")
Back to Journals » OncoTargets and Therapy » Volume 10
Comparison of a mouse and a novel human scFv-SNAP-auristatin F drug conjugate with potent activity against EGFR-overexpressing human solid tumor cells
Authors Woitok M, Klose D, Di Fiore S, Richter W, Stein C, Gresch G, Grieger E, Barth S , Fischer R, Kolberg K, Niesen J
Received 26 April 2017
Accepted for publication 23 May 2017
Published 6 July 2017 Volume 2017:10 Pages 3313—3327
DOI https://doi.org/10.2147/OTT.S140492
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Carlos E Vigil
Mira Woitok,1,2 Diana Klose,1 Stefano Di Fiore,1 Wolfgang Richter,3 Christoph Stein,1 Gerrit Gresch,1 Elena Grieger,1 Stefan Barth,1 Rainer Fischer,1,2 Katharina Kolberg,1,* Judith Niesen1,*
1Fraunhofer Institute for Molecular Biology and Applied Ecology (IME), Aachen, Germany; 2Institute of Molecular Biotechnology (Biology VII), RWTH Aachen University, Aachen, Germany; 3Tube Pharmaceuticals GmbH, Vienna, Austria
*These authors contributed equally to this work
Abstract: Antibody–drug conjugates (ADCs) can deliver toxins to specific targets such as tumor cells. They have shown promise in preclinical/clinical development but feature stoichiometrically undefined chemical linkages, and those based on full-size antibodies achieve only limited tumor penetration. SNAP-tag technology can overcome these challenges by conjugating benzylguanine-modified toxins to single-chain fragment variables (scFvs) with 1:1 stoichiometry while preserving antigen binding. Two (human and mouse) scFv-SNAP fusion proteins recognizing the epidermal growth factor receptor (EGFR) were expressed in HEK 293T cells. The purified fusion proteins were conjugated to auristatin F (AURIF). Binding activity was confirmed by flow cytometry/immunohistochemistry, and cytotoxic activity was confirmed by cell viability/apoptosis and cell cycle arrest assays, and a novel microtubule dynamics disassembly assay was performed. Both ADCs bound specifically to their target cells in vitro and ex vivo, indicating that the binding activity of the scFv-SNAP fusions was unaffected by conjugation to AURIF. Cytotoxic assays confirmed that the ADCs induced apoptosis and cell cycle arrest at nanomolar concentrations and microtubule disassembly. The SNAP-tag technology provides a platform for the development of novel ADCs with defined conjugation sites and stoichiometry. We achieved the stable and efficient linkage of AURIF to human or murine scFvs using the SNAP-tag technology, offering a strategy to improve the development of personalized medicines.
Keywords: epidermal growth factor receptor, EGFR, antibody–drug conjugate, ADC, SNAP-tag technology, single-chain fragment variable, scFv, BG-modified auristatin F, AURIF
Introduction
Antibody–drug conjugates (ADCs) provide an effective treatment strategy for both hematological malignancies and, increasingly, solid tumors. An ideal ADC combines the unique ability of an antibody to specifically bind a tumor-associated antigen that is minimally or in best case not expressed on healthy cells, a biodegradable linker that is stable in circulation, and the potent activity of a cytotoxic reagent such as a small-molecule drug.1,2 The first cytotoxic reagents in ADCs included the drugs doxorubicin and 5-fluorouracil, but these have been replaced more recently with two major classes of reagents with subnanomolar intracellular cytotoxicity, namely DNA-damaging drugs and microtubule inhibitors.1,3,4 Most of the cytotoxic ADC components in preclinical/clinical development are inhibitors of tubulin polymerization, including the maytansinoids (DM1/DM4) and monomethyl auristatin analogs E and F (MMAE/MMAF). They inhibit cell division by binding to tubulin, which blocks tubulin assembly, arrests the target cells in the G2/M phase of the cell cycle, and ultimately induces apoptosis.3,5 MMAE and MMAF are synthetic analogs of the antimitotic natural product dolastatin 10 from the sea hare Dolabella auricularia.4 In contrast to MMAE, conjugation with MMAF is possible with noncleavable linkers, and MMAF is also more hydrophobic and thus less membrane permeable than MMAE.3,4
The first ADC approved by the US Food and Drug Administration (FDA) (in 2000) was gemtuzumab ozogamicin (Mylotarg®). It binds to CD33 and was approved as a monotherapy for acute myeloid leukemia (AML) patients unsuitable for chemotherapy. However, 10 years later, it was voluntarily withdrawn from the market due to its low efficacy and an unsuccessful second Phase III study with observed toxicity.2,6,7 Two ADCs are currently approved by the FDA: brentuximab vedotin (Adcetris®), a CD30-specific MMAE conjugate approved in 2011, and ado-trastuzumab emtansine T-DM1 (Kadcyla®), an HER2/neu-specific DM1 conjugate approved in 2013.1,6,8 Kadcyla® is currently the only ADC approved for nonhematological malignancies, including metastatic breast cancer. About 120 ADCs are currently undergoing clinical trials, most of them targeting hematological malignancies and a few targeting solid tumors. The relative scarcity of solid tumor ADCs reflects the limited number of receptors on the tumor cell surface, but ADCs with a variety of mechanisms of action are changing this assumption,9 and ~50 open Phase I/II studies are currently addressing solid tumors.1,2,4 For example, glembatumomab vedotin (CDX-011) targets nonmetastatic melanoma protein B (NMB), a glycoprotein overexpressed in many breast tumors. This ADC comprises a human immunoglobulin class G (IgG)2 antibody and MMAE as the cytotoxic payload. CDX-011 is currently undergoing Phase II clinical trials in patients with advanced triple-negative breast cancer (TNBC), resulting thus far in 33% progression-free survival.4,10 CDX-011 has also been tested in Phase I/II trials for advanced melanoma, with a promising objective response rate and a maximum tolerated dose of 1.88 mg/kg once every 3 weeks.4,11 Other ADCs target the epidermal growth factor receptor (EGFR), a well-known tumor antigen that is overexpressed in a variety of solid tumors (including ~50% of TNBC patients) but minimally expressed in healthy tissues.12–14 TNBC is difficult to treat due to the absence of the three typical therapeutic targets (HER2/neu, progesterone receptor, and estrogen receptor), and better treatment options are therefore required.15–17 EGFR is also overexpressed in ~70% of tumors in the rare childhood muscle cancer embryonal rhabdomyosarcoma (ERMS).18,19 Recently, EGFR-specific immunotoxins and a human cytolytic fusion protein have shown promising results in vitro against ERMS cell lines among others.20–22
Here, we describe the development of next-generation ADCs in which a single-chain fragment variable (scFv) instead of a full-length monoclonal antibody (mAb) is used, and the scFv is combined with a SNAP-tag to facilitate covalent coupling to auristatin F (AURIF) via benzylguanine (BG). AURIF is a BG-modified version of MMAF suitable for SNAP-tag coupling. The SNAP-tag is an engineered version of the human DNA repair enzyme O6-alkylguanine-DNA-alkyltransferase, which allows the covalent coupling of BG-modified components with a defined 1:1 stoichiometry.23,24 The toxic payload of conventional ADCs is often chemically linked to cysteine or lysine residues in mAbs, resulting in a heterogeneous mixture of products with a undefined drug-to-antibody ratio (DAR) and varying conjugation sites.25 We used the SNAP-tag to overcome these limitations, and as proof of concept for the development of scFv-SNAP ADCs targeting EGFR, we compared two different scFvs, namely 425(scFv) derived from the mAb425 as an internal standard and 1711(scFV) derived from the FDA-approved human mAb panitumumab (the latter achieving high-affinity binding compared to its parental mAb).26–28 These next-generation ADCs achieved potent cytotoxicity and proapoptotic activity against ERMS and TNBC solid tumor cells and also showed specific binding to biopsies from cancer patients, confirming their suitability for further preclinical development. We also confirmed the suitability of a newly established microtubule dynamics assay for the characterization of novel ADCs.
Materials and methods
Cell lines and cell culture conditions
The human embryonic kidney cell line HEK 293T (CRL-11268), the melanoma cell line A2058 (CCL-136), the mammary gland cell line MDA-MB-468 (HTB-132), the human ERMS cell line RD (CCL-136), and the epithelial cell lines A431 (CRL-1555) and A549 (CCL-185) were obtained from the American Type Culture Collection (ATCC, Manassas, VA, USA). All cell lines except A549 were cultivated in Roswell Park Memorial Institute 1640 medium supplemented with GlutaMAX™ (Thermo Fisher Scientific, Waltham, MA, USA), 10% (v/v) fetal calf serum, and 100 μg/mL penicillin and streptomycin. A549 cells were cultivated in Dulbecco’s Modified Eagle’s Medium (DMEM) with high glucose (Thermo Fisher Scientific), also supplemented with fetal calf serum and penicillin and streptomycin as above. A549 cells transfected with pSNAP-tubulin B3 (TubB3) were cultivated in medium supplemented with 800 μg/mL G418.29 All cell lines were cultivated at 37°C in a humidified atmosphere with 5% CO2. Transfected HEK 293T cells were selected by supplementing the medium with 100 μg/mL Zeocin. All cell lines were authenticated by the analysis of short tandem repeats (STRs).
Expression of scFv-SNAP fusion proteins
The development of the EGFR-specific 425(scFv)-SNAP and 1711(scFv)-SNAP fusion proteins, as well as the mock-SNAP control (CD64-specific scFv-SNAP), is described elsewhere.21,24 The fusion proteins were expressed in HEK 293T cells, cultivated in CellBIND® Surface HYPERFlask® M Cell Culture Vessels (Corning Inc, Corning, NY, USA) and purified by immobilized metal ion affinity chromatography (IMAC) as previously described.21,24,30
Generation of AURIF-based ADCs
The AURIF-based ADCs were generated as previously described.31 Briefly, the purified scFv-SNAP proteins were incubated with a twofold molar excess of BG-modified AURIF (Tube Pharmaceuticals GmbH, Vienna, Austria) for 2 h at room temperature. The remaining unconjugated AURIF was removed using Zeba Spin columns with a molecular weight cutoff of 40 kDa (Thermo Fisher Scientific) according to the manufacturer’s protocol. Successful conjugation was verified by postincubation with a twofold molar excess of the BG-modified fluorophore VistaGreen (NEB, Ipswich, MA, USA) for 10 min in the dark, followed by sodium dodecyl sulfate-polyacrylamide gel electrophoresis and visualization of the fluorescence signal using a Gel Doc XR System (Bio-Rad Laboratories Inc, Hercules, CA, USA).
Evaluation of binding activity
The cell-binding activities of the ADCs were evaluated by flow cytometry as described elsewhere.22,31 Briefly, 4×105 cells were resuspended in phosphate-buffered saline (PBS) and incubated with 1 μg of each fusion protein for 20 min on ice. After washing the cells with PBS, bound protein was detected with a secondary His6-specific phycoerythrin (PE)-labeled antibody (diluted 1:100 in PBS) (Miltenyi Biotec, Bergisch-Gladbach, Germany) for 20 min on ice in the dark. After washing with PBS, the cells were resuspended in 300 μL of PBS and analyzed using a BD FACSVerse instrument (BD, Franklin Lakes, NJ, USA) and the corresponding software. The EGFR− cell line A2058 and mock-SNAP protein served as negative controls.
In vitro characterization of the ADCs
Cell viability assay
The cytotoxic activity of the ADCs was determined using an XTT-based assay as previously described.22,31 Briefly, 5×103 cells per well were seeded into 96-well microtiter plates (Greiner Bio-One, Kremsmünster, Austria), and after 24 h, they were treated with different concentrations of scFv-SNAP, scFv-SNAP-AURIF, or unconjugated AURIF for 72 h at 37°C, 5% CO2 and 100% humidity. Cells treated with PBS were used as negative controls, and cells treated with Zeocin served as positive controls. Cytotoxicity was determined by adding 50 μL of the calorimetric substrate XTT/phenazine methosulfate (Serva and Sigma, Steinheim, Germany) to each well, and the plates were incubated for up to 4 h under the same conditions. Substrate conversion by viable cells was confirmed by measuring the absorbance at 450 and 630 nm using an Epoch microplate reader (BioTek Instruments, Winooski, VT, USA). The experiments were carried out independently in duplicates at least three times. The concentration required to achieve a 50% reduction of cell viability (EC50 value) relative to the PBS-treated control cells was calculated using GraphPad Prism v5 and the Hill equation (GraphPad Software Inc, La Jolla, CA, USA).
Apoptosis assay
The proapoptotic activity of the EGFR-specific ADCs against MDA-MB-468 and RD cells after incubation with 10 nM (500 ng/mL) scFv-SNAP-AURIF or free AURIF for 48 h was determined by annexin V-enhanced green fluorescent protein/propidium iodide (PI) staining as previously described.22,31,32 All experiments were carried out independently in duplicates at least three times, and the data are presented as mean ± standard error of the mean (SEM). Significance was determined by one-way analysis of variance (ANOVA) followed by the Bonferroni post hoc test using GraphPad Prism v5 (**P≤0.01, ***P≤0.001).
Cell cycle analysis
The ability of the ADCs to arrest MDA-MB-468 and RD cells was evaluated by PI staining as previously described.33 Briefly, 5×104 cells per well were plated in 12-well plates and treated with 10 nM scFv-SNAP-AURIF for 48 h at 37°C, 5% CO2 and 100% humidity. After 48 h, the cells were fixed with 70% (v/v) ethanol and stained with 50 μg/mL PI (Carl Roth, Karlsruhe, Germany) supplemented with 1 mg/mL RNase I (Thermo Fisher Scientific). Cell cycle arrest was analyzed by flow cytometry using the BD FACSVerse instrument.
Microtubule disassembly
The effects on microtubule dynamics were visualized as previously described,29 using A549 cells stably expressing a recombinant SNAP-TubB3. Briefly, 7×103 cells were plated in a flat bottom black 96-well half-area microplate (μclear; Greiner Bio-One) in a total volume of 50 μL cell culture medium without G418. The recombinant tubulin structure was stained directly by adding 50 μL/well of a solution in growth medium containing 30 nM SNAP-Cell TMR-Star (NEB) for labeling the tubulin and 1 μg/mL 4′,6-diamidino-2-phenylindole (Thermo Fisher Scientific) for nuclei counterstaining. The cells were incubated overnight at 37°C, 5% CO2 and 100% humidity, before incubation with 50, 150, or 250 nM of the EGFR-specific ADCs. After incubation for up to 48 h at 37°C, changes in the microtubule network were recorded by automated microscopy, using an Opera High Content Screening System (PerkinElmer Inc, Waltham, WA, USA).
Ex vivo binding to tumor biopsies
Paraffin was removed from formalin-fixed paraffin-embedded (FFPE) tumor biopsy samples as previously described.20 After drying, circles were drawn around the tissue sections on the slides using a Dako Pen (Dako Denmark A/S, Glostrup, Denmark). After blocking in PBS supplemented with 1% (v/v) bovine serum albumin (BSA), the slides were incubated at 4°C overnight with the following constructs in PBS: 425(scFv)-SNAP, 1711(scFv)-SNAP, and mock-SNAP, all conjugated with AURIF. After washing with PBS, the slides were incubated with the M2D11 anti-SNAP antibody34 for up to 4 h at room temperature, followed by another washing step. The alkaline phosphatase (AP)-conjugated goat antimouse IgG (GAMAP) (Dianova, Hamburg, Germany) diluted 1:50 in blocking solution was used as secondary antibody, and the slides were incubated overnight. After washing with PBS, AP activity was detected as previously described.20,21 All slides were counterstained using hematoxylin and eosin (Sigma-Aldrich, St Louis, MI, USA). Images were captured using a Leica DMR-HC light microscope with the Leica QWin software (Leica Microsystems, Wetzlar, Germany). In accordance with the Declaration of Helsinki of 1975, the human tumor breast cancer biopsies (primary tissue samples) were obtained during routine clinical practice at University Hospital Aachen, Aachen, Germany, and were provided by the RWTH Aachen University Centralized Biomaterial Bank (cBMB) according to its regulations, following RWTH Aachen University, Medical Faculty Ethics Committee approval (decision EK 206/09).
Results
Generation of EGFR-specific AURIF-based ADCs
Both EGFR-specific scFv-SNAP fusion proteins 425(scFv)-SNAP and 1711(scFv)-SNAP as well as mock-SNAP were transiently expressed in HEK 293T cells. The secreted recombinant proteins were purified from the cell culture supernatant by IMAC using the C-terminal His6 tag as previously described.21,24,30 The scFv-SNAP-based ADCs were generated by conjugating a BG-modified version of MMAF (AURIF) to the purified fusion proteins as previously described, and yields of up to 20 mg/L were achieved.31 The SNAP-tag technology as conjugation strategy and the mechanism of action of the novel ADCs in the target cancer cells are shown in Figure 1. We were able to demonstrate SNAP-tag activity and saturation of the fusion protein with the BG-modified effector molecule by postincubation of the conjugated SNAP-tagged proteins with the BG-modified fluorophore VistaGreen (Figure 2A and B).
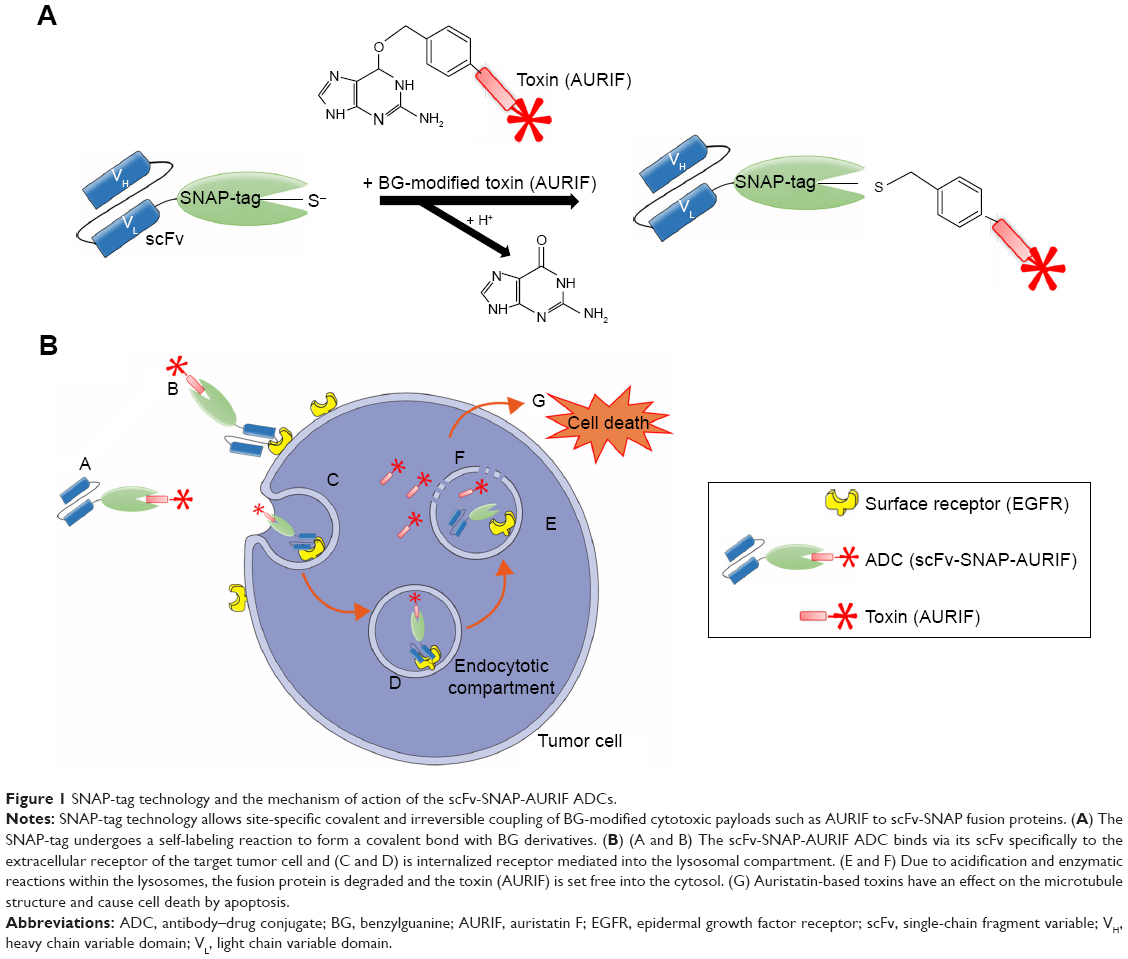 | Figure 1 SNAP-tag technology and the mechanism of action of the scFv-SNAP-AURIF ADCs. |
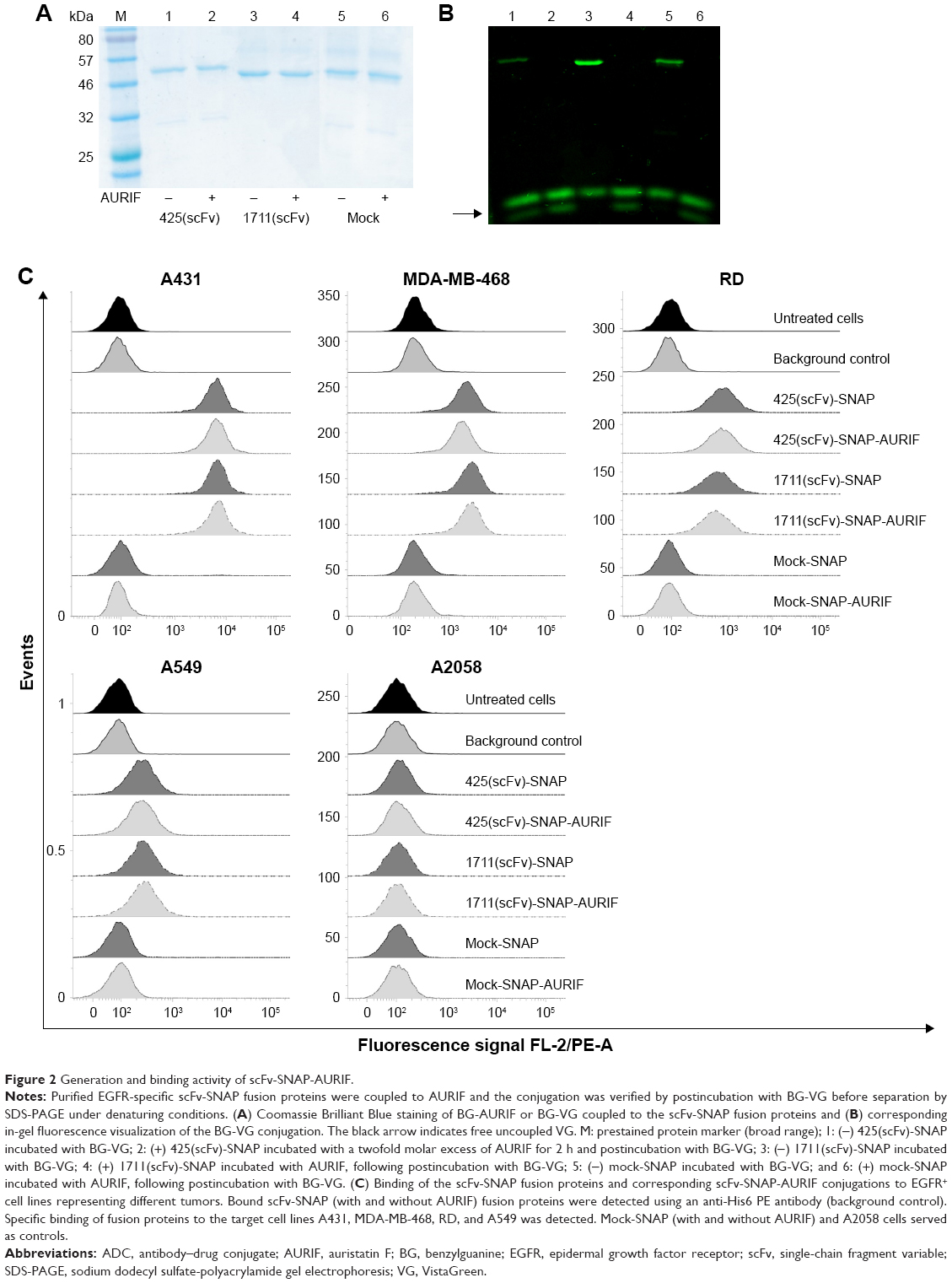 | Figure 2 Generation and binding activity of scFv-SNAP-AURIF. |
Binding analysis of EGFR-specific ADCs
The binding activity of the EGFR-specific ADCs was evaluated by flow cytometry using four EGFR+ cancer cell lines derived from different tumors and the EGFR− melanoma cell line A2058 as a control. ADC binding was detected using a PE-labeled His6-specific detection antibody. Specific binding of the scFv-SNAP fusion proteins to all EGFR+ cell lines was observed after incubation for 30 min on ice, but no binding to the EGFR− cells was observed (Figure 2C). The fluorescence signals generated by the bound ADCs were not different from the signals generated by the corresponding nonconjugated fusion proteins, indicating that conjugation to AURIF had no impact on binding activity. The absence of a signal in the mock-SNAP and mock-SNAP-AURIF controls demonstrates no unspecific binding activity. The mean fluorescence intensity (MFI) for 425(scFv)-SNAP and 1711(scFv)-SNAP with and without AURIF was ~10–100 times higher on the EGFR+ target cell lines A431, MDA-MB-468, and RD than the background. EGFR+ A549 cells showed only moderate levels of receptor expression, resulting in MFI values only ~1.5–3 times higher than the background, but binding was still specific.
Toxicity analysis of EGFR-specific ADCs
Having confirmed the specific binding of the EGFR-specific ADCs, we next measured their dose-dependent cytotoxicity using an XTT-based cell viability assay. A431, MDA-MB-468, RD, and A2058 cells were incubated with decreasing concentrations of AURIF alone, scFv-SNAP, or scFv-SNAP-AURIF. After incubation for 72 h, the proliferation of the treated cells was compared with the PBS-treated controls (Figures 3 and S1). Incubation of the EGFR+ cell lines with scFv-SNAP-AURIF caused a reduction in viability with EC50 values ranging from 4 to 12 nM (Table 1). MDA-MB-468 and RD cells were sensitive to both ADCs, with EC50 values of 4 and 4–8 nM, respectively. As expected, EGFR− A2058 cells were unaffected by any of the scFv-SNAP-AURIF constructs and the mock-SNAP-AURIF caused only a marginal reduction in the viability of all cell lines (Figure S1). Incubation of the cells with AURIF alone caused an expected reduction in cell viability with the EC50 values of 8–26 nM.
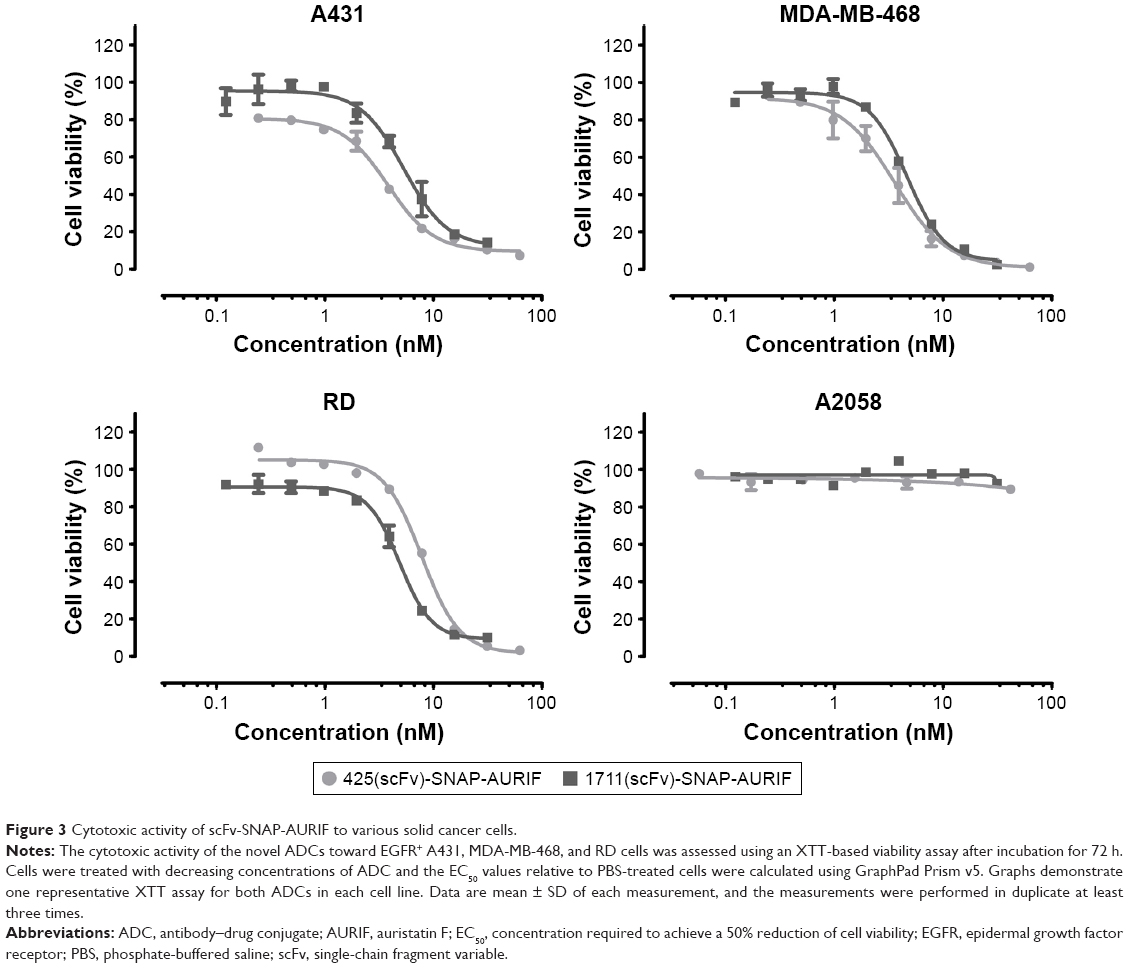 | Figure 3 Cytotoxic activity of scFv-SNAP-AURIF to various solid cancer cells. |
 | Table 1 EC50 values for AURIF and EGFR-specific ADCs |
Specific induction of apoptosis by cell cycle arrest
After showing the cytotoxic potential of the novel EGFR-specific ADCs, we also investigated the induction of apoptosis in MDA-MB-468 and RD cells using an annexin V/PI assay and measured this using flow cytometry. The cells were incubated with 10 nM of scFv-SNAP, scFv-SNAP-AURIF, or free AURIF for 72 h at 37°C. Cells incubated with PBS and camptothecin were used as negative and positive controls, respectively. The proportion of apoptotic cells in the EGFR+ MDA-MB-468 cell population increased significantly after treatment with 425(scFv)-SNAP-AURIF or 1711(scFv)-SNAP-AURIF (Figure 4), whereas RD cells were only significantly affected by 1711(scFv)-SNAP-AURIF (Figure 4C). The significance was measured against mock-SNAP-AURIF. Apoptosis was induced in more than ~70% of the MDA-MB-468 cells treated with each of the EGFR-specific ADCs (Figure 4A). In the RD cell line, apoptosis was induced in ~58% of the cells treated with 1711(scFv)-SNAP-AURIF and ~32% of those treated with 425(scFv)-SNAP-AURIF (Figure 4C). None of the EGFR-specific ADCs induced apoptosis in the control cell line A2058 (Figure 4E). As expected, the nonbinding mock-SNAP-AURIF did not induce apoptosis in any of the cell lines.
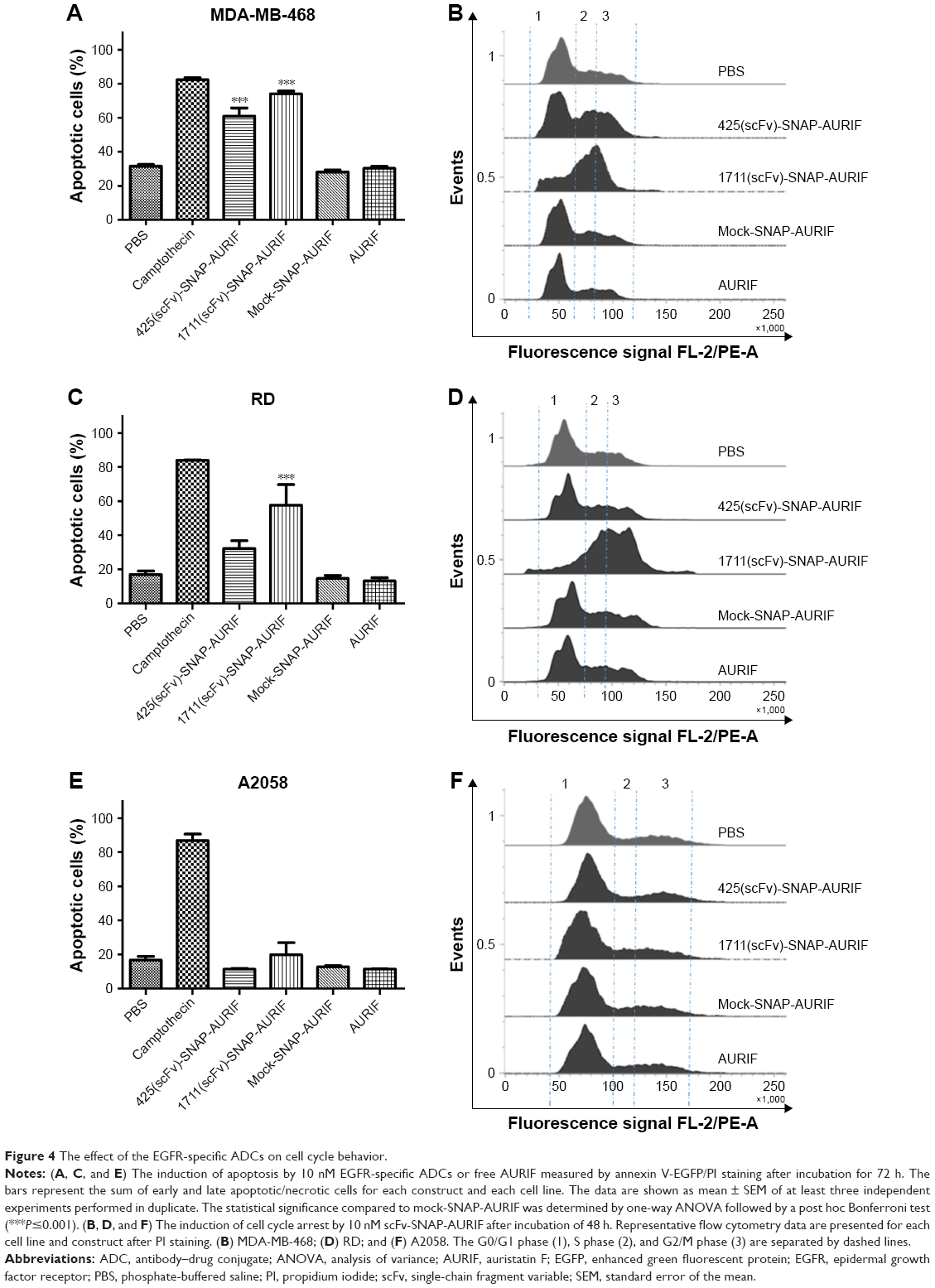 | Figure 4 The effect of the EGFR-specific ADCs on cell cycle behavior. |
To investigate whether the induction of apoptosis resulted from cell cycle arrest, we stained the cells with PI after incubation for 48 h with 10 nM of the ADCs or free AURIF. Cells treated with PBS were used as controls with normal cell cycle phases. EGFR+ MDA-MB-468 cells treated with the EGFR-specific ADCs showed an accumulation of arrested cells in G2/M phase, whereby RD cells were only affected by incubation with 1711(scFv)-SNAP-AURIF. The proportion of G2/M cells in the A2058 cell line did not change when treated with the EGFR-specific ADCs (Figure 4B and D). As expected, no changes in the distribution of cell cycle phases were observed for any of the cell lines treated with mock-SNAP-AURIF.
Analysis of the impact on microtubule dynamics
Having confirmed the specific induction of apoptosis and cell cycle arrest by the EGFR-specific ADCs, we investigated the effects on the tubulin cytoskeleton by microscopy using EGFR+ A549 cells stably transfected with SNAP-TubB3, which facilitates visualization of the tubulin network by covalent labeling with BG fluorophores.29 This cell line was used instead of the former used cell lines, because it was evaluated before and used as model cell line as described by Berges et al.29 First, we determined the cytotoxic activity of the ADCs against A549 cells using an XTT-based cell viability assay, and representative results showing cells treated with 1711(scFv)-SNAP-AURIF are presented in Figure 5A. A concentration-dependent reduction of cell viability was observed when the cells were incubated with 50, 150, and 250 nM of the fusion protein: there was no effect at 50 nM and cell viability declined by ~15% at 250 nM (Figure 5A). The cells treated with 250 nM 1711(scFv)-SNAP-AURIF for 48 h showed evidence of changes in the microtubule mass and structure (Figure 5B) when compared with the cells treated with lower concentrations.
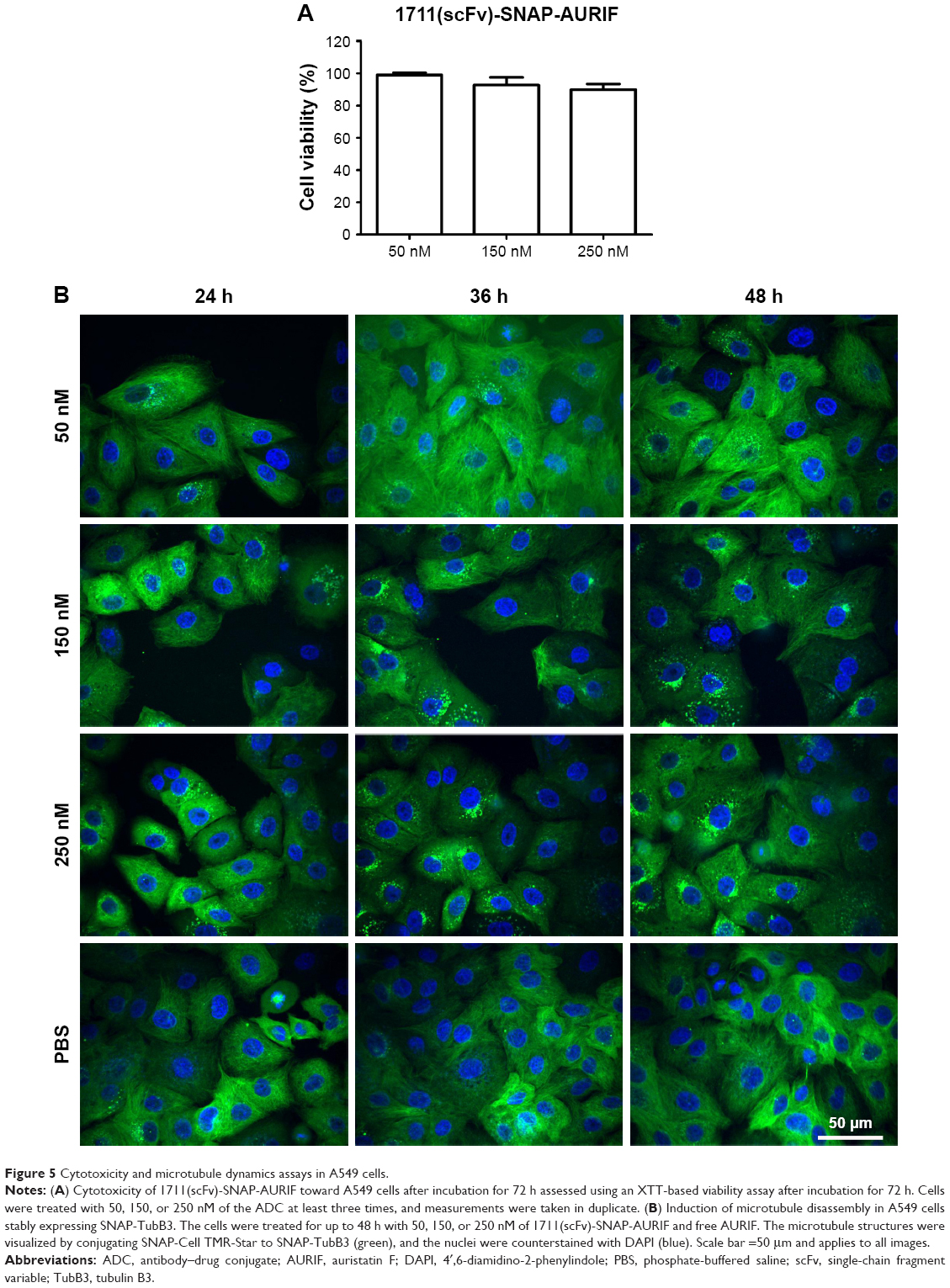 | Figure 5 Cytotoxicity and microtubule dynamics assays in A549 cells. |
Binding of EGFR-specific ADCs to breast cancer tumor biopsies
Clinically relevant information about the novel EGFR-specific ADCs was obtained by investigating their ability to bind primary cells from FFPE sections of breast cancer tumor biopsies. The specific binding of our two EGFR-specific ADCs was confirmed ex vivo by new fuchsin staining, with hematoxylin and eosin counterstaining to verify the presence of tumor cells. No signal was detected when the biopsies were incubated with the detection antibody alone in the absence of the ADCs, or incubated with mock-SNAP-AURIF, confirming the specificity of the novel ADCs (Figure 6).
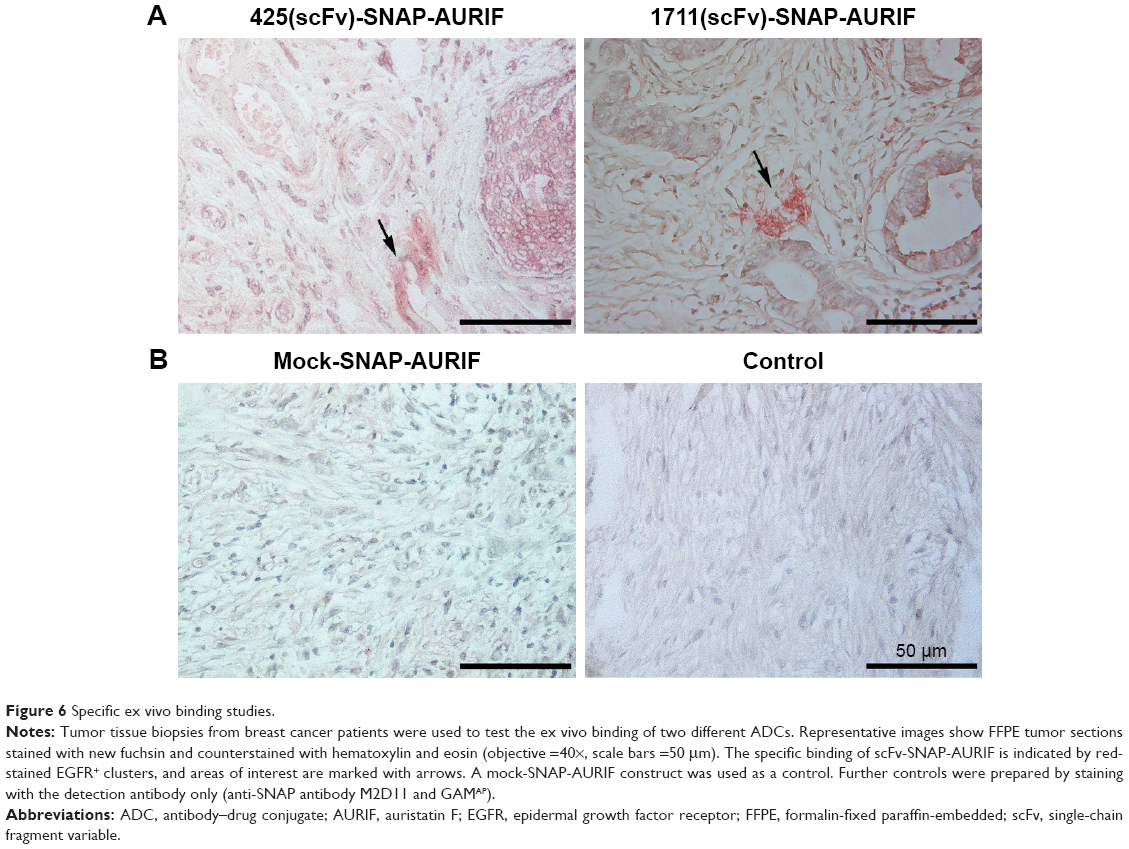 | Figure 6 Specific ex vivo binding studies. |
Discussion
The tumor-associated cell surface antigen EGFR is strongly overexpressed in various types of cancer, including TNBC, rhabdomyosarcoma, and other epidermal cancers.12,14–16,18,19 Several novel EGFR-specific ADCs are currently undergoing preclinical or clinical development.4 One example is ABT-414, an EGFR-specific ADC consisting of MMAF and a human IgG1, which has shown promising results in Phase I/II trials for the treatment of squamous cell carcinoma and as a monotherapy for recurrent glioblastoma.1,4,35,36 Another example is IMGN289, an EGFR-specific ADC conjugated with DM1, which is undergoing Phase I testing.1,4 Furthermore, HTI-1511 is a novel EGFR-specific ADC carrying MMAE, which is indicated for the treatment of solid tumors, including those with KRAS and BRAF mutations. Preclinical studies have shown that this ADC is well tolerated in monkeys.37
Most classic ADCs have been generated by the conjugation of toxic molecules to the cysteine or lysine residues of full-length mAbs, resulting in a heterogeneous product with unpredictable pharmacokinetic properties.38–40 Furthermore, full-length mAbs and the ADCs derived from them have a relatively high molecular weight of ~150 kDa, which makes it difficult to treat solid tumors due to the limited penetration and prolonged retention in nontarget tissues.41 Therefore, our aim was to generate novel EGFR-specific ADCs that are small enough to penetrate tumors effectively and that benefit from efficient conjugation with a defined stoichiometry. We tested two different EGFR-specific scFvs fused to the SNAP-tag, which allows the defined coupling of BG-modified toxins. Such scFv-SNAP fusion proteins can be produced in a range of heterologous expression systems, including bacteria, yeast, and higher eukaryotic cells,41–43 with yields of 5–20 mg/L in the supernatant of eukaryotic cell cultures,24,30,31 with no loss of functional attributes such as antigen specificity.24,30,31,44 We were able to produce all three fusion proteins with comparable yields of up to 20 mg/L in HEK 293T cells. The defined coupling strategy using the SNAP-tag is advantageous compared to the one of classic ADCs. Several other site-directed conjugation methods have been described for full-size mAbs, resulting in ADCs with superior pharmacokinetic profiles.45,46 However, these methods require the integration or modification of unnatural amino acids in the mAb sequence to generate unique conjugation sites.5,46,47 In contrast, the combination of scFvs with the self-labeling SNAP-tag produces scFv-SNAP fusion proteins with no unnatural amino acids, an ideal molecular weight of ~50 kDa to facilitate tumor penetration, and a much shorter retention time in nontarget tissues.48,49 The conjugation of a BG-modified auristatin analog (AURIF) resulted in the generation of the following two EGFR-targeting ADCs based on scFvs of murine or human origin: 425(scFv) derived from the murine mAb42526,50 and 1711(scFv) derived from the FDA-approved human mAb panitumumab.21,22
The EGFR-specific 425(scFv)-SNAP-AURIF and the HER2-specific αHER2(scFv)-SNAP-AURIF have each been shown to kill breast cancer cells efficiently.31 Here, we aimed to expand the SNAP-tag platform technology by using EGFR-specific scFvs derived from approved mAbs, given that EGFR is a major target for the treatment of solid cancers.14,15,18 The combination of scFv-SNAP fusion proteins with AURIF (Figure 2A and B) allows the production of homogenous ADCs with a defined stoichiometry. These ADCs could be generated in an easy, fast, and efficient manner with high expression yields, thus providing an inexpensive strategy for the development of novel ADCs.30,31,51 The conjugation of AURIF did not influence the binding behavior of the fusion proteins we investigated, as previously reported for other constructs (Figure 2C).31 Both ADCs, 425(scFv)-SNAP(-AURIF) and 1711(scFv)-SNAP(-AURIF), bound efficiently to all EGFR+ cell lines (Figure 2). Similarly, scFv-SNAP alone has no influence on the cell lines and showed neither unspecific cytotoxic effects nor unspecific apoptotic effects.21 Free AURIF demonstrated antiproliferative effects on RD cells with an EC50 value of 26 nM, on A431 cells with an EC50 value of 19 nM, and on MDA-MB-468 cells with an EC50 value of 26 nM (Figure 3B and Table 1). In contrast, both ADCs, 425(scFv)-SNAP-AURIF and 1711(scFv)-SNAP-AURIF, displayed potent EC50 values of 4–12 nM (~0.2–0.6 μg/mL) against all cell lines superior to free AURIF (Table 1). Interestingly, 1711(scFv)-SNAP-AURIF shows slightly better cytotoxic effects on RD cells (4 nM) than 425(scFv)-SNAP-AURIF (8 nM). When both scFvs were part of an EGFR-specific immunotoxin (scFv1711-ETA′ and 425(scFv)ETA′), the scFv1711-based immunotoxin showed also a better cytotoxic effect on RD cells, even if RD cells have only a moderate EGFR expression level.21 The affinity constant of 1711(scFv)-SNAP was good with ~4 nM, thus the binding strength should not have had impact on the receptor-mediated internalization and therefore the cytotoxicity.28 The parental mAb panitumumab of 1711(scFv) binds epitopes belonging to epitope bin III/B. The 425(scFv) and the parental mAb425 (IgG2a) bind to an epitope close to the active natural epidermal growth factor ligand-binding site on the extracellular EGFR domain.21,28,52 This could be a reason explaining the weaker and different cytotoxic and apoptotic effects of both scFvs as part of ADCs. Comparable cytotoxic effects against different EGFR+ cell lines were recently demonstrated with an EGFR-specific ADC consisting of zalutumumab or nimotuzumab conjugated to the microtubule-disrupting agent duostatin-3, resulting in the more effective killing of A431 cells, which strongly overexpress EGFR, compared to SK-OV-3 cells, which express the same protein at lower levels.14 Nevertheless, by coupling AURIF to both EGFR-specific scFv-SNAP fusion proteins, it was possible to demonstrate specific delivery of AURIF to the target cells since no unspecific binding or toxicity to EGFR− A2058 could be shown (Figure 3).
The in vitro cytotoxic efficacy of ADCs also correlates with the number of toxic molecules on each antibody. For example, the site-directed conjugation of maytansine to trastuzumab achieves a DAR of 1.8 and an EC50 value of ~11 ng/mL, whereas the commercial ADC ado-trastuzumab maytansine has a DAR of 3.5 and thus a much lower EC50 value of ~5 ng/mL.53 Accordingly, multiple AURIF derivatives could be coupled to one BG molecule and, thus, conjugated via the SNAP-tag. This could potentially improve the efficacy of the novel ADCs without influencing their antigen-binding specificity by increasing the DAR from one to higher but nonetheless specific DARs.
Various tubulin inhibitors have been tested as the cytotoxic components of ADCs and approximately one-third of novel ADCs are armed with auristatin analogs, such as MMAF.46,54 Cells treated with microtubule-disrupting agents build monopolar mitotic spindles instead of normal bipolar spindles and arrest during mitosis before undergoing apoptosis.55 We therefore confirmed the ability of our novel ADCs to induce cell cycle arrest and apoptosis/necrosis in the EGFR+ target cells (Figure 4). Up to 74% of the MDA-MB-468 cells and up to 60% of the RD cells were positive for annexin V/PI staining after incubation with 10 nM (~0.5 μg/mL) of 1711(scFv)-SNAP-AURIF, confirming the efficient induction of apoptosis/necrosis. In comparison, 70% of the MDA-MB-468 cells but only 32% of the RD cells underwent apoptosis when treated with 425(scFv)-SNAP-AURIF. As expected, none of the ADCs affected the cell cycle behavior of A2058 cells and none of the cell lines were influenced by the mock-SNAP-AURIF. Other reports have also confirmed that MMAF is suitable for the generation of potent ADCs.56 The conjugation of MMAF to a B-cell maturation antigen (BCMA)-specific mAb increased its specific toxicity against target cells such that incubation with ~1 μg/mL of the BCMA-specific ADC increased the proportion of late apoptotic/necrotic cells from 24 to 75%, similar to the levels achieved using our novel EGFR-specific ADCs.56 Interestingly, only 55% of the cells entered apoptosis when the mAb was conjugated to MMAE instead of MMAF, indicating the importance of synergy between the antibody and the toxin. The delivery of AURIF conjugated to EGFR-specific scFv-SNAP fusion proteins solely to EGFR+ cells was confirmed by analyzing the prevalence of cell cycle arrest induced by the ADCs. EGFR+ cell lines treated with the EGFR-specific ADCs accumulated cells in G2/M phase, whereas mock-SNAP-AURIF did not change the relative proportions of the different cell cycle stages (Figure 4). Similar proportions of cell cycle arrest have also been observed for other auristatin-based ADCs, such as brentuximab vedotin.57 We inspected the effect of the ADCs by confocal microscopy in EGFR+ A549 cells stably expressing SNAP-TubB3. This transfected cell line allows the covalent coupling of BG fluorophores to tubulin so that microtubule dynamics can be analyzed in the living cells.29 The treatment of these cells with the EGFR-specific ADCs resulted in destabilization of the microtubules, which would also fit with the effect of auristatin derivatives such as MMAF (Figure 5B).58 In addition, the assumption of the destabilization of microtubules with not only auristatin alone but also as part of a novel ADC could be proved.
Conclusion
We generated a novel human EGFR-specific ADC by the site-specific conjugation of AURIF using SNAP-tag technology, resulting in a defined DAR of 1. The novel ADC was compared to the previously described construct 425(scFv)-SNAP-AURIF.31 Both ADCs demonstrated specific cytotoxicity in the lower nanomolar range, depending on the cell line. Moreover, we demonstrated to the best of our knowledge for the first time interference of our novel human scFv-SNAP-based ADC with microtubule dynamics in vitro using a formerly established assay with A549-SNAP-TubB3 cells. This novel ADC (1711[scFv]-SNAP-AURIF), which was based on the approved human mAb panitumumab, also shows best in vitro efficacy, making this construct ideal for further in vivo testing and clinical development, also depending on its fully human origin. Accordingly, we confirmed the specific ex vivo binding of our ADCs to breast cancer biopsies, indicating their potential suitability for the treatment of breast cancer, and also other EGFR-overexpressing tumors.
Acknowledgments
Mira Woitok was supported by the RWTH Aachen University scholarship of Young Researchers at the RWTH Aachen University (RFwN). The data in this publication were produced during the TheraSECOURE project, which was funded by the Federal Ministry of Education and Research (Bundesministerium für Bildung und Forschung [BMBF]) under project number 03V0348. Responsibility for the content of this publication lies with the authors. We thank Frank Schmelter for providing A549-SNAP-TubB3 cells. The authors would like to thank Kai Fuhrmann for excellent technical support and Dr Richard M Twyman for critical reading of the manuscript.
The present address of Stefan Barth is University of Cape Town, South African Research Chair in Cancer Biotechnology, Institute of Infectious Disease and Molecular Medicine (IDM), Faculty of Health Sciences, Department of Integrative Biomedical Sciences, Anzio Road, Observatory, 7925 Cape Town, South Africa. The present adress of Judith Niesen is Research Institute Children’s Cancer Center, University Medical Center Hamburg-Eppendorf, Hamburg, Germany.
Disclosure
Wolfgang Richter is an employee of Tube Pharmaceuticals GmbH. The authors report no other conflicts of interest in this work.
References
Diamantis N, Banerji U. Antibody-drug conjugates – an emerging class of cancer treatment. Br J Cancer. 2016;114(4):362–367. | ||
Lambert JM. Antibody-drug conjugates: targeted delivery and future prospects. Ther Deliv. 2016;7(5):279–282. | ||
Parslow A, Parakh S, Lee F-T, Gan H, Scott A. Antibody–drug conjugates for cancer therapy. Biomedicines. 2016;4(3):14. | ||
Kim EG, Kim KM. Strategies and advancement in antibody-drug conjugate optimization for targeted cancer therapeutics. Biomol Ther (Seoul). 2015;23(6):493–509. | ||
Panowski S, Bhakta S, Raab H, Polakis P, Junutula JR. Site-specific antibody drug conjugates for cancer therapy. MAbs. 2014;6(1):34–45. | ||
Schumacher D, Hackenberger CP, Leonhardt H, Helma J. Current status: site-specific antibody drug conjugates. J Clin Immunol. 2016;36(suppl 1):100–107. | ||
O’Hear C, Rubnitz JE. Recent research and future prospects for gemtuzumab ozogamicin: could it make a comeback? Expert Rev Hematol. 2014;7(4):427–429. | ||
Chudasama V, Maruani A, Caddick S. Recent advances in the construction of antibody-drug conjugates. Nat Chem. 2016;8(2):114–119. | ||
Sapra P, Hooper AT, O’Donnell CJ, Gerber HP. Investigational antibody drug conjugates for solid tumors. Expert Opin Investig Drugs. 2011;20(8):1131–1149. | ||
Yardley DA, Weaver R, Melisko ME, et al. EMERGE: a randomized phase II study of the antibody-drug conjugate glembatumumab vedotin in advanced glycoprotein NMB-expressing breast cancer. J Clin Oncol. 2015;33(14):1609–1619. | ||
Ott PA, Hamid O, Pavlick AC, et al. Phase I/II study of the antibody-drug conjugate glembatumumab vedotin in patients with advanced melanoma. J Clin Oncol. 2014;32(32):3659–3666. | ||
Yewale C, Baradia D, Vhora I, Patil S, Misra A. Epidermal growth factor receptor targeting in cancer: a review of trends and strategies. Biomaterials. 2013;34(34):8690–8707. | ||
Tebbutt N, Pedersen MW, Johns TG. Targeting the ERBB family in cancer: couples therapy. Nat Rev Cancer. 2013;13(9):663–673. | ||
de Goeij BE, Satijn D, Freitag CM, et al. High turnover of tissue factor enables efficient intracellular delivery of antibody-drug conjugates. Mol Cancer Ther. 2015;14(5):1130–1140. | ||
Ueno NT, Zhang D. Targeting EGFR in triple negative breast cancer. J Cancer. 2011;2:324–328. | ||
Jamdade VS, Sethi N, Mundhe NA, Kumar P, Lahkar M, Sinha N. Therapeutic targets of triple negative breast cancer: a review. Br J Pharmacol. 2015;172(17):4228–4237. | ||
Masuda H, Zhang D, Bartholomeusz C, Doihara H, Hortobagyi GN, Ueno NT. Role of epidermal growth factor receptor in breast cancer. Breast Cancer Res Treat. 2012;136(2):331–345. | ||
Ricci C, Landuzzi L, Rossi I, et al. Expression of HER/erbB family of receptor tyrosine kinases and induction of differentiation by glial growth factor 2 in human rhabdomyosarcoma cells. Int J Cancer. 2000;87(1):29–36. | ||
Ganti R, Skapek SX, Zhang J, et al. Expression and genomic status of EGFR and ErbB-2 in alveolar and embryonal rhabdomyosarcoma. Mod Pathol. 2006;19(9):1213–1220. | ||
Niesen J, Brehm H, Stein C, et al. In vitro effects and ex vivo binding of an EGFR-specific immunotoxin on rhabdomyosarcoma cells. J Cancer Res Clin Oncol. 2015;141(6):1049–1061. | ||
Niesen J, Stein C, Brehm H, et al. Novel EGFR-specific immunotoxins based on panitumumab and cetuximab show in vitro and ex vivo activity against different tumor entities. J Cancer Res Clin Oncol. 2015;141(12):2079–2095. | ||
Niesen J, Hehmann-Titt G, Woitok M, et al. A novel fully-human cytolytic fusion protein based on granzyme B shows in vitro cytotoxicity and ex vivo binding to solid tumors overexpressing the epidermal growth factor receptor. Cancer Lett. 2016;347(2):229–240. | ||
Kampmeier F, Niesen J, Koers A, et al. Rapid optical imaging of EGF receptor expression with a single-chain antibody SNAP-tag fusion protein. Eur J Nucl Med Mol Imaging. 2010;37(10):1926–1934. | ||
Kampmeier F, Ribbert M, Nachreiner T, et al. Site-specific, covalent labeling of recombinant antibody fragments via fusion to an engineered version of 6-O-alkylguanine DNA alkyltransferase. Bioconjug Chem. 2009;20(5):1010–1015. | ||
Bouchard H, Viskov C, Garcia-Echeverria C. Antibody-drug conjugates-a new wave of cancer drugs. Bioorg Med Chem Lett. 2014;24(23):5357–5363. | ||
Rodeck U, Herlyn M, Herlyn D, et al. Tumor growth modulation by a monoclonal antibody to the epidermal growth factor receptor: immunologically mediated and effector cell-independent effects. Cancer Res. 1987;47(14):3692–3696. | ||
Bruell D, Stocker M, Huhn M, et al. The recombinant anti-EGF receptor immunotoxin 425(scFv)-ETA′ suppresses growth of a highly metastatic pancreatic carcinoma cell line. Int J Oncol. 2003;23(4):1179–1186. | ||
Niesen J, Sack M, Seidel M, et al. SNAP-tag technology: a useful tool to determine affinity constants and other functional parameters of novel antibody fragments. Bioconjug Chem. 2016;27(8):1931–1941. | ||
Berges N, Arens K, Kreusch V, Fischer R, Di Fiore S. Toward discovery of novel microtubule targeting agents: a SNAP-tag based high-content screening assay for the analysis of microtubule dynamics and cell cycle progression. SLAS Discov. 2017;22(4):387–398. | ||
Hussain AF, Kampmeier F, von Felbert V, Merk HF, Tur MK, Barth S. SNAP-tag technology mediates site specific conjugation of antibody fragments with a photosensitizer and improves target specific phototoxicity in tumor cells. Bioconjug Chem. 2011;22(12):2487–2495. | ||
Woitok M, Klose D, Niesen J, et al. The efficient elimination of solid tumor cells by EGFR-specific and HER2-specific scFv-SNAP fusion proteins conjugated to benzylguanine-modified auristatin F. Cancer Lett. 2016;381(2):323–330. | ||
Brehm H, Niesen J, Mladenov R, et al. A CSPG4-specific immunotoxin kills rhabdomyosarcoma cells and binds to primary tumor tissues. Cancer Lett. 2014;352(2):228–235. | ||
Darzynkiewicz Z, Bruno S, Del Bino G, et al. Features of apoptotic cells measured by flow cytometry. Cytometry. 1992;13(8):795–808. | ||
Puettmann C, Kolberg K, Hagen S, et al. A monoclonal antibody for the detection of SNAP/CLIP-tagged proteins. Immunol Lett. 2013;150(1–2):69–74. | ||
Phillips AC, Boghaert ER, Vaidya KS, et al. ABT-414, an antibody-drug conjugate targeting a tumor-selective EGFR epitope. Mol Cancer Ther. 2016;15(4):661–669. | ||
Van Den Bent MJ, Gan HK, Lassman AB, et al. Efficacy of a novel antibody-drug conjugate (ADC), ABT-414, as monotherapy in epidermal growth factor receptor (EGFR) amplified, recurrent glioblastoma (GBM). J Clin Oncol. 2016;18(Suppl 4):iv44. | ||
Huang L, Veneziale B, Frigerio M, et al. Preclinical evaluation of a next-generation, EGFR targeting ADC that promotes regression in KRAS or BRAF mutant tumors. In: AACR 2016: Abstracts 1-2696; New Orleans, LA; Philadelphia, PA; 2016. | ||
Junutula JR, Raab H, Clark S, et al. Site-specific conjugation of a cytotoxic drug to an antibody improves the therapeutic index. Nat Biotechnol. 2008;26(8):925–932. | ||
Wang L, Amphlett G, Blattler WA, Lambert JM, Zhang W. Structural characterization of the maytansinoid-monoclonal antibody immunoconjugate, huN901-DM1, by mass spectrometry. Protein Sci. 2005;14(9):2436–2446. | ||
Hamblett KJ, Senter PD, Chace DF, et al. Effects of drug loading on the antitumor activity of a monoclonal antibody drug conjugate. Clin Cancer Res. 2004;10(20):7063–7070. | ||
Ahmad ZA, Yeap SK, Ali AM, Ho WY, Alitheen NB, Hamid M. scFv antibody: principles and clinical application. Clin Dev Immunol. 2012;2012:980250. | ||
Colombo M, Mazzucchelli S, Montenegro JM, et al. Protein oriented ligation on nanoparticles exploiting O6-alkylguanine-DNA transferase (SNAP) genetically encoded fusion. Small. 2012;8(10):1492–1497. | ||
Keppler A, Gendreizig S, Gronemeyer T, Pick H, Vogel H, Johnsson K. A general method for the covalent labeling of fusion proteins with small molecules in vivo. Nat Biotechnol. 2003;21(1):86–89. | ||
von Felbert V, Bauerschlag D, Maass N, et al. A specific photoimmunotheranostics agent to detect and eliminate skin cancer cells expressing EGFR. J Cancer Res Clin Oncol. 2016;142(5):1003–1011. | ||
Kline T, Steiner AR, Penta K, Sato AK, Hallam TJ, Yin G. Methods to make homogenous antibody drug conjugates. Pharm Res. 2015;32(11):3480–3493. | ||
Polakis P. Antibody drug conjugates for cancer therapy. Pharmacol Rev. 2016;68(1):3–19. | ||
Agarwal P, Bertozzi CR. Site-specific antibody-drug conjugates: the nexus of bioorthogonal chemistry, protein engineering, and drug development. Bioconjug Chem. 2015;26(2):176–192. | ||
Robinson MK, Doss M, Shaller C, et al. Quantitative immuno-positron emission tomography imaging of HER2-positive tumor xenografts with an iodine-124 labeled anti-HER2 diabody. Cancer Res. 2005;65(4):1471–1478. | ||
Sundaresan G, Yazaki PJ, Shively JE, et al. 124I-labeled engineered anti-CEA minibodies and diabodies allow high-contrast, antigen-specific small-animal PET imaging of xenografts in athymic mice. J Nucl Med. 2003;44(12):1962–1969. | ||
Bruell D, Bruns CJ, Yezhelyev M, et al. Recombinant anti-EGFR immunotoxin 425(scFv)-ETA′ demonstrates anti-tumor activity against disseminated human pancreatic cancer in nude mice. Int J Mol Med. 2005;15(2):305–313. | ||
Hussain AF, Kruger HR, Kampmeier F, et al. Targeted delivery of dendritic polyglycerol-doxorubicin conjugates by scFv-SNAP fusion protein suppresses EGFR+ cancer cell growth. Biomacromolecules. 2013;14(8):2510–2520. | ||
Voigt M, Braig F, Gothel M, et al. Functional dissection of the epidermal growth factor receptor epitopes targeted by panitumumab and cetuximab. Neoplasia. 2012;14(11):1023–1031. | ||
Pillow TH, Tien J, Parsons-Reponte KL, et al. Site-specific trastuzumab maytansinoid antibody-drug conjugates with improved therapeutic activity through linker and antibody engineering. J Med Chem. 2014;57(19):7890–7899. | ||
Perez HL, Cardarelli PM, Deshpande S, et al. Antibody-drug conjugates: current status and future directions. Drug Discov Today. 2014;19(7):869–881. | ||
Orth JD, Tang Y, Shi J, et al. Quantitative live imaging of cancer and normal cells treated with Kinesin-5 inhibitors indicates significant differences in phenotypic responses and cell fate. Mol Cancer Ther. 2008;7(11):3480–3489. | ||
Tai YT, Mayes PA, Acharya C, et al. Novel anti-B-cell maturation antigen antibody-drug conjugate (GSK2857916) selectively induces killing of multiple myeloma. Blood. 2014;123(20):3128–3138. | ||
Francisco JA, Cerveny CG, Meyer DL, et al. cAC10-vcMMAE, an anti-CD30-monomethyl auristatin E conjugate with potent and selective antitumor activity. Blood. 2003;102(4):1458–1465. | ||
Waight AB, Bargsten K, Doronina S, Steinmetz MO, Sussman D, Prota AE. Structural basis of microtubule destabilization by potent auristatin anti-mitotics. PLoS One. 2016;11(8):e0160890. |
Supplementary material
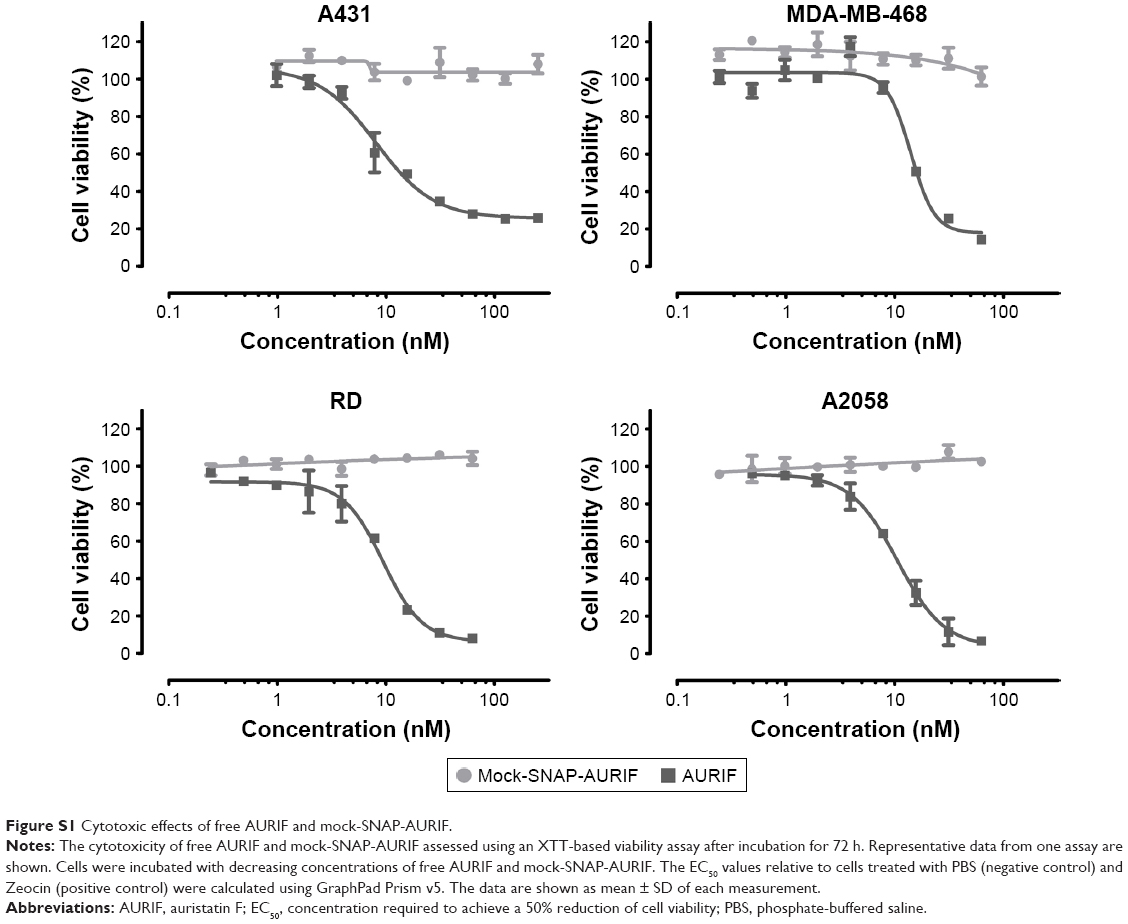 | Figure S1 Cytotoxic effects of free AURIF and mock-SNAP-AURIF. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.