Back to Journals » Blood and Lymphatic Cancer: Targets and Therapy » Volume 7
Complications and management of coagulation disorders in leukemia patients
Authors Lad DP, Jain A ![]() , Varma S
, Varma S
Received 6 June 2017
Accepted for publication 26 August 2017
Published 18 September 2017 Volume 2017:7 Pages 61—72
DOI https://doi.org/10.2147/BLCTT.S125121
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor David Dingli
Deepesh Lad, Arihant Jain, Subhash Varma
Department of Internal Medicine, Postgraduate Institute of Medical Education and Research, Chandigarh, India
Abstract: Patients with leukemia are predisposed to various coagulation abnormalities. Thrombosis and bleeding continue to be a major cause of morbidity and mortality in leukemias. The pathophysiology of these disorders is unique, and not only the disease but also the treatment and other factors play a role. There has been an increase in the understanding of these disorders in leukemias. However, it is still difficult to predict when and which patients will have these complications. The evidence for the management of coagulation abnormalities in leukemias is still evolving and not as established as in solid malignancies. The management of these disorders is complex, and making clinical decisions is often challenging. In the era of specialization, where there are different hematologists looking after benign- and malignant-hematology patients, opinions of thrombosis experts are often sought by leukemia specialists. This review aims to bridge the gap in the knowledge of these disorders between these specialists.
Keywords: leukemia, coagulation, thrombosis, bleeding
Introduction
Coagulation disorders are common in leukemia patients. The incidence of thromboembolism (TE) in hematologic malignancies (including lymphoma and myeloma) stands at 4% per year. This is the fourth most common cause after pancreatic (11%), brain (8%), and lung cancers (4.4%).1 The incidence of bleeding is the highest of all malignancies by virtue of the disease-related thrombocytopenia itself. Coagulation disorders seen in lymphoma, myeloma, and other solid malignancies are beyond the scope of this review. The reader may refer to excellent reviews and guidelines on these topics.2,3 For the purpose of simplicity, we deal with each leukemia type with a case vignette, followed by epidemiology, pathophysiology, and management issues. All patients provided written informed consent for their case details to be published.
Coagulation disorders in acute lymphoblastic leukemia
Case vignette
GK, a 39-year-old woman diagnosed with pre-B-cell acute lymphoblastic leukemia (ALL), standard risk was started on the Berlin–Frankfurt–Münster (BFM)-90 protocol. Her blood counts at presentation were hemoglobin 45 g/L, platelets 18×109/L, and white-cell count 1.2×109/L. She received native Escherichia coli l-asparaginase as part of the induction therapy. In the fourth week after completion of the l-asparaginase course, she presented with generalized tonic–clonic seizures and right-sided hemiparesis. Computed tomography suggested bifrontal hemorrhage. Magnetic resonance venography of brain confirmed superior sagittal sinus thrombosis. Her blood counts were hemoglobin 80 g/L, platelets 85×109/L, and white-cell count 2.8×109/L. Her fibrinogen levels were normal throughout the course of l-asparaginase. She was started on a therapeutic dose of low-molecular-weight heparin (LMWH). LMWH was dose-modified for thrombocytopenia during the intensive phase. She was not rechallenged with l-asparaginase during the reinduction phase. She remains in complete remission, with minimal residual disease negative at end of induction. LMWH was continued for 6 months. Her focal neurological deficit recovered, and she had no recurrence of thrombosis.
Epidemiology
The reported incidence of TE in childhood ALL varies from 1.1% to 36.7%.4 This wide variation is due to differences in TE diagnosis and treatment protocols. This incidence is even higher in adolescent and young adult (34%) and adult patients (43%) on l-asparaginase.5 The commonest sites for TE are central nervous system (CNS; 50%) and deep vein thrombosis (43%). The overall mortality of TE in ALL varies from 0 to 4.8%.4 On the contrary, deaths due to bleeding in ALL are less common than in acute myeloid leukemia (AML).6
Pathophysiology
The clinical presentation of TE or bleeding is an outcome of the delicate balance between procoagulant and anticoagulant forces at play in a patient (Figure 1). There are several studies showing increased thrombin generation shown by thrombin–antithrombin (AT) complexes at diagnosis and in initial therapy for ALL. Factor VIII, von Willebrand
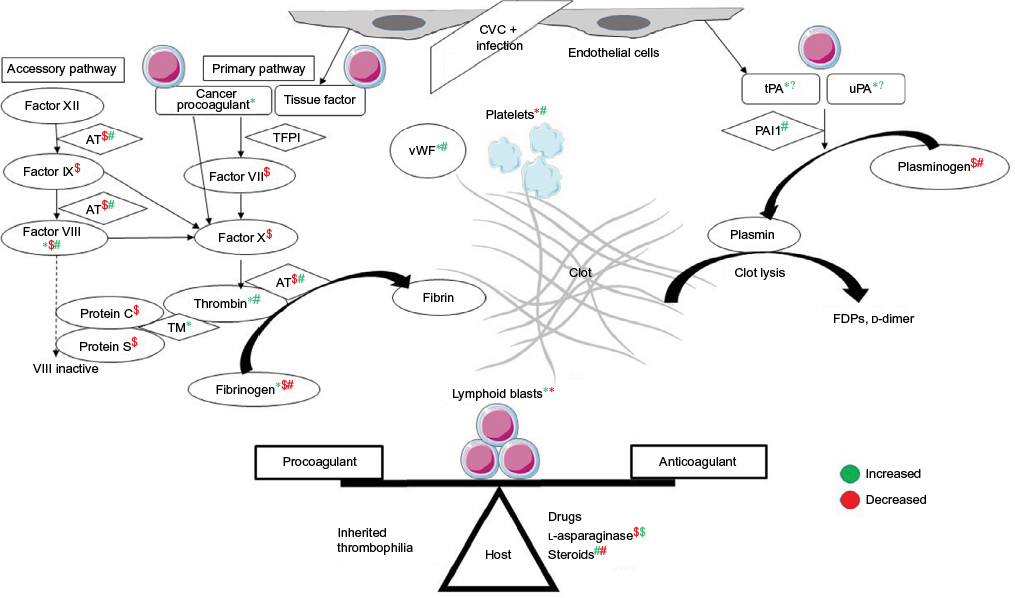 | Figure 1 Balance between procoagulant and anticoagulant forces in ALL and interactions among host, disease, and treatment related factors. Notes: *Effect of lymphoid blasts; $effect of l-asparaginase; #effect of steroids, on coagulation parameters; green color is increased effect and red color is decreased effect. Abbreviations: ALL, acute lymphoblastic leukemia; CVC, central venous catheter; TFPI, tissue-factor pathway inhibitor; vWF, von Willebrand factor; AT, antithrombin; TM, thrombomodulin; FDPs, fibrin-degradation products. |
factor (vWF), and fibrinogen are positive acute phase reactants.7 Besides this, ALL cells also express cancer procoagulant (CP).8 CP activates factor X directly, independently of factor VII.9 Disseminated intravascular coagulation (DIC) is underrecognized in ALL, with prevalence of around 14% at diagnosis and doubling to 27% during induction therapy.10
Chemotherapy in ALL augments the thrombogenic potential. Of these, l-asparaginase, which depletes the ALL cells of the essential amino acid asparagine, also ends up depleting the procoagulant (fibrinogen, factors V, VII, VIII, IX, X, and XI) and anticoagulant (AT, plasminogen, protein C/S) proteins. l-Asparaginase may in fact cause a prothrombotic state, as it reverses the procoagulant proteins earlier than anticoagulant proteins.11 E. coli asparaginase, which is more potent than Erwinia asparaginase, has more effect on the coagulation parameters.12 Steroids add to the prothrombotic state by increasing plasma levels of prothrombin, factor VIII/vWF, and PAI1 and decreasing plasminogen and fibrinogen.13 Dexamethasone may be associated with less risk of TE than prednisone.14 Central venous lines are the other reason for TE, with incidence as high as 30%, depending on the technique and site of insertion.15 A variable 18%–40% of cases may have inherited thrombophilia.16 Use of granulocyte/granulocyte-monocyte colony-stimulating factors during episodes of neutropenia is also associated with thrombosis at an incidence of 1.2% and 4.2%, respectively. This is through increased tissue-factor (TF) and factor-VIII levels.17 T-cell ALL patients are at higher risk of both TE and hemorrhagic complications, due to hyperleukocytosis.18 Thrombocytopenia – at diagnosis or therapy-related – is one of the major factors contributing to bleeding in ALL. Infections can tilt the balance either way of systemic inflammatory response syndrome or consumptive coagulopathy.
Investigations
Recommended tests include activated partial thromboplastin time (aPTT), international normalized ratio (INR), and fibrinogen levels (AT optional) prior to, during native asparaginase dosing, and 1 week after polyethylene glycol–asparaginase therapy. For prevention of TE, research using AT prophylaxis have shown reduced incidence of TE, but this was not powered for efficacy, and hence the role of AT replacement remains inconclusive.19 Though prophylactic fresh frozen plasma/cryoprecipitate to replace AT is associated with no CNS TE events, neither it nor LMWH lower the incidence of TE on l-asparaginase.20,21 Some centers use both anticoagulation and AT replacement for AT levels <60%.22 At our center, we give cryoprecipitate for fibrinogen <100 mg/dL.
Treatment
Development of TE while on l-asparaginase used to lead to discontinuation of asparaginase in up to 75% patients, which led to inferior event-free survival outcomes.23,24 However, with current strategies of rechallenging asparaginase and continued anticoagulation, outcomes have been similar.5 Current expert recommendations are to discontinue asparaginase only for CTCAE (Common Terminology Criteria for Adverse Events) grade 4 CNS thrombosis or bleed. For all grade 3 events and grade 4 non-CNS events, asparaginase needs to be withheld till resolution of clinical signs and either anticoagulation or coagulant therapy is stable. Asparaginase may be resumed at lower doses and/or longer intervals. Asparaginase need not be withheld for asymptomatic lab abnormalities.25 As regards prophylactic platelet transfusions, the restrictive strategy with the standard trigger set at 10×109/L has remained unchanged over the past decade.26
There have been no prospective randomized control trials on anticoagulant choice, dose, and duration in patients with leukemia. Most recommendations have been derived from extrapolation from studies done in patients with solid-organ malignancies. These guidelines endorse the use of LMWH as first-line therapy during the first 3–6 months after the diagnosis of malignancy-associated venous TE (VTE) based on the 2003 CLOT study.27–30 There have been recent expert recommendations on managing thrombosis in the setting of leukemia with thrombocytopenia.31–33 The consensus for anticoagulation dose modification in the setting of thrombocytopenia is as given in Tables 1 and 2. For catheter-related VTE, recommendations are to keep the catheter and continue anticoagulation till the catheter is in place or at least 3 months, whichever is longer. Catheter removal is only required if the device is dysfunctional, infected, or not required. For non-catheter-related TE, anticoagulation is recommended for at least 6 months and indefinitely for active malignancy or persistence of risk factors for recurrence.3 The decision to stop anticoagulation beyond 6 months in patients under treatment of a malignancy that is in complete remission is unclear. This is on a case-by-case basis after taking the risk:benefit ratio into consideration. To summarize, in this case, the patient had a grade 4 CNS vascular event on l-asparaginase, despite normal fibrinogen levels. She was managed with dose-modified anticoagulation as mentioned, and not re-challenged with l-asparaginase.
 | Table 1 Acute thrombosis (0–30 days after diagnosis of VTE) Note: aTransfusion to platelet count ≥20×109/L to allow at least 50% anticoagulant dose. Abbreviations: VTE, venous thromboembolism; LMWH, low-molecular-weight heparin. |
 | Table 2 Chronic thrombosis (>30 days after diagnosis of VTE) Note: aPlatelet transfusion not indicated for anticoagulant dose. Abbreviations: VTE, venous thromboembolism; SC, subcutaneously; LMWH, low-molecular-weight heparin. |
Coagulation disorders in acute myeloid leukemia
Case vignette
RM, a 28-year-old lady, presented with a 4-day history of gangrenous changes in both hands (left > right) and feet. On clinical examination, she had absent ulnar, dorsalis pedis, and posterior tibial artery pulse bilaterally. Her investigations revealed hemoglobin 63 g/L, platelets 39×109/L, and white cell count 33.2×109/L with 85% blasts + promyelocytes. Her coagulogram showed PT 20 seconds, control 14 seconds, INR 1.4, aPTT 31 seconds, control 25–32 seconds, and elevated d-dimer and fibrin-degradation products. Her fibrinogen levels were normal, however. She was started on treatment with all-trans retinoic acid (ATRA) and arsenic on suspicion of acute promyelocytic leukemia (APML) and DIC. This is the first line at our center, even in high-risk APML.34 She was given prophylactic steroids plus hydroxyurea to prevent differentiation syndrome. Computed tomography angiography confirmed the presence of arterial thrombosis in the distal ulnar and posterior tibial arteries. This is a rare presentation of APML, but included here to emphasize the management issues in this case. Given her state of established gangrene with line of demarcation and DIC, she could not be started on any vascular intervention or anticoagulation/antiplatelets. ATRA + arsenic and platelet support was continued. She attained complete remission at the end of 4 weeks. She underwent bilateral below-knee amputation with left phalangeal disarticulation for her dry gangrene 6 months later. She continues to be in remission 4 years later. This outcome is similar to what has been described in case reports.35
Epidemiology
Most cases of AML-M3/APML have DIC at diagnosis. The incidence of DIC in AML-non-M3 is 10%–50%, depending upon the subtype of leukemia and diagnostic criteria for DIC.36,37 The incidence of thrombosis in AML-non-M3 and APML at diagnosis is 3.2% and 9.6%, respectively. The same at follow-up is 1.7%–8.5% and 8.4%–11% respectively.38,39 In APML, most cases occur before or during induction therapy with ATRA. Arterial events are commoner than venous events. Thrombosis and severe hemorrhage co-occur in 15% of cases.40 The incidence of isolated severe bleeding remains high in APML – around 21%. The case-fatality rate of these episodes can be as high as 50%. The incidence of early-death rates in APML has remained unchanged over the years: at 5%–10%, even in the ATRA era.41 The incidence of thrombosis in APML at our center is 7.4% (n=136), with equal numbers of arterial and venous TE. The incidence of severe bleeds is 28%, with the early-death rate due to bleeding being 7.4% (Varma et al, unpublished data). Predictors of thrombosis include elevated white-cell count, Bcr3 isoform, FLT3 internal tandem duplication, and expression of CD2 and CD15 on promyelocytes.42 Though not a coagulation defect in principle, hyperleukocytosis can be seen in AML when the white-cell count is >100×109/L. This leads to leukostasis and vascular occlusion in any vital organ.43
Pathophysiology
The DIC seen in APML is a double-edged sword for thrombosis and bleeding (Figure 2). Procoagulant forces are CP expressed on leukemic promyelocytes.44 TF from endothelial cells is exposed by cytokines (IL1 and TNFα) released from apoptotic leukemic cells.45–47 TF is the site of activation of factor VII. TF-expressing microparticles in APML have procoagulant activity.48 Anticoagulant forces are elevated uPA and tPA and low PAI1.49,50 Leukemic promyelocytes express annexin II, which accelerates the conversion of plasminogen to plasmin and thus causes primary fibrinolysis.51 Cerebral endothelial cells also express this annexin II, which explains the high incidence of intracerebral bleeds seen in APML.52 Emerging evidence points to podoplanin expression on leukemic promyelocytes causing platelet aggregation as a novel mechanism for bleeding.53 The role of proteases like elastase and chymotrypsin released from leukemic promyelocytes on fibrinolysis, if any, is minor.54 ATRA can reverse these changes through various mechanisms of reduced expression of TF, CP, and annexin II and counteracting the effect of cytokines.55 ATRA may in fact tilt the balance to a prothrombotic state, as it reverses the procoagulant proteins earlier than anticoagulant proteins, similar to l-asparaginase.56 A novel cell-death pathway termed “ETosis” has been described recently in APML blasts treated with ATRA. This process releases intact chromatin in the extracellular space. This extracellular chromatin has multifold effects of increasing thrombin and plasmin generation and causes fibrinolysis resistance and endothelial activation.57
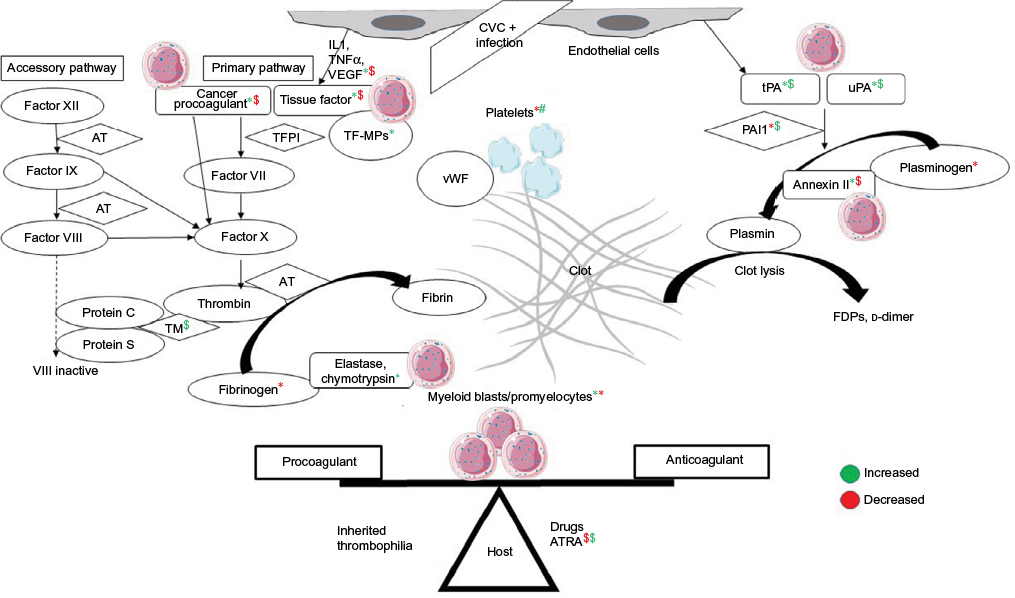 | Figure 2 Pathophysiology of disseminated intravascular coagulation in APML and the role of ATRA in counteracting it. Notes: *Effect of myeloid blasts/promyelocytes; $effect of ATRA on coagulation parameters; green color is increased effect and red color is decreased effect. Abbreviations: APML, acute promyelocytic leukemia; ATRA, all-trans retinoic acid; CVC, central venous catheter; TFPI, tissue-factor pathway inhibitor; TF-MPs; TF-expressing microparticles; vWF, von Willebrand factor; AT, antithrombin; TM, thrombomodulin; FDPs, fibrin-degradation products. |
Investigations
Routine coagulation tests may reveal overt DIC with prolongation of PT, variable aPTT, low fibrinogen levels and platelet counts, and elevated d-dimer and fibrin-degradation products.58 However, these variables do not correlate well with the severity of bleeding or thrombosis.41 No single test can confirm or rule out DIC. The International Society on Thrombosis and Haemostasis overt DIC score may miss subclinical DIC in acute leukemia.10 A recent study showed that a high d-dimer level >4 mg/L was most predictive for thrombosis in AML-non-M3 patients.59 Other tests of activated clotting like prothrombin fragment 1+2, thrombin–AT complex, and fibrinopeptide A may be helpful, but are not routinely available. Elevated white-cell count (>20×109/L) is an independent predictor of early hemorrhagic death in APL.60
Treatment
Both ATRA and arsenic trioxide (ATO) can reverse the coagulopathy of APML within 4–11 days. This underscores the importance of starting ATRA in the emergency department whenever a diagnosis of APML is suspected. The incidence of thrombosis in the ATRA era is higher than in the pre-ATRA era.40 However, the incidence of TE in ATRA + ATO is lower than in ATRA + chemotherapy in non-high-risk APML patients.61 European Leukemia Net guidelines recommend fresh frozen plasma, fibrinogen, and/or cryoprecipitate to maintain the fibrinogen concentration above 100–150 mg/dL. In APML, the platelet-transfusion threshold is more liberal, and the aim is to maintain a count >30–50×109/L. The role of heparin, tranexamic acid, or other anticoagulant or antifibrinolytic therapy is unclear and not recommended as part of routine practice.62 On the contrary, combining ATRA with tranexamic acid may lead to fatal thromboembolism.63 There is insufficient evidence to recommend thrombopoietin mimetics, recombinant factor VIIa, antifibrinolytics, desmopressin, activated protein C, or recombinant human soluble thrombomodulin to prevent or treat bleeding in hematological malignancies.64–66 The role of leukapheresis in hyperleukocytosis to prevent early mortality is also unclear; in fact, it may lower platelet counts and fibrinogen and prolong prothrombin time.67,68 Early cytoreductive chemotherapy and supportive care with platelets, fresh frozen plasma, cryoprecipitate, or fibrinogen concentrates are the only recommended measures to prevent or treat bleeding associated with DIC in AML.69 In summary, the patient had APML with DIC manifesting as acute arterial thrombosis of the extremities. She was managed with ATRA + ATO and supportive transfusions without anticoagulation. She survived the leukemia, but lost her limbs to gangrene.
Coagulation disorders in myeloproliferative neoplasms
Case vignette
MS, a 19-year-old man, presented with a 1-month history of abdominal pain. He had a massive splenomegaly with thrombosis of the splenoportal axis. His blood counts at presentation were hemoglobin 108 g/L, platelets 668×109/L, and white cells 5.6×109/L, with a normal differential count. His bone marrow suggested a diagnosis of essential thrombocytosis (ET). His JAK2V617F-mutation status was positive. The final diagnosis was high-risk ET with splanchnic vessel thrombosis. On upper gastrointestinal endoscopy, he had no esophageal varices, but had multiple small gastric varices at diagnosis. He was given LMWH and bridged to warfarin titrated to an INR of 2–3. Aspirin was withheld, due to gastric varices. He was also started on hydroxyurea 500 mg orally twice a day titrated to a normal platelet count. He had an episode of hematemesis a year later, requiring interruption of warfarin and glue injection of the gastric varices. Given the risk of gastric variceal bleeding with aspirin, and considering the risk:benefit ratio of anticoagulation, he was restarted on warfarin. The plan is to continue indefinite anticoagulation, with periodic upper gastrointestinal endoscopy for varices.
Epidemiology
Of the chronic myeloproliferative neoplasms (MPNs), polycythemia vera (PV), ET, and primary myelofibrosis (PMF) are most associated with thrombosis and bleeding. The prevalence of TE at the time of diagnosis in PV, ET, PMF is 4%–11%, 2%–8%, and 3%–7%, respectively. The incidence of TE thereafter is 2%, 0.6%, and 0.6% per year, respectively.70 Arterial thrombosis (70%) is more common than venous TE (30%). MPNs are also the etiology in abdominal splanchnic vessel thrombosis (extra- + intrahepatic veins leading to Budd–Chiari syndrome [BCS], portal vein thrombosis, and mesenteric vein thrombosis). The prevalence of latent or manifest MPNs is 40% in BCS and 30% in portal vein thrombosis. JAK2V617F mutation without manifest MPNs is seen in 17.1% of BCS and 15.4% of portal vein thrombosis.71 In chronic myeloid leukemia (CML), tyrosine-kinase inhibitors increase the risk of arterial (4.78%) and venous (0.72%) TE. The risk is higher with newer tyrosine-kinase inhibitors (0.96%) than imatinib (0.27%).72 In contrast to thrombosis, serious bleeding occurs in 5.5% of MPN patients.73 The incidence of dasatinib-induced platelet dysfunction leading to serious bleeding in CML patients is 7%.74 The incidence of thrombosis in PV at our center is 25%, with equal numbers of arterial and venous events.75 The incidence of thrombosis and bleeding in ET at our center is 33.3% and 8.3%, respectively. A quarter of thrombotic events are arterial (Varma et al, unpublished data).
Pathophysiology
The mechanism of thrombosis in MPNs is multifactorial (Figure 3). Besides thrombocytosis, platelets aggregate due to upregulation of p-selectin, thrombospondin and GPIIB/IIIA receptors.76 Besides leukocytosis, activated leukocytes form aggregates with platelets.77,78 Erythrocytosis causes hyperviscosity; erythrocytes also show increased adherence to vascular endothelia.79 Other mechanisms of thrombosis include microparticles and inflammation-mediated activation of endothelia.80,81 Using novel gene-expression meta-analysis, certain coagulation-related genes have been identified to be associated with thrombosis in MPN. These include IGF2R, PROS1, SELPLG, and ITGB2.82 The etiology of bleeding in MPN is multifactorial. Acquired von Willebrand syndrome is seen in patients with platelet counts >10×109/L.83 At these high counts, vWF binds to platelets and undergoes proteolysis with a resultant depletion of large multimers.84 Besides this, there is evidence of platelet dysfunction in ET.85 Other reasons for bleeding include treatment for disease and TE, thrombocytopenia due to disease progression, hypersplenism, liver dysfunction, and acquired hemophilia.86
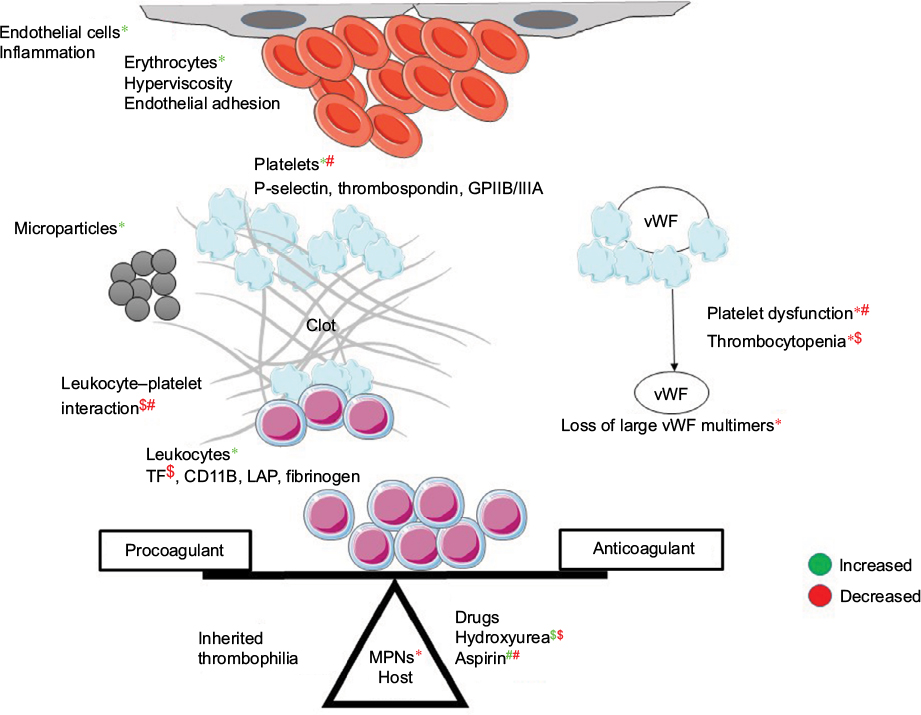 | Figure 3 Pathophysiology of coagulation disorders in MPNs. Notes: *Effect of MPN; $effect of hydroxyurea; #effect of aspirin on coagulation parameters; green color is increased effect and red color is decreased effect. Abbreviations: MPNs, myeloproliferative neoplasms; vWF, von Willebrand factor. |
Investigations
Acquired von Willebrand syndrome should be ruled out in all patients with platelet counts >10×109/L before starting these patients on aspirin. Diagnosis of acquired von Willebrand syndrome involves showing a reduced function:antigen ratio (vWF:Act/Ag, vWF:Act or ristocetin-cofactor assay<30%) and loss of high-von Willebrand multimers.87
Treatment
Risk factors for recurrent thrombosis in PV and ET are age >60 years, history of thrombosis, JAK2/MPL-mutation status, JAK2-allele burden, and cardiovascular (tobacco use, hypertension or diabetes mellitus, leukocytosis >11×109/L) in ET.88 The goal of treatment in PV/ET is to prevent thrombohemorrhagic complications. Phlebotomy to achieve a hematocrit level <45% is standard treatment in all PV patients.89 All low-risk PV and ET patients, except those with acquired von Willebrand syndrome, should get low-dose aspirin (40–100 mg/day). For high-risk PV or ET patients, hydroxyurea is recommended to target a platelet count in the normal range. All these measures reduce the risk of vascular events, including thrombosis and bleeding.90 Anagrelide is falling out of favor, due to its increased risk of arterial thrombosis and major bleeding.91 The requirement of phlebotomy on hydroxyurea correlates with the risk of thrombosis in PV patients.92 Both aspirin and hydroxyurea reduce platelet–leukocyte complex formation and downregulation of leukocyte TF expression by hydroxyurea.78,93 Ruxolitinib, a JAK2 inhibitor, is approved to treat MF and hydroxyurea-intolerant PV. It causes thrombocytopenia in 68% of patients and grade 3–4 thrombocytopenia in 13% of patients.94 A meta-analysis has shown that ruxolitinib reduces the risk of both arterial and venous thrombosis in PV and PMF patients. This is believed to be due to inhibition of JAK-mediated inflammation by ruxolitinib.95
As regards treatment of VTE events, standard LMWH and vitamin K antagonists (VKAs) are used. The risk:benefit ratio of bleeding versus recurrent thrombosis needs to considered on a case-by-case basis. The combination of LMWH/VKAs and aspirin increases the risk of bleeding to 2.8% per patient-year.73 The decision to stop anticoagulant therapy at 3–6 months is influenced by the preferences of an informed patient and risk of thrombosis. Recurrent VTE/cerebral or hepatic vein thrombosis, life-threatening VTE, and progressive MPNs uncontrolled with cytoreductive therapy would require indefinite treatment.96 Because the risk of recurrent thrombosis is doubled on stopping anticoagulation and the risk of bleeding on VKA is the same as the non-MPN population, secondary prophylaxis should be continued for an indefinite period.97 Aspirin should be continued in patients stopping anticoagulation. There are sparse data on the use of direct oral anticoagulants in hematological malignancies. These show no significant risk reduction in major bleeding compared to LMWH.98 However, there are ongoing randomized trials addressing this issue.99 If their use is considered, attention must be given to interactions with drugs used for treatment of the underlying disease (Table 3).100 To summarize, the young patient had high-risk JAK2 + ET and splenoportal vein thrombosis. He was managed with hydroxyurea and VKAs. He could not be given aspirin, due to the presence of gastric varices. He will need indefinite anticoagulation, given the risk of recurrent thrombosis in this population.
 | Table 3 Drug interactions between DOACs and TKIs Note: Risk category C: monitor therapy for enhanced anticoagulant effect of anticoagulants. Abbreviations: DOACs, direct oral anticoagulants; TKIs, tyrosine-kinase inhibitors. |
Coagulation disorders in chronic lymphocytic leukemia
Case vignette
HK, a 62-year-old lady, with known chronic lymphocytic leukemia (CLL) with 11q deletion and unmutated IGHV after early relapse on bendamustine–rituximab, was started on ibrutinib 420 mg daily for bulky lymphadenopathy in April 2016. Her complete blood counts at baseline were hemoglobin 100 g/L, platelets 79×109/L, and absolute lymphocyte count 231×109/L. Her baseline coagulogram was normal (PT 15 seconds, INR 1.2, aPTT 32 seconds, control 32 seconds). Within 2 weeks, her absolute lymphocyte count had risen to 412×109/L and platelets had dropped to 44×109/L. She had gum bleeding with a spontaneous hematoma of 10×15 cm in her left gluteal region. HK had grade 1 thrombocytopenia at baseline. She developed grade 3 thrombocytopenia with major but non-life-threatening bleeding. Her repeat coagulogram was normal (PT 14 seconds, control 14 seconds, INR 1, aPTT 31 seconds, control 32 seconds). This drop in platelet counts was due to ibrutinib, and was held till the resolution of hematoma and platelet count >50×109/L. Ibrutinib was restarted at the same dose. There was no recurrence of bleeding. At 1-year follow-up, her platelet count is up to 100×109/L.
Epidemiology
The incidence of at least one episode of severe bleeding in CLL is reported to be as high as 18%.101 This is often seen with autoimmune or disease-related thrombocytopenia. Acquired hemophilia with inhibitors to factor VIII are rare causes of bleeding.102 Bleeding assumes much more importance in the era of ibrutinib. While early studies showed major bleeding episodes occurred in 9% and 4% of CLL patients on anticoagulants and antiplatelets, later studies have this incidence at 2% and 1%, respectively.103,104 CLL has not been associated with a high incidence of thrombosis. However, recent studies have shown incidence rates may be as high as other leukemias – 5%–11%.105,106 The risk factors for VTE is these studies were age >60 years, advanced stage of CLL, CLL treatment, and inherited thrombophilia.
Pathophysiology
At baseline, CLL patients have impaired platelet function assessed by platelet-function assay using epinephrine and adenosine diphosphate. Collagen-induced platelet aggregation is further impaired on initiating ibrutinib. Though factor VIII and vWF levels are elevated at baseline in most patients, their levels decline with ibrutinib. Low levels of factor VIII, vWF, and epinephrine-closure time on platelet-function assay predict bleeding in patients taking ibrutinib. There is no difference in platelet counts in patients who do and do not bleed. Bleeding risk decreases over time.107
Investigations
Recommended tests include PT, aPTT, INR, and platelet counts at baseline. Bone-marrow examination should be done at baseline in thrombocytopenic patients to confirm or rule out a diagnosis of autoimmune thrombocytopenia. PT, aPTT, INR, and platelet counts should be repeated whenever there is clinical bleeding. Platelet counts should be repeated before each monthly cycle.
Treatment
Patients need to be told to report any bleeding episodes and hold ibrutinib for any medical or dental procedures. Ibrutinib needs to be held for at least 3–7 days pre- and postsurgery, depending upon the surgery and risk of bleeding.108 While there are no guidelines to hold ibrutinib if thrombocytopenia is due to disease, ibrutinib may be held for drug-related grade 4 thrombocytopenia (<25×109/L) or grade 3 thrombocytopenia (<50×109/L) with bleeding. It is restarted when platelet count rises to ≥50×109/L.109 Treatment of idiopathic thrombocytopenic purpura, disease-related thrombocytopenia, and acquired hemophilia involves steroids or immunosuppression to treat underlying CLL. Anticoagulation is required in 5%–10% of patients on ibrutinib who develop atrial fibrillation.110 Using direct oral anticoagulants in this setting needs interactions with ibrutinib to be taken in to consideration (Table 3).111 Patients on concurrent anticoagulants or antiplatelets should be watched for bleeding.112 The CLL patient mentioned in the vignette had grade 4 thrombocytopenia with non-life-threatening bleeding on ibrutinib. This mandated holding ibrutinib till platelet recovery. Subsequently, her platelet counts rose to >100×109/L, as has been suggested to happen in 68% of patients on ibrutinib by 6 months.107
Conclusion
In summary, coagulation disorders are common in leukemia. Thrombosis is as common as in solid malignancies. It is difficult to predict which patients will have bleeding and/or thrombosis. We need better predictors and coagulation tests to identify such patients. More research with prospective randomized control studies is needed on the role of novel oral anticoagulants, anticoagulant choice, dosing, and duration in hematologic malignancies.
Acknowledgments
The authors acknowledge the assistance of Dr Pankaj Malhotra, Dr Parathan Karunakaran, and Dr Ram Nampoothiri in providing the data related to the clinical vignettes and institutional data on coagulation disorders in leukemia patients.
Disclosure
The authors report no conflicts of interest in this work.
References
Horsted F, West J, Grainge MJ. Risk of venous thromboembolism in patients with cancer: a systematic review and meta-analysis. PLoS Med. 2012;9(7):e1001275. | ||
Lyman GH, Bohlke K, Khorana AA, et al. Venous thromboembolism prophylaxis and treatment in patients with cancer: American Society of Clinical Oncology clinical practice guideline update 2014. J Clin Oncol. 2015;33(6):654–656. | ||
National Comprehensive Cancer Network. Cancer-associated venous thromboembolic disease: version 1. 2016. Available from: https://www.nccn.org/professionals/physician_gls/pdf/vte.pdf. Accessed June 1, 2017. | ||
Athale UH, Chan AK. Thrombosis in children with acute lymphoblastic leukemia – part I: epidemiology of thrombosis in children with acute lymphoblastic leukemia. Thromb Res. 2003;111(3):125–131. | ||
Grace RF, Dahlberg SE, Neuberg D, et al. The frequency and management of asparaginase-related thrombosis in paediatric and adult patients with acute lymphoblastic leukaemia treated on Dana-Farber Cancer Institute consortium protocols. Br J Haematol. 2011;152(4):452–459. | ||
Athale UH, Chan AK. Hemorrhagic complications in pediatric hematologic malignancies. Semin Thromb Hemost. 2007;33(4):408–415. | ||
Athale UH, Chan AK. Thrombosis in children with acute lymphoblastic leukemia – part II: pathogenesis of thrombosis in children with acute lymphoblastic leukemia – effects of the disease and therapy. Thromb Res. 2003;111(4–5):199–212. | ||
Alessio MG, Falanga A, Consonni R, et al. Cancer procoagulant in acute lymphoblastic leukemia. Eur J Haematol. 1990;45(2):78–81. | ||
Gordon SG, Franks JJ, Lewis B. Cancer procoagulant A: a factor X activating procoagulant from malignant tissue. Thromb Res. 1975;6(2):127–137. | ||
Dixit A, Chatterjee T, Mishra P, et al. Disseminated intravascular coagulation in acute leukemia at presentation and during induction therapy. Clin Appl Thromb Hemost. 2007;13(3):292–298. | ||
Zakarija A, Kwaan HC. Adverse effects on hemostatic function of drugs used in hematologic malignancies. Semin Thromb Hemost. 2007;33(4):355–364. | ||
Duval M, Suciu S, Ferster A, et al. Comparison of Escherichia coli-asparaginase with Erwinia-asparaginase in the treatment of childhood lymphoid malignancies: results of a randomized European Organisation for Research and Treatment of Cancer – Children’s Leukemia Group phase 3 trial. Blood. 2002;99(8):2734–2739. | ||
Isacson S. Effect of prednisolone on the coagulation and fibrinolytic systems. Scand J Haematol. 1970;7(3):212–216. | ||
Nowak-Göttl U, Ahlke E, Fleischhack G, et al. Thromboembolic events in children with acute lymphoblastic leukemia (BFM protocols): prednisone versus dexamethasone administration. Blood. 2003;101(7):2529–2533. | ||
Male C, Chait P, Andrew M, Hanna K, Julian J, Mitchell L. Central venous line-related thrombosis in children: association with central venous line location and insertion technique. Blood. 2003;101(11):4273–4278. | ||
Athale UH, Chan AK. Thrombosis in children with acute lymphoblastic leukemia – part III: pathogenesis of thrombosis in children with acute lymphoblastic leukemia – effects of host environment. Thromb Res. 2003;111(6):321–327. | ||
Barbui T, Finazzi G, Grassi A, Marchioli R. Thrombosis in cancer patients treated with hematopoietic growth factors: a meta-analysis. Thromb Haemost. 1996;75(2):368–371. | ||
Giordano P, Del Vecchio GC, Santoro N, et al. Thrombin generation in children with acute lymphoblastic leukemia: effect of leukemia immunophenotypic subgroups. Pediatr Hematol Oncol. 2000;17(8):667–672. | ||
Mitchell LG, Andrew M, Hanna K, et al. A prospective cohort study determining the prevalence of thrombotic events in children with acute lymphoblastic leukemia and a central venous line who are treated with l-asparaginase: results of the Prophylactic Antithrombin Replacement in Kids with Acute Lymphoblastic Leukemia Treated with Asparaginase (PARKAA) study. Cancer. 2003;97(2):508–516. | ||
Lauw MN, van der Holt B, Middeldorp S, Meijers JC, Cornelissen JJ, Biemond BJ. Venous thromboembolism in adults treated for acute lymphoblastic leukaemia: effect of fresh frozen plasma supplementation. Thromb Haemost. 2013;109(4):633–642. | ||
Sibai H, Seki JT, Wang TQ, et al. Venous thromboembolism prevention during asparaginase-based therapy for acute lymphoblastic leukemia. Curr Oncol. 2016;23(4):e355–e361. | ||
Hunault-Berger M, Chevallier P, Delain M, et al. Changes in antithrombin and fibrinogen levels during induction chemotherapy with l-asparaginase in adult patients with acute lymphoblastic leukemia or lymphoblastic lymphoma: use of supportive coagulation therapy and clinical outcome – the CAPELAL study. Haematologica. 2008;93(10):1488–1494. | ||
Silverman LB, Gelber RD, Dalton VK, et al. Improved outcome for children with acute lymphoblastic leukemia: results of Dana-Farber consortium protocol 91–01. Blood. 2001;97(5):1211–1218. | ||
Ott N, Ramsay NK, Priest JR, et al. Sequelae of thrombotic or hemorrhagic complications following L-asparaginase therapy for childhood lymphoblastic leukemia. Am J Pediatr Hematol Oncol. 1988;10(3):191–195. | ||
Stock W, Douer D, DeAngelo DJ, et al. Prevention and management of asparaginase/pegasparaginase-associated toxicities in adults and older adolescents: recommendations of an expert panel. Leuk Lymphoma. 2011;52(12):2237–2253. | ||
Schiffer CA, Anderson KC, Bennett CL, et al. Platelet transfusion for patients with cancer: clinical practice guidelines of the American Society of Clinical Oncology. J Clin Oncol. 2001;19(5):1519–1538. | ||
Lyman GH, Khorana AA, Falanga A, et al. American Society of Clinical Oncology guideline: recommendations for venous thromboembolism prophylaxis and treatment in patients with cancer. J Clin Oncol. 2007;25(34):5490–5505. | ||
Kearon C, Akl EA, Comerota AJ, et al. Antithrombotic therapy for VTE disease: Antithrombotic Therapy and Prevention of Thrombosis, 9th ed – American College of Chest Physicians evidence-based clinical practice guidelines. Chest. 2012;141(2 Suppl):e419S–e494S. | ||
Estcourt LJ, Stanworth SJ, Doree C, Hopewell S, Trivella M, Murphy MF. Comparison of different platelet count thresholds to guide administration of prophylactic platelet transfusion for preventing bleeding in people with haematological disorders after myelosuppressive chemotherapy or stem cell transplantation. Cochrane Database Syst Rev. 2015;(11):CD010983. | ||
Lee AY, Levine MN, Baker RI, et al. Low-molecular-weight heparin versus a coumarin for the prevention of recurrent venous thromboembolism in patients with cancer. N Engl J Med. 2003;349(2):146–153. | ||
Carrier M, Khorana AA, Zwicker J, Noble S, Lee AY. Management of challenging cases of patients with cancer-associated thrombosis including recurrent thrombosis and bleeding: guidance from the SSC of the ISTH. J Thromb Haemost. 2013;11(9):1760–1765. | ||
Easaw JC, Shea-Budgell MA, Wu CM, et al. Canadian consensus recommendations on the management of venous thromboembolism in patients with cancer: part 2 – treatment. Curr Oncol. 2015;22(2):144–155. | ||
Mantha S, Miao Y, Wills J, Parameswaran R, Soff GA. Enoxaparin dose reduction for thrombocytopenia in patients with cancer: a quality assessment study. J Thromb Thrombolysis. 2017;43(4):514–518. | ||
Varma S, Yanamandra U, Khadwal A, et al. High risk APML treated successfully with four cycles of ATO and ATRA combination in resource constrained settings. Blood. 2015;126(23):3322. | ||
Kalk E, Goede A, Rose P. Acute arterial thrombosis in acute promyelocytic leukaemia. Clin Lab Haematol. 2003;25(4):267–270. | ||
Yanada M, Matsushita T, Suzuki M, et al. Disseminated intravascular coagulation in acute leukemia: clinical and laboratory features at presentation. Eur J Haematol. 2006;77(4):282–287. | ||
Uchiumi H, Matsushima T, Yamane A, et al. Prevalence and clinical characteristics of acute myeloid leukemia associated with disseminated intravascular coagulation. Int J Hematol. 2007;86(2):137–142. | ||
De Stefano V, Sorà F, Rossi E, et al. The risk of thrombosis in patients with acute leukemia: occurrence of thrombosis at diagnosis and during treatment. J Thromb Haemost. 2005;3(9):1985–1992. | ||
Vu K, Luong NV, Hubbard J, et al. A retrospective study of venous thromboembolism in acute leukemia patients treated at the University of Texas MD Anderson Cancer Center. Cancer Med. 2015;4(1):27–35. | ||
Rashidi A, Silverberg ML, Conkling PR, Fisher SI. Thrombosis in acute promyelocytic leukemia. Thromb Res. 2013;131(4):281–289. | ||
Tallman MS, Abutalib SA, Altman JK. The double hazard of thrombophilia and bleeding in acute promyelocytic leukemia. Semin Thromb Hemost. 2007;33(4):330–338. | ||
Breccia M, Avvisati G, Latagliata R, et al. Occurrence of thrombotic events in acute promyelocytic leukemia correlates with consistent immunophenotypic and molecular features. Leukemia. 2007;21(1):79–83. | ||
Zuckerman T, Ganzel C, Tallman MS, Rowe JM. How I treat hematologic emergencies in adults with acute leukemia. Blood. 2012;120(10):1993–2002. | ||
Falanga A, Alessio MG, Donati MB, Barbui T. A new procoagulant in acute leukemia. Blood. 1988;71(4):870–875. | ||
Nawroth PP, Handley DA, Esmon CT, Stern DM. Interleukin 1 induces endothelial cell procoagulant while suppressing cell-surface anticoagulant activity. Proc Natl Acad Sci U S A. 1986;83(10):3460–3464. | ||
Bevilacqua MP, Pober JS, Majeau GR, Fiers W, Cotran RS, Gimbrone MA. Recombinant tumor necrosis factor induces procoagulant activity in cultured human vascular endothelium: characterization and comparison with the actions of interleukin 1. Proc Natl Acad Sci U S A. 1986;83(12):4533–4537. | ||
Greeno EW, Bach RR, Moldow CF. Apoptosis is associated with increased cell surface tissue factor procoagulant activity. Lab Invest. 1996;75(2):281–289. | ||
Kwaan HC, Rego EM. Role of microparticles in the hemostatic dysfunction in acute promyelocytic leukemia. Semin Thromb Hemost. 2010;36(8):917–924. | ||
Bennett B, Booth NA, Croll A, Dawson AA. The bleeding disorder in acute promyelocytic leukaemia: fibrinolysis due to u-PA rather than defibrination. Br J Haematol. 1989;71(4):511–517. | ||
Sakata Y, Murakami T, Noro A, Mori K, Matsuda M. The specific activity of plasminogen activator inhibitor-1 in disseminated intravascular coagulation with acute promyelocytic leukemia. Blood. 1991;77(9):1949–1957. | ||
Menell JS, Cesarman GM, Jacovina AT, McLaughlin MA, Lev EA, Hajjar KA. Annexin II and bleeding in acute promyelocytic leukemia. N Engl J Med. 1999;340(13):994–1004. | ||
Kwaan HC, Wang J, Weiss I. Expression of receptors for plasminogen activators on endothelial cell surface depends on their origin. J Thromb Haemost. 2004;2(2):306–312. | ||
Lavallée VP, Marquis M, Bordeleau ME, et al. Transcriptional landscape of APL identifies aberrant podoplanin expression as a defining feature and missing link for the bleeding disorder of this disease. Blood. 2016;128(22):1075. | ||
Oudijk EJ, Nieuwenhuis HK, Bos R, Fijnheer R. Elastase mediated fibrinolysis in acute promyelocytic leukemia. Thromb Haemost. 2000;83(6):906–908. | ||
Franchini M, Di Minno MN, Coppola A. Disseminated intravascular coagulation in hematologic malignancies. Semin Thromb Hemost. 2010;36(4):388–403. | ||
Runde V, Aul C, Heyll A, Schneider W. All-trans retinoic acid: not only a differentiating agent, but also an inducer of thromboembolic events in patients with M3 leukemia. Blood. 1992;79(2):534–535. | ||
Cao M, Li T, He Z, et al. Promyelocytic extracellular chromatin exacerbates coagulation and fibrinolysis in acute promyelocytic leukemia. Blood. 2017;129(13):1855–1864. | ||
Barbui T, Falanga A. Disseminated intravascular coagulation in acute leukemia. Semin Thromb Hemost. 2001;27(6):593–604. | ||
Libourel EJ, Klerk CP, van Norden Y, et al. Disseminated intravascular coagulation at diagnosis is a strong predictor for thrombosis in newly diagnosed acute myeloid leukemia. Blood. 2016;128(14):1854–1861. | ||
Mantha S, Goldman DA, Devlin SM, et al. Determinants of fatal bleeding during induction therapy for acute promyelocytic leukemia in the ATRA era. Blood. 2017;129(13):1763–1767. | ||
Platzbecker U, Avvisati G, Cicconi L, et al. Improved outcomes with retinoic acid and arsenic trioxide compared with retinoic acid and chemotherapy in non-high-risk acute promyelocytic leukemia: final results of the randomized Italian-German APL0406 trial. J Clin Oncol. 2017;35(6):605–612. | ||
Sanz MA, Grimwade D, Tallman MS, et al. Management of acute promyelocytic leukemia: recommendations from an expert panel on behalf of the European LeukemiaNet. Blood. 2009;113(9):1875–1891. | ||
Hashimoto S, Koike T, Tatewaki W, et al. Fatal thromboembolism in acute promyelocytic leukemia during all-trans retinoic acid therapy combined with antifibrinolytic therapy for prophylaxis of hemorrhage. Leukemia. 1994;8(7):1113–1115. | ||
Desborough M, Hadjinicolaou AV, Chaimani A, et al. Alternative agents to prophylactic platelet transfusion for preventing bleeding in people with thrombocytopenia due to chronic bone marrow failure: a meta-analysis and systematic review. Cochrane Database Syst Rev. 2016;10:CD012055. | ||
Estcourt LJ, Desborough M, Brunskill SJ, et al. Antifibrinolytics (lysine analogues) for the prevention of bleeding in people with haematological disorders. Cochrane Database Syst Rev. 2016;3:CD009733. | ||
Martí-Carvajal AJ, Anand V, Solà I. Treatment for disseminated intravascular coagulation in patients with acute and chronic leukemia. Cochrane Database Syst Rev. 2015;(6):CD008562. | ||
Oberoi S, Lehrnbecher T, Phillips B, et al. Leukapheresis and low-dose chemotherapy do not reduce early mortality in acute myeloid leukemia hyperleukocytosis: a systematic review and meta-analysis. Leuk Res. 2014;38(4):460–468. | ||
Van de Louw A. Effect of leukapheresis on blood coagulation in patients with hyperleukocytic acute myeloid leukemia. Transfus Apher Sci. 2017;56(2):214–219. | ||
Thachil J, Falanga A, Levi M, Liebman H, Di Nisio M. Management of cancer-associated disseminated intravascular coagulation: guidance from the SSC of the ISTH. J Thromb Haemost. 2015;13(4):671–675. | ||
Casini A, Fontana P, Lecompte TP. Thrombotic complications of myeloproliferative neoplasms: risk assessment and risk-guided management. J Thromb Haemost. 2013;11(7):1215–1227. | ||
Smalberg JH, Arends LR, Valla DC, Kiladjian JJ, Janssen HL, Leebeek FW. Myeloproliferative neoplasms in Budd-Chiari syndrome and portal vein thrombosis: a meta-analysis. Blood. 2012;120(25):4921–4928. | ||
Haguet H, Douxfils J, Mullier F, Chatelain C, Graux C, Dogné JM. Risk of arterial and venous occlusive events in chronic myeloid leukemia patients treated with new generation BCR-ABL tyrosine kinase inhibitors: a systematic review and meta-analysis. Expert Opin Drug Saf. 2017;16(1):5–12. | ||
De Stefano V, Za T, Rossi E, et al. Recurrent thrombosis in patients with polycythemia vera and essential thrombocythemia: incidence, risk factors, and effect of treatments. Haematologica. 2008;93(3):372–380. | ||
Quintás-Cardama A, Kantarjian H, Ravandi F, et al. Bleeding diathesis in patients with chronic myelogenous leukemia receiving dasatinib therapy. Cancer. 2009;115(11):2482–2490. | ||
Varma S, Sharma A, Malhotra P, Kumari S, Jain S, Varma N. Thrombotic complications of polycythemia vera. Hematology. 2008;13(6):319–323. | ||
Jensen MK, Brown PN, Lund BV, Nielsen OJ, Hasselbalch HC. Increased platelet activation and abnormal membrane glycoprotein content and redistribution in myeloproliferative disorders. Br J Haematol. 2000;110(1):116–124. | ||
Falanga A, Marchetti M, Evangelista V, et al. Polymorphonuclear leukocyte activation and hemostasis in patients with essential thrombocythemia and polycythemia vera. Blood. 2000;96(13):4261–4266. | ||
Falanga A, Marchetti M, Vignoli A, Balducci D, Barbui T. Leukocyte-platelet interaction in patients with essential thrombocythemia and polycythemia vera. Exp Hematol. 2005;33(5):523–530. | ||
Wautier MP, El Nemer W, Gane P, et al. Increased adhesion to endothelial cells of erythrocytes from patients with polycythemia vera is mediated by laminin α5 chain and Lu/BCAM. Blood. 2007;110(3):894–901. | ||
Duchemin J, Ugo V, Ianotto JC, Lecucq L, Mercier B, Abgrall JF. Increased circulating procoagulant activity and thrombin generation in patients with myeloproliferative neoplasms. Thromb Res. 2010;126(3):238–242. | ||
Barbui T, Carobbio A, Finazzi G, et al. Inflammation and thrombosis in essential thrombocythemia and polycythemia vera: different role of C-reactive protein and pentraxin 3. Haematologica. 2011;96(2):315–318. | ||
Jha PK, Vijay A, Sahu A, Ashraf MZ. Comprehensive gene expression meta-analysis and integrated bioinformatic approaches reveal shared signatures between thrombosis and myeloproliferative disorders. Sci Rep. 2016;6:37099. | ||
Budde U, van Genderen PJ. Acquired von Willebrand disease in patients with high platelet counts. Semin Thromb Hemost. 1997;23(5):425–431. | ||
Budde U, Dent JA, Berkowitz SD, Ruggeri ZM, Zimmerman TS. Subunit composition of plasma von Willebrand factor in patients with the myeloproliferative syndrome. Blood. 1986;68(6):1213–1217. | ||
Moore SF, Hunter RW, Harper MT, et al. Dysfunction of the PI3 kinase/Rap1/integrin αIIbβ3 pathway underlies ex vivo platelet hypoactivity in essential thrombocythemia. Blood. 2013;121(7):1209–1219. | ||
Appelmann I, Kreher S, Parmentier S, et al. Diagnosis, prevention, and management of bleeding episodes in Philadelphia-negative myeloproliferative neoplasms: recommendations by the Hemostasis Working Party of the German Society of Hematology and Medical Oncology (DGHO) and the Society of Thrombosis and Hemostasis Research (GTH). Ann Hematol. 2016;95(5):707–718. | ||
Tiede A, Priesack J, Werwitzke S, et al. Diagnostic workup of patients with acquired von Willebrand syndrome: a retrospective single-centre cohort study. J Thromb Haemost. 2008;6(4):569–576. | ||
Tefferi A, Barbui T. Polycythemia vera and essential thrombocythemia: 2017 update on diagnosis, risk-stratification, and management. Am J Hematol. 2017;92(1):94–108. | ||
Marchioli R, Finazzi G, Specchia G, et al. Cardiovascular events and intensity of treatment in polycythemia vera. N Engl J Med. 2013;368(1):22–33. | ||
Barbui T, Barosi G, Birgegard G, et al. Philadelphia-negative classical myeloproliferative neoplasms: critical concepts and management recommendations from European LeukemiaNet. J Clin Oncol. 2011;29(6):761–770. | ||
Harrison CN, Campbell PJ, Buck G, et al. Hydroxyurea compared with anagrelide in high-risk essential thrombocythemia. N Engl J Med. 2005;353(1):33–45. | ||
Alvarez-Larrán A, Pérez-Encinas M, Ferrer-Marín F, et al. Risk of thrombosis according to need of phlebotomies in patients with polycythemia vera treated with hydroxyurea. Haematologica. 2017;102(1):103–109. | ||
Maugeri N, Giordano G, Petrilli MP, et al. Inhibition of tissue factor expression by hydroxyurea in polymorphonuclear leukocytes from patients with myeloproliferative disorders: a new effect for an old drug? J Thromb Haemost. 2006;4(12):2593–2598. | ||
Verstovsek S, Mesa RA, Gotlib J, et al. A double-blind, placebo-controlled trial of ruxolitinib for myelofibrosis. N Engl J Med. 2012;366(9):799–807. | ||
Samuelson BT, Vesely SK, Chai-Adisaksopha C, Scott BL, Crowther M, Garcia D. The impact of ruxolitinib on thrombosis in patients with polycythemia vera and myelofibrosis: a meta-analysis. Blood Coagul Fibrinolysis. 2016;27(6):648–652. | ||
Kreher S, Ochsenreither S, Trappe RU, et al. Prophylaxis and management of venous thromboembolism in patients with myeloproliferative neoplasms: consensus statement of the Haemostasis Working Party of the German Society of Hematology and Oncology (DGHO), the Austrian Society of Hematology and Oncology (ÖGHO) and Society of Thrombosis and Haemostasis Research (GTH e.V.). Ann Hematol. 2014;93(12):1953–1963. | ||
Barbui T, De Stefano V. Management of venous thromboembolism in myeloproliferative neoplasms. Curr Opin Hematol. 2017;24(2):108–114. | ||
Carrier M, Cameron C, Delluc A, Castellucci L, Khorana AA, Lee AY. Efficacy and safety of anticoagulant therapy for the treatment of acute cancer-associated thrombosis: a systematic review and meta-analysis. Thromb Res. 2014;134(6):1214–1219. | ||
Young A, Phillips J, Hancocks H, et al. Anticoagulation therapy in selected cancer patients at risk of recurrence of venous thromboembolism. Thromb Res. 2016;140 Suppl 1:S172–S173. | ||
Lexicomp: drug interactions. Available from: https://www.uptodate.com/drug-interactions/?source=responsive_home#di-analyze. Accessed September 10, 2017. | ||
Gifkins DM, Matcho A, Yang, H, Xu Y, Gooden MA, Wildgust M. Incidence of major hemorrhage among CLL and MCL patients compared to the general elderly population: an analysis of the US SEER-Medicare linked database. Blood. 2015;126(23):3268. | ||
Mahipal A, Bilgrami S. Acquired hemophilia in chronic lymphocytic leukemia. Leuk Lymphoma. 2007;48(5):1026–1028. | ||
Byrd JC, Furman RR, Coutre SE, et al. Targeting BTK with ibrutinib in relapsed chronic lymphocytic leukemia. N Engl J Med. 2013;369(1):32–42. | ||
Byrd JC, Brown JR, O’Brien S, et al. Ibrutinib versus ofatumumab in previously treated chronic lymphoid leukemia. N Engl J Med. 2014;371(3):213–223. | ||
Whittle AM, Allsup DJ, Bailey JR. Chronic lymphocytic leukaemia is a risk factor for venous thromboembolism. Leuk Res. 2011;35(3):419–421. | ||
Simkovič M, Vodárek P, Motyčková M, et al. Venous thromboembolism in patients with chronic lymphocytic leukemia. Thromb Res. 2015;136(6):1082–1086. | ||
Lipsky AH, Farooqui MZ, Tian X, et al. Incidence and risk factors of bleeding-related adverse events in patients with chronic lymphocytic leukemia treated with ibrutinib. Haematologica. 2015;100(12):1571–1578. | ||
Imbruvica (ibrutinib) [package insert]. Horsham (PA): Janssen Biotech; 2017. | ||
BC Cancer Agency. BCCA protocol summary for treatment of previously untreated chronic lymphocytic leukemia or small lymphocytic lymphoma with chromosome 17 p deletion using ibrutinib. 2017. Available from: http://www.bccancer.bc.ca/chemotherapy-protocols-site/Documents/Lymphoma-Myeloma/ULYFIBRU_Protocol.pdf. Accessed August 30, 2017. | ||
Leong DP, Caron F, Hillis C, et al. The risk of atrial fibrillation with ibrutinib use: a systematic review and meta-analysis. Blood. 2016;128(1):138–140. | ||
Chai KL, Rowan G, Seymour JF, Burbury K, Carney D, Tam CS. Practical recommendations for the choice of anticoagulants in the management of patients with atrial fibrillation on ibrutinib. Leuk Lymphoma. 2017;58(12):2811–2814. | ||
Jones JA, Hillmen P, Coutre S, et al. Use of anticoagulants and antiplatelet in patients with chronic lymphocytic leukaemia treated with single-agent ibrutinib. Br J Haematol. 2017;178(2):286–291. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.