Back to Journals » Journal of Inflammation Research » Volume 14
Complement Profiles in Patients with Amyotrophic Lateral Sclerosis: A Prospective Observational Cohort Study
Authors Kjældgaard AL, Pilely K, Olsen KS, Øberg Lauritsen A, Wørlich Pedersen S, Svenstrup K, Karlsborg M, Thagesen H, Blaabjerg M, Theódórsdóttir Á, Gundtoft Elmo E, Torvin Møller A ![]() , Pedersen NA, Kirkegaard N, Møller K, Garred P
, Pedersen NA, Kirkegaard N, Møller K, Garred P ![]()
Received 31 December 2020
Accepted for publication 16 February 2021
Published 23 March 2021 Volume 2021:14 Pages 1043—1053
DOI https://doi.org/10.2147/JIR.S298307
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Ning Quan
Anne-Lene Kjældgaard,1,2 Katrine Pilely,1 Karsten Skovgaard Olsen,2 Anne Øberg Lauritsen,2 Stephen Wørlich Pedersen,3 Kirsten Svenstrup,3,4 Merete Karlsborg,4 Helle Thagesen,5 Morten Blaabjerg,5 Ásta Theódórsdóttir,6 Elisabeth Gundtoft Elmo,3 Anette Torvin Møller,7 Niels Anker Pedersen,8 Niels Kirkegaard,8 Kirsten Møller,2,9 Peter Garred1,9
1Laboratory of Molecular Medicine, Department of Clinical Immunology, Section 7631, Diagnostic Centre, Rigshospitalet, Copenhagen, Denmark; 2Department of Neuroanaesthesiology Neuroscience Centre, Rigshospitalet, Copenhagen, Denmark; 3Department of Neurology, Neuroscience Centre, Rigshospitalet, Copenhagen, Denmark; 4Department of Neurology, Bispebjerg Hospital, Copenhagen, Denmark; 5Department of Neurology, Roskilde University Hospital, Roskilde, Denmark; 6Department of Neurology, Odense University Hospital, Odense, Denmark; 7Department of Neurology, Aarhus Hospital, Copenhagen, Denmark; 8Department of Anaesthesiology, Private Hospital Gildhøj, Brondby, Denmark; 9Institute of Clinical Medicine, Faculty of Health and Medical Sciences, University of Copenhagen, Copenhagen, Denmark
Correspondence: Anne-Lene Kjældgaard
Laboratory of Molecular Medicine, Department of Clinical Immunology, Section 7631, Diagnostic Centre, Rigshospitalet, Ole Maaloesvej 26, Copenhagen, DK-2200, Denmark
Tel +45 35457631
Fax +45 35398766
Email [email protected]
Background: The complement system has been suggested to be involved in the pathophysiology of amyotrophic lateral sclerosis (ALS), a progressive motor neuron disease. In the present study, we compared levels of selected complement markers to clinical outcome in ALS patients.
Methods: This observational, explorative cohort study included 92 ALS patients, 61 neurological controls (NCs) admitted for suspected aneurysmal subarachnoid haemorrhage, and 96 neurologically healthy controls (NHCs). Peripheral blood and cerebrospinal fluid (CSF) were obtained for the measurement of ficolin-1, − 2, and − 3; collectin-11, MBL, MASP-3, MAP-1, C4, C3, PTX-3, and complement activation products C4c, C3bc, and sC5b-9. We recorded clinical outcomes of ALS patients for 24 to 48 months after inclusion in order to analyse the effects of the complement markers on survival time.
Results: Compared with both control groups, ALS patients exhibited increased collectin-11, C4 and sC5b-9 in plasma, as well as increased ficolin-3 in CSF. Ficolin-2 was significantly decreased in plasma of the ALS patients compared with NHCs, but not with NCs. The concentration of collectin-11, C3 and C3bc correlated negatively with the revised ALS functional rating scale (ALSFRS-R). No association was found between levels of complement markers and survival as estimated by hazard ratios.
Conclusion: ALS patients exhibit aberrant expression of selected mediators of the lectin complement pathway as well as increased activation of the terminal complement pathway, corroborating the notion that the complement system might be involved in the pathophysiology of ALS.
Keywords: amyotrophic lateral sclerosis, innate immunity, complement, lectin pathway, cerebrospinal fluid, pathophysiology, observational cohort study
Background
Amyotrophic lateral sclerosis (ALS) is a rare, rapidly progressing disease with a mean life expectancy of 30 months from the time of diagnosis.1 The hallmark of this disease is continual degeneration of the motor neurons, which leads to progressive dysfunction of extremity, trunk and bulbar skeletal muscles.
Several findings have suggested an essential role of the complement system in the pathophysiology of ALS.2 From being regarded as part of the host defence, recent research has shown a role for the complement system in development processes and homeostatic mechanisms of different tissues,3 including the brain, where complement components are crucial for neuronal migration and synaptic remodeling,4 and for regulating memory.5 Moreover, a dysregulated complement system is associated with the pathophysiology of many systemic inflammatory diseases such as sepsis, systemic lupus erythematosus, rheumatoid arthritis, anti-neutrophilic cytoplasmic autoantibodies (ANCA) vasculitis, inflammatory myopathies, and microangiopathies such as atypical hemolytic uremic syndrome.6 Increased complement activity also seems to be implicated in disorders of neurodevelopmental origin, such as schizophrenia, autism spectrum disorder, and Rett syndrome, as well as the neuroinflammatory diseases, neuromyelitis optica and myasthenia gravis.4,5
Dysfunctional complement activation also seems to be present during the progression of ALS in the surroundings of the diseased motor neurons in the central nervous system (CNS)7–19 as well as systemically.20–24 Several studies based on animal ALS models suggest that the complement system plays a significant pathophysiological role during the initial phase of ALS, as activated complement proteins appear in the surroundings of the neuromuscular junctions (NMJs) concomitantly with the first pathological changes that appear in the NMJs of the failing motor neurons.16,25 A terminal complement inhibitor, ravulizumab, is currently being tested on ALS patients in a Phase III trial (ClinicalTrials.gov Identifier: NCT04248465).
Recently, Xu et al conducted a mass spectrometry analysis of plasma from ALS patients and reported upregulation not only of the complement pathway in general but also ficolin-3 expression.21 The latter finding validated a previous observation by the same research group21,26 and indicates a contribution of the lectin pathway of complement in the pathophysiology of ALS. Lectin pathway components are present in the CNS of healthy individuals and appear to be involved in normal embryogenesis, but have so far not been studied in conjunction with ALS. In the present study, we measured levels of selected markers of the lectin complement pathway as well as complement activation products in peripheral blood and cerebrospinal fluid (CSF) from ALS and non-ALS patients. We hypothesised that these mediators are upregulated both intrathecally and systemically during ALS and that more extreme changes are associated with more severe disease and shorter survival times.
Materials and Methods
This observational, exploratory cohort study, which is reported according to the STROBE guidelines,27 was approved by the Committees on Health Research Ethics in the Capital Region of Denmark (Approval number H-16017145) and the Danish Data Protection Agency (File number 2012-58-0004), registered with Clinicaltrials.gov (NCT02869048) and conducted in accordance with the Declaration of Helsinki. After receiving written and verbal information, all participants gave their written consent prior to enrollment.
In accordance with the Global Data Protection Regulation of the European Union, data are not publicly available but may be available upon reasonable request if a mutual data sharing agreement is approved by the relevant authorities.
Participants
As previously described,28 we included adults (> 18 years) into the following groups:
- ALS cohort: Patients who were referred to or followed at five large Danish motor-neuron disease outpatient clinics were recruited if they fulfilled the El Escorial Revised criteria29 for probable or definite ALS.
- Neurologically healthy controls (NHCs): Healthy patients (American Society of Anesthesiologists score, 1–2) scheduled for planned orthopaedic surgery in spinal anaesthesia at a large elective surgical centre were included.
- Non-ALS neurological controls (NCs): Patients who underwent emergency admission to hospital because of suspected aneurysmal subarachnoid haemorrhage (SAH), and who underwent a lumbar puncture following computed tomography (CT) scanning of the brain; were included if the SAH diagnosis was refuted.
For both control groups (NHCs and NCs), patients with a history or symptoms of motor neuron disease, acute or chronic inflammatory disease, or autoimmune disease were excluded.
Samples
Peripheral venous blood and CSF were sampled at inclusion sites (Figure 1). The samples were immediately centrifugated (2000 g). Samples of EDTA plasma, serum, and CSF were kept at −20 °C during transportation and stored at −80 °C until analysis.28
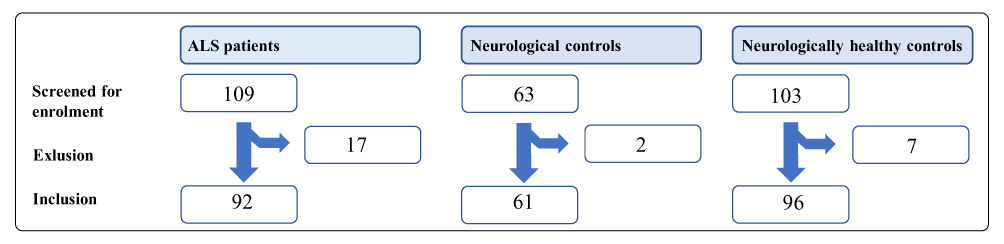 |
Figure 1 Inclusion of patients. Abbreviation: ALS, amyotrophic lateral sclerosis. |
The level of the complement lectin pathway components ficolin-1, ficolin-2, ficolin-3, collectin-11, mannose-binding lectin (MBL), MBL/ficolin/collectin-associated serine protease 3 (MASP-3), MBL/ficolin/collectin associated protein 1 (MAP-1), the acute phase marker pentraxin-3 (PTX-3), the complement components C4 and C3 and the activation products C4c, C3bc and sC5b-9 were measured in plasma and serum as well as in CSF as described below.
Complement Measurements
Complement related proteins ficolin-1, −2, and −3; collectin-11, MBL, MASP-3, MAP-1, PTX-3, and complement activation products C4c, C3bc, and sC5b-9 were measured in EDTA plasma and CSF samples by in-house sandwich enzyme-linked immunosorbent assays (ELISA). The measurements were conducted as previously described.30–41 The antibodies used for each of the ELISA analyses are listed in Supplementary Table 1. The serum concentrations of C3 and C4 were estimated using an automated turbidimetric protein analyser (SPAPLUS®, The Binding Site group LDT, Birmingham, UK). An overview of the performed analyses is found in Table 1.
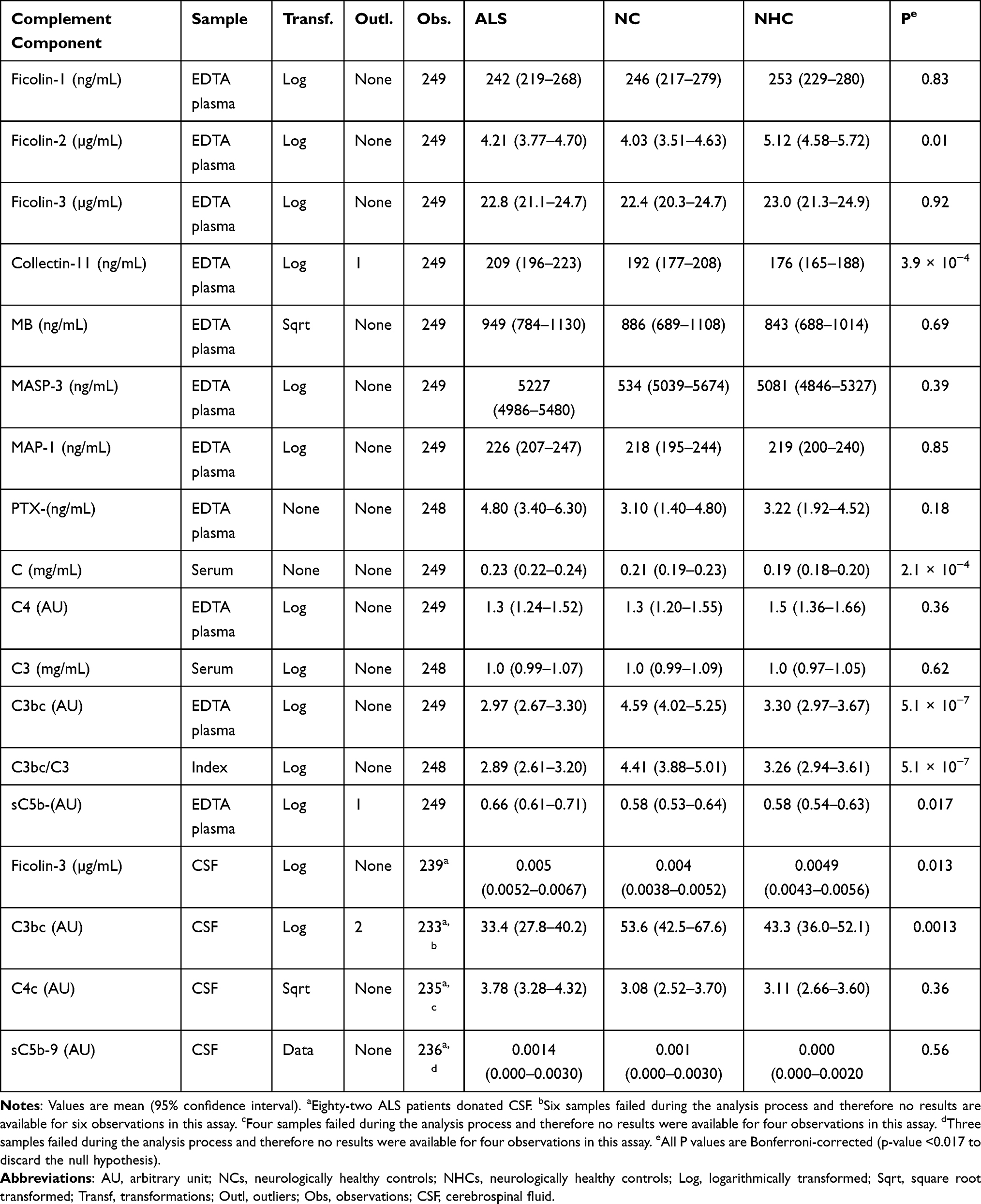 |
Table 1 Complement Markers and Mediators |
Statistical Analyses
The ALS group and the two control groups (NCs and NHCs) were compared using Bonferroni-corrected, stepwise three-way analysis of variance (ANOVA). For this comparison, analysis testing for differences was done, using the following covariates: subject category (ALS, NC, NHC), sex, and age. Data which did not follow a Gaussian distribution were log- or square-root transformed. To further analyse the significant differences between the ALS patients, the NHCs, and the NCs in the subject category, Bonferroni corrected, one-way ANOVA and t-testing were used. Missing observations were omitted from the statistical analyses on a listwise basis and were assumed to be missing at random.
The potential association between complement marker levels and clinical severity of ALS was assessed by a univariate regression model (Pearson’s correlation). For clinical severity, the revised ALS functional rating scale (ALSFRS-R) was used. ALSFRS-R quantifies the level of daily living activities in ALS.42
To evaluate the association between complement activation and survival, markers for which the level in ALS patients differed from both or one of the control groups were analyzed as continuous variables in a univariate Cox’ proportional hazards regression model. Moreover, we calculated the median survival time of the ALS cohort as we defined the primary end point as either death or the date for the initiation of invasive mechanical ventilatory support at censor date (15th of June 2020). ALS patients that had reached primary end point (invasive mechanical ventilation) prior to the test date were not included in the survival analysis.
Results
Patients
Between June 2016 and June 2018, 109 patients were screened for enrollment into the ALS cohort, of whom 92 were enrolled; we excluded 17 because the ALS diagnosis was dismissed. Of 63 potential NCs, two were excluded due to autoimmune disease and neuroinfection, respectively. Finally, of 101 possible NHCs, five were excluded due to the cancellation of surgery.
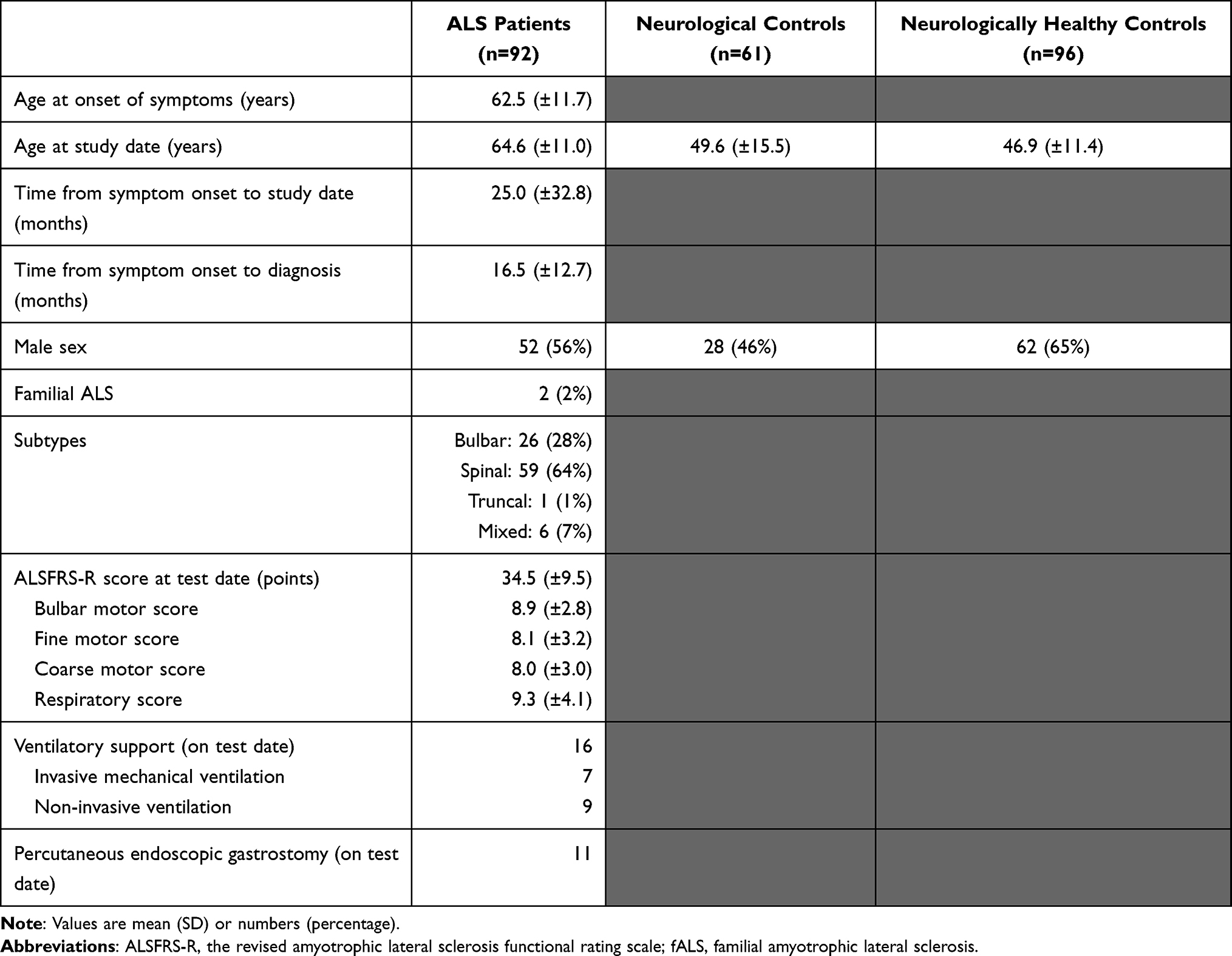 |
Table 2 Demographic Data |
Plasma and Serum Levels in ALS Patients, NCs, and NHCs
According to the ANOVA, the levels of ficolin-2, collectin-11, C4, C3bc, sC5b-9 and the C3bc/C3 index varied significantly between patients with ALS, NCs, and NHCs in plasma and serum samples (Figure 2 and Table 1). Following Bonferroni-corrected t-tests, ficolin-2 levels remained lower in ALS patients and NCs compared to NHCs, with no difference between ALS patients and NCs. Moreover, ALS patients had higher collectin-11, C4, and sC5b-9 levels than NHCs, but not compared to NCs. Finally, C3bc and consequently C3bc/C3 index were markedly higher in NCs than in ALS patients and NHCs, with no difference between the latter groups.
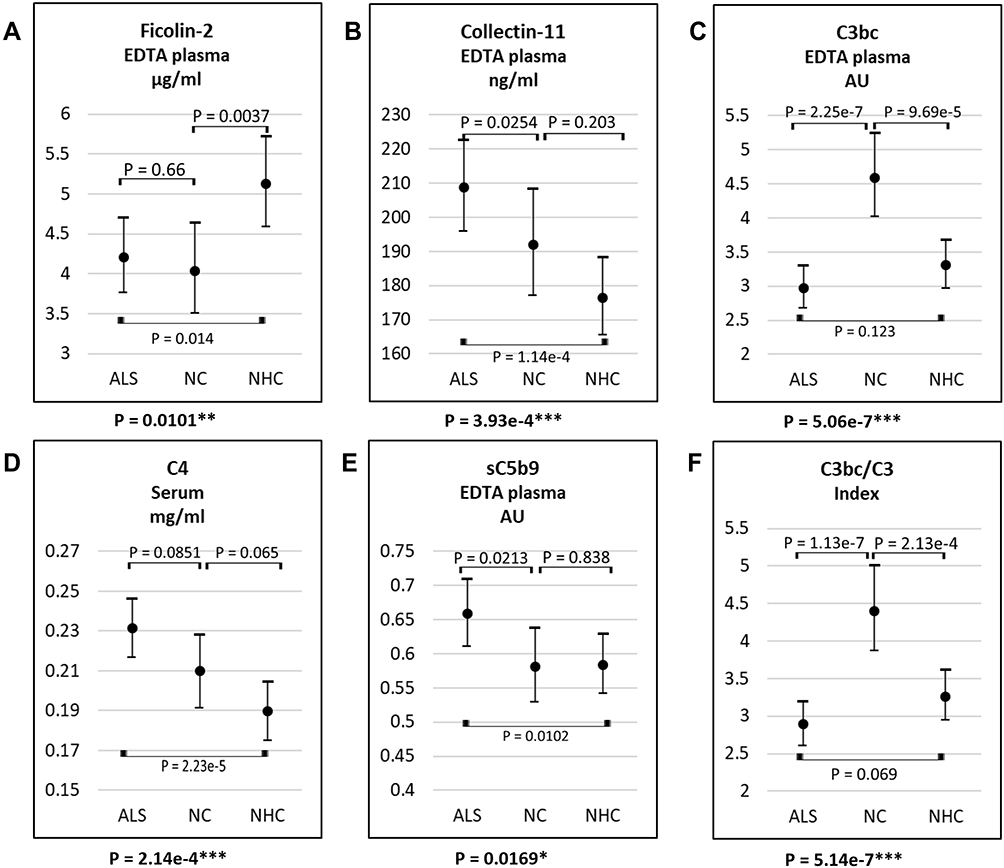 |
Figure 2 Plasma or serum level of ficolin-2 (A), collectin-11 (B), C3bc (C), C4 (D), sC5b-9 (E), and C3bc/C3 (F) in samples from patients with ALS, NC, and NHC. The lower p-value of each figure describes that there is a significant difference in complement levels among the covariate groups. The remaining p-values describe whether there is a difference between the two, marked groups. *P-values = 0.01–0.05; **P-values = 0.01–0.001; ***P-values <0.0001. Abbreviations: ALS: amyotrophic lateral sclerosis, NC: neurological controls, NHC: neurologically healthy controls. |
There were no significant differences between the three groups concerning ficolin-1, ficolin-3, MBL, MASP-3, MAP-1, C4c, PTX-3 (Table 1). MASP-3 levels were lower in males (mean value, 5031 (95% confidence interval, 4842–5227) ng/mL) than in females (5432 (5198–5677) ng/mL; p = 0.011); apart from MASP-3, no influence on mediator levels was exerted by age or sex.
CSF Levels in ALS Patients, NCs, and NHCs
In CSF, only ficolin-3 and the complement activation products C4c, C3bc, and sC5b-9 were measured (Table 1). ALS patients had higher ficolin-3 and lower C3bc levels compared to NCs and NHCs (Figure 3A and C); levels of C4c and C5b-9 did not differ between groups. For ALS patients, blood levels of ficolin-3 and C3bc correlated positively with CSF levels (Figure 3B and D); no correlation was found for C4c and C5b-9.
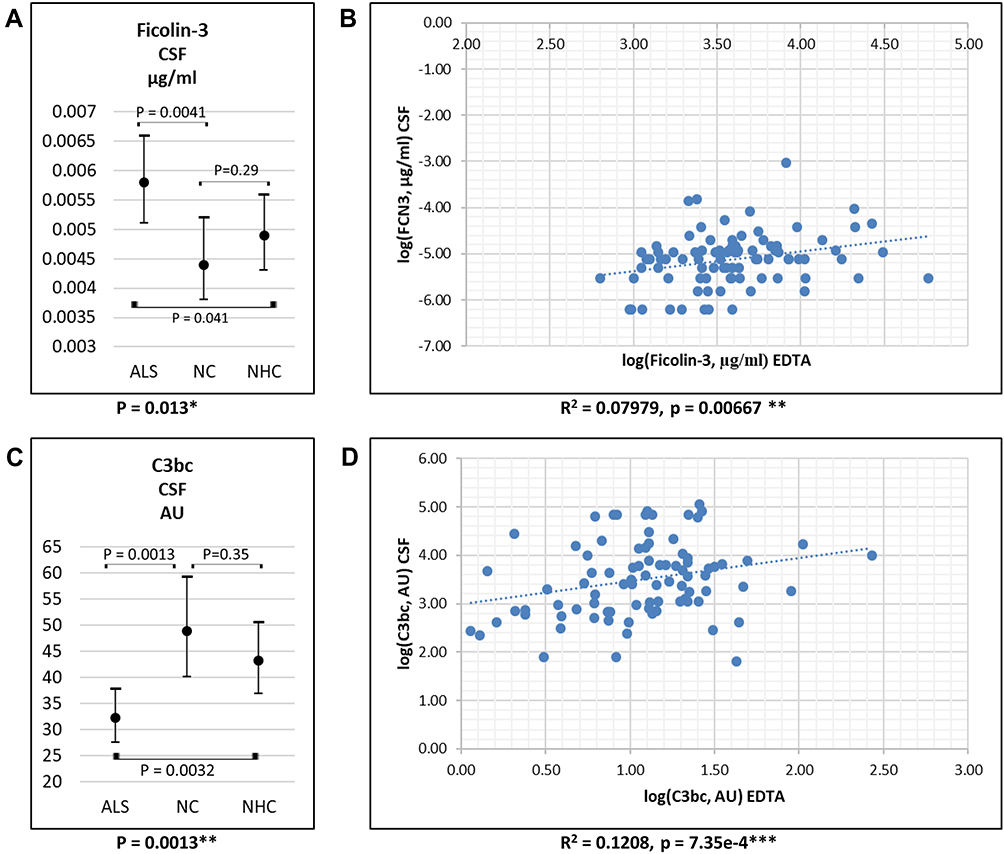 |
Figure 3 CSF levels of ficolin-3 (A) and C3bc (C) in CSF in patients with ALS, NC and NHC. Values are mean (95% confidence interval). Correlation between plasma/serum levels and CSF levels of ficolin-3 (B) and C3bc (D) in ALS patients. *P-values = 0.01–0.05; **P-values = 0.01–0.001; ***P-values <0.0001. Abbreviations: ALS, amyotrophic lateral sclerosis; CSF, cerebrospinal fluid; NC, neurological controls; NHC, neurologically healthy controls. |
Association Between Complement Markers and Clinical Score
Collectin-11, C3 and C3bc correlated negatively with the ALSFRS-S score (Figure 4). The remaining biomarkers (ficolin-1, ficolin-2, ficolin-3, MBL, MASP-3, MAP-1, PTX3, C4, C4c, and sC5b-9) were not associated with the ALSFRS-R score (Supplementary Table 2, Supplementary Figures 1 and 2).
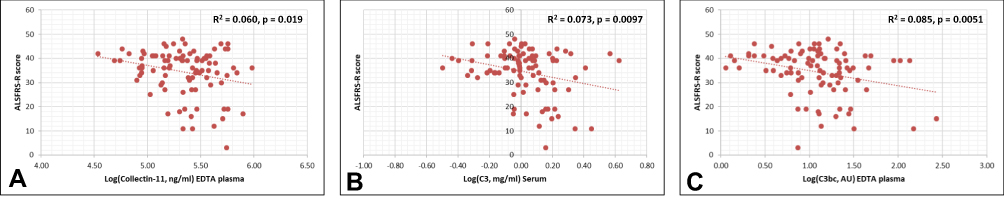 |
Figure 4 Univariate regression analysis (Pearson’s correlation). The measured complement protein levels of collectin-11 (A), C3 (B), C3bc (C) were found to be significantly associated with ALSFRS-R. Abbreviations: ALSFRS-R, amyotrophic lateral sclerosis functional rating scale – revised; AU, arbitrary units; Log, logarithmically transformed. |
Association Between Complement Markers and Survival
Eighty-five ALS patients were included for the survival analysis; the remaining seven patients were already receiving invasive mechanical ventilatory support on the test date (Supplementary Table 3). Among the included ALS patients, 70 patients reached the primary end-point (initiation of invasive mechanical ventilatory treatment or death) at censor date. Of those, the median survival time from onset of symptoms until the primary end point was 34 months. The median survival time from the time of diagnosis until the primary end point was 18 months. According to the Cox proportional hazard model, none of the complement markers was associated with survival time.
Discussion
In the present study, we analysed several markers and activation products of the lectin complement system and found that several of these were markedly changed as assessed by variance analysis among the groups of ALS patients, neurological controls and neurologically healthy controls. Even though correction for multiple testing rendered some differences non-significant when these changes were further compared between groups, the point estimates for the circulating levels of collectin-11, C4, and sC5b-9 were increased in the ALS group compared to both control groups, whereas ficolin-2 was decreased both in the ALS and neurological controls.
Even though sC5-9 complex was increased in the ALS patients compared to the two control groups, as previously reported,20 we observed no difference between the three groups regarding C4c, an activation marker which is supposedly increased by either classical or lectin pathway activation. However, the activation marker C3bc was increased only in neurological patients compared to ALS and neurologically healthy controls. These differences between the three different complement activation markers are challenging to explain. Still, it could indicate that the terminal complement route is activated by a non-canonical mechanism not involving the traditional activation routes. Apart from the activation by the classical, lectin, and alternative pathways direct activation of C5 has been shown, resulting in the generation of sC5b-9 without the involvement of C3 complement convertases.43 This has been documented by the action of thrombin on C5,44 which is not very likely in ALS, but other proteases might be involved. C3 activation without downstream generation of an efficient C5 convertase and generation of sC5b-9 has been demonstrated and might explain why we only observed increased levels of C3bc in neurological controls.45 In these patients, complement activation might be related to the alternative route and not the classical and lectin pathway, explaining the absence of C4c activation. However, to further complicate the picture, C3 activation via the lectin pathway enzymes without the involvement of C4 and C2 has recently been reported.46
In CSF, we were able to detect ficolin-3, C4c, and C3bc. Trace amounts of sC5-9 were only detected in some individuals, while the other components were undetectable by the methods used. In contrast to the results found in plasma, the ficolin-3 level was significantly increased in the CSF from ALS patients compared with the two control groups. As for plasma, CSF C4c did not differ between ALS and controls, while the same difference as was observed in plasma was also observed for C3bc in CSF. The level of sC5b-9 in CSF was too low to allow a meaningful comparison between the groups.
Previously, increased levels of ficolin-3 have been detected by mass spectrometry in ALS patients and confirmed by ELISA in a small group of patients.26,47 We could not confirm these observations in our study. There might be several reasons for this. ALS is a heterogeneous disease, and protein levels might differ over time. The strength of our study was that we carefully selected the control groups to be able to detect differences specific for ALS and not just a random difference from the diseased individuals compared with healthy controls. Thus, the former difference might have been due to a type 1 error.
Nevertheless, we did observe an increased concentration of ficolin-3 in CSF compared with the controls. Whether this is because ficolin-3 might be differentially regulated in the different compartments at different stages of the disease cannot be answered from our study, but is indeed an indication of the involvement of ficolin-3 at least as a biological marker in ALS. The decreased level of ficolin-2 in both ALS and neurological controls is relevant since it is consistent with findings that ficolin-2 is either consumed or down-regulated during certain inflammatory processes.48
Two novel findings of this study were the selective increase of collectin-11 and C4 in plasma. The knowledge of the biological role of collectin-11 is not well established, but there is an emerging acceptance that it plays an important role in organ development during embryogenesis and in tissue homeostatic mechanisms.49 C4 has been suggested to be important for the development of the brain, as increased C4 gene copy numbers are associated with the development of neuropsychiatric diseases like schizophrenia.50 Thus, it would be interesting to investigate if this phenomenon is also associated with ALS.
This study has several limitations. First, we obtained the samples when it was appropriate according to the patients’ clinical workup after they had consented to the study. For the majority of patients, this was during the diagnostic workup, whereas a few patients with confirmed ALS were also included. This potentially increases the heterogeneity of the population. Second, the controls were not matched with regard to age and sex of the patients, mainly for practical purposes; since the characteristics of the lectin-complement system change both with age and sex,51 this makes any comparison between patients and controls harder. Along the same line of reasoning, it would have been interesting to compare the ALS patients to other patients with chronic neurological disease. As controls with neurological disease, we included only patients suspected of an acute neurological disease (subarachnoid hemorrhage), which provided an acute indication for lumbar puncture and as such was logistically attractive. By contrast, including patients with chronic neurological disease was not clinically feasible. Third, the neurologically healthy patients’ did not undergo specific testing to exclude neurological disease, but were simply assumed to be healthy based on the available medical information on the day of surgery. Fourth, we included patients only with definite or probable ALS; this may have excluded patients during the onset or early stages of ALS and thus obviated our chances of studying inflammatory processes as part of the pathogenesis of ALS. On the other hand, any association between inflammation and progression of ALS should be more marked in patients with established ALS. The present findings do not support the presence of a persistent inflammatory process to drive the progression of ALS, at least not one that includes the lectin-complement system. They do, however, not exclude temporary or ‘pulsatile’ inflammatory phenomena that were not present at the time of sampling. Fifth, we did not obtain data regarding general systemic or neuroinflammation, such as blood or CSF white blood cell and differential counts or plasma C-reactive protein. Finally, ALS may be associated with cognitive impairment; we did not attempt to distinguish between patients with and without cognitive difficulties. Neither did we attempt to characterize the functional level of patients beyond that which was comprised in the ALSFRS-R scale; a more granular clinical staging could potentially have been obtained using tools such as the Kings ALS clinical staging.52
Conclusion
This explorative study substantiates that the terminal part of the complement system is activated in ALS patients in a non-canonical manner. Collectin-11, C3 and C3bc correlated negatively with the ALSFRS-S score. We also observed that collectin-11 and C4 were significantly increased in plasma compared to controls, but not after correction for multiple comparisons. Thus, further analysis in independent cohorts is necessary to validate these observations. Nevertheless, this study supports the notion that the complement system might be involved in the pathophysiology of ALS.
Abbreviations
ALS, amyotrophic lateral sclerosis; ANOVA, analysis of variance; AU, arbitrary unit; AUROC, area under the ROC curve; CNS, central nervous system; CSF, cerebrospinal fluid; EDTA, ethylenediamine tetraacetic acid; ELISA, enzyme-linked immunosorbent assay; fALS, familial amyotrophic lateral sclerosis; g, the relative centrifugal force; Log, logarithmic; MAP-1, MBL/ficolin/collectin associated protein 1 (MAP-1); MASP-3, MBL/ficolin/collectin-associated serine protease 3; MBL, Mannose-binding lectin; NMJ, neuromuscular junction; NCs, neurological controls; NHCs, neurologically healthy controls; NPV, negative predictive value; PPV, positive predictive value; PTX-3, pentraxin-3; ROC, receiver operating characteristic; SAH: aneurysmal subarachnoid haemorrhage; sALS, sporadic amyotrophic lateral sclerosis; Sqrt, Square root.
Acknowledgments
We gratefully acknowledge the clinical staff of the participating neurological departments as well as the staff at Gildhøj Private Hospital; their outstanding support and effort made this study possible. We thank the trial participants that have consented to be part of this study, with special thanks and warm thoughts to all the Danish ALS patients. Many thanks to the researchers and lab technicians at the Laboratory of Molecular Medicine, Rigshospitalet. Finally, we wish to acknowledge Anders Hedegaard Jessen, MSc, PhD for invaluable, significant statistical support.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agreed to be accountable for all aspects of the work.
Funding
This work was funded by The Jascha Foundation, Aase and Ejnar Danielsen’s Foundation, The Danish Research Council for Independent Research (DFF-6110-00489), The Danish Heart Association (15-R99-A5943-22922), The Svend Andersen Research Foundation, Rigshospitalet, and the Novo Nordisk Research Foundation.
Disclosure
The authors report no conflicts of interest for this work.
References
1. Hardiman O, Al-Chalabi A, Brayne C, et al. The changing picture of amyotrophic lateral sclerosis: lessons from European registers. J Neurol Neurosurg Psychiatry. 2017;88(7):557–563. doi:10.1136/jnnp-2016-314495
2. Kjældgaard AL, Pilely K, Olsen KS, et al. Amyotrophic lateral sclerosis: the complement and inflammatory hypothesis. Mol Immunol. 2018;102:14–25. doi:10.1016/j.molimm.2018.06.007
3. Hawksworth OA, Coulthard LG, Woodruff TM. Complement in the fundamental processes of the cell. Mol Immunol. 2017;84:17–25. doi:10.1016/j.molimm.2016.11.010
4. Magdalon J, Mansur F, Teles ESAL, de Goes VA, Reiner O, Sertié AL. Complement system in brain architecture and neurodevelopmental disorders. Front Neurosci. 2020;14:23. doi:10.3389/fnins.2020.00023
5. Wang C, Yue H, Hu Z, et al. Microglia mediate forgetting via complement-dependent synaptic elimination. Science (New York, NY). 2020;367(6478):688–694. doi:10.1126/science.aaz2288
6. Vignesh P, Rawat A, Sharma M, Singh S. Complement in autoimmune diseases. Clin Chim Acta. 2017;465:123–130. doi:10.1016/j.cca.2016.12.017
7. Kawamata T, Akiyama H, Yamada T, McGeer PL. Immunologic reactions in amyotrophic lateral sclerosis brain and spinal cord tissue. Am J Pathol. 1992;140(3):691–707.
8. Ferraiuolo L, Heath PR, Holden H, Kasher P, Kirby J, Shaw PJ. Microarray analysis of the cellular pathways involved in the adaptation to and progression of motor neuron injury in the SOD1 G93A mouse model of familial ALS. J Neurosci. 2007;27(34):9201–9219. doi:10.1523/JNEUROSCI.1470-07.2007
9. Annunziata P. Complement alterations in the CSF of patients with amyotrophic lateral sclerosis. J Neurol Neurosurg Psychiatry. 1995;58(3):393. doi:10.1136/jnnp.58.3.393-b
10. Ganesalingam J, An J, Shaw CE, Shaw G, Lacomis D, Bowser R. Combination of neurofilament heavy chain and complement C3 as CSF biomarkers for ALS. J Neurochem. 2011;117(3):528–537. doi:10.1111/j.1471-4159.2011.07224.x
11. Tsuboi Y, Yamada T. Increased concentration of C4d complement protein in CSF in amyotrophic lateral sclerosis. J Neurol Neurosurg Psychiatry. 1994;57(7):859–861. doi:10.1136/jnnp.57.7.859
12. Grewal RP, Morgan TE, Finch CE. C1qB and clusterin mRNA increase in association with neurodegeneration in sporadic amyotrophic lateral sclerosis. Neurosci Lett. 1999;271(1):65–67. doi:10.1016/S0304-3940(99)00496-6
13. Lee JD, Kamaruzaman NA, Fung JN, et al. Dysregulation of the complement cascade in the hSOD1G93A transgenic mouse model of amyotrophic lateral sclerosis. J Neuroinflammation. 2013;10(1):119. doi:10.1186/1742-2094-10-119
14. Lee JD, Kumar V, Fung JN, Ruitenberg MJ, Noakes PG, Woodruff TM. Pharmacological inhibition of complement C5a-C5a1 receptor signalling ameliorates disease pathology in the hSOD1 G93A mouse model of amyotrophic lateral sclerosis. Br J Pharmacol. 2017;174(8):689–699. doi:10.1111/bph.13730
15. Lobsiger CS, Boillee S, Cleveland DW. Toxicity from different SOD1 mutants dysregulates the complement system and the neuronal regenerative response in ALS motor neurons. Proc Natl Acad Sci U S A. 2007;104(18):7319–7326. doi:10.1073/pnas.0702230104
16. Heurich B, El Idrissi NB, Donev RM, et al. Complement upregulation and activation on motor neurons and neuromuscular junction in the SOD1 G93A mouse model of familial amyotrophic lateral sclerosis. J Neuroimmunol. 2011;235(1–2):104–109. doi:10.1016/j.jneuroim.2011.03.011
17. Humayun S, Gohar M, Volkening K, et al. The complement factor C5a receptor is upregulated in NFL-/- mouse motor neurons. J Neuroimmunol. 2009;210(1–2):52–62. doi:10.1016/j.jneuroim.2009.01.028
18. Woodruff TM, Costantini KJ, Crane JW, et al. The complement factor C5a contributes to pathology in a rat model of amyotrophic lateral sclerosis. J Immunol. 2008;181(12):8727–8734. doi:10.4049/jimmunol.181.12.8727
19. Woodruff TM, Lee JD, Noakes PG. Role for terminal complement activation in amyotrophic lateral sclerosis disease progression. Proc Natl Acad Sci U S A. 2014;111(1):E3–4. doi:10.1073/pnas.1321248111
20. Mantovani S, Gordon R, Macmaw JK, et al. Elevation of the terminal complement activation products C5a and C5b-9 in ALS patient blood. J Neuroimmunol. 2014;276(1–2):213–218. doi:10.1016/j.jneuroim.2014.09.005
21. Xu Z, Lee A, Nouwens A, Henderson RD, McCombe PA. Mass spectrometry analysis of plasma from amyotrophic lateral sclerosis and control subjects. Amyotroph Lateral Scler Frontotemporal Degener. 2018;1–15.
22. Chen X, Feng W, Huang R, et al. Evidence for peripheral immune activation in amyotrophic lateral sclerosis. J Neurol Sci. 2014;347(1–2):90–95. doi:10.1016/j.jns.2014.09.025
23. Goldknopf IL, Sheta EA, Bryson J, et al. Complement C3c and related protein biomarkers in amyotrophic lateral sclerosis and Parkinson’s disease. Biochem Biophys Res Commun. 2006;342(4):1034–1039. doi:10.1016/j.bbrc.2006.02.051
24. Apostolski S, Nikolic J, Bugarski-Prokopljevic C, Miletic V, Pavlovic S, Filipovic S. Serum and CSF immunological findings in ALS. Acta Neurol Scand. 1991;83(2):96–98. doi:10.1111/j.1600-0404.1991.tb04656.x
25. Bahia El Idrissi N, Bosch S, Ramaglia V, Aronica E, Baas F, Troost D. Complement activation at the motor end-plates in amyotrophic lateral sclerosis. J Neuroinflammation. 2016;13(1):72. doi:10.1186/s12974-016-0538-2
26. Mohanty L, Henderson RD, McCombe PA, Lee A. Levels of clusterin, CD5L, ficolin-3, and gelsolin in ALS patients and controls. Amyotroph Lateral Scler Frontotemporal Degener. 2020;1–4.
27. von Elm E, Altman DG, Egger M, Pocock SJ, Gøtzsche PC, Vandenbroucke JP. The Strengthening the Reporting of Observational Studies in Epidemiology (STROBE) statement: guidelines for reporting observational studies. PLoS Med. 2007;4(10):e296. doi:10.1371/journal.pmed.0040296
28. Kjældgaard AL, Pilely K, Olsen KS, et al. Amyotrophic lateral sclerosis and the innate immune system: protocol for establishing a biobank and statistical analysis plan. BMJ Open. 2020;10(8):e037753. doi:10.1136/bmjopen-2020-037753
29. Brooks BR, Miller RG, Swash M, Munsat TL. El Escorial revisited: revised criteria for the diagnosis of amyotrophic lateral sclerosis. Amyotroph Lateral Scler Other Motor Neuron Disord. 2000;1(5):293–299. doi:10.1080/146608200300079536
30. Bayarri-Olmos R, Kirketerp-Moller N, Perez-Alos L, Skjodt K, Skjoedt MO, Garred P. Development of a quantitative assay for the characterization of human collectin-11 (CL-11, CL-K1). Front Immunol. 2018;9:2238. doi:10.3389/fimmu.2018.02238
31. Bastrup-Birk S, Skjoedt MO, Munthe-Fog L, Strom JJ, Ma YJ, Garred P. Pentraxin-3 serum levels are associated with disease severity and mortality in patients with systemic inflammatory response syndrome. PLoS One. 2013;8(9):e73119. doi:10.1371/journal.pone.0073119
32. Hansen S, Selman L, Palaniyar N, et al. Collectin 11 (CL-11, CL-K1) is a MASP-1/3-associated plasma collectin with microbial-binding activity. J Immunol. 2010;185(10):6096–6104. doi:10.4049/jimmunol.1002185
33. Skjoedt MO, Hummelshoj T, Palarasah Y, et al. Serum concentration and interaction properties of MBL/ficolin associated protein-1. Immunobiology. 2011;216(5):625–632. doi:10.1016/j.imbio.2010.09.011
34. Pilely K, Skjoedt MO, Nielsen C, et al. A specific assay for quantification of human C4c by use of an anti-C4c monoclonal antibody. J Immunol Methods. 2014;405:87–96. doi:10.1016/j.jim.2014.01.011
35. Garred P, Mollnes TE, Lea T. Quantification in enzyme-linked immunosorbent assay of a C3 neoepitope expressed on activated human complement factor C3. Scand J Immunol. 1988;27(3):329–335. doi:10.1111/j.1365-3083.1988.tb02354.x
36. Mollnes TE, Lea T, Froland SS, Harboe M. Quantification of the terminal complement complex in human plasma by an enzyme-linked immunosorbent assay based on monoclonal antibodies against a neoantigen of the complex. Scand J Immunol. 1985;22(2):197–202. doi:10.1111/j.1365-3083.1985.tb01871.x
37. Munthe-Fog L, Hummelshoj T, Honore C, et al. Variation in FCN1 affects biosynthesis of ficolin-1 and is associated with outcome of systemic inflammation. Genes Immun. 2012;13:515–522.
38. Munthe-Fog L, Hummelshøj T, Hansen BE, et al. The impact of FCN2 polymorphisms and haplotypes on the Ficolin-2 serum levels. Scand J Immunol. 2007;65(4):383–392. doi:10.1111/j.1365-3083.2007.01915.x
39. Munthe-Fog L, Hummelshøj T, Ma YJ, et al. Characterization of a polymorphism in the coding sequence of FCN3 resulting in a Ficolin-3 (Hakata antigen) deficiency state. Mol Immunol. 2008;45(9):2660–2666. doi:10.1016/j.molimm.2007.12.012
40. Garred P, Madsen HO, Kurtzhals JA, et al. Diallelic polymorphism may explain variations of the blood concentration of mannan-binding protein in Eskimos, but not in black Africans. Eur J Immunogen. 1992;19(6):403–412. doi:10.1111/j.1744-313X.1992.tb00083.x
41. Skjoedt MO, Palarasah Y, Munthe-Fog L, et al. MBL-associated serine protease-3 circulates in high serum concentrations predominantly in complex with Ficolin-3 and regulates Ficolin-3 mediated complement activation. Immunobiology. 2010;215:921–931.
42. Cedarbaum JM, Stambler N, Malta E, et al. The ALSFRS-R: a revised ALS functional rating scale that incorporates assessments of respiratory function. BDNF ALS Study Group (Phase III). J Neurol Sci. 1999;169:13–21.
43. Huber-Lang M, Sarma JV, Zetoune FS, et al. Generation of C5a in the absence of C3: a new complement activation pathway. Nature Med. 2006;12(6):682–687. doi:10.1038/nm1419
44. Krisinger MJ, Goebeler V, Lu Z, et al. Thrombin generates previously unidentified C5 products that support the terminal complement activation pathway. Blood. 2012;120(8):1717–1725. doi:10.1182/blood-2012-02-412080
45. Bhakdi S, Fassbender W, Hugo F, et al. Relative inefficiency of terminal complement activation. J Immunol. 1988;141:3117–3122.
46. Yaseen S, Demopulos G, Dudler T, et al. Lectin pathway effector enzyme mannan-binding lectin-associated serine protease-2 can activate native complement C3 in absence of C4 and/or C2. FASEB J. 2017;31(5):2210–2219. doi:10.1096/fj.201601306R
47. Xu Z, Lee A, Nouwens A, Henderson RD, McCombe PA. Mass spectrometry analysis of plasma from amyotrophic lateral sclerosis and control subjects. Amyotroph Lateral Scler Frontotemporal Degener. 2018;19(5–6):362–376. doi:10.1080/21678421.2018.1433689
48. Hansen CB, Bayarri-Olmos R, Kristensen MK, Pilely K, Hellemann D, Garred P. Complement related pattern recognition molecules as markers of short-term mortality in intensive care patients. The J Infect. 2020;80:378–387.
49. Gajek G, Świerzko AS, Cedzyński M. Association of polymorphisms of MASP1/3, COLEC10, and COLEC11 genes with 3MC syndrome. Int J Mol Sci. 2020;21(15):21. doi:10.3390/ijms21155483
50. Sekar A, Bialas AR, de Rivera H, et al. Schizophrenia risk from complex variation of complement component 4. Nature. 2016;530(7589):177–183. doi:10.1038/nature16549
51. Gaya da Costa M, Poppelaars F, van Kooten C, et al. Age and sex-associated changes of complement activity and complement levels in a healthy Caucasian population. Front Immunol. 2018;9.
52. Balendra R, Al Khleifat A, Fang T, Al-Chalabi A. A standard operating procedure for King’s ALS clinical staging. Amyotroph Lateral Scler Frontotemporal Degener. 2019;20(3–4):159–164. doi:10.1080/21678421.2018.1556696
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.