")
Back to Journals » International Journal of Nanomedicine » Volume 10 » Issue 1
Comparison of two self-assembled macromolecular prodrug micelles with different conjugate positions of SN38 for enhancing antitumor activity
Authors Liu Y, Piao H, Gao Y, Xu C, Tian Y, Wang L, Liu J, Tang B, Zou M, Cheng G
Received 22 November 2014
Accepted for publication 26 January 2015
Published 23 March 2015 Volume 2015:10(1) Pages 2295—2311
DOI https://doi.org/10.2147/IJN.S77957
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Professor Lei Yang
Yi Liu,1 Hongyu Piao,1 Ying Gao,1 Caihong Xu,2 Ye Tian,1 Lihong Wang,1 Jinwen Liu,1 Bo Tang,1 Meijuan Zou,1 Gang Cheng1
1Department of Pharmaceutics, Shenyang Pharmaceutical University, Shenyang, Liaoning Province, People’s Republic of China; 2Department of Food Science, Shenyang Normal University, Shenyang, Liaoning Province, People’s Republic of China
Abstract: 7-Ethyl-10-hydroxycamptothecin (SN38), an active metabolite of irinotecan (CPT-11), is a remarkably potent antitumor agent. The clinical application of SN38 has been extremely restricted by its insolubility in water. In this study, we successfully synthesized two macromolecular prodrugs of SN38 with different conjugate positions (chitosan-(C10-OH)SN38 and chitosan-(C20-OH)SN38) to improve the water solubility and antitumor activity of SN38. These prodrugs can self-assemble into micelles in aqueous medium. The particle size, morphology, zeta potential, and in vitro drug release of SN38 and its derivatives, as well as their cytotoxicity, pharmacokinetics, and in vivo antitumor activity in a xenograft BALB/c mouse model were studied. In vitro, chitosan-(C10-OH)SN38 (CS-(10s)SN38) and chitosan-(C20-OH)SN38 (CS-(20s)SN38) were 13.3- and 25.9-fold more potent than CPT-11 in the murine colon adenocarcinoma cell line CT26, respectively. The area under the curve (AUC)0–24 of SN38 after intravenously administering CS-(10s)SN38 and CS-(20s)SN38 to Sprague Dawley rats was greatly improved when compared with CPT-11 (both P<0.01). A larger AUC0–24 of CS-(20s)SN38 was observed when compared to CS-(10s)SN38 (P<0.05). Both of the novel self-assembled chitosan-SN38 prodrugs demonstrated superior anticancer activity to CPT-11 in the CT26 xenograft BALB/c mouse model. We have also investigated the differences between these macromolecular prodrug micelles with regards to enhancing the antitumor activity of SN38. CS-(20s)SN38 exhibited better in vivo antitumor activity than CS-(10s)SN38 at a dose of 2.5 mg/kg (P<0.05). In conclusion, both macromolecular prodrug micelles improved the in vivo conversion rate and antitumor activity of SN38, but the prodrug in which C20-OH was conjugated to macromolecular materials could be a more promising platform for SN38 delivery.
Keywords: self-assembled prodrug micelles, in vitro cytotoxicity, pharmacokinetics, in vivo antitumor activity
Introduction
7-Ethyl-10-hydroxycamptothecin (SN38), a potent inhibitor of topoisomerase I, is a semisynthetic analog of camptothecins. It is well known as a very potent anticancer agent that exhibits 100- to 1,000-fold higher in vitro cytotoxicity against various cancer cell lines than irinotecan (CPT-11).1,2 However, the clinical application of SN38 has been extremely restricted by its insolubility in water as well as in most pharmaceutically acceptable solvents.3 To overcome these disadvantages, a water-soluble prodrug of SN38, CPT-11, was developed for clinical application. However, the clinical use of CPT-11 is accompanied by severe diarrhea and myelosuppression.4 Moreover, the conversion of CPT-11 to SN38 exhibits interindividual variability because of patient differences in enzyme or protein expression,5,6 and the conversion rate is only 2%–8% after administration.7 In order to improve the drug solubility and enhance the antitumor activity of SN38, various preparations have been developed in recent years, such as liposome-based formulations,8,9 polymeric micelles, and complex compounds.10–15 Unfortunately, these preparations are accompanied by inherent shortcomings, especially the need for complex preparation methods, low drug loading, and inefficient delivery. To overcome these problems, macromolecular prodrug systems that self-assemble in aqueous medium have been investigated in recent years.16–18 The use of macromolecular prodrugs is an attractive potential approach to improve solubility, produce sustained release, and obtain tumor localization by enhanced permeability and retention (EPR).19–21
Like other camptothecins, SN38 is comprised of a (3,4-β) quinoline moiety, an indolizinone, and an α-hydroxy-δ-lactone ring. The lactone ring is necessary for topoisomerase inhibition.22,23 The active lactone form of SN38 is converted into the inactive carboxylate form at alkaline pH (Figure 1). However, the activity of the lactone form is much greater than the carboxylate form. Previous research has shown that modifications of hydroxycamptothecin at C10-OH or C20-OH can influence the antitumor activity and stability of the lactone ring.24–26 Although several trials have been carried out by conjugating SN38 to macromolecular materials, there are no significant reports of any differences between C10-OH and C20-OH conjugated to hydrophilic materials as the reaction group.
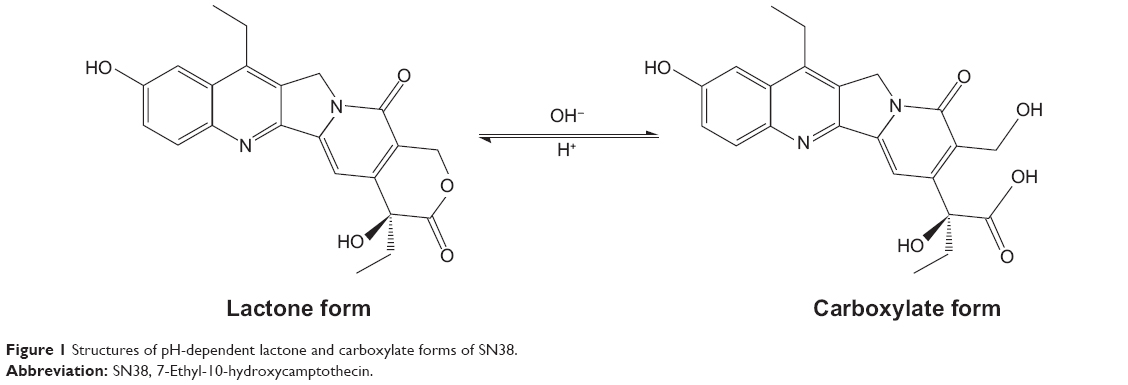 | Figure 1 Structures of pH-dependent lactone and carboxylate forms of SN38. |
In order to develop new self-assembled macromolecular prodrugs for SN38 delivery and to investigate the differences between the two conjugated positions of SN38, a suitable macromolecular polymer must be chosen as the hydrophilic vehicle. Water-soluble chitosan with a high degree of deacetylation and a low molecular weight has been extensively studied due to its outstanding biocompatibility,27 abundant biomass, low cost, biodegradability, and low toxicity.28 To date, polymeric micelles based on chitosan remain an excellent choice for anticancer drug delivery.29–32 In our study, water-soluble chitosan was chosen as a hydrophilic vehicle for the delivery of SN38.
In this study, we illustrate the outstanding advantage of self-assembled chitosan-SN38 prodrug micelles. Both of the novel self-assembled chitosan-SN38 prodrug micelles exhibited better in vitro cytotoxicity and in vivo antitumor activity than CPT-11 on CT26. The in vitro and in vivo differences in CS-(10s)SN38 and CS-(20s)SN38 macromolecular prodrug micelles were investigated for the first time in this study.
Materials and methods
Materials
SN38 (98% pure) and CPT-11 (98% pure) were purchased from Knowshine Pharmachemicals Inc. (Shanghai, People’s Republic of China). Chitosan (average molecular weight =30,000, degree of deacetylation =97%) and coumarin-6 were supplied by Sigma-Aldrich Co. (St Louis, MO, USA). 1-Ethyl-3-(3-dimethylaminopropyl) carbodiimide hydrochloride (EDC), N-hydroxysuccinimide (NHS), 4-dimethylamiopyridine (DMAP), N,N-diisopropylethylamine (DIPEA), succinic acid anhydrous (SA), and di-tert-butyl dicarbonate (DIBOC) were obtained from Tokyo Chemical Industry Co. (Tokyo, Japan). All other chemicals and reagents were analytical grade after dehydration.
Animals and cell line
Pathogen-free Sprague Dawley (SD) rats (male, weighing 200±20 g) and BALB/c mice (male, weighing 20±2 g) were purchased from the Laboratory Animal Center of Shenyang Pharmaceutical University. All animals were housed under standard conditions and the studies were carried out according to the regulations for animal experimentation issued by the State Committee of Science and Technology of the People’s Republic of China.
The murine colon adenocarcinoma cell line CT26 was purchased from the National Platform of Experimental Cell Resource for Sci-Tech. CT26 cells were maintained in RPMI 1640 with 10% fetal bovine serum (FBS). Ascites of the mouse liver cancer cell line H22 were supplied by the Pharmacology Laboratory of Shenyang Pharmaceutical University.
Synthesis of CS-SN38 macromolecular prodrugs with different conjugate positions
Synthesis of succinyl-(C10-OH)SN38 and succinyl-(C20-OH)SN38
The entire synthesis strategy is shown in Figure 2. These derivatives of SN38 were synthesized according to the reported references with modifications.33–35
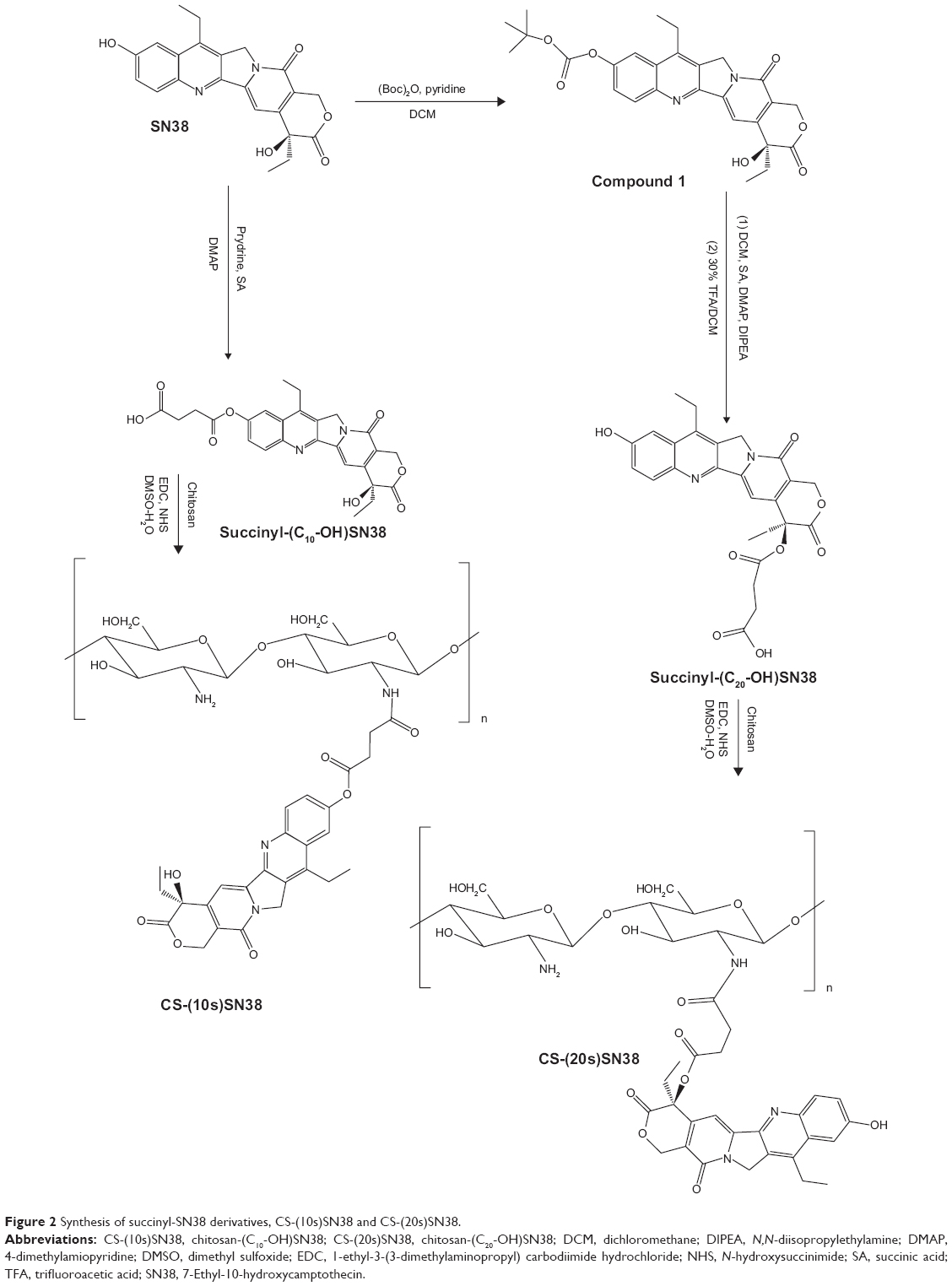 | Figure 2 Synthesis of succinyl-SN38 derivatives, CS-(10s)SN38 and CS-(20s)SN38. |
SN38 (1 g, 2.55 mmol) and DIBOC (0.61 g, 2.81 mmol) were stirred in anhydrous dichloromethane (DCM) with a catalytic amount of anhydrous pyridine for 14 hours at room temperature. The reaction mixture was then washed several times with 0.5% NaHCO3 and 0.1 mol/L HCl. Crude compound 1 was obtained by evaporating the solvent.
Compound 1 (200 mg, 0.41 mmol), SA (162.7 mg, 1.63 mmol), DIPEA (0.032 mL, 0.30 mmol), and DMAP (24.9 mg, 0.21 mmol) were reacted in 6 mL anhydrous DCM under N2 protection for 7 hours at 40°C, and then deprotected with 30% trifluoroacetic acid (TFA)/DCM at room temperature. The mixture was precipitated and purified to obtain succinyl-(C10-OH)SN38.
SN38 (1.00 g, 2.55 mmol) and SA (0.33 g, 3.32 mmol) were stirred in 10 mL of anhydrous pyridine for 48 hours at 40°C. The mixture was precipitated in water with excess HCl. After filtration and vacuum drying, the product was purified by column chromatography to obtain succinyl-(C20-OH)SN38.
Synthesis of CS-(10s)SN38- and CS-(20s)SN38-conjugated macromolecular prodrugs
To obtain CS-(10s)SN38 and CS-(20s)SN38, succinyl-(C10-OH)SN38 and succinyl-(C20-OH)SN38 were reacted with EDC and NHS in dimethyl sulfoxide (DMSO) for 4 hours, then transferred to a chitosan/H2O/DMSO solution. The temperature of the mixture was raised to 70°C for 5 hours, and the mixture was stirred at room temperature for another 24 hours. The mixture was then poured into a methanol/ammonia solution and the resulting precipitate was collected by centrifugation. After washing several times with ethanol until no SN38 or derivatives were detected, the yellow precipitate was dissolved in acidic distilled water, and then dialyzed against distilled water in regenerated cellulose dialysis bags to remove any water-soluble byproducts. The final CS-(10s)SN38 and CS-(20s)SN38 were lyophilized (freeze dryer model FD-1C-50; Boyikang Experimental Instrument Co. Ltd., Beijing, People’s Republic of China).
1H nuclear magnetic resonance and mass spectrometry analysis
Proton nuclear magnetic resonance (1H NMR) spectra of intermediates and CS-SN38 macromolecular prodrugs were determined using a Bruker AVANCE Spectrometer (AV-600; Bruker Corporation, Billerica, MA, USA) operated at 400 MHz and 600 MHz, respectively. Intermediates were dissolved in deuterated DMSO (DMSO−d6) and the prodrugs were dissolved in a mixture of deuterated water (D2O) with 1% tetradeuteroacetic acid (CD3COOD). The mass spectrometry (MS) experiments on the intermediates were performed on a mass spectrometer (micrOTOF-Q; Bruker Corporation).
Fourier transform infrared analysis
In order to investigate the chemical interaction between SN38 and chitosan of the two different prodrugs, Fourier transform infrared (FT-IR) spectra were recorded on an FT-IR spectrometer (Bruker Corporation). The samples were ground with KBr crystals, and then pressed into pellets for infrared (IR) measurement over the range of 400–4,000 cm−1.
Differential scanning calorimetry analysis
Five samples, including SN38, chitosan, the physical mixture of SN38 and chitosan, CS-(10s)SN38, and CS-(20s)SN38, were weighed and sealed in aluminum pans. Differential scanning calorimetry (DSC) studies were then carried out using a differential scanning calorimeter (STAR® DSC System; Mettler Toledo, Greifensee, Switzerland) under dynamic nitrogen flow and a heating–cooling rate of 10°C/minute from 30°C to 240°C.
Drug-loading measurement
To permit the complete release of SN38 from the macromolecular prodrug, a certain amount of prodrug was dissolved in a 0.1 mol/L NaOH solution and incubated for 30 minutes. An excess amount of HCl was then added to transform the carboxylate form of SN38 to the lactone form. The samples were diluted and analyzed. Samples were tested in triplicate, and the drug loading (%) was determined by dividing the weight of SN38 by the weight of the prodrug. Information about the recovery rate of this method can be found in Figure S1.
Particle size and zeta potential measurement of CS-SN38 micelles
In aqueous medium, both macromolecular prodrugs could self-assemble into micelles. The size and the zeta potential of the micelles were determined by laser light scattering with a Malvern Zetasizer (Zetasizer Nano S90; Malvern Instruments, Malvern, UK) at 25°C.
Transmission electron microscopy micrographs
A transmission electron microscope (TEM; JEM-2100; JEOL, Tokyo, Japan) was used for the morphological analysis of the self-assembled prodrug micelles following negative staining with a phosphotungstic acid solution (2% weight per volume) at an accelerating voltage of 200 kV. The samples were air-dried before the observations were carried out.
Measurement of the critical micelle concentration
To determine the critical micelle concentration (CMC) of CS-(20s)SN38 and CS-(10s)SN38, a known amount of coumarin-6 in chloroform was added to a series of vials. After the chloroform was evaporated, a series of concentrations of the macromolecular prodrug solution was added to each vial, then ultrasonicated for 30 minutes and stirred overnight in the dark. The fluorescence emission intensity was measured using a spectrofluorophotometer (F-2500 FL Spectrophotometer; Hitachi Ltd., Tokyo, Japan) at 466 nm (emission wavelength) and 504 nm (lasing wavelength). The CMC of the self-assembled macromolecular prodrug micelle was determined at the intersection of the tangents to the two linear portions of the fluorescence intensity curve.36
In vitro drug release studies
The behavior of the SN38 in vitro release from both of the self-assembled macromolecular prodrugs was studied by dialysis at 37°C±1°C in different release media for 48 hours.37 Briefly, CS-(10s)SN38 and CS-(20s)SN38 were dissolved in 1 mL pH 6.5 phosphate-buffered saline (PBS), pH 7.4 PBS, 25% liver homogenate, and 50% plasma, where the concentration of the prodrugs was 250 μg/mL (equivalent to SN38), respectively. The solutions were then put into dialysis tubes (molecular weight cut-off: 10,000–12,000 kDa) and the tubes were immersed in 20 mL of PBS (pH 6.5 for the first tube and pH 7.4 for the others). Experiments were performed at 37°C in a shaking water bath at 100 rpm. Samples were collected at 1, 3, 5, 8, 12, 24, 36, and 48 hours and the release media were replaced at certain intervals, so that the media volume could meet the sink condition. The concentration of SN38 was determined by high-performance liquid chromatography (HPLC) (LC-10AT; Shimadzu Corporation, Kyoto, Japan) at 385 nm with an ultraviolet (UV) detector (SPD-10A; Shimadzu Corporation) using a C18 reversed-phase column (Dikma Technologies Inc., Lake Forest, CA, USA). The mobile phase consisted of acetonitrile and acetate buffer (48:52 volume per volume).
In vitro cytotoxicity assays
CT26 was used to determine the in vitro cytotoxicity. Briefly, adherent cells (about 5,000 cells per well) were plated in 96-well plates and incubated for 24 hours at 37°C with 5% CO2. After incubation, the cells were treated with serial dilutions of CS-(10s)SN38, CS-(20s)SN38, succinyl-(C10-OH)SN38, succinyl-(C20-OH)SN38, CPT-11, and SN38, and then further incubated for 48 hours. At the end of this time, 3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2-H-tetrazolium bromide (MTT) was added and the cells were incubated for another 2 hours. After removing surplus MTT, the colored formazan product was assayed using an enzyme-linked immunosorbent assay (ELISA) analyzer (Tecan, Männedorf, Switzerland) at a test wavelength of 492 nm.
Pharmacokinetic and statistical analysis
Male SD rats were used for the pharmacokinetic study. All animals were fasted overnight before starting the experiment. CS-(10s)SN38 (10 mg/kg), CS-(20s)SN38 (10 mg/kg), and CPT-11 (10 mg/kg) were administrated to rats intravenously. All preparations were equivalent to SN38. Blood samples were collected into heparinized tubes at the following times: 5, 10, 20, and 40 minutes, and 1, 1.5, 2, 3, 4, 6, 8, 12, and 24 hours after administration. The plasma was then obtained from the blood samples by centrifugation at 12,000× g for 10 minutes and then stored at −60°C until required for analysis.
Analysis of the free SN38 (chitosan-unbound SN38) and CPT-11 was performed as follows: 100 μL of plasma, 10 μL of internal standard solution (hydroxycamptothecin, 5 μg/mL), and 5 μL 0.2 mol/L HCl were mixed for 30 seconds; the samples were then extracted with 2 mL of ethyl acetate for 5 minutes at room temperature. After centrifugation at 3,000× g for 10 minutes, the supernatants were transferred into glass tubes and then dried under nitrogen at 40°C. The samples were redissolved until the HPLC analysis was carried out.
The concentration of chitosan-bound SN38 (SN38 remaining bound to chitosan) was calculated as follows: the concentration of whole SN38 minus the concentration of free SN38 is equal to the concentration of chitosan-bound SN38. The concentration of whole SN38 was detected by the following method: 50 μL of plasma was incubated with 5 μL 1 mol/L NaOH for 30 minutes at 37°C. After incubation, 2 mol/L HCl (5 μL), 10 μL of internal standard (hydroxycamptothecin, 5 μg/mL), and 200 μL of methanol were added. The samples were centrifuged at 12,000× g for 10 minutes before being analyzed by HPLC at 385 nm with a UV detector.
In vivo antitumor activity and pathological examination
CT26 tumor xenograft models were established to investigate in vivo antitumor activity. Approximately 2×106 CT26 cells were injected into the right axillary region of male BALB/c mice (weight: 18~22 g).
When the tumor volumes (TV) reached 100–200 mm3, the mice were divided into six groups (n=8) as follows: (A) saline; (B) CPT-11 at 2.5 mg/kg; (C) CS-(10s)SN38 at 1.25 mg/kg; (D) CS-(20s)SN38 at 1.25 mg/kg; (E) CS-(10s)SN38 at 2.5 mg/kg; and (F) CS-(20s)SN38 at 2.5 mg/kg (all preparations were equivalent to SN38). Drugs were given to the mice by intravenous injection into the tail vein every 2 days for 2 consecutive weeks. The TV and body weights were measured every 2 days and the TV (mm3) was calculated using the following formula:38
| (1) |
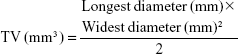
At the end of the experiment, the mice were euthanized and the tumors were isolated and weighed. Differences in TV between the treatment groups and the control group were analyzed. The inhibition ratio of the tumors (IRTs) was calculated according to the following equation:38
|
|

At day 14, tumors from the mice that received different treatments were excised. The tumors were fixed in 10% formalin and embedded in paraffin. Sections with a thickness of 4 μm were then prepared. The sections were stained with hematoxylin and eosin (H&E) for microscopic observation.
Results and discussion
Characterization of succinyl-(C10-OH)SN38 and succinyl-(C20-OH)SN38
Characterization of succinyl-(C10-OH)SN38: 1H NMR (400 MHz, DMSO−d6, δ): 0.95 (t, 3H, CH3), 1.27 (t, 3H, CH3), 2.14 (q, 2H, CH2), 2.77 (m, 2H, CH2), 3.00 (q, 2H, CH2), 5.18 (d, 2H, J=18.8, CH2), 5.48 (s, 2H, CH2), 5.75 (s, 1H, OH), 7.00 (s, 1H, ArH), 7.32 (d, 1H, J=2.4, ArH), 7.38 (dd, 1H, J=8.8, J=2.4, ArH), 7.97 (d, 1H, J=8.8, ArH); MS (ESI-TOF) m/z: [M + H]+ calculated for C22H20N2O5, 493.1611; found, 493.1605.
Characterization of succinyl-(C20-OH)SN38: 1H NMR (400 MHz, DMSO−d6, δ): 0.92 (t, 3H, CH3), 1.26 (t, 3H, CH3), 2.14 (q, 2H, CH2), 2.47 (m, 2H, CH2), 2.73 (m, 2H, CH2), 3.07 (q, 2H, CH2), 5.27 (d, 2H, CH2), 5.47 (s, 2H, CH2), 7.01 (s, 1H, ArH), 7.39 (d, 1H, J=2.4Hz, ArH), 7.42 (dd, 1H, J=9.2Hz, J=2.4Hz, ArH), 8.03 (d, 1H, J=9.2Hz, ArH), 10.33 (s, 1H, OH); MS (ESI-TOF) m/z: [M + H]+ calculated for C22H20N2O5, 493.1611; found, 493.1605.
Succinyl-(C10-OH)SN38 and succinyl-(C20-OH)SN38 were characterized by 1H NMR and MS, as shown earlier; the original spectrum can be found in Figure S2. The absorption peaks of succinyl-linker H atoms were observed in both of the derivatives, and the chemical shift was 2.4–2.8 ppm. For succinyl-(C10-OH)SN38, the absorption peak of C10-OH disappeared due to the esterification of SA with the phenolic hydroxyl of SN38. The chemical shift of C20-OH was 5.75 ppm. In the case of succinyl-(C20-OH)SN38, the chemical shift of C10-OH was seen at 10.33 ppm. The disappearance of the absorption peak of C20-OH indicated that the succinyl-linker was conjugated to SN38 at C20-OH. Since these two derivatives are isomers, the same molecular weight was found in the MS study.
Characterization of CS-(10s)SN38 and CS-(20s)SN38
After purification, CS-(10s)SN38, CS-(20s)SN38, and chitosan were characterized by their 1H NMR spectra. A typical 1H NMR spectrum of chitosan is presented in Figure 3A. As shown in Figures 3B and C, the proton peaks at 0.8–1.2 ppm, which are attributed to the methyl protons of SN38, were observed in the spectra of CS-(10s)SN38 and CS-(20s)SN38. Furthermore, no such peaks were present at the same chemical shifts for chitosan. The protons corresponding to the methylene of SN38 appeared between chemical shifts 2.1–2.8 ppm, which is in agreement with a previous study.15,18 These results indicated that CS-(10s)SN38 and CS-(20s)SN38 had been synthesized successfully.
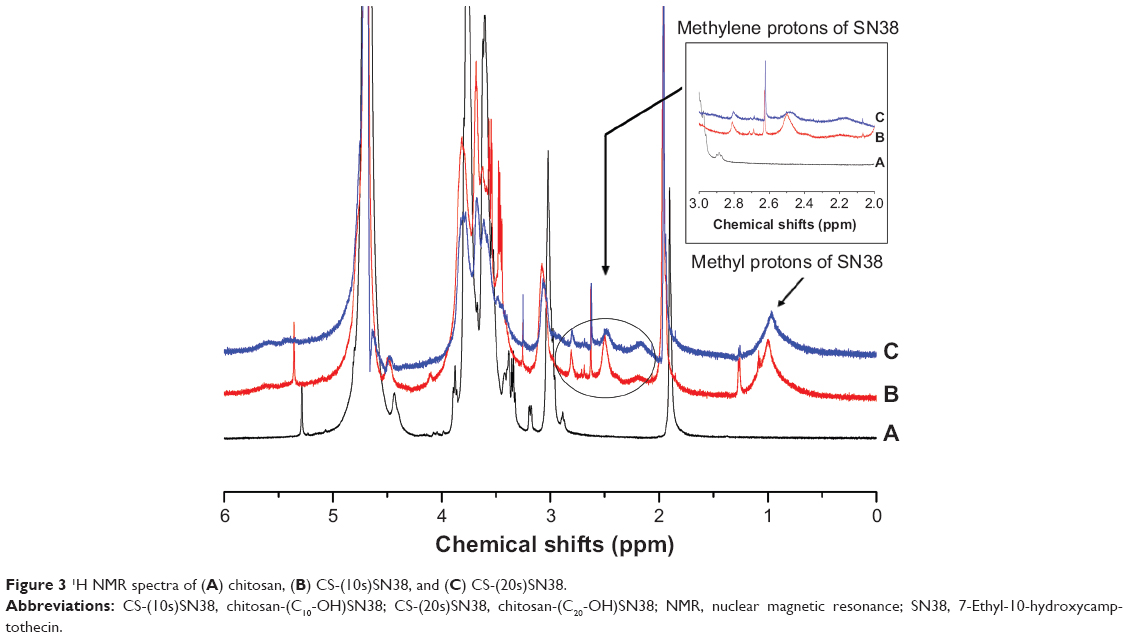 | Figure 3 1H NMR spectra of (A) chitosan, (B) CS-(10s)SN38, and (C) CS-(20s)SN38. |
The FT-IR spectra of chitosan, SN38, the mixture of chitosan and SN38, CS-(10s)SN38, and CS-(20s)SN38 are shown in Figure 4. The spectrum of CS indicated that the peaks at 1,633 cm−1 and 1,520 cm−1 were due to the C–O stretching vibration of the carbonyl group and the N–H bending vibration of the secondary amine (amide I and II), respectively (original spectra of chitosan could be found in Figure S3). Compared with CS, the new signals at 1,656 cm−1 and 1,566 cm−1 were assigned to the new amide link for amide I and II. In addition, peaks at 1,598 cm−1 and 1,600 cm−1 were observed in the spectra of CS-(10s)SN38 and CS-(20s)SN38, representing the characteristic vibration of the benzene skeleton. All of the results showed that the grafting of SN38 onto the amino groups of CS had been successful.
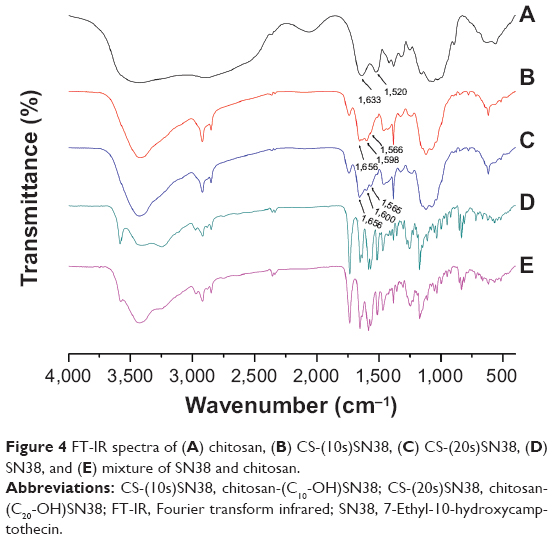 | Figure 4 FT-IR spectra of (A) chitosan, (B) CS-(10s)SN38, (C) CS-(20s)SN38, (D) SN38, and (E) mixture of SN38 and chitosan. |
To further confirm that SN38 had been covalently linked to chitosan, a DSC experiment was performed. As illustrated in Figure 5, the endothermic peak that was related to the melting point of SN38 in its crystalline form appeared at 208°C–210°C. For the physical mixtures, all the characteristic peaks of every component showed only a slight shift. Both macromolecular prodrugs exhibited no sharp peaks, which suggested that SN38 was present in an amorphous state in the prodrugs.
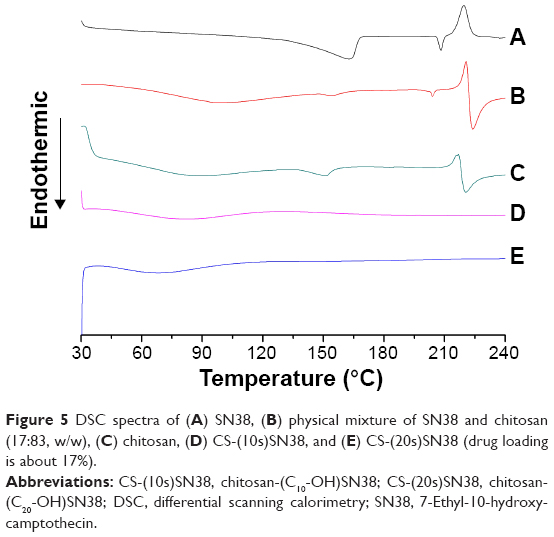 | Figure 5 DSC spectra of (A) SN38, (B) physical mixture of SN38 and chitosan (17:83, w/w), (C) chitosan, (D) CS-(10s)SN38, and (E) CS-(20s)SN38 (drug loading is about 17%). |
The drug loading was found to be 16.5%±0.7% for CS-(10s)SN38 and 17.4%±0.8% for CS-(20s)SN38 as found using HPLC with a UV detector, and this was much higher than the amphiphilic block copolymer micelles loaded with SN38.13,39 In our study, the water-insoluble drug was directly linked to chitosan by a covalent bond. The force of the covalent bond was stronger than the hydrophobic interactions between SN38 and the hydrophobic group of the amphiphilic block copolymers, and this could be an important reason for the high drug-loading efficiency. The solubility of both macromolecular prodrugs was more than 40 mg/mL (corresponding to 7.2 mg/mL for SN38) while SN38 had a very poor solubility of 11~38 μg/mL in water.11 These observations further confirm that SN38 was covalently bound to chitosan.
Characterization and morphology of self-assembled micelles
Both CS-(10s)SN38 and CS-(20s)SN38 were SN38-loaded macromolecular micelles produced in aqueous medium by the self-assembly of amphiphilic graft copolymers and the CMC values were calculated as 15 μg/mL and 12 μg/mL by the method described earlier (Figure 6). The CMC value reflects the threshold concentration of the self-aggregation formation by the hydrophilic and hydrophobic groups of the macromolecular prodrugs.40 Such low CMC values for CS-(10s)SN38 and CS-(20s)SN38 indicate their excellent self-aggregation behavior in aqueous medium and stabilization following dilution. The TEM image and particle size are shown in Figure 7. The shape of both macromolecular prodrugs was almost spherical and the mean particle size was 128.0±1.8 nm for CS-(10s)SN38 micelles (Figure 7A) and 122.4±1.1 nm for CS-(20s)SN38 micelles (Figure 7B) (n=3). The particle sizes were similar when the drug loading of the two prodrug micelles was similar. This indicated that particle size could mainly be determined by the degree of grafting of the hydrophobic groups and the characteristics of the hydrophilic groups, rather than the conjugate position of SN38. The mean zeta potential of CS-(10s)SN38 was 21.1±0.5 mV (n=3), whereas the mean zeta potential of CS-(20s)SN38 was 22.3±0.4 mV (n=3). These relatively high zeta potentials of the micelles play an important role in the mutual repulsion and stability against aggregation, which could be due to the ionized amino groups of chitosan.
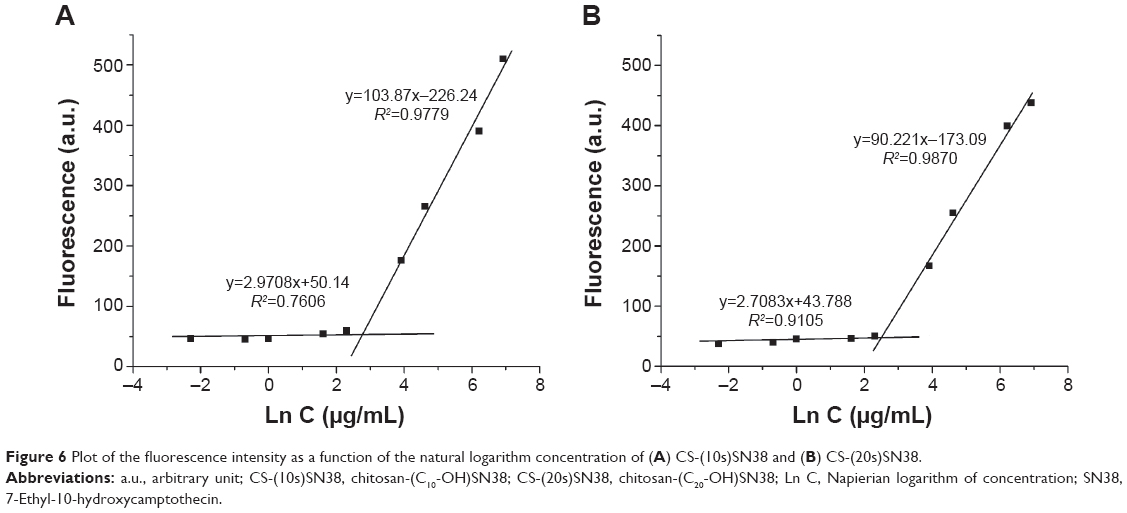 | Figure 6 Plot of the fluorescence intensity as a function of the natural logarithm concentration of (A) CS-(10s)SN38 and (B) CS-(20s)SN38. |
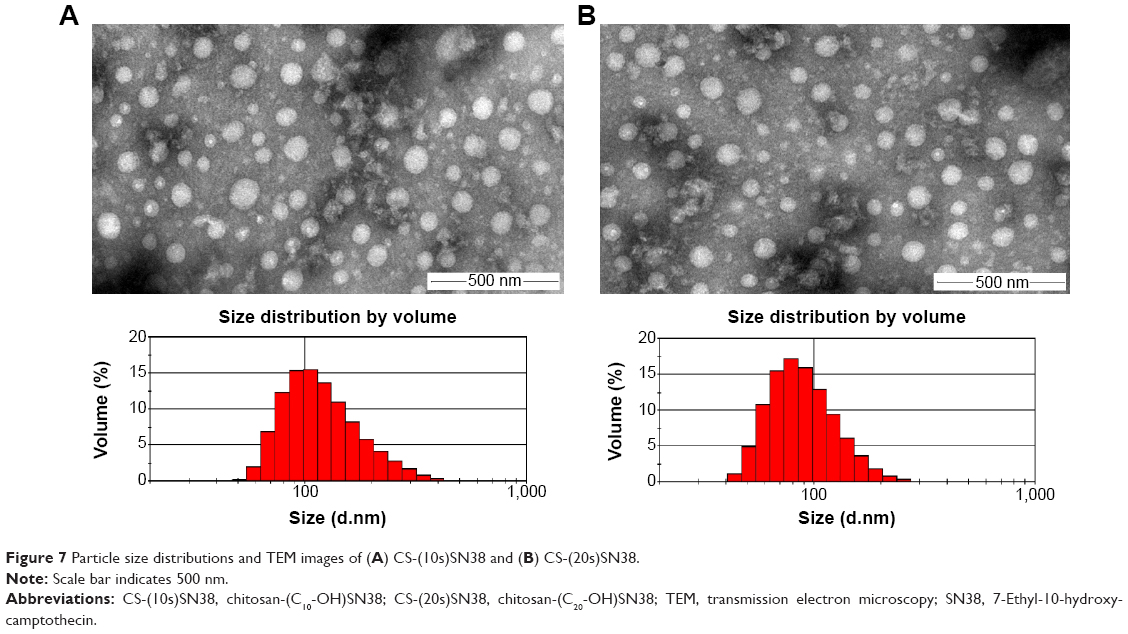 | Figure 7 Particle size distributions and TEM images of (A) CS-(10s)SN38 and (B) CS-(20s)SN38. |
Drug release studies
The release of SN38 from the self-assembled macromolecular prodrugs was performed at 37°C. We chose releasing media to simulate body fluids (pH 7.4 PBS), the tumor extracellular environment (pH 6.5 PBS), blood circulation (50% plasma), and liver circulation (25% liver homogenate).
When SN38 is bound to chitosan by a succinyl linker, the macromolecular prodrug may have two different pathways to release the active metabolite, as illustrated in Figure 8: 1) rupture at the amide bond, followed by hydrolysis to release SN38; or 2) direct hydrolysis of the ester bond to obtain SN38. Thus, we investigated the cumulative profiles of both SN38 and succinyl-SN38 derivatives of the two prodrug micelles. SN38 was released from CS-(10s)SN38 and CS-(20s)SN38 in different media, as shown in Figures 9A and B. For both prodrugs, the SN38 release rates were relatively higher in the 50% plasma and 25% liver homogenate than in PBS. The ester bond between the drug and the spacer could be cleaved by esterases, but in PBS, the release was only due to nonspecific hydrolysis. The release of succinyl-(C10-OH)SN38 is shown in Figure 9C, and that of succinyl-(C20-OH)SN38 is shown in Figure 9D. The low cumulative curves of the two succinyl-SN38 derivatives in 50% plasma and 25% liver homogenate reflected fast in vivo elimination. The succinyl-SN38s had a fluctuating release profile in different media, which could be due to a dynamic transformation from succinyl-SN38 to free SN38. Comparing the cumulative profiles of the two macromolecular prodrugs, CS-(20s)SN38 showed slower drug release by 48 hours in different media (P<0.05), which indicates better stability of CS-(20s)SN38 in vitro for the controlled release of SN38. Esterification at C20-OH could enhance steric hindrance, prevent the ring-opening of SN38, and protect the prodrug micelles from hydrolysis, which could be an explanation for the slower release of CS-(20s)SN38.
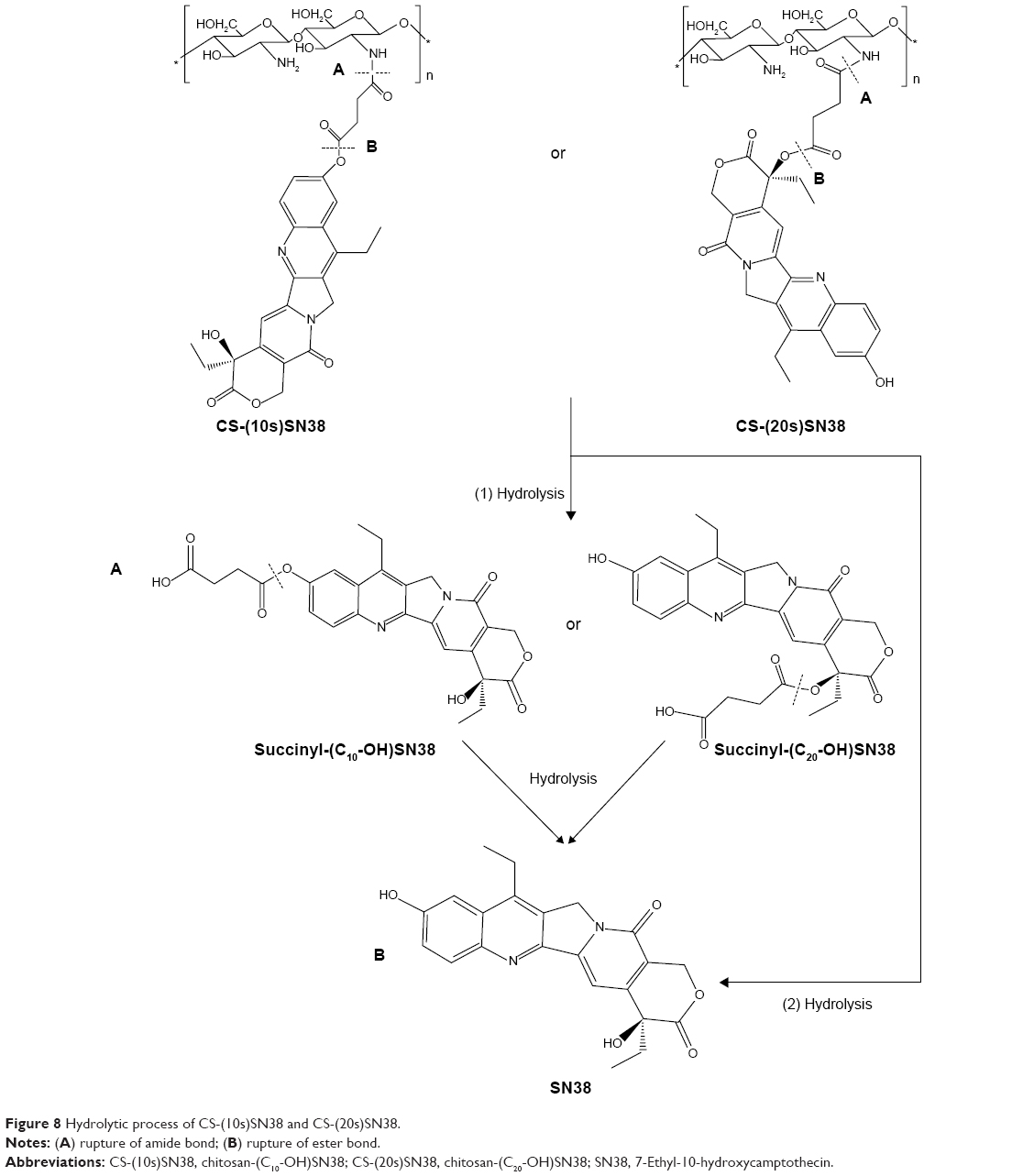 | Figure 8 Hydrolytic process of CS-(10s)SN38 and CS-(20s)SN38. |
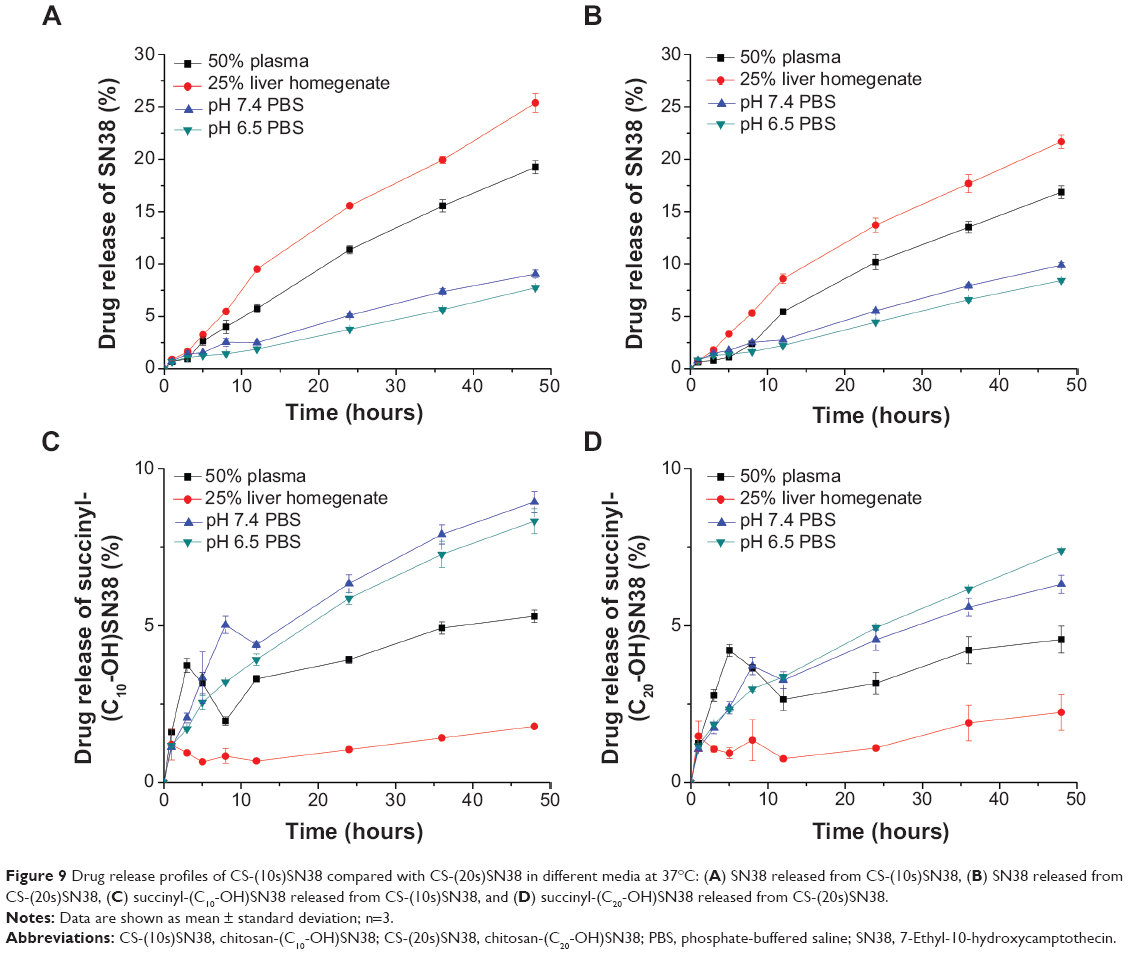 | Figure 9 Drug release profiles of CS-(10s)SN38 compared with CS-(20s)SN38 in different media at 37°C: (A) SN38 released from CS-(10s)SN38, (B) SN38 released from CS-(20s)SN38, (C) succinyl-(C10-OH)SN38 released from CS-(10s)SN38, and (D) succinyl-(C20-OH)SN38 released from CS-(20s)SN38. |
Cytotoxicity study
In the toxicity study, both the CS-(10s)SN38 and CS-(20s)SN38 prodrugs, succinyl-SN38 derivatives, CPT-11, and SN38 were incubated with CT26 cells for 48 hours in order to compare their in vitro cytotoxicity (Table 1). SN38 showed the highest activity against the CT26 cell line with a half maximal inhibitory concentration (IC50) value of 20.4 nmol/L. CPT-11 is the main clinical form of SN38, and its IC50 value was (46.7±8.4)×103 nmol/L. The IC50 values of CS-(10s)SN38 and CS-(20s)SN38 were (3.5±0.2)×103 nmol/L and (1.8±0.4)×103 nmol/L, respectively, which were 13.3- and 25.9-fold more sensitive than CPT-11. The in vitro cytotoxicity of all SN38 derivatives and prodrugs was highly potent when compared with that of CPT-11. Moreover, it was observed that CS-(20s)SN38 and succinyl-(C20-OH)SN38 were more cytotoxic than CS-(10s)SN38 and succinyl-(C10-OH)SN38 (both P<0.05). This suggests that the C20-OH position derivatives might have superior anticancer activity to C10-OH position derivatives, which could perhaps be due to the stabilization of the lactone ring of SN38.41 Only the lactone form is active against topoisomerase I, and the activity is reduced to 1/10 when the lactone form is transformed into the carboxylate form under basic conditions.42 Also, modification at C20-OH could reduce the hydrogen-bonding interaction between C20-OH and the carbonyl groups.43
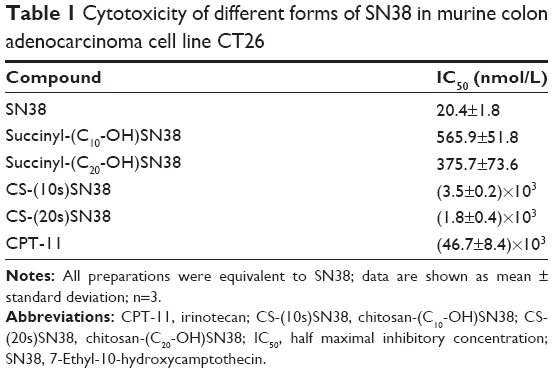 | Table 1 Cytotoxicity of different forms of SN38 in murine colon adenocarcinoma cell line CT26 |
Pharmacokinetic analysis of CS-(20s)SN38, CS-(10s)SN38, and CPT-11
To investigate the in vivo pharmacokinetic behaviors of CS-(10s)SN38, CS-(20s)SN38, and CPT-11, free SN38 and the prodrugs (equivalent to SN38) were examined by HPLC.
The mean plasma concentration–time curves of SN38 after intravenous administration of the macromolecular prodrug micelles and CPT-11 are shown in Figure 10. The pharmacokinetic parameters of SN38 were calculated using noncompartmental analysis with DAS 2.0 software (Table 2). The area under the curve (AUC)0–24 values of free SN38 after intravenous administration of CS-(10s)SN38 and CS-(20s)SN38 were 7,011.18±969.76 μg·h/L and 8,925.92±1,907.46 μg·h/L respectively, which were statistically insignificant (P>0.05). However, when compared with CPT-11, both AUC0–24 values were much higher than that of CPT-11 (P<0.05). For CS-(10s)SN38 and CS-(20s)SN38, the half-life (T1/2) and mean residence time (MRT) of plasma SN38 were prolonged, while the renal clearance (CL) was reduced when compared with CPT-11 (P<0.05). The nonspecificity of hydrolysis could be an explanation for the higher conversion rate observed for SN38 of the prodrugs when compared with CPT-11. The AUC0–24 for free SN38 of CS-(10s)SN38 was slightly higher than that of CS-(20s)SN38, which might be due to the faster release of SN38 from CS-(10s)SN38 in plasma. The lactone form of SN38 is necessary for topoisomerase inhibition, and the determination of lactone form of SN38 is valuable for macromolecular prodrug study. We would like to discuss the percent of lactone form of SN38 in vivo in our future work.
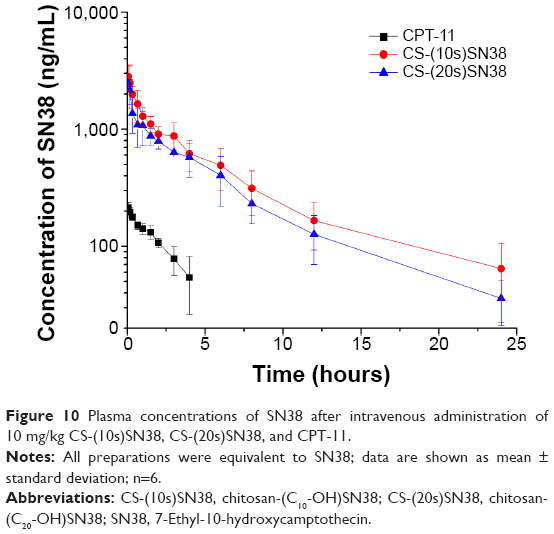 | Figure 10 Plasma concentrations of SN38 after intravenous administration of 10 mg/kg CS-(10s)SN38, CS-(20s)SN38, and CPT-11. |
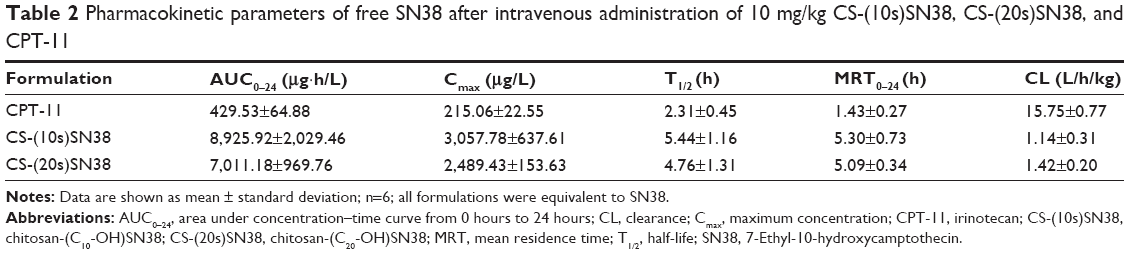 | Table 2 Pharmacokinetic parameters of free SN38 after intravenous administration of 10 mg/kg CS-(10s)SN38, CS-(20s)SN38, and CPT-11 |
The plasma concentration of the two different macromolecular prodrugs and CPT-11 are shown in Figure 11. The calculated parameters of the plasma prodrugs are given in Table 3. CS-(20s)SN38 has a higher AUC0–24 than CS-(10s)SN38 (P<0.05). The Cmax values of the two prodrugs were not significantly different. The existence of SN38 bound to the macromolecular prodrug might take advantage of the EPR effect and a better in vivo antitumor effect was thus expected.
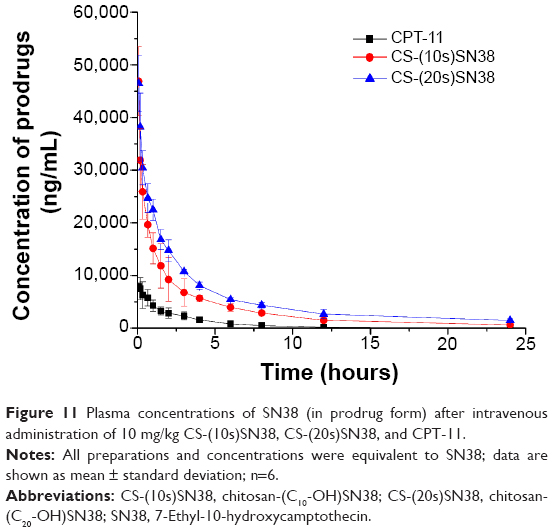 | Figure 11 Plasma concentrations of SN38 (in prodrug form) after intravenous administration of 10 mg/kg CS-(10s)SN38, CS-(20s)SN38, and CPT-11. |
 | Table 3 Pharmacokinetic parameters of prodrugs after intravenous administration of 10 mg/kg CS-(10s)SN38, CS-(20s)SN38, and CPT-11 |
In vivo antitumor activity and pathological examination
In order to further evaluate the in vivo antitumor activity of the two macromolecular prodrug micelles, an experiment was performed using a CT26 xenograft BALB/c mouse model. The results obtained are shown in Table 4 and Figure 12A. The growth in TV was very fast in the saline group (group A), while the TV growth of mice injected with a low dose of CS-(10s)SN38 and CS-(20s)SN38 (groups C and D) showed no statistically significant difference. The tumor growth was significantly inhibited at high doses of CS-(10s)SN38 and CS-(20s)SN38 (groups E and F), especially for CS-(20s)SN38. The average tumor weight of the saline group was 3.27±0.28 g. At the low dose, the IRT (%) of the CS-(20s)SN38 group (31%) was not significantly different from that of the CS-(10s)SN38 group (27.7%) (P>0.05). At the high dose of the CS-(20s)SN38 group, the IRT (%) (64.7%) was much higher than that of CPT-11 or CS-(10s)SN38 at the same dose (both P<0.05). Compared with CPT-11, both prodrug micelles could increase in vivo antitumor activity. The reason for this could be due to the fact that both prodrug micelles increased the solubility of SN38 and promoted the tumor accumulation of SN38. In addition, another reason for the better in vivo antitumor activity of CS-(20s)SN38 could be the following: firstly, the CS-(20s)SN38 micelles had a better circulation effect in vivo than did CS-(10s)SN38, which could enhance the EPR effect; secondly, occupation at the C20-OH of SN38 might increase the lactone ring stability of SN38 and lead to antitumor activity.
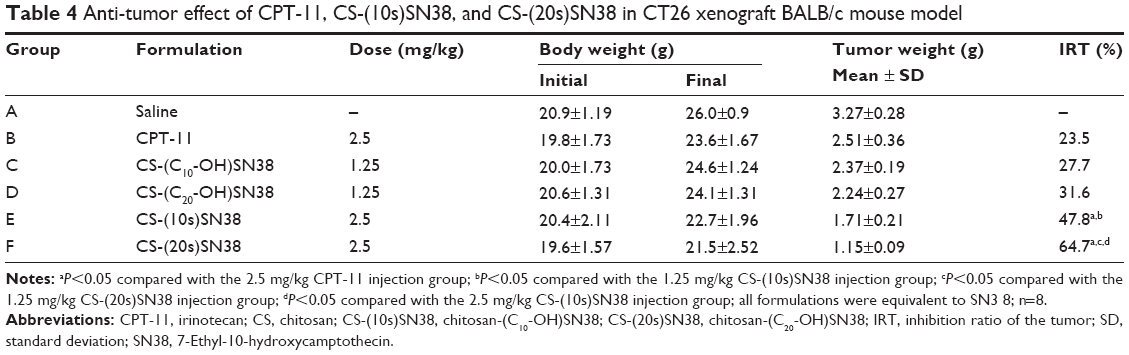 | Table 4 Anti-tumor effect of CPT-11, CS-(10s)SN38, and CS-(20s)SN38 in CT26 xenograft BALB/c mouse model |
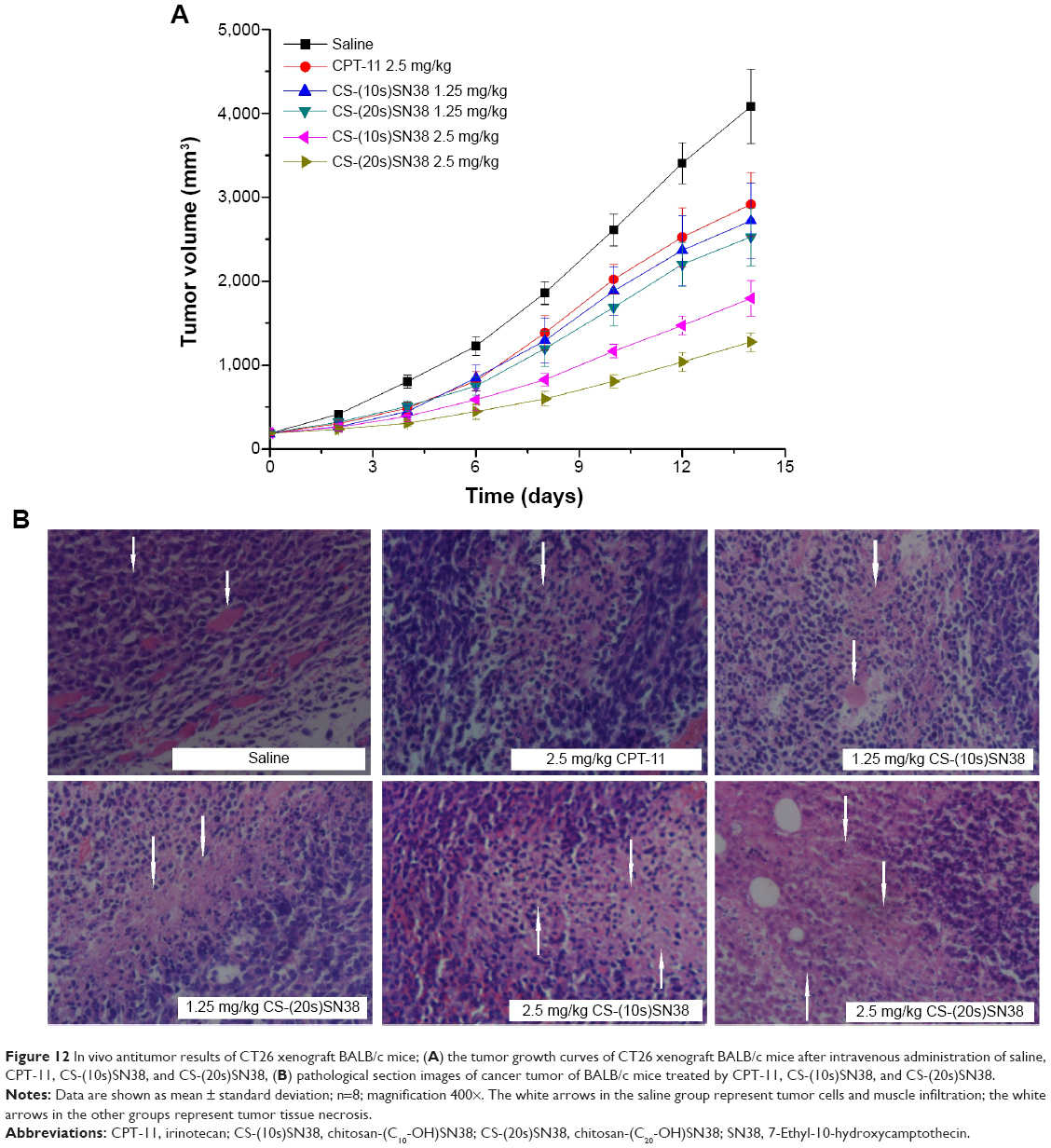 | Figure 12 In vivo antitumor results of CT26 xenograft BALB/c mice; (A) the tumor growth curves of CT26 xenograft BALB/c mice after intravenous administration of saline, CPT-11, CS-(10s)SN38, and CS-(20s)SN38, (B) pathological section images of cancer tumor of BALB/c mice treated by CPT-11, CS-(10s)SN38, and CS-(20s)SN38. |
Additionally, body weight was used as a toxicity marker (Figure 13). In the saline group, body weight significantly increased by 24% by the end of the experiment. Similar results were shown for the low-dose group of the macromolecular prodrugs. In the high-dose macromolecular prodrug groups, the body weights were slightly increased with excellent antitumor activities (Figure 12A). However, there was no statistically significant difference between the prodrug groups and the saline group (P>0.05). This might be due to the good biocompatibility and low toxicity of the prodrug micelles. Therefore, these prodrug micelles demonstrated excellent antitumor activity and good safety.
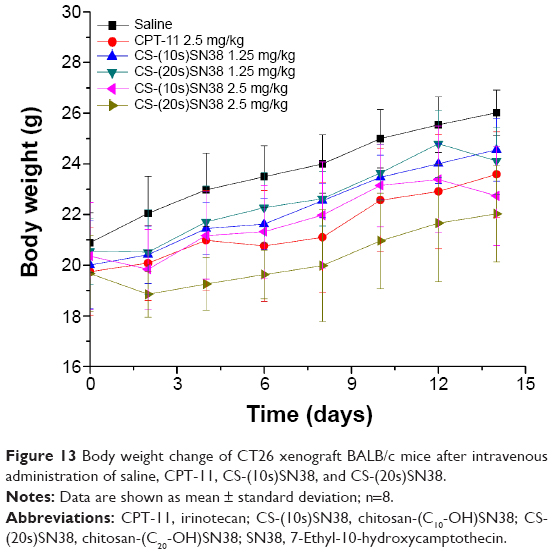 | Figure 13 Body weight change of CT26 xenograft BALB/c mice after intravenous administration of saline, CPT-11, CS-(10s)SN38, and CS-(20s)SN38. |
To further confirm therapeutic efficacy, a histological section examination was performed. Nuclei were stained violet and the extracellular matrix and cytoplasm were stained pink using H&E staining. As shown in Figure 12B, a large number of tumor cells and muscle infiltration were observed in the saline group. Partial tumor tissue necrosis was found in the group treated with 2.5 mg/kg of CPT-11, 1.25 mg/kg of CS-(10s)SN38, and 1.25 mg/kg of CS-(20s)SN38. By contrast, extensive tissue necrosis was found in the group treated with 2.5 mg/kg of CS-(10s)SN38 and 2.5 mg/kg CS-(20s)SN38. The most severe damage in the tumor tissue was observed in the high-dose CS-(20s)SN38 group. These findings revealed that the delivery of SN38 by both macromolecular prodrugs produced superior anticancer efficacy at the high dose, especially for CS-(20s)SN38, due to the destruction and reduction of tumor cell proliferation.
Conclusion
In this study, we successfully synthesized two SN38 prodrugs in the form of macromolecular prodrug micelles. These prodrugs can self-assemble into micelles in aqueous medium and release SN38 in vitro. Both prodrugs increased the AUC0–24 of SN38 and antitumor activity against CPT-11. The in vitro cytotoxicity and in vivo antitumor activity of CS-(20s)SN38 were better than those of CS-(10s)SN38. Macromolecular biomaterial involving conjugated SN38 with C20-OH could be a more promising strategy to increase the antitumor activity of SN38, and this should be a subject for further study.
Acknowledgments
We would like to thank Professor Mingyu Xia and Master Qiang Li for their support with the cell experiments. English language editing of this manuscript was provided by Journal Prep.
Disclosure
The authors report no conflicts of interest in this work.
References
Ulukan H, Swaan PW. Camptothecins: a review of their chemotherapeutic potential. Drugs. 2002;62(14):2039–2057. | ||
Zhang Y, Zhang H, Wu W, et al. Folate-targeted paclitaxel-conjugated polymeric micelles inhibits pulmonary metastatic hepatoma in experimental murine H22 metastasis models. Int J Nanomedicine. 2014;9:2019–2030. | ||
Kawato Y, Aonuma M, Hirota Y, Kuga H, Sato K. Intracellular roles of SN38, a metabolite of the camptothecin derivative CPT-11, in the antitumor effect of CPT-11. Cancer Res. 1991;51(16):4187–4191. | ||
Pommier Y. Topoisomerase I inhibitors: camptothecins and beyond. Nat Rev Cancer. 2006;6(10):789–802. | ||
Roger E, Lagarce F, Benoit JP. Development and characterization of a novel lipid nanocapsule formulation of Sn38 for oral administration. Eur J Pharm Biopharm. 2011;79(1):181–188. | ||
Gupta E, Lestingi TM, Mick R, Ramirez J, Vokes EE, Ratain MJ. Metabolic fate of irinotecan in humans: correlation of glucuronidation with diarrhea. Cancer Res. 1994;54(14):3723–3725. | ||
Mathijssen RH, van Alphen RJ, Verweij J, et al. Clinical pharmacokinetics and metabolism of irinotecan (CPT-11). Clin Cancer Res. 2001; 7(8):2182–2194. | ||
Hoff DV. Future directions of clinical research with CPT-11 (irinotecan). Eur J Cancer. 1996;32(Suppl 3):S9–S12. | ||
Chabot GG. Clinical pharmacokinetics of irinotecan. Clin Pharmacokinet. 1997;33(4):245–259. | ||
Peikov V, Ugwu S, Parmar M, Zhang A, Ahmad I. pH-dependent association of SN-38 with lipid bilayers of a novel liposomal formulation. Int J Pharm. 2005;299(1–2):92–99. | ||
Zhang JA, Xuan T, Parmar M, et al. Development and characterization of a novel liposome-based formulation of SN-38. Int J Pharm. 2004;270(1–2):93–107. | ||
Chen M, Liu Y, Yang W, et al. Preparation and characterization of self-assembled nanoparticles of 6-O-cholesterol-modified chitosan for drug delivery. Carbohydr Polym. 2011;84(4):1244–1251. | ||
Duan K, Zhang X, Tang X, et al. Fabrication of cationic nanomicelle from chitosan-graft-polycaprolactone as the carrier of 7-ethyl-10-hydroxy-camptothecin. Colloids Surf B Biointerfaces. 2010;76(2):475–482. | ||
Ebrahimnejad P, Dinarvand R, Sajadi SA, Atyabi F, Ramezani F, Jaafari MR. Preparation and characterization of poly lactide-co-glycolide nanoparticles of SN-38. PDA J Pharm Sci Technol. 2009;63(6):512–520. | ||
Sepehri N, Rouhani H, Tavassolian F, et al. SN38 polymeric nanoparticles: in vitro cytotoxicity and in vivo antitumor efficacy in xenograft babl/c model with breast cancer versus irinotecan. Int J Pharm. 2014;471(1–2):485–497. | ||
Vangara KK, Liu J, Palakurthi S. Hyaluronic acid-decorated PLGA-PEG nanoparticles for targeted delivery of SN-38 to ovarian cancer. Anticancer Res. 2013;33(6):2425–2434. | ||
Manaspon C, Nittayacharn P, Vejjasilpa K, Fongsuk C, Nasongkla N. SN-38: β-cyclodextrin inclusion complex for in situ solidifying injectable polymer implants. Conf Proc IEEE Eng Med Biol Soc. 2011;2011:3241–3244. | ||
Vijayalakshmi N, Ray A, Malugin A, Ghandehari H. Carboxyl-terminated PAMAM-SN38 conjugates: synthesis, characterization, and in vitro evaluation. Bioconjug Chem. 2010;21(10):1804–1810. | ||
Koizumi F, Kitagawa M, Negishi T, et al. Novel SN-38-incorporating polymeric micelles, NK012, eradicate vascular endothelial growth factor-secreting bulky tumors. Cancer Res. 2006;66(20):10048–10056. | ||
Saito Y, Yasunaga M, Kuroda J, Koga Y, Matsumura Y. Antitumour activity of NK012, SN-38-incorporating polymeric micelles, in hypovascular orthotopic pancreatic tumour. Eur J Cancer. 2010;46(3):650–658. | ||
Daman Z, Ostad S, Amini M, Gilani K. Preparation, optimization and in vitro characterization of stearoyl-gemcitabine polymeric micelles: a comparison with its self-assembled nanoparticles. Int J Pharm. 2014;468(1–2):142–151. | ||
Huang L, Song J, Chen B. A novel targeting drug carrier to deliver chemical bonded and physical entrapped anti-tumor drugs. Int J Pharm. 2014;466(1–2):52–57. | ||
Mao L, Wang H, Tan M, Qu L, Kong D, Yang Z. Conjugation of two complementary anti-cancer drugs confers molecular hydrogels as a co-delivery system. Chem Commun (Camb). 2012;48(3):395–397. | ||
Greenwald RB, Pendri A, Conover C, Gilbert C, Yang R, Xia J. Drug delivery systems. 2. Camptothecin 20-O-poly(ethylene glycol) ester transport forms. J Med Chem. 1996;39(10):1938–1940. | ||
Wani MC, Nicholas AW, Manikumar G, Wall ME. Plant antitumor agents. 25. Total synthesis and antileukemic activity of ring A rubstituted camptothecin analogues. Structure-activity correlations. J Med Chem. 1987;30(10):1774–1779. | ||
Wang CY, Pan XD, Liu HY, Fu ZD, Wei XY, Yang LX. Synthesis and antitumor activity of 20-O-linked succinate-based camptothecin ester derivatives. Bioorg Med Chem. 2004;12(13):3657–3662. | ||
Lee KY, Ha WS, Park WH. Blood compatibility and biodegradability of partially N-acylated chitosan derivatives. Biomaterials. 1995;16(16):1211–1216. | ||
Hirano S, Seino H, Akiyama Y, Nonaka I. Chitosan: a biocompatible material for oral and intravenous administrations.In: Gebelein CG, Dunn RL, editors. Progress in Biomedical Polymers. New York, NY: Plenum Publishing Co.; 1990:283–290. | ||
Duhem N, Rolland J, Riva R, et al. Tocol modified glycol chitosan for the oral delivery of poorly soluble drugs. Int J Pharm. 2012;423(2):452–460. | ||
Wang XY, Zhang L, Wei XH, Wang Q. Molecular dynamics of paclitaxel encapsulated by salicylic acid-grafted chitosan oligosaccharide aggregates. Biomaterials. 2013;34(7):1843–1851. | ||
Zhang C, Qineng P, Zhang H. Self-assembly and characterization of paclitaxel-loaded N-octyl-O-sulfate chitosan micellar system. Colloids Surf B Biointerfaces. 2004;39(1–2):69–75. | ||
Zhang J, Chen XG, Li YY, Liu CS. Self-assembled nanoparticles based on hydrophobically modified chitosan as carriers for doxorubicin. Nanomedicine. 2007;3(4):258–265. | ||
Moon SJ, Govindan SV, Cardillo TM, D’Souza CA, Hansen HJ, Goldenberg DM. Antibody conjugates of 7-ethyl-10-hydroxycamptothecin (SN-38) for targeted cancer chemotherapy. J Med Chem. 2008;51(21):6916–6926. | ||
Jon S, Lee E, Lee JJ, Lee I, inventor; Gwangju Institute of Science and Technology, assignee. Transmucosal delivery of pharmaceutical active substances. United States patent US 20070292387. 2007 Dec 20. | ||
Angenault S, Thirot S, Schmidt F, Monneret C, Pfeiffer B, Renard P. Cancer chemotherapy: a SN-38 (7-ethyl-10-hydroxycamptothecin) glucuronide prodrug for treatment by a PMT (prodrug monotherapy) strategy. Bioorg Med Chem Lett. 2003;13(5):947–950. | ||
Zhang H, Wang J, Mao W, et al. Novel SN38 conjugate-forming nanoparticles as anticancer prodrug: in vitro and in vivo studies. J Control Release. 2013;166(2):147–158. | ||
Hu Q, Liang B, Sun Y, et al. Preparation of bufalin-loaded pluronic polyetherimide nanoparticles, cellular uptake, distribution, and effect on colorectal cancer. Int J Nanomedicine. 2014;9:4035–4041. | ||
Kawato Y, Furuta T, Aonuma M, Yasuoka M, Yokokura T, Matsumoto K. Antitumor activity of a camptothecin derivative, CPT-11, against human tumor xenografts in nude mice. Cancer Chemother Pharmacol. 1991;28(3):192–198. | ||
Guo M, Rong WT, Hou J, et al. Mechanisms of chitosan-coated poly(lactic-co-glycolic acid) nanoparticles for improving oral absorption of 7-ethyl-10-hydroxycamptothecin. Nanotechnology. 2013;24(24):245101. | ||
Wang J, Sun J, Chen Q, et al. Star-shape copolymer of lysine-linked di-tocopherol polyethylene glycol 2000 succinate for doxorubicin delivery with reversal of multidrug resistance. Biomaterials. 2012;33(28):6877–6888. | ||
He X, Lu W, Jiang X, Cai J, Zhang X, Ding J. Synthesis and biological evaluation of bis and monocarbonate prodrugs of 10-hydroxycamptothecins. Bioorg Med Chem. 2004;12(15):4003–4008. | ||
Burke TG, Mi Z. Preferential binding of carboxylate form of camptothecin by human serum albumin. Anal Biochem. 1993;212(1):285–287. | ||
Rahier NJ, Eisenhauer BM, Gao R, Jones SH, Hecht SM. Water-soluble camptothecin derivatives that are intrinsic topoisomerase I poisons. Org Lett. 2004;6(3):321–324. |
Supplementary materials
 | Figure S1 The recovery rate of the lactone form of SN38 at 37°C. |
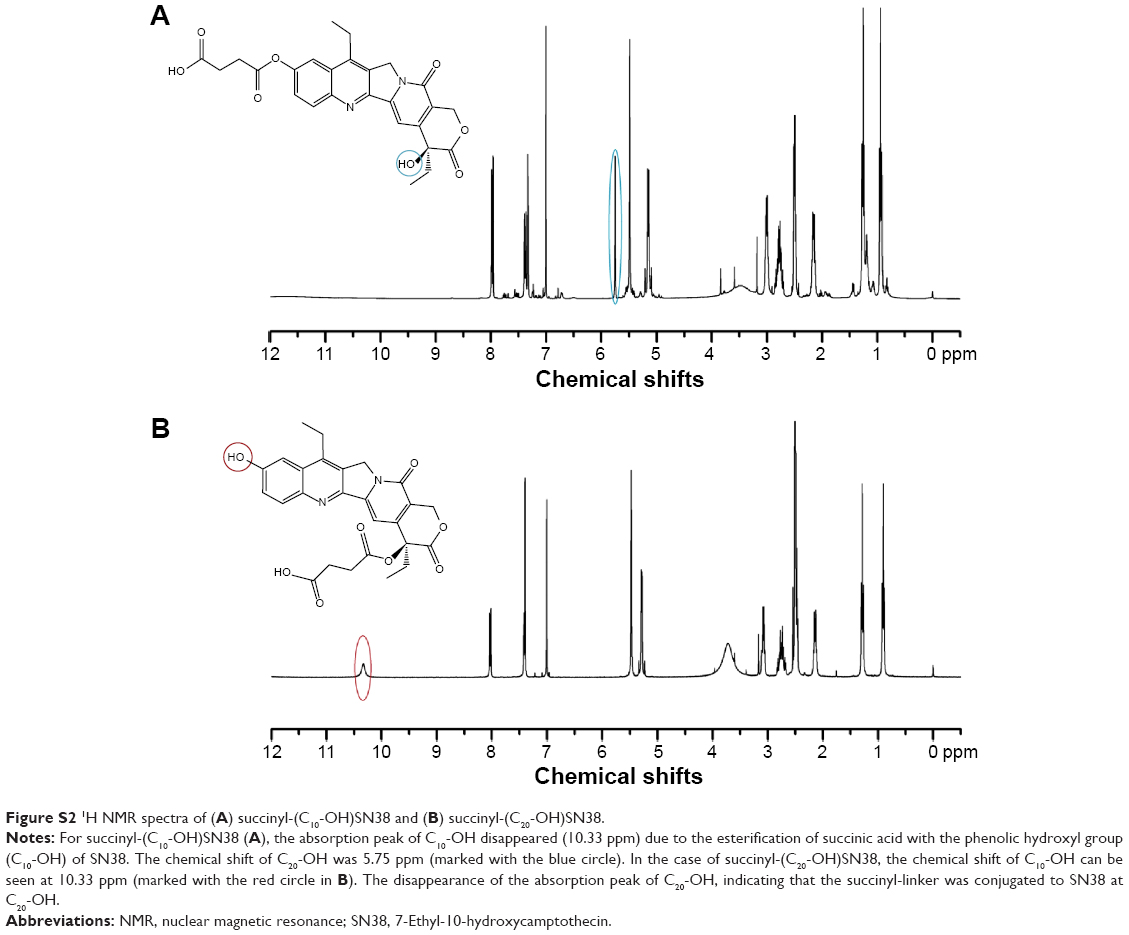 | Figure S2 1H NMR spectra of (A) succinyl-(C10-OH)SN38 and (B) succinyl-(C20-OH)SN38. |
 | Figure S3 Scanned images of FT-IR spectra of chitosan: (A) old FT-IR spectrum and (B) new FT-IR spectrum. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.