiRNA/DEmRNA between CSCC tumour tissues and normal cervix tissues. |
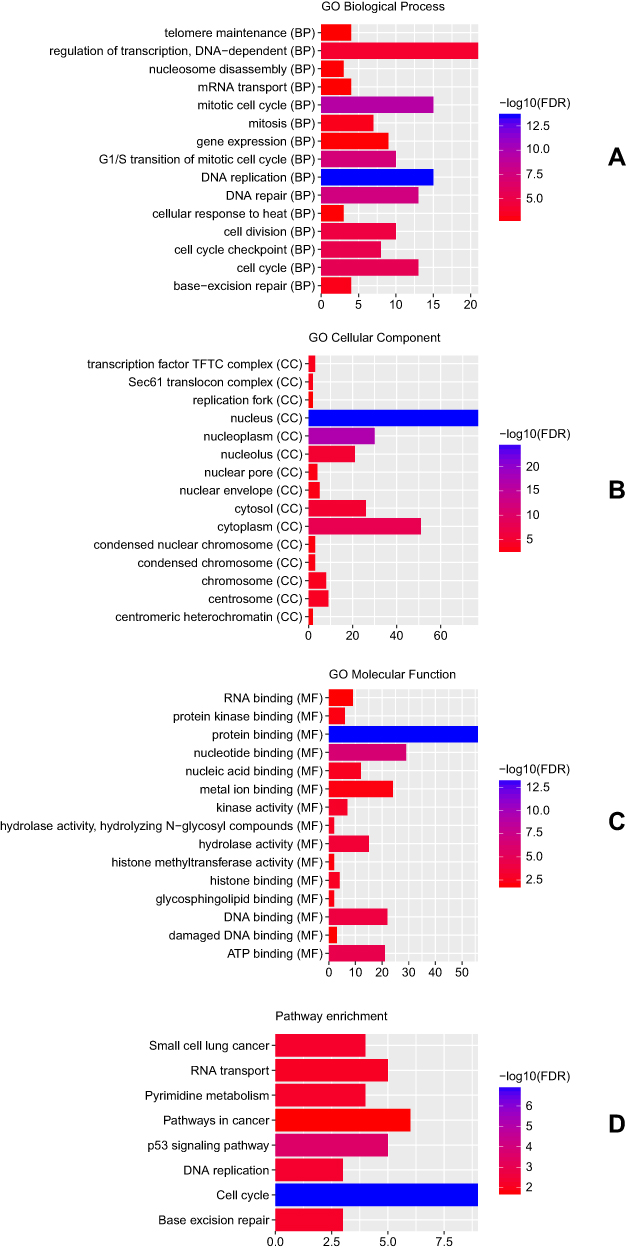 |
Figure 4 Significantly enriched GO terms and KEGG pathways of DEmRNAs in ceRNA regulatory network. (A) BP, biological process; (B) CC, cellular component; (C) MF, molecular function; (D) KEGG pathways. The x-axis shows counts of DEmRNAs in ceRNA regulatory network enriched in GO terms or KEGG pathways and the y-axis shows GO terms or KEGG pathways. The colour scale represented -lg FDR. |
Validation in qRT-PCR and TCGA Database
Three DEmRNAs (CCNB2, RRM2 and CDKN2A), three DEmiRNAs (hsa-miR-199b-5p, hsa-miR-125b-5p and hsa-let-7c-5p) and one DEcircRNA (hsa-circ-0000069) were selected to perform validation in qRT-PCR and TCGA. Based on our integrated analysis, CCNB2, RRM2, CDKN2A and hsa-circ-0000069 were up-regulated while hsa-miR-199b-5p, hsa-miR-125b-5p and hsa-let-7c-5p were down-regulated in CSCC. The qRT-PCR results were consistent with those of our integrated analysis (Figure 5). In addition, the validation of CDKN2A, CCNB2, hsa-let-7c-5p and hsa-miR-125b-5p in TCGA database was consistent with our integrated analysis as well (Figure S1). There was no data available for hsa_circ_0020594 and hsa_circ_0000069 in the TCGA datasets. Based on the results of survival analysis, the expression of hsa-let-7c-5p (p < 0.0001) and hsa-miR-125b-5p (p = 0.00045) was significantly correlated with the overall survival time of CSCC patients (Figure S2).
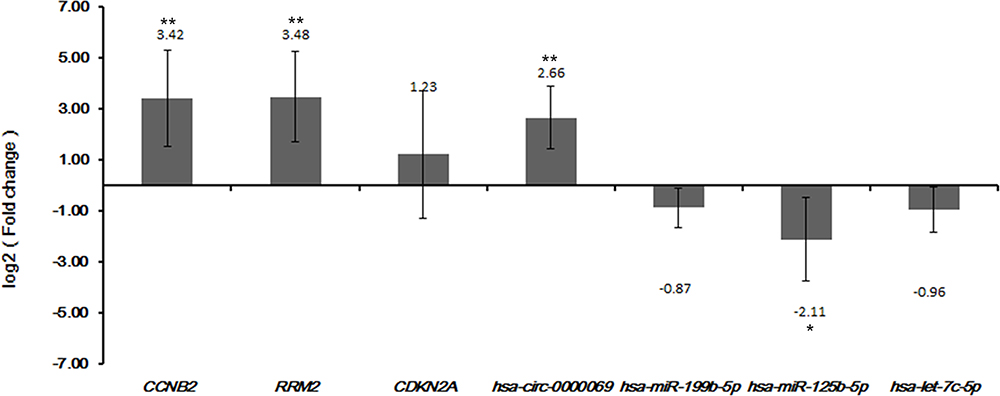 |
Figure 5 The quantitative real-time polymerase chain reaction (qRT-PCR) results of the DEmRNAs, DEmiRNAs and DEcircRNAs in CSCC tumour tissues. The x-axis represents the DEmRNAs/DEmiRNAs/DEcircRNAs and the y-axis represents log2 (fold change). The p-value was assessed by one-way analysis of variance (ANOVA). *Indicates p < 0.05 and **Indicates that p < 0.01. |
Discussion
CSCC, with a high recurrence rate and poor prognosis, seriously affects women’s health worldwide.10 Despite the rapid development of study on CSCC, the five-year survival rate remains not optimistic.11 As the specific molecular mechanism remains unclear, it is of great clinical diagnostic significance for illuminating the specific role of circRNAs by serving as ceRNAs in CSCC.
CDKN2A (full name: cyclin-dependent kinase inhibitor 2A), mapped to chromosome 9, codes for the p16INK4A and p14ARF proteins, and can induce cell cycle arrest.12 Liu et al linked overexpression of p16INK4A with poor prognosis of CC.13 Niu et al found that up-regulation of CDKN2A was observed in both high-grade squamous intraepithelial lesions (HSILs) and squamous cell carcinomas (SCCs), which suggested that CDKN2A may contribute to the development of CC.2 Our analysis, qRT-PCR validation and TCGA validation results consistently showed higher expression levels of CDKNA2 in CSCC, which indicated its critical role in CSCC.
The B-type cyclins, including B1 (CCNB1) and B2 (CCNB2), belong to cyclin family and are essential for cell cycle regulation. Increased CCNB2 is associated with various types of cancer, including CC.14 Compared with normal and CIN1/CIN2, CCNB2 was identified to be highly expressed in tumours and CIN3/CIS by using a microarray technique, which indicated the crucial role of CCNB2 in the early phase of tumorigenesis.15 A previous study reported that overexpressed CCNB2, belonging to the mitosis pathway, was involved in cell cycle and closely associated with CC.14 Similarly, a comparative analysis suggested that up-regulated CCNB2 promoted cell cycle transition in CC as well.16 In this analysis, up-regulated CCNB2 was detected in bioinformatics analysis, qRT-PCR validation, and TCGA validation results, which suggested the importance of CCNB2 in CSCC.
It has been reported that hsa-miR-125b-5p showed controversial properties in different tumours. For instance, compared with adjacent tissues, hsa-miR-125b-5p showed up-regulated trend in stage I lung adenocarcinoma tissues.17 Down-regulation of hsa-miR-125b-5p was observed in bladder cancer, which may be involved in the development of bladder cancer.18 Decreased hsa-miR-125b-5p was observed in CC cells.19 In contrast, hsa-let-7c-5p showed consistent trends in a variety of tumours. Down-regulated hsa-let-7c-5p was observed in endometrioid endometrial carcinoma.20 Significant decrease hsa-let-7c-5p was detected in prostatic cancer.21 QRT-PCR analysis showed that hsa-let-7c-5p was down-regulated in grade 3 endometrial cancer.22 Let-7c showed a down-regulated trend in cervical intraepithelial lesions.23 In addition, both hsa-miR-125b-5p and hsa-let-7c-5p were indicated to be related to the prognosis of colon cancer.24 Dysregulated hsa-miR-125b-5p and hsa-let-7c-5p were detected by three approaches, including integrated analysis, validation in qRT-PCR and validation with TCGA, in the current study, which indicated their crucial roles in CSCC. In addition, both hsa-miR-125b-5p and hsa-let-7c-5p were determined to be significantly correlated with the overall survival time of CSCC patients.
In 2016, Guo et al reported that overexpressed hsa_circ_0000069 was observed in colorectal cancer and correlated to patients’ age and TNM stage.25 As expected, knockdown of hsa_circ_0000069 could inhibit cell proliferation, migration, and invasion in vitro.25 However, there is no previous report on hsa_circ_0020594.
In this analysis, CDKN2A was targeted by hsa-miR-125b-5p, and CCNB2 was targeted by hsa_circ_0020594, respectively. Then, we found that hsa_circ_0000069 and hsa_circ_0020594 may act as ceRNAs to capture hsa-miR-125b-5p and hsa-let-7c-5p. Therefore, we concluded that the regulation of the hsa_circ_0000069/hsa-miR-125b-5p/CDKN2A and hsa_circ_0020594/hsa-let-7c-5p/CCNB2 axes may be involved in CSCC. This study laid the foundation for further research on ceRNAs of CSCC, and these findings need to be verified in the future. Our study also had a limitation. The sample size used for qRT-PCR validation in this study was small. Although the results were validated by qRT-PCR and TAGA analysis and shown to generally exhibit the same pattern as those in our integrated analysis, studies with larger sample sizes are needed to confirm these findings.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Yi Y, Liu Y, Wu W, Wu K, Zhang W. Reconstruction and analysis of circRNAmiRNAmRNA network in the pathology of cervical cancer. Oncol Rep. 2019;41(4):2209–2225. doi:10.3892/or.2019.7028
2. Niu G, Wang D, Pei Y, Sun L. Systematic identification of key genes and pathways in the development of invasive cervical cancer. Gene. 2017;618:28–41. doi:10.1016/j.gene.2017.03.018
3. Bava SV, Thulasidasan AK, Sreekanth CN, Anto RJ. Cervical cancer: a comprehensive approach towards extermination. Ann Med. 2016;48(3):149–161. doi:10.3109/07853890.2016.1145796
4. Zhou J, Liu X, Wang C, Li C. The correlation analysis of miRNAs and target genes in metastasis of cervical squamous cell carcinoma. Epigenomics. 2018;10(3):259–275. doi:10.2217/epi-2017-0104
5. Huang M, Zhong Z, Lv M, Shu J, Tian Q, Chen J. Comprehensive analysis of differentially expressed profiles of lncRNAs and circRNAs with associated co-expression and ceRNA networks in bladder carcinoma. Oncotarget. 2016;7(30):47186–47200. doi:10.18632/oncotarget.9706
6. Salmena L, Poliseno L, Tay Y, Kats L, Pandolfi PP. A ceRNA hypothesis: the rosetta stone of a hidden RNA language? Cell. 2011;146(3):353–358. doi:10.1016/j.cell.2011.07.014
7. Wang Y, Wu N, Liu J, Wu Z, Dong D. FusionCancer: a database of cancer fusion genes derived from RNA-seq data. Diagn Pathol. 2015;10:131. doi:10.1186/s13000-015-0310-4
8. Wang Y, Huang L, Li D, et al. Hsa_circ_0101996 combined with hsa_circ_0101119 in peripheral whole blood can serve as the potential biomarkers for human cervical squamous cell carcinoma. Int J Clin Exp Pathol. 2017;10(12):11924–11931.
9. Ma HB, Yao YN, Yu JJ, Chen XX, Li HF. Extensive profiling of circular RNAs and the potential regulatory role of circRNA-000284 in cell proliferation and invasion of cervical cancer via sponging miR-506. Am J Transl Res. 2018;10(2):592–604.
10. Ma D, Cheng Y, Zhang Y, Guo Y, Li Z, Li G. Expression of CDC42 in cervical squamous cell carcinoma and its correlation with clinicopathologic characteristics. Chin J Cancer Res. 2013;25(6):656–661. doi:10.3978/j.issn.1000-9604.2013.11.04
11. Pan XB, Lu Y, Huang JL, Long Y, Yao DS. Prognostic genes in the tumor microenvironment in cervical squamous cell carcinoma. Aging. 2019;11(22):10154–10166. doi:10.18632/aging.102429
12. Kanao H, Enomoto T, Ueda Y, et al. Correlation between p14(ARF)/p16(INK4A) expression and HPV infection in uterine cervical cancer. Cancer Lett. 2004;213(1):31–37. doi:10.1016/j.canlet.2004.03.030
13. Liu HQ, Wang YH, Wang LL, Hao M. P16INK4A and survivin: diagnostic and prognostic markers in cervical intraepithelial neoplasia and cervical squamous cell carcinoma. Exp Mol Pathol. 2015;99(1):44–49. doi:10.1016/j.yexmp.2015.04.004
14. Espinosa AM, Alfaro A, Roman-Basaure E, et al. Mitosis is a source of potential markers for screening and survival and therapeutic targets in cervical cancer. PLoS One. 2013;8(2):e55975. doi:10.1371/journal.pone.0055975
15. Rajkumar T, Sabitha K, Vijayalakshmi N, et al. Identification and validation of genes involved in cervical tumourigenesis. BMC Cancer. 2011;11:80. doi:10.1186/1471-2407-11-80
16. Cheng J, Lu X, Wang J, Zhang H, Duan P, Li C. Interactome analysis of gene expression profiles of cervical cancer reveals dysregulated mitotic gene clusters. Am J Transl Res. 2017;9(6):3048–3059.
17. Zeybek A, Oz N, Kalemci S, et al. Diagnostic value of MiR-125b as a potential biomarker for stage I lung adenocarcinoma. Curr Mol Med. 2019;19(3):216–227. doi:10.2174/1566524019666190314113800
18. Canturk KM, Ozdemir M, Can C, et al. Investigation of key miRNAs and target genes in bladder cancer using miRNA profiling and bioinformatic tools. Mol Biol Rep. 2014;41(12):8127–8135. doi:10.1007/s11033-014-3713-5
19. Jin XJ, Chen XJ, Zhang ZF, et al. Long noncoding RNA SNHG12 promotes the progression of cervical cancer via modulating miR-125b/STAT3 axis. J Cell Physiol. 2019;234(5):6624–6632. doi:10.1002/jcp.27403
20. Xiong H, Li Q, Liu S, et al. Integrated microRNA and mRNA transcriptome sequencing reveals the potential roles of miRNAs in stage I endometrioid endometrial carcinoma. PLoS One. 2014;9(10):e110163. doi:10.1371/journal.pone.0110163
21. Knyazev EN, Fomicheva KA, Mikhailenko DS, et al. Plasma levels of hsa-miR-619-5p and hsa-miR-1184 differ in prostatic benign hyperplasia and cancer. Bull Exp Biol Med. 2016;161(1):108–111. doi:10.1007/s10517-016-3357-7
22. Ye F, Tang QL, Ma F, et al. Analysis of the circular RNA transcriptome in the grade 3 endometrial cancer. Cancer Manag Res. 2019;11:6215–6227. doi:10.2147/CMAR.S197343
23. Malta M, Ribeiro J, Monteiro P, Loureiro J, Medeiros R, Sousa H. Let-7c is a candidate biomarker for cervical intraepithelial lesions: a Pilot Study. Mol Diagn Ther. 2015;19(3):191–196. doi:10.1007/s40291-015-0145-4
24. Zhou XG, Huang XL, Liang SY, et al. Identifying miRNA and gene modules of colon cancer associated with pathological stage by weighted gene co-expression network analysis. Onco Targets Ther. 2018;11:2815–2830. doi:10.2147/OTT.S163891
25. Guo JN, Li J, Zhu CL, et al. Comprehensive profile of differentially expressed circular RNAs reveals that hsa_circ_0000069 is upregulated and promotes cell proliferation, migration, and invasion in colorectal cancer. Onco Targets Ther. 2016;9:7451–7458. doi:10.2147/OTT.S123220
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.