Back to Journals » ImmunoTargets and Therapy » Volume 9
Complement Inhibition for the Treatment of Myasthenia Gravis
Authors Mantegazza R ![]() , Vanoli F, Frangiamore R, Cavalcante P
, Vanoli F, Frangiamore R, Cavalcante P
Received 3 October 2020
Accepted for publication 2 December 2020
Published 15 December 2020 Volume 2020:9 Pages 317—331
DOI https://doi.org/10.2147/ITT.S261414
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Professor Michael Shurin
Renato Mantegazza, Fiammetta Vanoli, Rita Frangiamore, Paola Cavalcante
Neurology IV - Neuroimmunology and Neuromuscular Diseases Unit, Fondazione IRCCS Istituto Neurologico Carlo Besta, Milan, Italy
Correspondence: Renato Mantegazza
Neurology IV - Neuroimmunology and Neuromuscular Diseases Unit, Fondazione IRCCS Istituto Neurologico Carlo Besta, Via Celoria 11, Milan 20133, Italy
Tel +39-02-23942471
Fax +39-02-23942413
Email [email protected]
Abstract: Generalized myasthenia gravis (gMG) is a rare autoimmune disorder affecting the neuromuscular junction (NMJ). Approximately 80– 90% of patients display antibodies directed against the nicotinic acetylcholine receptor (AChR). A major drive of AChR antibody-positive MG pathology is represented by complement activation. The role of the complement cascade has been largely demonstrated in patients and in MG animal models. Complement activation at the NMJ leads to focal lysis of the post-synaptic membrane, disruption of the characteristic folds, and reduction of AChR. Given that the complement system works as an activation cascade, there are many potential targets that can be considered for therapeutic intervention. Preclinical studies have confirmed the efficacy of complement inhibition in ameliorating MG symptoms. Eculizumab, an antibody directed towards C5, has recently been approved for the treatment of AChR antibody-positive gMG. Other complement inhibitors, targeting C5 as well, are currently under phase III study. Complement inhibitors, however, may present prohibitive costs. Therefore, the identification of a subset of patients more or less prone to respond to such therapies would be beneficial. For such purpose, there is a critical need to identify possible biomarkers predictive of therapeutic response, a field not yet sufficiently explored in MG. This review aims to give an overview of the complement cascade involvement in MG, the evolution of complement-inhibiting therapies and possible biomarkers useful to tailor and monitor complement-directed therapies.
Keywords: myasthenia gravis, complement system, biological drugs, C5, biomarkers
Introduction
Myasthenia Gravis (MG) is a rare autoimmune disorder that targets the neuromuscular junction (NMJ). It is caused by B-cell activation with subsequent production of autoantibodies targeting different proteins of the postsynaptic endplate. About 80–90% of patients have antibodies directed against the nicotinic acetylcholine receptor (AChR), which are rarely present in healthy subjects. The remaining 10–20% can have antibodies against the muscle-specific tyrosine kinase (MuSK) or the lipoprotein-related protein 4 (LRP4) or have no specific antibodies at all.1,2 Pathogenic features mostly depend on serological profile. In fact, as treated more in depth further on, in AChR-positive MG the most critical pathogenic mechanism is complement activation by AChR antibodies, which are of IgG1 and IgG3 subclass. Anti-LRP4 antibodies also act through complement activation, being mainly of IgG1 subclass, and through inhibition of LRP4–agrin interaction which is fundamental for AChR clustering.3 Anti-MuSK antibodies are predominantly of IgG4 subclass and are therefore unable to activate the complement cascade, yet they are able to interfere with AChR clustering.4
MG clinical hallmarks are weakness and fatigability involving ocular, bulbar, and skeletal muscles.5 Ocular involvement is often the first to appear, and in most cases, patients progress to generalized MG (gMG) within 3 years.2 Pharmacological treatment of MG comprehends cholinesterase inhibitors and immunosuppressive therapy (IST), such as chronic corticosteroids or other ISTs used as second-line therapy. Plasmapheresis (PE) or intravenous immunoglobulins (IVIg) are recommended for MG crisis or exacerbation.6–8 However, in MG there is a great variability in treatment response and about 10–15% of patients are refractory to treatment.5–9 Moreover, prolonged treatment with ISTs also comes with several other issues such as chronic immunosuppression, adverse events, and comorbidities. Therefore, there is an urgent need of new drugs, with a more specific and effective action.
In this review we will provide an extensive overview of the complement cascade and its role in AChR-positive gMG, focusing on preclinical and clinical data encouraging the application of complement inhibitors as a new therapeutic approach in gMG.
The Complement Cascade
The complement is a protein cascade, composed of over 50 proteins, key arm of the innate immune system. Its main function is to recognize and destroy invading microorganisms through the ultimate formation of the membrane attack complex (MAC) (Figure 1).10 The complement also plays a crucial role in adaptive immunity, by boosting the antibody response, and is implicated in the clearance of dead cells and immune complexes.11 Other beneficial effects are post-injury tissue regeneration,12 synaptic pruning in developmental stages,13 and a possible important role in the modulation of T cell responses.14
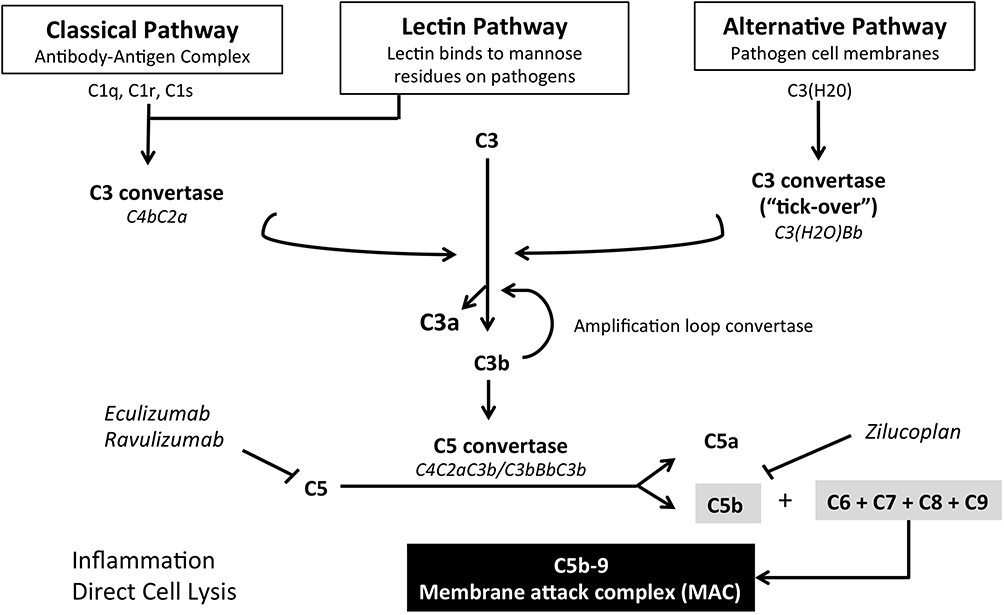 |
Figure 1 Schematic representation of the complement cascade and therapeutic targets of current complement inhibitors. |
The system can be divided into three main pathways depending on the modality of complement activation: i) the classical pathway, which occurs when C1 recognition molecule is activated by the binding of an antibody to a specific surface; ii) the mannose-binding lectin (MBL) pathway, activated by mannose residues found on the bacterial surface; iii) the alternative pathway, characterized by spontaneous formation of C3b. All pathways converge on the formation of C3 convertase, which converts many molecules of C3 into C3a and C3b. C3b has two main key roles: opsonization of pathogens, with following destruction by complement receptor 3 (CR3) expressing phagocytes, and formation of C5 convertase, through its binding with C3 convertase. C5 convertase initiates the terminal pathway by converting C5 molecules into C5a and C5b. C5a works as a chemotactic protein and, with C3a, is also involved in the anaphylactic reaction. On the other hand, C5b leads to the formation of MAC, composed of C5b, C6, C7, C8, and polymeric C9, which together form a lytic pore in the cellular membrane causing cell destruction.15 As aforementioned, the alternative pathway is characterized by spontaneous and continuous activation of the complement. This process is known as “tick-over” and represents an important part of innate immunity. In fact, meta-stable C3 can interact with water to form C3 (H2O), which operates as partly functional C3. Preformed C3b or circulating C3 (H2O) fixate on to tissues and, thanks to the activity of factor B and properdin factor D, enhance complement activation by creating an alternative C3 convertase (C3bBb). High levels of local C3b encourage the formation of alternative pathway C5 convertase (C3bBC3b).16,17
The complement cascade can be spontaneously activated and this eventuality is held back by a network of inhibitory regulators, which act on different points of the cascade, blocking its activation on self-tissues. Plasma proteins (eg, C4bp, C4b-binding protein, and FH, factor H) and “intrinsic” membrane proteins (eg, CD55, decay-accelerating Factor or DAF, and CD46, membrane co-factor protein or MCP), which are expressed on almost all cell surfaces and are particularly concentrated at the NMJ, function by decay accelerating activity, inactivating C3/C5 convertases through the dissociation of their enzymatic subunits, Bb or C2a. Another target of the regulatory proteins, specifically CD59 (also known as MAC-IP, MAC-inhibitory protein, another “intrinsic” protein), is the inhibition of MAC formation. Just before the formation of the lytic pore, when C8 binds to the complex, CD59 inhibits the C9 polymerization, thus preventing the formation of the MAC complex.18–20 Interestingly, the expression of “intrinsic” complement regulators was found to be decreased at the extraocular muscle (EOM) junction, possibly explaining the higher susceptibility of EOM to MG compared to skeletal muscles.21
Complement Role in MG and AChR Ab-Mediated Animal Models
Nowadays, the role of complement in anti-AChR antibody-positive (AChR+) MG is well established. In fact, together with AChR internalization, complement activation represents the main effector of AChR+ MG.22 Other involved pathogenic mechanisms are steric interference of ACh-AChR binding,23,24 and sodium entry blocking.25
In AChR+ MG, the complement-mediated damage involves the post-synaptic surface of the NMJ and is triggered by the activity of antibodies (primarily of IgG1 and IgG3 subclasses, and to a lesser extent IgG2) directed towards the AChR.26 These antibodies activate the classical pathway by binding C1q on the Fc domain, with activation of C1r and subsequently of C1s. C1s activates C4 into C4a and C4b and also C2 into C2a and C2b, with consequent formation of C3 convertase (C4b2a). C3 convertase, cleaving C3 into C3a and C3b, leads to the formation of C5 convertase (C4b2a3b), which initiates the terminal pathway that leads to MAC formation (C5b6789). The final effect is a focal lysis of the NMJ with loss of AChR and disruption of the postsynaptic folds.27
The first evidence that suggested complement involvement in AChR+ MG was the observation of C3 and MAC deposition at the NMJ, in the absence of inflammation.28–30 Other following important findings were: persistent simplified structure and detritus at the intrasynaptic space of NMJ consistent with complement-mediated damage, reduction of C3 and C4 levels with increase of complement terminal components in MG sera, and complement-mediated destruction of cultured myotubes provoked by sera.31–33 These findings were also mirrored in the animal models of MG, allowing to further investigate the role of complement in MG pathogenesis and to elaborate new possible therapeutic strategies.
Modelling of MG can be obtained by the passive transfer MG (PT-MG) model and the experimental autoimmune MG (EAMG) model. PT-MG is induced by the administration of anti-AChR antibodies and is characterized by simple disease induction, via a single antibody injection, and rapid development of weakness, within 24 h from injection. This model is very useful to investigate agents that block the antibody binding or the complement cascade. However, in PT-MG there is no cellular activation that drives the antibody production and also the postsynaptic injury induced by antibodies is accompanied by a considerable inflammatory infiltration, which does not happen in human MG.34 EAMG (or active EAMG) has been studied since the 1970s and is caused by immunization with purified AChR or small fragments of AChR.35–37 Immunization results into tolerance break, which best resembles the cellular mechanisms involved in human MG pathogenesis. Autoreactive B-cells, with the help of deregulated T-cells, produce IgG1 and IgG3 class antibodies directed towards the exogenous antigen. Ultimately, antibodies targeting self-AChR can be found, witnessing the occurrence of tolerance loss. The main limit of this model is the time of weakness onset, which, varying among species, occurs from weeks to months.32 Thus, defining the right time window to intervene with a preventive or therapeutic approach is quite difficult when using active EAMG models.
The first studies to investigate complement activation in EAMG concerned complement depletion. The administration of cobra venom, before anti-AChR antibodies administration in PT-MG and in the acute phase of active EAMG models, hydrolyzes C3 and C4 inducing a functional depletion of the complement, with subsequent marked reduction of MG severity.38 A step further in research was possible thanks to genetically modified rodent strains. C3 and C4-depleted mice also demonstrated weakness reduction, compared to wild type, while C3 and C4 knockout (KO) mice showed no weakness at all, with maintained postsynaptic folds and AChR density.25 Interestingly, C3 and C4-deficient rodents had low AChR IgG2b levels as well as low B-cell expression. This evidence was found both in the non-immunized and in EAMG mice, suggesting a role of complement in cellular immunity.39 Conversely, C5 KO mice develop similar anti-AChR levels to their C5-sufficient littermates. Moreover, C5 KO mice present C3 and IgG, but no MAC deposits. Nonetheless, C5 KO mice do not present weakness nor junctional damage.40 C6-deficient mice, after anti-AChR antibodies injection, did not build the MAC complex, while exogenous administration of C6 restored MAC assembly.41 Finally, DAF KO mice displayed higher susceptibility to disease induction, especially compared with the CD59 KO model.42,43
Together these findings greatly encouraged the research of novel therapeutic approaches targeting different levels of the complement cascade.
Complement Inhibition in MG Therapeutics
Preclinical Studies
In accordance with the evidence collected throughout the years of complement involvement in MG, many EAMG trials investigating different complement inhibitors have been conducted. Therapeutic strategies included recombinant proteins, chemicals, soluble isoforms of the complement receptor, monoclonal antibodies, and small interfering RNA (siRNA). These studies focused on either inhibiting a branch of the complement system (terminal or classical pathway) or acting on complement regulators (Table 1).
 |
Table 1 Preclinical Studies of Potential Complement Inhibitors in MG |
Terminal Pathway Inhibitors
One of the first studies to be conducted involved an anti-C6 antibody to prevent MAC formation. The administration of anti-C6 antibody before passive administering of anti-AChR antibody prevented EAMG induction. C6-blocking inhibited the accumulation of components from C6 to C9, preventing MAC formation at the NMJ. Serum levels of C3 and C5 remained normal, indicating that passive EAMG prevention depended solely on the inhibition of MAC formation.44
Similarly, treatment with anti-C5-antibody before passive administration of anti-AChR prevented EAMG induction. In fact, anti-C5 pretreated rats displayed no weakness after 48 h, in contrast to their untreated littermates which required euthanasia due to severe weakness.45 Moreover, a subsequent set of experiments conducted by the same group demonstrated that treatment with anti-C5 antibody 24 h after passive EAMG induction was able to restore strength in over two-thirds of the rats. Treated rats presented normo-structured endplates and low C9 deposition at the NMJ. Consistent with anti-C5 functioning downstream of C3, C3b depositions were still found at the NMJ.45 Likewise, a recombinant C5 inhibitor, rEV576, was found to be effective in the prevention and treatment of myasthenic symptoms both in the passive and active EAMG model. In both models, rats generally developed a milder phenotype, with less weakness and longer survival. Treated rats displayed low complement activity (CH50), reduced C9 deposition at the NMJ, while total IgG concentration remained the same. Interestingly, levels of complement fixing IgG1 were reduced in rEV576-treated rats, suggesting a role of C5 in adaptive immunity as well.46
A more recent study exploited siRNA, ALN-CC5, to silence liver expression of C5. Subcutaneous administration of ALN-CC5, both in a rodent and non-human primate MG model, resulted in potent and prolonged suppression of liver C5 expression. siRNA-C5-treated animals displayed significantly reduced levels of circulating C5, which demonstrated having a key role in pathology at the NMJ in this study. Moreover, in the muscle, no C5 mRNA expression was identified, suggesting that C5-depending NMJ pathology is probably liver derived. C5 silencing greatly reduced disease severity in PTMG animals, with minimal evidence of weakness. Similarly, disease severity in active EAMG animals improved independently of whether ALN-CC5 was administered before or after onset of weakness.47
Classical Pathway Inhibitors
Giving the core role of complement in the defense against invading pathogens, major disadvantage of terminal pathway inhibition is an increased susceptibility to infections. This problem can be overcome by acting on the classical pathway, primarily involved in AChR antibody-related MG, leaving the lectin and alternative pathways intact. The administration of anti-C1q antibody before or after AChR immunization is able to reduce the incidence and severity of EAMG induction. To these beneficial effects correspond a reduced NMJ deposition of C3, IgG and MAC, decreased IL-6 production, and reduced serum levels of anti-AChR IgG2b. However, treated mice displayed renal glomerular deposition of IgG and C3, proving the importance of C1q in the clearance of immune complexes.48 To reduce the problem of complex deposition, the same group developed a siRNA silencing C2. In fact, C2 deficiency does not entail immune complex disorders as much as C1q or C4 deficiencies.49 Moreover, C2 represents a valid target also because it is expressed at very low levels and does not have any primary immunological function other than MAC formation. The administration of C2-siRNA in established EAMG mice determined improvement of muscle strength and survival. The clinical amelioration was accompanied by reduced C3 and MAC deposits and increased AChR levels at the NMJ. Serum C3 levels were comparable between the C2-siRNA treated and untreated group, confirming that the alternative pathway was unaffected.50
Complement Regulators
Administration of human complement receptor 1 (sCR1), which targets both the classical and alternate pathway, reduced symptom severity, and weight loss in PT-MG rats.51 Another regulator that has been investigated is complement receptor 1–related gene/protein y (CRRY), a specific murine complement inhibitor with functions corresponding to human DAF and MCP (ie, convertases inhibition). The application of CRRY regulator, coupled to rat IgG2a Fc to lengthen circulating half-life, prevented passive induction of EAMG and markedly reduced the deposition of C3 and MAC at the NMJ.52 Lastly, DAF-regulator protein associated to a single-chain antibody directed towards the AChR α-subunit, in order to be delivered at the NMJ, produced significantly less muscle weakness and low MAC deposition when administered as treatment in EAMG rats.53
Clinical Trials
Eculizumab
Eculizumab (Soliris) is the first complement-targeting drug approved for complement-mediated diseases. It is a humanized IgG2/4 monoclonal antibody which binds to C5 with high affinity, inhibiting its cleavage into C5a and C5b, thereby blocking MAC formation. The efficacy of eculizumab in the inhibition of MAC formation was previously assessed in paroxysmal nocturnal hemoglobinuria (PNH) and atypical hemolytic uremic syndrome (aHUS).54,55 Eculizumab also demonstrated to be effective in reducing relapse risk in neuromyelitis optica spectrum disorder (NMOSD).56 The efficacy of eculizumab in gMG was initially assessed in AChR+ treatment refractory gMG patients in a pilot randomized, double-blind, placebo-controlled Phase II trial (Study C08-001) sponsored by Alexion Pharmaceuticals.57 The study involved 14 patients with severe refractory disease randomized to either eculizumab or placebo for 16 weeks (Period 1) with a crossover to the opposite group after a washout period of 5 weeks (Period 2). Primary efficacy endpoint of a ≥ 3-point reduction in QMG score was met in 6 (86%) of eculizumab-treated patients, compared to 4 (56%) of the placebo group, suggesting a therapeutic effect of the drug in gMG. Interestingly, before entering Period 2, patients treated with eculizumab did not return to their baseline QMG despite the 5-week washout period. These encouraging data led the way to the randomized, double-blind, placebo-controlled, multicenter, phase III REGAIN study (ECU-MG-301)58 and its following open-label extension (OLE) study (ECU-MG-302).59 The REGAIN study enrolled 125 AChR+ patients with refractory gMG, defined as persistent weakness albeit treatment with at least two immunosuppressive therapies (ISTs) or the need of chronic plasma exchange or IVIg in association with at least 1 IST. Patients also required an MG-ADL score of at least 6 points and an MGFA class between II and IV. Patients were randomized 1:1 to either eculizumab or placebo for 26 weeks. One-hundred-seventeen patients completed the 26-week double-blind phase and were eligible to enter the OLE phase. Primary efficacy endpoint was a change in MG-ADL score from baseline to week 26 measured by worst-rank analysis of covariance (ANCOVA). Secondary endpoints included: change in QMG total score form baseline, improvement of at least 3 points in MG-ADL score and 5 points in QMG score, and changes form baseline in MGC and MG Quality of Life 15 (MG-QoL15) total scores. Primary endpoint was not met (p=0.0698), but the pre-specified secondary outcome of at least 3-point improvement in MG-ADL was significant. Seemingly, QMG achieved statistical significance (p=0.0129) as well as MG-QoL15 (p = 0.0281). MGC score, on the other hand, showed no statistically significant change. Despite not reaching its primary endpoint, the success in the secondary endpoints demonstrated a potential benefit of eculizumab, which was maintained in the OLE phase. Clinical efficacy appeared in the treatment group the week after the first infusion, reached its peak around week 12 and was maintained throughout the 130-weeks of the OLE phase. Eculizumab reduced the frequency of exacerbation by 75% (p = 0.0001) and 56% of patients reached the state of pharmacological remission or minimal manifestations. In the REGAIN study, by week 12 in the eculizumab-treated arm, 67.3% of patients reached at least a 3-point variation in MG-ADL score, and 56.1% reached at least a 5-point variation in QMG score. At the end of the OLE phase, 84.7% of patients improved in the MG-ADL criteria and 71.4% improved in the QMG criteria.58–60
Currently, eculizumab is approved for AChR+ gMG in USA,61 for refractory AChR+ gMG in the EU62, and for AChR+ gMG patients that do not respond to IVIg/PE in Japan.63,64 Every trial cited so far was conducted on adult patients (≥18 years of age). Currently, recruitment is open in USA and Japan for a phase III OLE study of eculizumab in pediatric participants (6 to <18 years of age) with refractory gMG (NCT03759366).
Noteworthy, patients with a history of thymoma or thymic neoplasm were excluded from the REGAIN trial; therefore, data on this subgroup of patients are lacking. However, recent evidences of case reports demonstrate that eculizumab represents a valid therapeutic approach also for thymoma-associated MG, which strongly associates with treatment-refractory disease.65,66
Ravulizumab
Ravulizumab (Ultomiris) is another recombinant humanized monoclonal antibody developed by Alexion Pharmaceuticals that binds the complement protein C5 with high affinity. Ravulizumab was primarily tested and then approved for PNH in the USA, following positive results of two phase III clinical trials, on December 2018. The drug was developed by re-engineering eculizumab to enhance its pharmacokinetic and pharmacodynamic profile. Ravulizumab has a longer duration of action, requiring intravenous administrations every 8 weeks, allowing to improve therapeutic efficacy while keeping a similar safety profile to eculizumab.67
Given the encouraging results obtained in other complement-mediated diseases, Alexion Pharmaceuticals started a phase III, randomized, double-blind, placebo-controlled multicenter trial to evaluate safety and efficacy of ravulizumab in complement-inhibitor-naïve adult gMG patients (NCT03920293). The trial started in 2019 and is currently underway in Europe, United States, Japan, and South Korea. Primary efficacy endpoint is MG-ADL change at Week 26 from Baseline.
Zilucoplan
Zilucoplan is a small macrocyclic peptide that inhibits MAC formation by a dual mechanism: prevention of downstream complement activation by allosterically inhibiting C5 cleavage and direct inhibition of the first step of MAC assembly (C5b-C6-binding). The binding site of zilucoplan on C5 is distinct from eculizumab, as demonstrated by its ability to bind C5 in blood samples of patients genetically resistant to eculizumab. Ra Pharmaceutical sponsored a Phase II double-blind, placebo-controlled trial on 44 AChR+ gMG patients (NCT03315130). Patients were randomized 1:1:1 to zilucoplan 0.1 mg/kg, 0.3 mg/kg, or placebo with daily subcutaneous self-administration for 12 weeks. Eligible patients could subsequently enter the OLE. Primary outcome measure was QMG score change from Baseline to week 12, and the secondary outcome measures included the MG-ADL, the MGC, and the MG-QOL15r. Analyses showed clinically meaningful and statistically significant improvements in primary and secondary endpoints in the treated group versus placebo.68 Of note, despite none of the participants randomized to the 0.3-mg/kg dose group worsened, and most participants improved greatly, 28% of participants did not improve by the minimal clinically important difference of 3 points on QMG. The variability in the degree of improvement, registered both in this study and the eculizumab studies, could depend on various factors, which need to be further investigated: previously undetected fixed weakness, genetic and epigenetic factors (further discussed in this review), or the subsequent predominant activity of other complement independent pathogenic mechanisms such as antigenic modulation and AChR blockade.68
A phase III study, named RAISE (NCT04115293) is currently underway for patients with gMG. Primary efficacy endpoint is MG-ADL change at week 12 from Baseline.
Regarding the safety of complement inhibitors, common adverse events were upper-tract infections and headaches, as observed also in PNH and aHUS trials.58–60 More importantly, the inhibition of C5 increases the risk of Neisseria Meningitidis infection. Therefore, it is mandatory for patients treated with anti-C5 inhibitors to be vaccinated with both quadrivalent and B-serotype vaccines. Patients must be correctly informed of the risks and symptoms of meningococcal meningitis, and should always carry an informational safety card to show whenever necessary. Vaccination should be administered at least 14 days prior to the initial dose of C5-inhibitor. If not possible, prophylactic antibiotic therapy is necessary.
No meningococcal infection occurred in all three concluded trials, except one non-fatal case following the completion of REGAIN OLE.58,59
Biomarkers for Tailoring and Monitoring Complement-Targeted Therapies
Complement inhibitors promise to increase specificity, efficacy, and safety of AChR+ gMG treatment in the next future. However, their introduction into the MG therapeutic algorithm may be limited by high costs. Considering the inter-individual variation in treatment response,5 the development of personalized medicine approaches based on biomarkers predictive of therapeutic success may be pivotal to improve the cost/effectiveness ratio of the therapy, and hence the sustainability in the health system. At present, complement-related biomarkers in MG are missing and need to be defined for their possible employment into the common medical practice. Their identification could allow to select patients with a greater benefit from complement-targeted therapies as well as those with lower responsiveness or tolerability. The best candidate biomarkers could be serological indicators of the complement activity, and/or individual molecular factors affecting complement activation and regulation, or the mechanism of action of the complement-targeted drugs (Table 2).
 |
Table 2 Potential Biomarkers for Tailoring and Monitoring Anti-Complement Therapies |
Complement-Related Serological Markers
Complement status prediction in the single patient could represent the first step towards anti-complement therapy selection as treatment option. It is mainly based on serological analyses to quantify complement components, activation products, and in vitro complement activity. It enables to reveal any deficiency of complement proteins due to consumption and abnormal disease-related complement activation.69
Different immunoassays are commercially available to determine the plasma/serum concentration of complement components. Recently, multiplex assays have been developed with the advantage of simultaneously determine more complement proteins in one sample, providing a comprehensive complement component profile. In addition, quantification of products generated during complement activation, including C3a, C5a, and soluble C5b9 (sC5b9 or sMAC), in properly collected serum/plasma samples,70 can be used to monitor the degree of complement activation during the disease course. Complement activity functional tests, mainly consisting in hemolytic assays measuring classical (CH50)71 and alternative (AH50)72 pathways, from initiation to the effector phase (MAC formation), provide further information regarding the patient-specific complement activation status, and can be therefore useful to monitor complement function during disease exacerbations or crisis. These assays could be tested for evaluating their potential value in predicting good clinical response to anti-complement therapy in patients showing largely altered values of complement-related parameters. Moreover, if applied during therapy, some of them could be important to monitor the functional drug effects in relationship with disease improvement.73 This would allow to adjust individual dosage or dosing intervals, possibly reducing costs and maximizing response. Among serological parameters, C5 function and total complement activity estimated by CH50 are the most promising biomarkers to monitor anti-complement therapies, being reported as eculizumab efficacy markers.74 A correct approach could be a combination of such assays. In fact, “C3:CH50 ratio” has been described as better marker of complement inhibition and disease activity in atypical aHUS eculizumab-treated patients than other single serological factors.75
Despite the clear involvement of complement in MG,3 serological complement parameters have never been investigated as possible immunological biomarkers and their role as reliable factors predictive of treatment response still needs to be evaluated. In a study by Liu and colleagues,76 C3 levels were found to be lower in AChR+ compared to AChR− gMG patients and healthy controls, according with C3 consumption due to complement activation at the NMJ. Interestingly, serum C3 concentration increased after prednisone or IVIg therapies in relationship with clinical improvement, suggesting its variation as possible biomarker of disease status and therapeutic efficacy in AChR+ gMG. In a case report, eculizumab administration in an AChR+ gMG patient with severe bulbar signs was able to decrease CH50 levels in association with symptoms’ improvement. Disease severity was correlated temporally with CH50 level during the therapy,77 supporting a role of CH50 as a marker of anti-complement drug-induced complement blockade, and an indicator of a potential worsening of symptoms during the treatment in MG. In line with this observation, in the phase II study on zilucoplan in gMG patients, the high dose (0.3-mg/kg) arm, resulting in clinically meaningful and statistically significant improvements, achieved rapid, sustained, and near-complete complement inhibition (>97%) as measured in the CH50 assay, whereas the low dose (0.1-mg/kg) arm, whose outcomes were less pronounced, did not achieve complete CH50 ablation (~88%).68
Genetic and Epigenetic Factors Associated with Complement Activation and Regulation
Genetic markers associated with anti-complement therapy efficacy or toxicity represent potent predictive tools for the development of individualized treatments, allowing to identify beforehand responders vs non-responders. Identification of such biomarkers is still an open research field that needs to be expanded in MG to minimize potentially ineffective use of such expensive drugs. Candidate genetic biomarkers for these drugs are variants able to affect complement activity or to interfere with the drug mechanism of action and effectiveness.
The term “complotype” has been proposed to describe the inherited set of variants affecting complement activity, and hence susceptibility to complement-related diseases, disease severity degree, and response to therapies.78 “Complotype” variants of interest for their possible usage as clinical biomarkers for personalized medicine mainly include polymorphisms in genes encoding complement proteins and regulators, that set an individual intrinsic complement activity by impacting on the delicate balance between activation and regulation.78 Of note, if a single polymorphism can cause only small changes, when combined the effects of “complotype” variants may be enormous. For this reason, analysis of complement-related gene panels should be considered as a useful pharmacogenetic tool. Targeted next-generation sequencing approaches can serve as a comprehensive, rapid, and reliable method to analyze common and novel pharmacogenetic “complotype” variants.
As regard to the variants directly affecting anti-complement drug effectiveness, to our knowledge few studies have been performed so far, mainly because of the recent development of these biological drugs. Genetic basis underlying resistance to eculizumab was described in Japanese PNH patients.79 Specifically, a rare missense C5 heterozygous sequence variant c.2654G → A (p.Arg885His), able to inhibit eculizumab binding to C5, was identified in 11 PNH patients (3.2% of the eculizumab-treated PNH population), all characterized by a poor response to the drug. An Asian PNH patient, also showing a poor response to eculizumab, had a very similar mutation in C5 (c.2653C→T).79 Both mutations caused a replacement of arginine by histidine or cysteine in C5, so that the arginine-binding pocket was too small to allow eculizumab to bind to the protein.80 The HindIII polymorphism of the complement regulatory gene CR1 was also reported to affect the response to eculizumab in PHN patients, likely by influencing the binding of C3.81 In MG patients, genetic studies associating complement-related gene variants with response to eculizumab or zilucoplan are lacking. Variants already identified in other complement-related disease patients could be interesting to be tested. However, population ethnicity needs to be taken into consideration, since the genetic profile associated with treatment response can differ depending on ethnicity.
Along with genetic factors, epigenetic modifications could also be associated with inter-individual differences in response to, or toxicity of, anti-complement drugs, pointing out the need of pharmacoepigenetic profiling for these biological drugs. The most promising pharmacoepigenetic factors to be investigated as biomarkers are microRNAs (miRNAs) because they are stable in body fluid, can be easily assessed, and their expression reflects specific pathophysiological conditions or diseases.82 Moreover, miRNAs are not only able to predict optimal patient-specific treatment, but their changes upon therapy may be predictive of the patient clinical response, that is particularly important for an early identification of non-responder patients to be directed towards other therapeutic options, with an optimization of costs. MiRNAs with the ability to modulate the expression of complement components or regulators may be candidate pharmacomiRs to be investigated for possible association with anti-complement drug activity. MiR-200b, miR-200c, and miR-217 have been implicated in regulation of complement-dependent cytotoxicity, via modulation of the complement membrane regulatory proteins CD46 and CD55, which act as complement inhibitors.83 MiR-150, miR-328, and miR-616 were also found to affect cell resistance to complement-dependent cytotoxicity by modifying the expression of CD46 and CD59, another complement regulatory protein.84 In addition, miR-19a and miR-20a were described as regulators of the expression of CD46.85 Nevertheless, knowledge in this field needs to be greatly enhanced. The advent of multiomics analyzes promises to significantly increase pharmacogenetic/miR data in the next years, with enormous implications on the development of innovative biomarker-based personalized medicine schemes for eculizumab and other complement-targeted drugs. To this purpose, biomarker validation for clinical usage on a large scale will be a critical step, for which well-designed retrospective or prospective clinical trials will be needed. Retrospective analyses could be performed on samples obtained from the clinical trials already completed,58,59,66 for which the outcome data are available. Collection of biological samples is an essential condition for allowing retrospective biological analyses; thus, it should be considered a key step during clinical trials. Prospective studies could highlight the potential of preliminary patient-selection, carried out using pharmacogenetic/miR biomarkers, in increasing the positive outcome rate.
What are the Prospects of the Complement Inhibitor Therapy?
The present treatment of MG is based on a multi-step approach in which three layers of decisions are taken: 1. improve neuromuscular transmission using cholinesterase inhibitors; 2. contrast the immune mechanisms of MG using ISTs or immunomodulating therapies; 3. modify the natural course of the disease by thymectomy.6 The choice of any of the above-described approaches is based on clinical parameters such as MG subgroups (eg, antibody stratification, presence of thymoma, age at onset), clinical severity, other comorbidities, and the need to limit side effects. Most MG patients start with symptomatic treatment, but in a considerable proportion corticosteroids and/or ISTs become necessary; IVIg and PLEX are mostly used as rescue therapies in case of clinical deterioration. Corticosteroid and other ISTs are proposed as chronic treatments and, though neurologists seek for the minimal effective dose (particularly for corticosteroids), patients are treated for very long periods of time leading to a severe burden of adverse events.
With the rapidly growing pharmacological innovation, as testified by the enormous number of clinical trials performed and still ongoing, it is possible to distinguish “global immunosuppression” from “specific immunosuppression”. Global ISTs include all ISTs employed in clinical practice so far, whereas the specific ones are characterized by a higher selective mechanism of action. Complement inhibition is part of treatment innovation and C5 inhibitors are particularly relevant for MG as their effect is highly specific by selectively blocking one of the mechanisms of action of the anti-AChR antibodies, thus fulfilling the characteristic of precision medicine.
Based on these considerations, use of complement inhibitors should address the following aspects in the pharmacological approach to MG:
- To assess their immunosuppressive effect in patients naïve to immunosuppression, ie, a formal evaluation of their effect in controlling MG symptoms when patients have not been treated with corticosteroids or other ISTs. Today the effect of complement inhibitors has been evaluated as an add-on therapy; hence, we have not yet appreciated their efficacy as a single drug. This is of particular relevance for those countries where the drug has been authorized only.
- To assess how rapidly they can exert a clinical improvement, ie, whether complement inhibitors are competitive with PLEX or high doses of immunoglobulins to serve as immunomodulating agents.
- To assess their clinical effect in the real-world setting, a condition significantly different from a controlled study and more adherent to the need of patients.
- To assess the cost-benefit of complement inhibitors, also through a pharmaco-economic analysis; introduction of new drugs should be attained taking into consideration the sustainability of the healthcare system.
- The introduction of compounds that target selectively the immune system will also offer a new opportunity to investigate immunological markers of disease activity and response to treatment. The topic of biomarkers in MG and other autoimmune disorders is not new but very little is known in case of biological treatments, including complement inhibitors.86 To our knowledge, pharmacogenetic profiling for biological drugs in MG has not been performed yet and genetic variants known to modulate responsiveness to these drugs in patients with other autoimmune diseases have not been investigated in MG patients. This field should be implemented in MG also in light of the usually high costs of the innovative therapies. Interestingly, the whole miRNome sequencing performed in MG patients has revealed pharmacomiRs possibly predicting or monitoring clinical response to immunosuppressive treatments.87
In very recent years the introduction of drugs targeting specific immune-pathological mechanisms, such as complement inhibition, has raised the expectation to change in the paradigm of immunosuppression in MG. Whether these compounds will effectively modify the algorithm of immunosuppression in MG is not known, but such a strong expectation will depend on their capacity to face the unmet clinical needs and to improve the quality of life of MG patients. Of note, however, is that currently we have very limited information to suggest that complement inhibition can influence antibody production. Therefore, it is possible that complement inhibiting therapies alone could not be able to lead to remission and that lifelong treatment may be necessary.
Disclosure
Renato Mantegazza has received compensation for partecipating on Advisory Boards in relation to MG clinical trial design, Congress partecipations and research support in the last 5 years from: Alexion Pharmaceuticals, ARGENX Pharma, and Biomarin. The other authors report no potential conflicts of interest for this work.
References
1. Vincent A, Huda S, Cao M, et al. Serological and experimental studies in different forms of myasthenia gravis. Ann N Y Acad Sci. 2018;1413:143–153. doi:10.1111/nyas.13592
2. Gilhus NE, Tzartos S, Evoli A, Palace J, Burns TM, Verschuuren JJGM. Myasthenia gravis. Nat Rev Dis Primers. 2019;5(1):30.
3. Tüzün E, Christadoss P. Complement associated pathogenic mechanisms in myasthenia gravis. Autoimmun Rev. 2013;12:904–911. doi:10.1016/j.autrev.2013.03.003
4. Huijbers MG, Zhang W, Klooster R, et al. MuSK IgG4 autoantibodies cause myasthenia gravis by inhibiting binding between MuSK and Lrp4. Proc Natl Acad Sci USA. 2013;110:20783–20788. doi:10.1073/pnas.1313944110
5. Gilhus NE. Eculizumab: a treatment option for myasthenia gravis? Lancet Neurol. 2017;16(12):947–948. doi:10.1016/S1474-4422(17)30363-0
6. Sanders DB, Wolfe GI, Benatar M, et al. International consensus guidance for management of myasthenia gravis: executive summary. Neurology. 2016;87:419–425. doi:10.1212/WNL.0000000000002790
7. Sanders DB, Wolfe GI, Narayanaswami P. MGFA task force on MG treatment guidance. Developing treatment guidelines for myasthenia gravis. Ann N Y Acad Sci. 2018;1412:95–101. doi:10.1111/nyas.13537
8. Mantegazza R, Bonanno S, Camera G, Antozzi C. Current and emerging therapies for the treatment of myasthenia gravis. Neuropsychiatr Dis Treat. 2011;7:151–160. doi:10.2147/NDT.S8915
9. Mantegazza R, Antozzi C. When myasthenia gravis is deemed refractory: clinical signposts and treatment strategies. Ther Adv Neurol Disord. 2018;11:1756285617749134. doi:10.1177/1756285617749134
10. Chamberlain JL, Huda S, Whittam DH, Matiello M, Morgan BP, Jacob A. Role of complement and potential of complement inhibitors in myasthenia gravis and neuromyelitis optica spectrum disorders: a brief review. J Neurol. 2019;3.
11. Botto M, Walport MJ. C1q, autoimmunity and apoptosis. Immunobiology. 2002;205(4–5):395–406. doi:10.1078/0171-2985-00141
12. Strey CW, Markiewski M, Matellos D, et al. The proinflammatory mediators C3a and C5a are essential for liver regeneration. J Exp Med. 2003;198:913–923. doi:10.1084/jem.20030374
13. Stephan AH, Barres BA, Stevens B. The complement system: an unexpected role in synaptic pruning during development and disease. Ann Rev Neurosci. 2012;35:369–389. doi:10.1146/annurev-neuro-061010-113810
14. Kolev M, LeFriec G, Kemper C. Complement – tapping into new sites and effector systems. Nat Rev Immunol. 2014;14:811–820. doi:10.1038/nri3761
15. Serna M, Giles JL, Morgan BP, Bubeck D. Structural basis of complement membrane attack complex formation. Nat Commun. 2016;7:1–7. doi:10.1038/ncomms10587
16. Ricklin D, Reis ES, Lambris JD. Complement in disease: a defence system turning offensive. Nat Rev Nephrol. 2016;12(7):383–401. doi:10.1038/nrneph.2016.70
17. Howard JF. Myasthenia gravis: the role of complement at the neuromuscular junction. Ann N Y Acad Sci. 2018;1412(1):113–128. doi:10.1111/nyas.13522
18. Kim DD, Song WC. Membrane complement regulatory proteins. Clin Immunol. 2006;118(2–3):127–136. doi:10.1016/j.clim.2005.10.014
19. Hourcade DE, Mitchell L, Kuttner-Kondo LA, Atkinson JP, Medof ME. Decay-accelerating factor (DAF), complement receptor 1 (CR1), and factor H dissociate the complement AP C3 convertase (C3bBb) via sites on the type A domain of Bb. J Biol Chem. 2002;277(2):1107–1112. doi:10.1074/jbc.M109322200
20. Navenot JM, Villanova M, Lucas-Héron B, Malandrini A, Blanchard D, Louboutin JP. Expression of CD59, a regulator of the membrane attack complex of complement, on human skeletal muscle fibers. Muscle Nerve. 1997;20(1):92–96. doi:10.1002/(SICI)1097-4598(199701)20:1<92::AID-MUS12>3.0.CO;2-3
21. Kaminski HJ, Li Z, Richmonds C, Lin F, Medof ME. Complement regulators in extraocular muscle and experimental autoimmune myasthenia gravis. Exp Neurol. 2004;189(2):333–342. doi:10.1016/j.expneurol.2004.06.005
22. Drachman DB, Angus CW, Adams RN, Michelson JD, Hoffman GJ. Myasthenic antibodies cross-link acetylcholine receptors to accelerate degradation. N Engl J Med. 1978;298(20):1116–1122. doi:10.1056/NEJM197805182982004
23. Hara H, Hayashi K, Ohta K, Itoh N, Nishitani H, Ohta M. Detection and characterization of blocking-type anti-acetylcholine receptor antibodies in sera from patients with myasthenia gravis. Clin Chem. 1993;39(10):2053–2057. doi:10.1093/clinchem/39.10.2053
24. Almon RR, Andrew CG, Appel SH. Serum globulin in myasthenia gravis: inhibition of alpha-bungarotoxin binding to acetylcholine receptors. Science. 1974;186(4158):55–57.
25. Lang B, Richardson G, Rees J, Vincent A, Newsom-Davis J. Plasma from myasthenia gravis patients reduces acetylcholine receptor agonist-induced Na+ flux into TE671 cell line. J Neuroimmunol. 1988;19(1–2):141–148. doi:10.1016/0165-5728(88)90043-4
26. Rødgaard A, Nielsen FC, Djurup R, Somnier F, Gammeltoft S. Acetylcholine receptor antibody in myasthenia gravis: predominance of IgG subclasses 1 and 3. Clin Exp Immunol. 1987;67(1):82–88.
27. Tüzün E, Scott BG, Goluszko E, Higgs S, Christadoss P. Genetic evidence for involvement of classical complement pathway in induction of experimental autoimmune myasthenia gravis. J Immunol. 2003;171:3847–3854.
28. Engel AG, Lambert EH, Gomez MR. A new myasthenic syndrome with end-plate acetylcholinesterase deficiency, small nerve terminals, and reduced acetylcholine release. Ann Neurol. 1977;1:315–330. doi:10.1002/ana.410010403
29. Sahashi K, Engel AG, Linstrom JM, Lambert EH, Lennon VA. Ultrastructural localization of immune complexes (IgG and C3) at the end-plate in experimental autoimmune myasthenia gravis. J Neuropathol Exp Neurol. 1978;37(2):212–223. doi:10.1097/00005072-197803000-00008
30. Nakano S, Engel AG. Myasthenia gravis: quantitative immunocytochemical analysis of inflammatory cells and detection of complement membrane attack complex at the end-plate in 30 patients. Neurology. 1993;43(6):1167–1172. doi:10.1212/WNL.43.6.1167
31. Romi F, Kristoffersen EK, Aarli JA, Gilhus NE. The role of complement in myasthenia gravis: serological evidence of complement consumption in vivo. J Neuroimmunol. 2005;158(1–2):191–194. doi:10.1016/j.jneuroim.2004.08.002
32. Barohn RJ, Brey RL. Soluble terminal complement components in human myasthenia gravis. Clin Neurol Neurosurg. 1993;95(4):285–290. doi:10.1016/0303-8467(93)90103-N
33. Ashizawa T, Appel SH. Complement-dependent lysis of cultured rat myotubes by myasthenic immunoglobulins. Neurology. 1985;35(12):1748–1753. doi:10.1212/WNL.35.12.1748
34. Kusner LL, Sengupta M, Kaminski HJ. Acetylcholine receptor antibody-mediated animal models of myasthenia gravis and the role of complement. Ann N Y Acad Sci. 2018;1413(1):136–142. doi:10.1111/nyas.13555
35. Tüzün E, Berrih-Aknin S, Brenner T, et al. Guidelines for standard preclinical experiments in the mouse model of myasthenia gravis induced by acetylcholine receptor immunization. Exp Neurol. 2015;270:11–17. doi:10.1016/j.expneurol.2015.02.009
36. Losen M, Martinez-Martinez P, Molenaar PC, et al. Standardization of the experimental autoimmune myasthenia gravis (EAMG) model by immunization of rats with Torpedo californica acetylcholine receptors–Recommendations for methods and experimental designs. Exp Neurol. 2015;270:18–28. doi:10.1016/j.expneurol.2015.03.010
37. Baggi F, Annoni A, Ubiali F, et al. Breakdown of tolerance to a self-peptide of acetylcholine receptor alpha-subunit induces experimental myasthenia gravis in rats. J Immunol. 2004;172(4):2697–2703. doi:10.4049/jimmunol.172.4.2697
38. Kock MA, Hew BE, Bammert H, Fritzinger DC, Vogel CW. Structure and function of recombinant cobra venom factor. J Biol Chem. 2004;279(29):30836–30843. doi:10.1074/jbc.M403196200
39. Arbore G, Kemper C, Kolev M. Intracellular complement - the complosome - in immune cell regulation. Mol Immunol. 2017;89:2–9. doi:10.1016/j.molimm.2017.05.012
40. Christadoss P. C5 gene influences the development of murine myasthenia gravis. J Immunol. 1988;140(8):2589–2592.
41. Chamberlain-Banoub J, Neal JW, Mizuno M, Harris CL, Morgan BP. Complement membrane attack is required for endplate damage and clinical disease in passive experimental myasthenia gravis in Lewis rats. Clin Exp Immunol. 2006;146(2):278–286. doi:10.1111/j.1365-2249.2006.03198.x
42. Kaminski HJ, Kusner LL, Richmonds C, Medof ME, Lin F. Deficiency of decay accelerating factor and CD59 leads to crisis in experimental myasthenia. Exp Neurol. 2006;202(2):287–293. doi:10.1016/j.expneurol.2006.06.003
43. Lin F, Kaminski HJ, Conti-Fine BM, Wang W, Richmonds C, Medof ME. Markedly enhanced susceptibility to experimental autoimmune myasthenia gravis in the absence of decay-accelerating factor protection. J Clin Invest. 2002;110(9):1269–1274. doi:10.1172/JCI0216086
44. Biesecker G, Gomez CM. Inhibition of acute passive transfer experimental autoimmune myasthenia gravis with Fab antibody to complement C6. J Immunol. 1989;142(8):2654–2659.
45. Zhou Y, Gong B, Lin F, Rother RP, Medof ME, Kaminski HJ. Anti-C5 antibody treatment ameliorates weakness in experimentally acquired myasthenia gravis. J Immunol. 2007;179:8562–8567. doi:10.4049/jimmunol.179.12.8562
46. Soltys J, Kusner LL, Young A, et al. Novel complement inhibitor limits severity of experimentally myasthenia gravis. Ann Neurol. 2009;65:67–75. doi:10.1002/ana.21536
47. Kusner LL, Yucius K, Sengupta M, et al. Investigational RNAi therapeutic targeting C5 is efficacious in pre-clinical models of myasthenia gravis. Mol Ther Methods Clin Dev. 2019;13:484–492. doi:10.1016/j.omtm.2019.04.009
48. Tüzün E, Li J, Saini SS, Yang H, Christadoss P. Pros and cons of treating murine myasthenia gravis with anti-C1q antibody. J Neuroimmunol. 2007;182:167–176. doi:10.1016/j.jneuroim.2006.10.014
49. Klint C, Gullstrand B, Sturfelt G, Truedsson L. Binding of immune complexes to erythrocyte CR1 (CD35): difference in requirement of classical pathway components and indication of alternative pathway-mediated binding in C2-deficiency. Scand J Immunol. 2000;52(1):103–108. doi:10.1046/j.1365-3083.2000.00752.x
50. Huda R, Tüzün E, Christadoss P. Complement C2 siRNA mediated therapy of myasthenia gravis in mice. J Autoimmun. 2013;42:94–104. doi:10.1016/j.jaut.2013.01.003
51. Piddlesden SJ, Jiang S, Levin JL, Vincent A, Morgan BP. Soluble complement receptor 1 (sCR1) protects against experimental autoimmune myasthenia gravis. J Neuroimmunol. 1996;71(1–2):173–177. doi:10.1016/S0165-5728(96)00144-0
52. Hepburn NJ, Chamberlain-Banoub JL, Williams AS, Morgan BP, Harris CL. Prevention of experimental autoimmune myasthenia gravis by rat Crry-Ig: a model agent for long-term complement inhibition in vivo. Mol Immunol. 2008;45(2):395–405. doi:10.1016/j.molimm.2007.06.144
53. Kusner LL, Satija N, Cheng G, Kaminski HJ. Targeting therapy to the neuromuscular junction: proof of concept. Muscle Nerve. 2014;49(5):749–756. doi:10.1002/mus.24057
54. Dmytrijuk A, Robie-Suh K, Cohen MH, Rieves D, Weiss K, Pazdur R. FDA report: eculizumab (Soliris) for the treatment of patients with paroxysmal nocturnal hemoglobinuria. Oncologist. 2008;13(9):993–1000. doi:10.1634/theoncologist.2008-0086
55. Legendre CM, Licht C, Muus P, et al. Terminal complement inhibitor eculizumab in atypical hemolytic-uremic syndrome. N Engl J Med. 2013;368(23):2169–2181. doi:10.1056/NEJMoa1208981
56. Pittock SJ, Berthele A, Fujihara K, et al. Eculizumab in aquaporin-4-positive neuromyelitis optica spectrum disorder. N Engl J Med. 2019;381:614–625. doi:10.1056/NEJMoa1900866
57. Howard JF, Barohn RJ, Cutter GR, et al. A randomized, double-blind, placebo-controlled phase II study of eculizumab in patients with refractory generalized myasthenia gravis. Muscle Nerve. 2013;48:76–84. doi:10.1002/mus.23839
58. Howard JF, Utsugisawa K, Benatar M, et al. Safety and efficacy of eculizumab in antiacetylcholine receptor antibody-positive refractory generalized myasthenia gravis (REGAIN): a Phase 3, randomised, double-blind, placebo-controlled, multicentre study. Lancet Neurol. 2017;16:976–986.
59. Muppidi S, Utsugisawa K, Benatar M, et al. Long-term safety and efficacy of eculizumab in generalized myasthenia gravis. Muscle Nerve. 2019;60:14–24.
60. Howard J, Karam C, Yountz M, O’Brien F, Mozaffar T. Long-term efficacy of eculizumab in refractory generalized myasthenia gravis: responder analyses. Muscle Nerve. 2019;60:S133.
61. Alexion Pharmaceuticals Inc. Soliris (eculizumab): US prescribing information; 2015. Available from: https://www.accessdata.fda.gov/drugsatfda_docs/label/2017/125166s422lbl.pdf.
62. Alexion Europe SAS. Soliris (eculizumab): summary of product characteristics; 2017. Available from: http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_Product_Information/human/000791/WC500054208.pdf.
63. Japan Ministry of Health Labour and Welfare. Soliris (eculizumab): Japanese prescribing information; 2017. Available from: https://www.businesswire.com/news/home/20171226005046/en/Soliris%C2%AE-Eculizumab-Receives-Marketing-Authorization-Japan-Treatment.
64. Murai H, Uzawa A, Suzuki Y, et al. Long-term efficacy and safety of eculizumab in Japanese patients with generalized myasthenia gravis: a subgroup analysis of the REGAIN open-label extension study. J Neurol Sci. 2019;407:116419. doi:10.1016/j.jns.2019.08.004
65. Vélez-Santamaría V, Nedkova V, Díez L, Homedes C, Alberti MA, Casasnovas C. Eculizumab as a promising treatment in thymoma-associated myasthenia gravis. Ther Adv Neurol Disord. 2020;(13):1756286420932035.
66. Amano E, Otsu S, Suzuki S, Machida A. Eculizumab improved weakness and taste disorder in thymoma-associated generalized myasthenia gravis with anti-striational antibodies: a case report. eNeurologicalSci. 2019;14:72–73. doi:10.1016/j.ensci.2019.01.006
67. Sheridan D, Yu ZX, Zhang Y, et al. Design and preclinical characterization of ALXN1210: a novel anti-C5 antibody with extended duration of action. PLoS One. 2018;13(4):e0195909. doi:10.1371/journal.pone.0195909
68. Howard JF, Nowak RJ, Wolfe GI, et al. Clinical effects of the self-administered subcutaneous complement inhibitor zilucoplan in patients with moderate to severe generalized myasthenia gravis: results of a phase 2 randomized, double-blind, placebo-controlled, multicenter clinical trial. JAMA Neurol. 2020;77(5):582–592. doi:10.1001/jamaneurol.2019.5125
69. Ekdahl KN, Persson B, Mohlin C, Sandholm K, Skattum L, Nilsson B. Interpretation of serological complement biomarkers in disease. Front Immunol. 2018;9:2237. doi:10.3389/fimmu.2018.02237
70. Mohebnasab M, Eriksson O, Persson B, et al. Current and future approaches for monitoring responses to anti-complement therapeutics. Front Immunol. 2019;10:2539. doi:10.3389/fimmu.2019.02539
71. Mayer MM. Complement and complement fixation. In: Kabat E, Mayer MM, editors. Experimental Immunochemistry. Springfield, Ill: C. C. Thomas; 1961:133–240.
72. Joiner KA, Hawinger A, Gelfand JA. A study of optimal reaction conditions for an assay of the human alternative complement pathway. Am J Clin Pathol. 1983;79:65–72. doi:10.1093/ajcp/79.1.65
73. Willrich MAV, Andreguetto BD, Sridharan M, et al. The impact of eculizumab on routine complement assays. J Immunol Methods. 2018;460:63–71. doi:10.1016/j.jim.2018.06.010
74. Wijnsma KL, Heine RT, Moes DJAR, et al. Pharmacology, pharmacokinetics and pharmacodynamics of eculizumab, and possibilities for an individualized approach to eculizumab. Clin Pharmacokinet. 2019;58(7):859–874.
75. Kerboua KE, Haiba F, Batouche D. C3:CH50 ratio as a proposed composite marker for eculizumab monitoring in atypical hemolytic uremic syndrome: preliminary results. J Immunoassay Immunochem. 2017;38(2):178–189. doi:10.1080/15321819.2016.1234485
76. Liu A, Lin H, Liu Y, Cao X, Wang X, Li Z. Correlation of C3 level with severity of generalized myasthenia gravis. Muscle Nerve. 2009;40(5):801–808. doi:10.1002/mus.21398
77. Yanagidaira M, Nishida Y, Yokota T. Temporal correlation between serum CH50 level and symptom severity of myasthenia gravis during eculizumab therapy. Clin Neurol Neurosurg. 2020;189:105630. doi:10.1016/j.clineuro.2019.105630
78. Harris CL, Heurich M, Rodriguez de Cordoba S, Morgan BP. The complotype: dictating risk for inflammation and infection. Trends Immunol. 2012;33(10):513–521. doi:10.1016/j.it.2012.06.001
79. Nishimura JI, Yamamoto M, Hayashi S, et al. Genetic variants in C5 and poor response to eculizumab. N Engl J Med. 2014;370(7):632–639. doi:10.1056/NEJMoa1311084
80. Schatz-Jakobsen JA, Zhang Y, Johnson K, Neill A, Sheridan D, Andersen GR. Structural basis for eculizumab-mediated inhibition of the complement terminal pathway. J Immunol. 2016;197(1):337–344. doi:10.4049/jimmunol.1600280
81. Rondelli T, Risitano AM, Peffault de Latour R, et al. Polymorphism of the complement receptor 1 gene correlates with the hematologic response to eculizumab in patients with paroxysmal nocturnal hemoglobinuria. Haematologica. 2014;99(2):262–266. doi:10.3324/haematol.2013.090001
82. Etheridge A, Lee I, Hood L, Galas D, Wang K. Extracellular microRNA: a new source of biomarkers. Mutat Res. 2011;717(1–2):85–90. doi:10.1016/j.mrfmmm.2011.03.004
83. Hillman Y, Mazkereth N, Farberov L, Shomron N, Fishelson Z. Regulation of complement-dependent cytotoxicity by microRNAs miR-200b, miR-200c, and miR-217. J Immunol. 2016;196(12):5156–5165. doi:10.4049/jimmunol.1502701
84. Hillman Y, Mardamshina M, Pasmanik-Chor M, et al. MicroRNAs affect complement regulator expression and mitochondrial activity to modulate cell resistance to complement-dependent cytotoxicity. Cancer Immunol Res. 2019;7(12):1970–1983. doi:10.1158/2326-6066.CIR-18-0818
85. Tan JR, Tan KS, Yong FL, et al. MicroRNAs regulating cluster of differentiation 46 (CD46) in cardioembolic and non-cardioembolic stroke. PLoS One. 2017;12(2):e0172131. doi:10.1371/journal.pone.0172131
86. Cavalcante P, Mantegazza R, Bernasconi P. Pharmacogenetic and pharmaco-miR biomarkers for tailoring and monitoring myasthenia gravis treatments. Expert Rev Precis Med Drug Dev. 2020;5:317–329. doi:10.1080/23808993.2020.1804865
87. Cavalcante P, Mizrachi T, Barzago C, et al. MicroRNA signature associated with treatment response in myasthenia gravis: a further step towards precision medicine. Pharmacol Res. 2019;148:104388. doi:10.1016/j.phrs.2019.104388
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.