Back to Journals » Drug Design, Development and Therapy » Volume 15
Comparison of Pharmacokinetic, Pharmacodynamic and Tolerability Profiles of CKD-11101, Darbepoetin Alfa (NESP®) Biosimilar, to Those of NESP® After a Single Subcutaneous or Intravenous Administration to Healthy Subjects
Authors Jeon I ![]() , Oh J
, Oh J ![]() , Kwon YK
, Kwon YK ![]() , Yoon SH, Cho JY
, Yoon SH, Cho JY ![]() , Jang IJ
, Jang IJ ![]() , Yu KS
, Yu KS ![]() , Lee S
, Lee S ![]()
Received 25 January 2021
Accepted for publication 6 April 2021
Published 28 April 2021 Volume 2021:15 Pages 1735—1747
DOI https://doi.org/10.2147/DDDT.S303772
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Manfred Ogris
Inseung Jeon,1,* Jaeseong Oh,1,* Yu-Kyung Kwon,2 Seo Hyun Yoon,1 Joo-Youn Cho,1 In-Jin Jang,1 Kyung-Sang Yu,1 SeungHwan Lee1
1Department of Clinical Pharmacology and Therapeutics, Seoul National University College of Medicine and Hospital, Seoul, Republic of Korea; 2Department of Clinical Research, Chong Kun Dang, Seoul, Republic of Korea
*These authors contributed equally to this work
Correspondence: SeungHwan Lee
Department of Clinical Pharmacology and Therapeutics, Seoul National University College of Medicine and Hospital, 103 Daehak-ro, Jongno-gu, Seoul, 03080, Republic of Korea
Tel +82 2 2072 2343
Fax +82 2 742 9252
Email [email protected]
Introduction: Darbepoetin alfa (NESP® and ARANESP®) has a sustained erythropoietic activity with a longer half-life than conventional recombinant human erythropoietin. CKD-11101 is under clinical development as a biosimilar of darbepoetin alfa. The purpose of this study was to compare the pharmacokinetic (PK), pharmacodynamic (PD), and tolerability profiles of CKD-11101 with those of reference drug in healthy subjects.
Methods: This study was performed in two parts for healthy subjects. In each period, CKD-11101 and reference, both at 60 μg, were administered via intravenous (IV) or subcutaneous (SC) route of administration.
Results: After both IV or SC dose, the geometric mean ratio (GMR) of CKD-11101 to reference drug and its 90% confidence intervals (CIs) for Cmax, AUC0–last and AUC0–∞ were all within 0.8– 1.25. No statistically significant differences were noted in the maximum baseline adjusted reticulocyte count or the area under the baseline adjusted reticulocyte count-time between the CKD-11101 and reference drug after IV or SC dose (all p-value> 0.05). Both CKD-11101 and reference drug were generally well tolerated.
Discussion: After a single IV or SC dose, the CKD-11101 was well tolerated and showed comparable PK and PD characteristics with reference drug.
Keywords: pharmacokinetics, pharmacodynamics, biosimilar, darbepoetin alfa
Introduction
Erythropoietin (EPO), a glycoprotein cytokine, is an essential factor for the erythropoiesis from erythropoietic stem cells within bone marrow.1 It is primarily produced from the kidney in response to hypoxic stimuli, and EPO deficiency is commonly observed in patients with chronic kidney disease (CKD).1 Recombinant human EPO (rhEPO) therapy has been widely used for the treatment of anemia due to CKD or due to cytotoxic chemotherapy.2–4 However, due to the short serum half-life (4 to 12 hours) of rhEPO, it has to be administered 3 times weekly in CKD patients.5
Darbepoetin alfa is a second-generation of rhEPO, co-developed by Amgen Inc. (ARANESP®, Amgen Inc. CA, USA) and Kyowa Hakko Kirin Co., Ltd. (NESP®, Kyowa Hakko Kirin Co., Ltd, Tokyo, Japan). Owing to the 5 N-linked oligosaccharide chains and to the larger molecular weight than rhEPO darbepoetin alfa has an enhanced serum half-life (2 to 3 times longer than rhEPO), which enables once weekly administration in CKD patients.6,7 It has same mechanism of action with rhEPO and was effective for the treatment of anemia in the CKD patients and patients receiving cytotoxic chemotherapy.8,9
A biosimilar is a biological product that has a highly similar quality, safety and efficacy profile to the original biological product which is approved in regulatory agencies.10,11 According to European Medicines Agency (EMEA) and US Food and Drug Administration guidelines, various evidence from analytical (structural and physicochemical characteristics), nonclinical and clinical studies should be considered collectively to demonstrate the biosimilarity between the two biological products.10,11
CKD-11101 is a darbepoetin alfa developed by Chong Kun Dang Pharmaceutical Corp. (Seoul, Republic of Korea) is developed as a biosimilar of darbepoetin alfa which used as a test drug in this study. CKD-11101 is produced by recombinant DNA technology in modified Chinese hamster ovary cells and it showed high structural similarity with reference drug in terms of amino acid sequence characteristics, peptide mapping characteristics, disulfide bond characteristics. The CKD-11101 also showed highly similar physicochemical and immunological characteristics with reference drug in various in-vitro tests. Although the CKD-11101 showed some minor difference with reference drug in the relative contents of neutral sugar, in-vitro and in-vivo biological activity of CKD-11101 was similar to reference drug. Therefore, CKD-11101 (Nesbell, Chong Kun Dang Pharmaceutical Corp., Seoul, Republic of Korea and Mylan EPD G.K., Tokyo, Japan) is approved by Korea’s Ministry of Food and Drug Safety (MFDS) in 2018 and by Japan’s Pharmaceuticals and Medical Devices Agency (PMDA) in 2019.
Based on these quality and nonclinical studies results, these clinical studies aimed to compare the pharmacokinetic (PK), pharmacodynamic (PD) and tolerability profiles of CKD-11101 with that of reference drug, after a single intravenous (IV) or after a subcutaneous (SC) administration in healthy subjects.
Materials and Methods
Study Design and Subjects
This study (ClinicalTrials.gov identifier: NCT01684605 and NCT01685671) was approved by Ministry of Food and Drug Safety of Republic of Korea and the Institutional Review Board of Seoul National University Hospital (Seoul, Republic of Korea). The study was conducted in accordance with the International Conference on Harmonization Good Clinical Practice and the ethical principles of the Declaration of Helsinki. All of the subjects provided written informed consent prior to the participation of this study.
The study was performed using randomized, double-blind, single-dose, two-way, two-period, two-sequence, crossover design which was referenced from various comparative PK studies.12–14 The study consisted of two parts, one with IV dose (Part 1, Figure 1) and the other with SC dose (Part 2, Figure 1). Male subjects aged 20 and 55 years weighing at least 55 kg and below 90 kg, and with a body mass index (BMI) between 18 and 27 kg/m2 were eligible for these studies if they were healthy, assessed by medical histories, physical examinations, vital sign measurements, 12-lead electrocardiograms (ECGs) and clinical laboratory tests within 4 weeks prior to administration of the study drug. Subjects with anemia or with a history of drug abuse or with a positive urine drug screening test result were excluded.
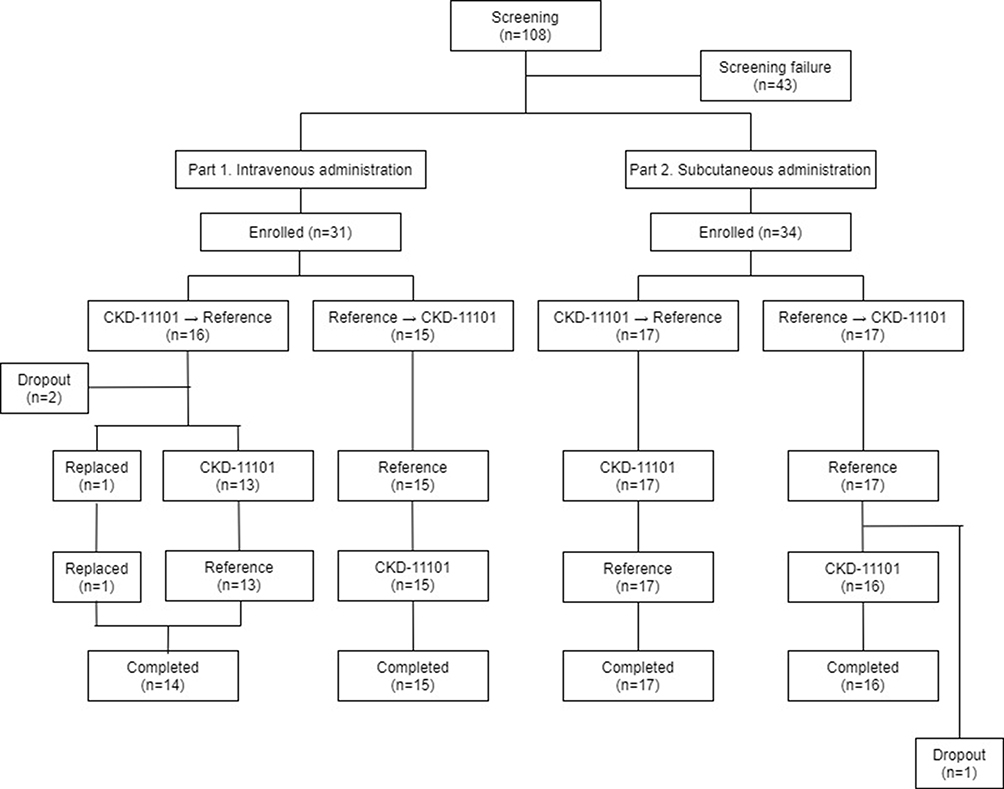 |
Figure 1 Subject disposition. |
Eligible subjects were randomly allocated to one of the treatment sequences and received a test drug (CKD-11101 60 μg prefilled syringe, Chong Kun Dang Pharmaceutical Corp., Seoul, Republic of Korea) in one period and a reference drug (60 μg prefilled syringe, Kyowa Hakko Kirin Co., Ltd, Tokyo, Japan) in the other period. To maintain double-blind, unblinded staffs were designated for drug administration and those staffs did not participate in any other study-related procedures. Three weeks of washout were set between each dose considering the half-life of darbepoetin alfa and the time for recovery of reticulocyte (PD endpoint) to the baseline.15–18
Bioanalytical Methods
The serum concentrations of darbepoetin alfa were determined using a validated enzyme-linked immunosorbent assay (ELISA) method. Human erythropoietin ELISA Kit (STEMCELL Technologies, Vancouver, Canada) was used to capture darbepoetin alfa. Serum PK samples were diluted and then added to the microplate with quality control and standards samples. Biotinylated anti-EPO Ab was added to the plate and incubated for one hour at 25°C. After washing the microplate, a streptavidin conjugated to horseradish peroxidase (HRP) was added to the plate and incubated for one hour at 25°C. For the color development, tetramethylbenzidine (TMB) substrate solution was added to the plate. The reaction was stopped by adding stop solution and a microplate reader was used to detect the absorbance at 450 nm with a reference wavelength at 650 nm. The lower limit of quantification was 0.18 μg/L. The precision and accuracy of the standard curve was <8.3% and 97%–108%, respectively. The precision and accuracy of intra- and inter-assay were all within the acceptance criteria.
Immunogenicity (anti-drug antibodies) of study drugs was assayed using a validated enzyme-linked immunosorbent assay (ELISA) with a sensitivity of 230.726 μg/L for CKD-11101 and 309.560 μg/L for reference drug. Serum samples and quality control samples were incubated for two hours in the CKD-11101 or reference drug coated microplate. After washing the microplate, an anti-human IgG conjugated to HRP was added to the microplate as a detection antibody and incubated for one hour at 25°C. For the color development, TMB substrate solution was added to the plate and the reaction was stopped by adding 2N sulfuric acid. The microplate reader was used to detect absorbance at 450 nm.
PK Samples and Analysis
Serum samples for PK analysis of darbepoetin alfa were collected at 0 (Pre-dose), 0.25, 0.5, 0.75, 1, 2, 3, 4, 6, 8, 12, 24, 36, 48, 72, 96, 120, 168, and 264-hour after IV dose at each period (Part 1) and at 0 (Pre-dose), 1, 2, 4, 6, 8, 12, 24, 36, 48, 60, 72, 96, 120, 168, 216, 264, and 360-hour after SC dose at each period (Part 2). The serum sampling time points were different in part 1 and part 2. It is because that the half-life of darbepoetin alfa was 25 hours when administered intravenously, while the half-life was 48 hours when administered subcutaneously. The PK parameters were calculated by a non-compartmental method using WinNonlin 8.0 software (Pharsight, CA, USA). The maximum serum concentration (Cmax) and the time to reach peak concentration (Tmax) were directly obtained from the serum concentration-time profiles. The area under the concentration–ime curve from zero time to the last observed concentration (AUC0–last) was determined by the trapezoidal method. The area under the curve from time zero to infinity (AUC0–∞) was estimated by the equation AUClast + Clast/λz, where λz was the terminal elimination constant. The terminal half-life (t1/2) was calculated by the equation In(2) divided λz. The clearance and apparent clearance (CL and CL/F) were calculated by Dose/AUClast, and the volume of distribution and apparent volume of distribution (Vz and Vz/F) were calculated as the CL and CL/F divided by λz.
PD Samples and Analysis
Whole blood samples for PD analysis were collected at 0 (Pre-dose), 8, 24, 48, 96, 168 and 264-hour after IV dose at each period (Part 1) and at 0 (Pre-dose), 8, 24, 48, 72, 120, 216 and 360-hour after SC dose at each period (Part 2). The time course of baseline-adjusted reticulocyte count (%) after administration of the test and reference drug was used for primary PD marker. The maximal baseline-adjusted reticulocyte count (ΔEmax) was directly obtained from the observed data. The area under the baseline-adjusted reticulocyte count–time curve from time 0 to the last detectable time point (ΔAUEC0–last) was determined by the trapezoidal method. Other exploratory PD markers including hemoglobin (g/L), hematocrit (%) and red blood cell (RBC) count (106/mm3) were baseline-adjusted and graphically compared between the treatments.
Immunogenicity Evaluation
Anti-drug antibodies to CKD-11101 or darbepoetin alfa were determined at pre-dose, 12 days (Part 1) or 16 days (Part 2) post-dose at each study period.
Tolerability Evaluation
The tolerability was assessed for the subjects who received at least one of the study drugs. Safety profiles were evaluated based on adverse events (AEs), physical examinations, assessment on injection site reactions, vital signs, 12-lead electrocardiograms (ECGs), clinical laboratory evaluations.
Statistical Analysis
All statistical analyses were performed using the SAS® Version 9.4 (SAS Institute Inc., Cary, NC, USA). The baseline characteristics of demographic parameters and anemia parameters such as reticulocyte count, hemoglobin, ferritin and transferrin were analyzed using either t-test or Mann–Whitney U-test depending on whether normality is satisfied. Descriptive statistics were used to summarize study data and a general linear mixed effect model was developed to compare the primary PK parameters (Cmax, AUC0-last and AUC0–∞) between test and reference drugs. Using the model, the geometric mean ratio (GMR) and its 90% CI of the test drug to reference drug were estimated for the primary PK parameters. If 90% confidence intervals (CIs) of the GMR for these parameters were within the range of 0.8-1.25, the PK characteristics of test and reference drugs were considered as equivalent. For the comparison of primary PD parameters (ΔEmax and ΔAUEC0-last of reticulocyte count), the mean difference and its p-values of the test drug and reference drug were estimated for the primary PD parameters using a general linear mixed effect model. If the p-values of the mean difference for these parameters were above 0.05 the PD characteristics of test and reference drugs were considered as equivalent.19 A P-value of less than 0.05 was considered as statistically significant.
Results
Subjects
In part 1, a total of 31 subjects were enrolled and 29 of whom completed the study as planned (Figure 1). In part 2, a total of 34 subjects were enrolled and 33 of whom completed the study as planned (Figure 1). The PK and PD analyses were done for 29 and 33 subjects who completed the study in part 1 and part 2, respectively. In part 1, one subject’s baseline of reticulocyte result was missed during the CKD-11101 (test drug) period and that data was excluded from PD analysis. Tolerability analysis was performed for 29 and 34 subjects who administered the study drug more than once in part 1 and part 2, respectively.
The baseline characteristics of subjects were comparable between part 1 and part 2 (Table 1). The mean ± standard deviation (SD) values for baseline characteristics of subjects who participated in part 1 were 27.6 ± 4.9 years for age, 22.3 ± 2.0 kg/m2 for BMI, 1.09 ± 0.29% for reticulocyte count, 15.9 ± 0.8 g/dL for hemoglobin, 84.9 ± 59.9 ng/mL for ferritin and 275.65 ± 36.65 mg/dL for transferrin. Those values of subjects who participated in part 2 were 27.1 ± 7.3 years for age, 22.5 ± 1.7 kg/m2 for BMI, 1.00 ± 0.30% for reticulocyte count, 15.7 ± 0.8 g/dL for hemoglobin, 88.9 ± 60.3 ng/mL for ferritin and 266.62 ± 38.58 mg/dL for transferrin. All the subjects were male in both part 1 and part 2 and both the baseline characteristics of subjects were not significantly different between the sequence group both in part 1 and part 2 (Table 1).
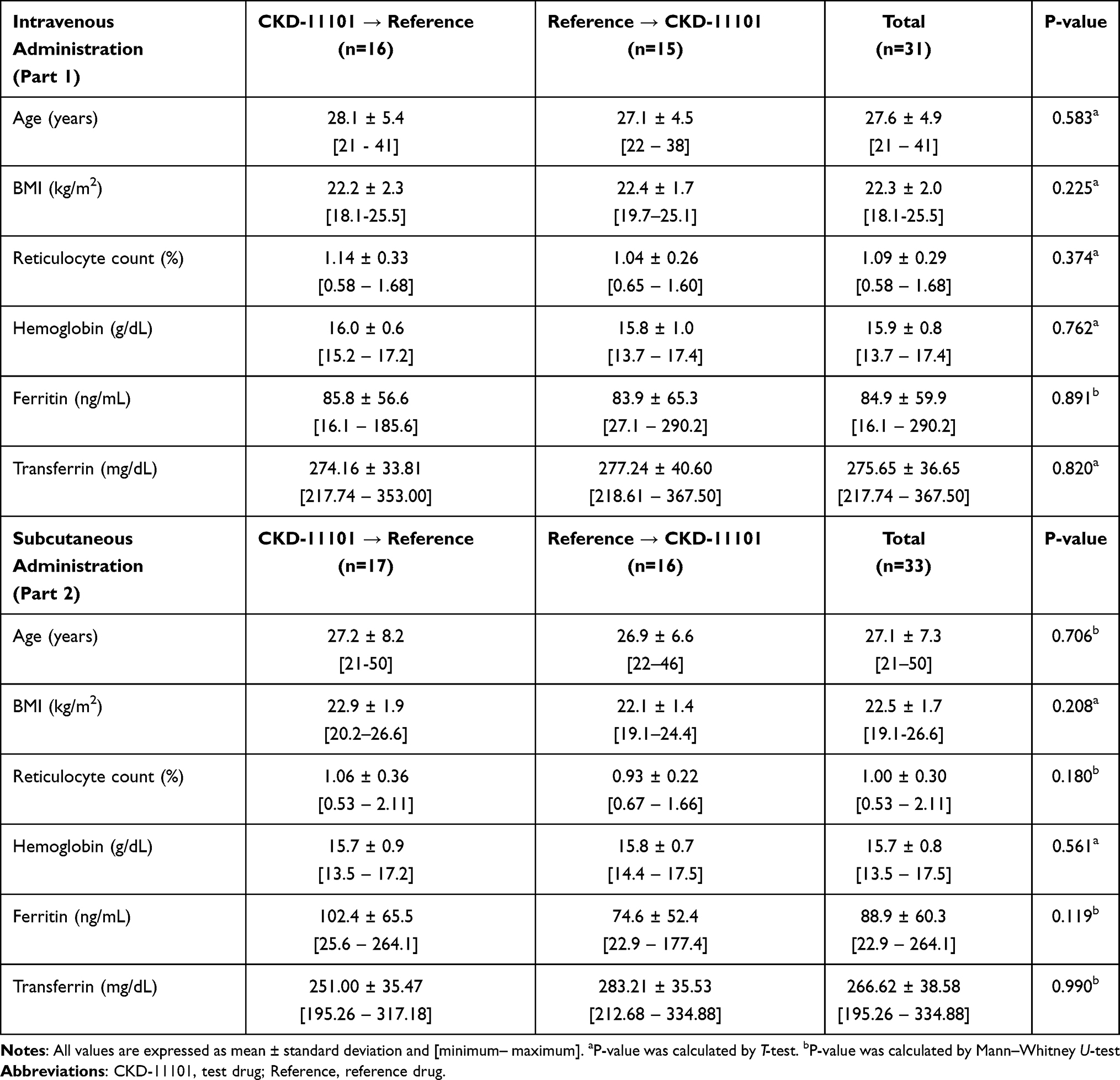 |
Table 1 Demographic Characteristics |
PK Analysis
The systemic exposure to darbepoetin alfa was similar after single IV or SC administration of the test (CKD-11101) or reference drug given as 60 μg prefilled syringe. The mean serum concentration–time profiles of darbepoetin alfa were superimposable both in part 1 and part 2 (Figure 2A and B, respectively).
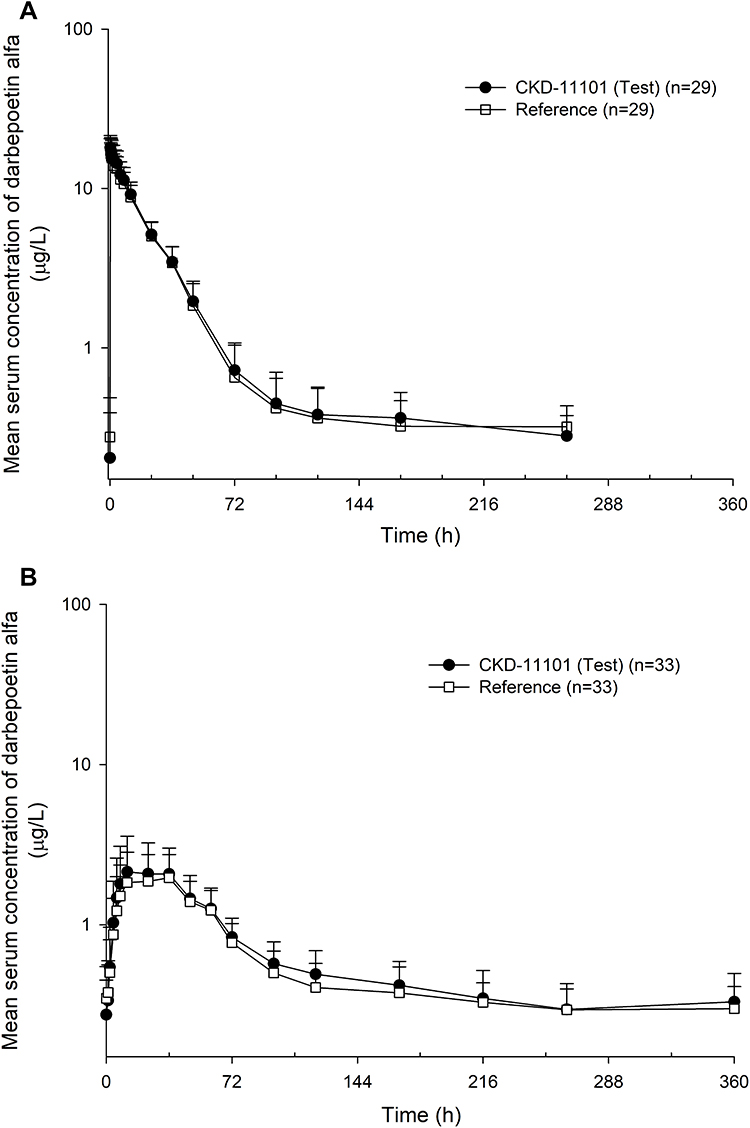 |
Figure 2 Mean serum concentration-time profiles of darbepoetin alfa after a single intravenous (A) or subcutaneous (B) administration of CKD-11101 60 μg (test drug) or reference drug 60 μg. |
In part 1, the mean serum concentration-time profiles were similar after a single IV administration of test or reference drug (Figure 2A). The serum concentration of darbepoetin alfa reached a maximal level at a median of 0.25-hours post-dose and eliminated following a multi-exponential decrease pattern in both test and reference drug (Figure 2A). The primary PK parameters were comparable between the test and reference drug (Figure 3A–C and Table 2). The GMRs of test drug to reference drug and its 90% CI for Cmax, AUC0-last and AUC0–∞ were 1.05 (1.03–1.07), 1.07 (1.04–1.11) and 1.03 (0.96–1.10), respectively; and all of the values were fell within the conventional bioequivalence range of 0.80 - 1.25 (Table 2).
 |
Figure 3 Comparison of primary pharmacokinetic parameters between CKD-11101 (test drug) or reference drug after a single intravenous (A–C) or subcutaneous (D–F) administration. The values are expressed as mean (SD). |
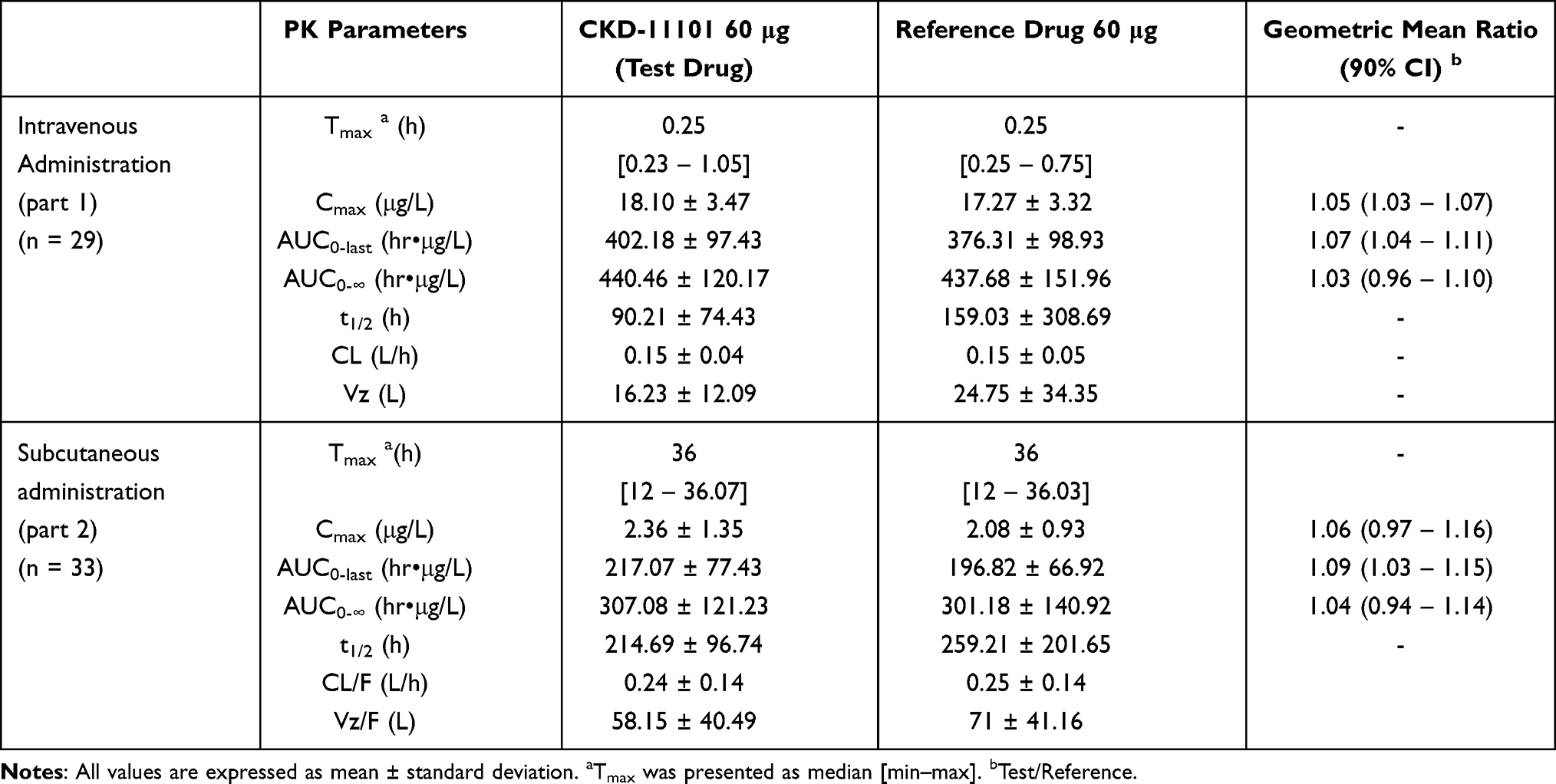 |
Table 2 Pharmacokinetic Parameters of Darbepoetin Alfa After a Single Intravenous or Subcutaneous Administration of CKD-11101 60 μg (Test Drug) or Reference Drug 60 μg |
In part 2, the mean serum concentration-time profiles were similar after a single SC administration of test or reference drug (Figure 2B). The serum concentration of darbepoetin alfa reached a maximal level at a median of 36-hour post-dose and eliminated following a multi-exponential decrease pattern in both test and reference drug (Figure 2B). The primary PK parameters were comparable between the test and reference drug (Figure 3D–F and Table 2). The GMRs of test drug to reference drug and its 90% CI for Cmax, AUC0-last and AUC0–∞ were 1.06 (0.97–1.16), 1.09 (1.03–1.15) and 1.03 (0.94–1.14), respectively; and all of the values were fell within the conventional bioequivalence range of 0.80 - 1.25 (Table 2).
PD Analysis
In both study part 1 and 2, the time trend of PD markers (change of reticulocyte, hemoglobin, hematocrit, and red blood cell count from baseline) was similar after a single administration of test or reference drug (Figure 4, Supplemental Figure 1). The primary PD parameters (Emax and AUEC0-last) were not significantly different between the test and reference drug (all p-value >0.05, Table 3). The exploratory PD markers such as hemoglobin (g/L), hematocrit (%) and red blood cell (RBC) count (106/mm3) parameters PD characteristics of darbepoetin alfa were similar to the known PK and PD characteristics of darbepoetin alfa.
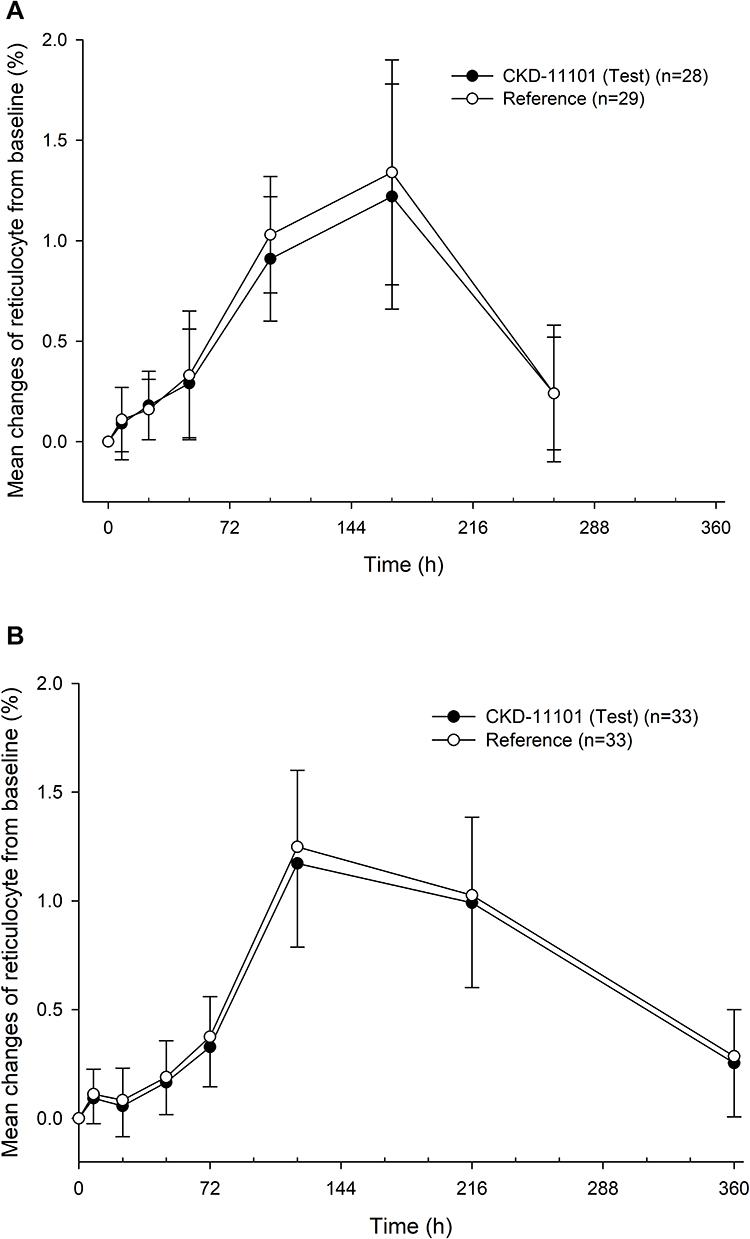 |
Figure 4 Mean changes of reticulocyte from baseline after a single intravenous (A) or subcutaneous (B) administration of CKD-11101 60 μg (test drug) or reference drug 60 μg. Bars represent standard deviations. |
 |
Table 3 Pharmacodynamic Parameters of Darbepoetin Alfa After a Single Intravenous or Subcutaneous Administration of CKD-11101 60 μg (Test Drug) or Reference Drug 60 μg |
The exposure-response relationship was also similar between the test and reference drug (Figure 5). The relationship between the mean serum concentration of darbepoetin alfa and the mean changes of reticulocyte from baseline showed a counterclockwise hysteresis pattern both in the test and the reference drug (Figure 5).
 |
Figure 5 Relationship between mean serum concentration of darbepoetin alfa and mean changes of reticulocyte from baseline after a single intravenous (A) or subcutaneous (B) administration of CKD-11101 60 μg (test drug) or reference drug 60 μg. |
Immunogenicity Evaluation
No subject was positive on the anti-CKD-11101 and anti-reference drug antibody assay before or after administration of the study drugs in part 1 and part 2.
Tolerability Evaluation
Both test (CKD-11101) and reference drug were well tolerated in healthy subjects after a single IV or SC administration. In part 1, a total of 26 treatment emergent AEs (TEAEs) were reported and 15 (51.7%) subjects experienced ≥1 TEAE. The most commonly reported AEs were itching and headache after test drug administration, while throat irritation, rhinorrhea and bruise were reported after reference drug administration (Supplemental Table 1). Among these people, 15 and 8 TEAEs were considered to be related to test and reference drug, respectively (Table 4). Itching, headache, and abdominal pain were observed after test drug administration and throat irritation and rhinorrhea were observed after reference drug administration of the drug-related AEs in the present study.
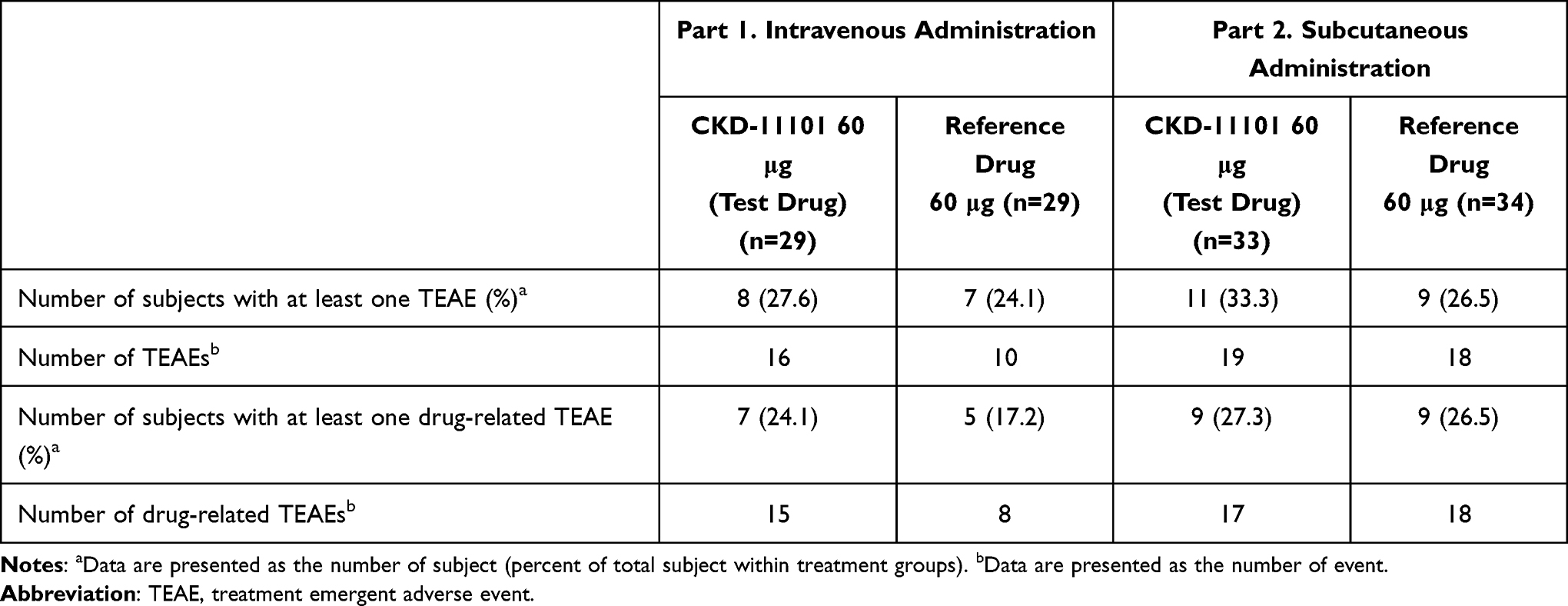 |
Table 4 Summary of All Treatment Emergent Adverse Events |
In part 2, a total of 37 TEAEs were reported and 15 (44.1%) subjects experienced ≥1 TEAE. Most common AEs were abdominal pain, diarrhea, headache, nasal congestion and upper respiratory tract infection for test drug treatment, while dyspepsia, dizziness, headache, skin nodule were commonly reported after reference drug administration (Supplemental Table 1). Among these people, 17 and 18 TEAEs were considered to be related to test and reference drug, respectively (Table 4). Headache was reported as a drug-related AE in both test and reference drug, and also abdominal pain and dizziness were reported. Most of the adverse events were mild in severity and there were no serious adverse events or unexpected adverse drug reaction. No clinically significant abnormalities were found in physical examinations, assessment on injection site reactions, vital signs, ECGs, or clinical laboratory evaluations.
Discussion
This clinical study indicates that CKD-11101, a proposed biosimilar of reference drug, has comparable PK, PD and tolerability profile after a single IV or SC administration in healthy subjects. The similarity between CKD-11101 and reference drug was demonstrated by following evidence. First, the mean serum concentration-time profiles were superimposable after a single IV or SC administration of CKD-11101 or reference drug (Figure 2). Also, the GMRs of CKD-11101 to reference drug and their 90% CIs for the Cmax, AUC0-last and AUC0–∞ were contained entirely within the conventional bioequivalence range of 0.80 - 1.25 (Table 2). Second, the time trend of PD markers was also similar between the CKD-11101 and reference drug treatments (Figure 4, Supplemental Figure 1), and no statistical difference was observed for primary PD parameters (Emax and AUEC0-last) between the CKD-11101 and reference drug treatments (all p-value >0.05, Table 3). Lastly, both of CKD-11101 and reference drug were well tolerated in these healthy subjects and there was no subject with the anti-drug antibody regardless of administration routes.
Although patients with CKD or patients receiving cytotoxic chemotherapy differ from the healthy subject with respect to their baseline hematologic status, this study was performed in healthy subjects to minimize possible confounding factors. However, if the PK and PD response in healthy subjects of two darbepoetin alfa are similar, efficacy in the CKD patients with anemia is expected similar. It is because the primary site of action for rhEPO is same regardless of disease status.20,21 As expected, based on the results of this study, CKD-11101 showed equivalent efficacy and safety profile compared with reference drug for the treatment of renal anemia in the two randomized double-blind confirmatory clinical trials for CKD patients who are on or not on hemodialysis.22,23
In the present study, the PK and PD characteristics of darbepoetin alfa were similar to the known PK and PD characteristics of darbepoetin alfa.16,18 In our study, the mean serum concentration of darbepoetin alfa was eliminated following a multi-exponential decrease pattern (Figure 2) and similar patterns were observed the previous clinical studies.16,18 The PK parameters of darbepoetin alfa observed in our study were similar to known PK parameters of darbepoetin alfa.16,18 The bioavailability of the SC dose calculated by the ratio of the AUC0–last of SC dose to that of IV dose was 52% to 54% and these values were also similar to a published clinical study result.18 The PK and PD relationship showed a counterclockwise hysteresis pattern (Figure 5) which indicates a time delay between the serum concentration of darbepoetin alfa and the PD response related to the maturation of erythroid progenitors in bone marrow, and it was in agreement with the previous clinical study result.18,24–27
The reticulocyte count was evaluated as a primary endpoint for PD analysis in this study. Reticulocytes are immature red blood cell and develop and mature in the bone marrow, then circulate for about a day in the bloodstream before developing in the mature RBC.25,28 Since the reticulocytes are produced primarily from EPO stimulation, it is physiologically appropriate that investigating the increase of reticulocytes to observe the activity of rhEPO as a PD marker.25,28 The EMEA also recommend the reticulocyte as a recommended PD marker for assessment of the activity of EPO in a single-dose study although it is not a suitable endpoint for clinical efficacy studies.29
Both the CKD-11101 and reference drug were well tolerated in this study. There were no serious TEAEs or unexpected adverse drug reaction, and that no subject developed anti-drug antibody after treatment. Itching, headache, throat irritation, rhinorrhea and abdominal pain were the drug-related AEs observed in the present study. There were 6 and 2 cases of injection site reactions (eg, erythema, infiltration, edema and induration) after a single SC or IV dose of study drugs, respectively. All of the TEAEs and the injection site reactions were recovered spontaneously during the study. No clinically significant changes were observed in the physical examinations, vital signs, ECGs or clinical laboratory tests.
Collectively, these study results suggest that the CKD-11101 can be an alternative therapeutic option of reference drug for the treatment of anemia in the CKD patients and patients receiving cytotoxic chemotherapy.
Conclusion
In conclusion, the CKD-11101 was well tolerated and showed comparable PK and PD characteristics with reference drug. The results of this study implies to support that the CKD-11101 and reference drug can be interchangeably used.
Data Sharing Statements
The individual de-identified participant data supporting published results are available with approval from the corresponding author on reasonable request, at any time after publication.
Acknowledgments
This study was sponsored by a research grant from Chong Kun Dang Pharmaceutical Corp., Seoul, Republic of Korea. The funders had no role in data collection and analysis or preparation of the manuscript. The authors thank the staff of the Seoul National University Hospital Clinical Trials Center for their generous cooperation. Inseung Jeon and Jaeseong Oh and are co-first authors.
Disclosure
Yu-Kyung Kwon is a full-time employee of Chong Kun Dang Pharmaceutical Corp., Seoul, Republic of Korea. The other authors report no conflicts of interest associated with this work.
References
1. Jelkmann W. Physiology and pharmacology of erythropoietin. Transfusion Med Hemother. 2013;40(5):302–309. doi:10.1159/000356193
2. Jones M, Ibels L, Schenkel B, Zagari M. Impact of epoetin alfa on clinical end points in patients with chronic renal failure: a meta–analysis. Kidney Int. 2004;65(3):757–767.
3. Leitgeb C, Pecherstorfer M, Fritz E, Ludwig H. Quality of life in chronic anemia of cancer during treatment with recombinant human erythropoietin. Cancer. 1994;73(10):2535–2542.
4. Wilson J, Yao GL, Raftery J, et al. A systematic review and economic evaluation of epoetin alpha, epoetin beta and darbepoetin alpha in anaemia associated with cancer, especially that attributable to cancer treatment. Health Technol Assess (Rockv). 2007;11(13):
5. Macdougall IC, Roberts DE, Coles GA, Williams JD. Clinical pharmacokinetics of epoetin (recombinant human erythropoietin). Clin Pharmacokinet. 1991;20(2):99–113.
6. Allon M, Kleinman K, Walczyk M, et al. Pharmacokinetics and pharmacodynamics of darbepoetin alfa and epoetin in patients undergoing dialysis. Clin Pharmacol Ther. 2002;72(5):546–555.
7. Powell J, Gurk-Turner C. Darbepoetin alfa (Aranesp). Proceedings. 2002;15(3):332–335.
8. Overbay DK, Manley HJ. Darbepoetin-alpha: a review of the literature. Pharmacotherapy. 2002;22(7):889–897.
9. Siddiqui MA, Keating GM. Darbepoetin alfa: a review of its use in the treatment of anaemia in patients with cancer receiving chemotherapy. Drugs. 2006;66(7):997–1012.
10. Christl LA, Woodcock J, Kozlowski S. Biosimilars: the US regulatory framework. Annu Rev Med. 2017;68(1):243–254. doi:10.1146/annurev-med-051215-031022
11. Wolff-Holz E, Tiitso K, Vleminckx C, Weise M. Evolution of the EU biosimilar framework: past and future. BioDrugs. 2019;33(6):621–634. doi:10.1007/s40259-019-00377-y
12. Hwang I, Park S-I, Lee S, et al. Pharmacokinetics of fixed-dose combination of rosuvastatin 20 mg and ezetimibe 10 mg compared to concurrent administration of individual tablets in healthy Korean subjects. Transl Clin Pharmacol. 2017;68(1):16–24. doi:10.12793/tcp.2018.26.1.16
13. Hwang JG, Yoo H, Lee JW, Song GS, Lee S, Kim M-G. Comparison of pharmacokinetic characteristics of two Tegoprazan (CJ-12420) formulations in healthy male subjects. Transl Clin Pharmacol. 2019;27(2):80–85.
14. Kim, Y-M, Jeon J-Y, Moon SJ, Jung J, Son H, Kim M-G. Pharmacokinetics comparison of solifenacin tartrate and solifenacin succinate: a randomized, open-label, single-dose, 2-way crossover study in healthy male volunteers. Transl Clin Pharmacol. 2018;26(2):73–78.
15. Cheung WK, Goon BL, Guilfoyle MC, Wacholtz MC. Pharmacokinetics and pharmacodynamics of recombinant human erythropoietin after single and multiple subcutaneous doses to healthy subjects. Clin Pharmacol Ther. 1998;64(4):412–423.
16. Agoram B, Sutjandra L, Sullivan JT. Population pharmacokinetics of darbepoetin alfa in healthy subjects. Br J Clin Pharmacol. 2007;63(1):41–52.
17. Yoon S, Rhee SJ, Heo SJ, et al. Comparable pharmacokinetics and pharmacodynamics of two epoetin alfa formulations Eporon((R)) and Eprex((R)) following a single subcutaneous administration in healthy male volunteers. Drug Des Devel Ther. 2017;11:3127–3135.
18. Kim S, Hong T, Ko JW, Huh W, Kim JR. Comparison of the pharmacokinetic-pharmacodynamic relationships of two darbepoetin alfa formulations in healthy male volunteers. BioDrugs. 2019;33(1):101–112.
19. Whelton PK, Carey RM, Aronow WS, et al. 2017 ACC/AHA/AAPA/ABC/ACPM/AGS/APhA/ASH/ASPC/NMA/PCNA guideline for the prevention, detection, evaluation, and management of high blood pressure in adults: a report of the American College of Cardiology/American Heart Association Task force on clinical practice guidelines. J Am Coll Cardiol. 2018;71(19):e127–e248.
20. Flaharty KK, Caro J, Erslev A, et al. Pharmacokinetics and erythropoietic response to human recombinant erythropoietin in healthy men. Clin Pharmacol Ther. 1990;47(5):557–564.
21. McGowan T, Vaccaro NM, Beaver JS, Massarella J, Wolfson M. Pharmacokinetic and pharmacodynamic profiles of extended dosing of epoetin alfa in anemic patients who have chronic kidney disease and are not on dialysis. Clin j Am Soc Nephrol. 2008;3(4):1006–1014.
22. Kim Y, Park SK, Cho WY, et al. Efficacy and safety of CKD-11101 (Proposed Biosimilar of Darbepoetin-Alfa) compared with darbepoetin-alfa in patients on hemodialysis: a randomized, double-blinded, Parallel-Group Phase III study. BioDrugs. 2019;34(1):99–110.
23. Lee JH, Ha Chung B, Joo KW, et al. Efficacy and safety of CKD-11101 (darbepoetin-alfa proposed biosimilar) compared with NESP in anaemic chronic kidney disease patients not on dialysis. Curr Med Res Opin. 2019;35(6):1111–1118.
24. Louizos C, Yanez JA, Forrest ML, Davies NM. Understanding the hysteresis loop conundrum in pharmacokinetic/pharmacodynamic relationships. J Pharm Pharmaceutical Sci. 2014;17(1):34–91.
25. Koepke JF, Koepke JA. Reticulocytes. Clin Lab Haematol. 1986;8(3):169–179.
26. Ramakrishnan R, Cheung WK, Wacholtz MC, Minton N, Jusko WJ. Pharmacokinetic and pharmacodynamic modeling of recombinant human erythropoietin after single and multiple doses in healthy volunteers. J Clin Pharmacol. 2004;44(9):991–1002.
27. Krzyzanski W, Jusko WJ, Wacholtz MC, Minton N, Cheung WK. Pharmacokinetic and pharmacodynamic modeling of recombinant human erythropoietin after multiple subcutaneous doses in healthy subjects. Eur J Pharm Sci. 2005;26(3–4):295–306.
28. Ovchynnikova E, Aglialoro F, von Lindern M, van den Akker E. The shape shifting story of reticulocyte maturation. Front Physiol. 2018;9:829.
29. Committee for Medicinal Products for Human Use (CHMP). Guideline on non-clinical and clinical development of similar biological medicinal products containing recombinant erythropoietins (Revision). Available from: http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2010/04/WC500089474.pdf. 2010.
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.