Back to Journals » Infection and Drug Resistance » Volume 12
Comparative genomic analysis and multi-drug resistance differences of Acinetobacter baumannii in Chongqing, China
Authors Pu L, Jian Z, Pan F, Geng Y, He M, Liao P
Received 22 May 2019
Accepted for publication 28 August 2019
Published 11 September 2019 Volume 2019:12 Pages 2827—2838
DOI https://doi.org/10.2147/IDR.S216745
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Joachim Wink
Ling Pu,1 Zuoyi Jian,2 Fen Pan,3 Yang Geng,4 Miao He,5 Pu Liao1,6
1Key Laboratory of Clinical Laboratory Diagnostics (Ministry of Education of China), College of Laboratory Medicine, Chongqing Medical University, Chongqing, People’s Republic of China; 2Department of Bioinformatics, Novogene Biotechnology Co., Ltd, Sichuan, People’s Republic of China; 3Department of Clinical Laboratory, Shanghai Children’s Hospital, Shanghai, People’s Republic of China; 4College of Life Sciences, Sichuan University, Sichuan, People’s Republic of China; 5Institute of Blood Transfusion, Chinese Academy of Medical Sciences, Chengdu, Sichuan, People’s Republic of China; 6Department of Clinical Laboratory, Chongqing People’s Hospital, Chongqing, People’s Republic of China
Correspondence: Pu Liao
College of Laboratory Medicine, Chongqing Medical University, No.1, Yixueyuan Road, Chongqing 400016, People’s Republic of China
Tel +86 0 236 848 5240
Fax +86 0 236 848 5240
Email [email protected]
Miao He
Institute of Blood Transfusion, Chinese Academy of Medical Sciences, No. 26, Huacai Road, Chengdu 610052, Sichuan, People’s Republic of China
Tel +86 0 286 164 8549
Fax +86 0 286 164 8556
Email [email protected]
Introduction: Multidrug-resistance in Acinetobacter baumannii has emerged as a serious problem to public health. There is still a significant gap in the understanding of the multidrug-resistance and the genome diversity evolutionary process of A. baumannii in China, especially in the central and western regions.
Methods: Ten A. baumannii strains were collected from three hospitals in Chongqing, China. Whole-genome re-sequencing was used to obtain differences in genomic levels among strains. The diversity were determined by multi-locus sequence typing method, and investigate the genetic relationship between the ten strains and others by phylogenetic analysis. Comparative analysis focused on resistance genes related to insertions and deletions (InDels) and single-nucleotide polymorphisms (SNPs) was performed.
Results: The overall G+C% content was 39.05%∼39.43%, the average sequencing depth was 273.95∼428.99, and the alignment ratio of the sequencing data was 92.93%∼99.27%. A total of 42 InDels and 11,387 SNPs were detected in the coding sequence region of the isolates. Phylogenetic tree shows that the 10 A. baumannii isolates were divided into four relative groups, and there exist the possibility of cross-regional spread pattern. A total number of 19 drug resistance genes had been found in each strain, and efflux pump-related genes accounted for the most. Only AacA4 underwent a change in InDel. Six types of drug resistance genes were found in the SNPs resistance gene-related loci, among which gene ANT(3’’)-II and QacE mutations were found in each strain.
Conclusion: In this study, the main mechanism of A. baumannii multi-drug resistance is due to the multi-drug efflux pump related genes. The point mutations at the SNPs sites of the six types of resistance genes are the main differences in A. baumannii between Chongqing and the eastern coastal areas of China.
Keywords: Acinetobacter baumannii, antimicrobial resistance, genome-wide re-sequencing, MLST, phylogenetic analysis, SNP
Introduction
Acinetobacter baumannii is an important opportunistic pathogen that accounts for one-tenth of all nosocomial infections, and susceptible individuals are those who are immune-compromised, especially those in intensive care units (ICUs).1 The generation and spread of drug resistance genes are essential contributors to the resistance of A. baumannii. The development of high-throughput sequencing technology has greatly facilitated the study of the molecular mechanism of drug resistance. The complement to high-throughput sequencing is whole-genome resequencing (WGRS), which is a technique for sequencing a genome and comparing it to the previously sequenced reference genome to distinguish sequence polymorphisms that exist in the two sequences.2 Comparative genomics research can help to assess the evolutionary evolution, resistance mechanisms, and pathogenicity of bacterial pathogens at the genome-wide level; it is also useful for the ensuing study of virulence factors involved in pathogenicity.3 Commonly used microbial genome comparison methods include next-generation DNA sequencing,4 polymerase chain reaction screening,5 and transcriptional genomics (ie, 16S rRNA high-throughput sequencing).6 In comparison, whole-genome resequencing can generate large amounts of higher resolution data quickly. At present, whole-genome resequencing is rarely used to investigate the differences in drug resistance between A. baumannii strains isolated in China. In this study, we conducted whole-genome resequencing to analyze 10 A. baumannii isolates and confirm their genetic diversity, including resistance gene-related InDels and SNPs that differed among the strains.
Thus far, various A. baumannii isolates from around the world have been found to possess the msrA/msrB genes, which are associated with macrolides resistance, the aacC1, armA, and aphA1 genes, which are associated with aminoglycosides resistance, and the tet(39), tet(A), and tet(B) genes, which are associated with tetracycline resistance.7–9 Despite extensive research on the epidemiology and population evolution of A. baumannii worldwide, the understanding of the evolutionary processes driving A. baumannii multidrug resistance and genome diversity in China remains incomplete, especially in the central and western regions of the country. Only in the eastern and coastal areas has a comprehensive genetic study of A. baumannii collected at different sites and at multiple times been conducted.10
In 2016, our group performed genome-wide sequencing of the multidrug-resistant A. baumannii isolate MDR-CQ (BioProject number: PRJNA360504), which has a genome size of 3,927,258 bp and a total of 3,682 coding sequences. On the basis of this prior work, 10 representative multidrug-resistant A. baumannii strains isolated at three teaching hospitals in Chongqing were subjected to genome-wide resequencing to assess the genetic diversity and gene content variations of the strains. This study had the following aims: to fill the gaps in the knowledge of genome diversity and the differentiation of drug resistance genes of A. baumannii in the central and western regions of China, to better understand the evolution of A. baumannii resistance genes in China, and to provide a foundation for the development of antibiotics effective against A. baumannii.
Materials and methods
Microbiology methods
The study was conducted from February 2017 to June 2017 at the Second Affiliated Hospital of Chongqing Medical University, Chongqing People’s Hospital, and Chongqing No.5 People’s Hospital. All three hospitals have more than 1,000 beds, of which more than 20 beds are in the intensive care unit (ICU), receiving more than 10,000 patients each year. Ten A. baumannii strains were part of the routine hospital laboratory procedure and were selected according to their similar resistance patterns: multidrug-resistance. They were isolated from adults between the ages of 25 and 65 who had not received antibiotic therapy prior to sample collection at the three hospitals in Chongqing, China. The selected isolates were collected from the intensive care unit, neurosurgery, respiratory medicine, and anorectal surgery of the above hospitals. The strains were stored in 40% sterile glycerol-broth medium at −80 °C in preparation for subsequent studies.
The departments and specimens from which the strains were obtained are shown in Table 1 (Hospital a: Chongqing People’s Hospital; Hospital b: Second Affiliated Hospital of Chongqing Medical University; Hospital c: Chongqing No.5 People’s Hospital).
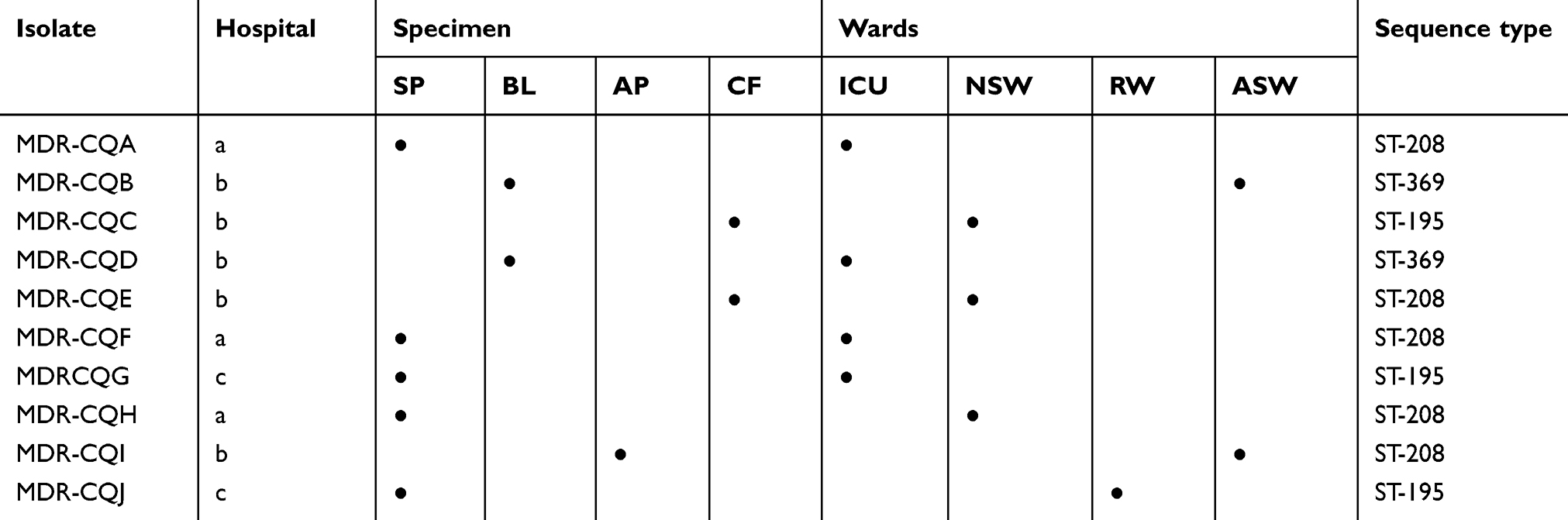 |
Table 1 List of sources of 10 Acinetobacter baumannii isolates |
The reference strain used for the whole-genome resequencing was MDR-CQ (accession number PRJNA360504), which was previously collected at the Second Affiliated Hospital of Chongqing Medical University and for which a complete genome-wide sequence was obtained by our group in 2016.The reference strains used for the research of drug resistance gene differences were MDR-TJ, HRAB-85, and MDR-ZJ06 under accession numbers CP003500, CP018143, and CP001937, respectively.
Bacterial identification and antibiotic susceptibility test
For the operation and isolation of all 10 strains of A. baumannii in this study we strictly followed the requirements set by the National Clinical Laboratory Procedures. Ten clinical isolates of A. baumannii frozen in the laboratory were taken out from the ultra-low temperature refrigerator at −80 °C, inoculated on blood plates and was placed in a 37 °C incubator overnight. In the next day, the single bacteria was added into distilled water to 0.5 Meek turbidity.
Bacterial species were identified by using VITEK-2 Compact bacterial instrument (BioMerieux, Lyons, France) following the manufacturer’s instruction.11 Three different methods performed antibiotic susceptibility testing: the sensitivity of piperacillin/tazobactam was determined by the K-B method; the sensitivity of tigecycline was determined by the E-test method (AB Biodisk, Solna, Sweden); the sensitivity of other antimicrobial agents was detected using the VITEK-GN13 drug susceptibility card. The criteria of the susceptibility of the GN13 card and piperacillin/tazobactam were adapted from the Clinical and Laboratory Standards Institute (CLSI; http://clsi.org/standards/).
DNA extraction and genome-wide resequencing protocols
The genomic DNA of the 10 A. baumannii isolates was extracted using the TIANamp bacterial DNA kit (Tiangen Biotech Co., Ltd., Beijing, China) according to the manufacturer’s instructions. The Illumina sequencing library was then prepared using the Nextera™ DNA Sample Preparation Kit (Illumina®-Compatible). Paired-end dual-index 2x90-bp sequencing was performed following the Illumina HiSeq 2000 (150-bp paired-end reading) protocol. Sequencing was performed by the Beijing Novogene Technology Co., Ltd. (Beijing, China). The sequencing data were uploaded to the public database at the National Center for Biotechnology Information (NCBI) (http://www.ncbi.nlm.nih.gov/) under the BioProject PRJNA360504.
High-quality sequence reads were mapped to the A. baumannii MDR-CQ reference genome, and BWA (version 0.7.12) was used to obtain sequencing coverage data to determine the closeness of the isolates to the reference strain.12 The isolate genomes were assembled using the SOAPdenovo (version 2.04) program, and the assembled sequences were aligned to the reference genome to detect the SNPs and InDels present in each strain. Afterwards, the SNPs and InDels were annotated and statistics were collected based on the reference genome annotation. BreakDancer (version 1.1.2) was utilized to detect structural variation (SV). For any SV resulting from insertion or deletion, the SV was excluded if any of the following criteria were met: 1) the number of PE reads supporting the SV was less than 10; 2) the SV length was less than 100 or more than 1,000,000; and 3) the quality value was less than 30. The CNVnator program (version 0.3) was run to detect copy number variation (CNV) for each strain, and the CNVs were annotated using a custom script.
BlastX (version 2.2.26) was used to search for the gene region in both the reference genome and the SwissProt database.13 Genes possibly associated with drug resistance were identified by the following criteria: identity ≥45%, e value <1e-6, and coverage ≥70%. Next, we used a custom Perl script to compare the genes containing InDels and SNPs with the predicted drug resistance genes to identify which predicted drug resistance genes contained InDels and SNPs.
We analyzed the resequencing results for the ten strains using a python script. Position information including SNP sites, InDels, SV, and CNV were extracted, and these information were then saved in a file delimited by a separator. To facilitate the visual display of the results, we used Circos (version 0.69.6) to visualize SNP location, SNP density, insertion, structural variation and other information.14
Multilocus sequence typing
In this study, BioNumerics 7.6 (Applied Maths, Belgium) was used to perform multi-site sequencing based on the genome-wide sequencing data of the 10 A. baumannii isolates.15 Automatic splicing was conducted after introduction of sequence data followed by the analysis of 7 housekeeping genes (including gltA, gyrB, gdhB, recA, cpn60, gpiF and rpoD) against the MLST database of A. baumannii (http://pubmlst.org/abaumannii). The alleles were automatically matched online with the STs type and then the MLST results were obtained.
Phylogenetic analysis
We used RealPhy (version 1.12) for SNP analysis and constructed a corresponding SNP phylogenetic tree. The original sequences of the 10 A. baumannii isolates in this study were used for mass filtration, first screening out low-quality linker sequences and then filtering the remaining sequences for high quality. The sequence was used as an input file for RealPhy. The reference genomes for RealPhy analysis were downloaded from GenBank and included the MDR strains AB0057, ACICU, AB5075-UW, AYE, MDR-TJ, and MDR-ZJ06, and the drug-sensitive strain ATCC 17978. RealPhy first compared all SNPs we entered against the selected reference genome using Bowtie2 (version 2.3.4) and then screened out the SNP sites. Finally, the software package PHYLIP (version 3.695), along with the bootstrap algorithm, was used to construct the maximum likelihood phylogenetic tree, and the phylogenetic tree was visualized in MEGA (version 7.0.2).
Detection of differences in the drug resistance genes
The comprehensive antibiotic resistance database (CARD) (https://card.mcmaster.ca/) was searched using the RGI program (version 4.2.2) to identify all drug resistance genes in the reference genome.16 We ran the BWA program to compare paired-end reads to the reference genome to determine a consensus sequence of the resistance gene region in each strain. The Muscle program (version 3.8.31) was used to perform multiple sequence alignments for the same drug resistance gene in each sample. From this comparison, the variation of drug resistance genes in each isolate was obtained, after which the variation was manually verified using the IGV program (version 2.0.1), and the mutation in the domain was verified using the InterPro database (http://www.ebi.ac.uk/interpro/).
Next, we compared the differences in A. baumannii resistance genes. Three multidrug-resistant A. baumannii strains, namely, MDR-TJ, MDR-ZJ06, and HRAB-85, which were all reported in China and for which there is valuable information for reference, were selected as reference strains for analyzing the drug resistance genes. We analyzed the genetic differences in InDels and SNPs among the drug-resistant strains and investigated the evolution of the resistance genes in multidrug-resistant A. baumannii in Chongqing.
Genbank accession number
The whole-genome resequencing for 10 A. baumannii clinical isolates have been deposited in GenBank under accession numbers: SNVT00000000, SNVS00000000, SNVR00000000, SNVQ00000000, SNVP00000000, SNVO00000000, SNVN00000000, SNVM00000000, SNVL00000000 and SNVK00000000, respectively.
Results
Antibiotic susceptibilities
The results of antimicrobial sensitivity are shown in Table 2. All 10 isolates are resistant to ≥3 antibiotics and can be identified as multidrug-resistant (MDR).17 It can be seen from the table that 8 strains are only susceptible to tigecycline. Besides, MDR-CQF is not only susceptible to tigecycline, but also susceptible to tetracycline, tobramycin and cotrimoxazole. MDR-CQG is susceptible to both tigecycline and cotrimoxazole.
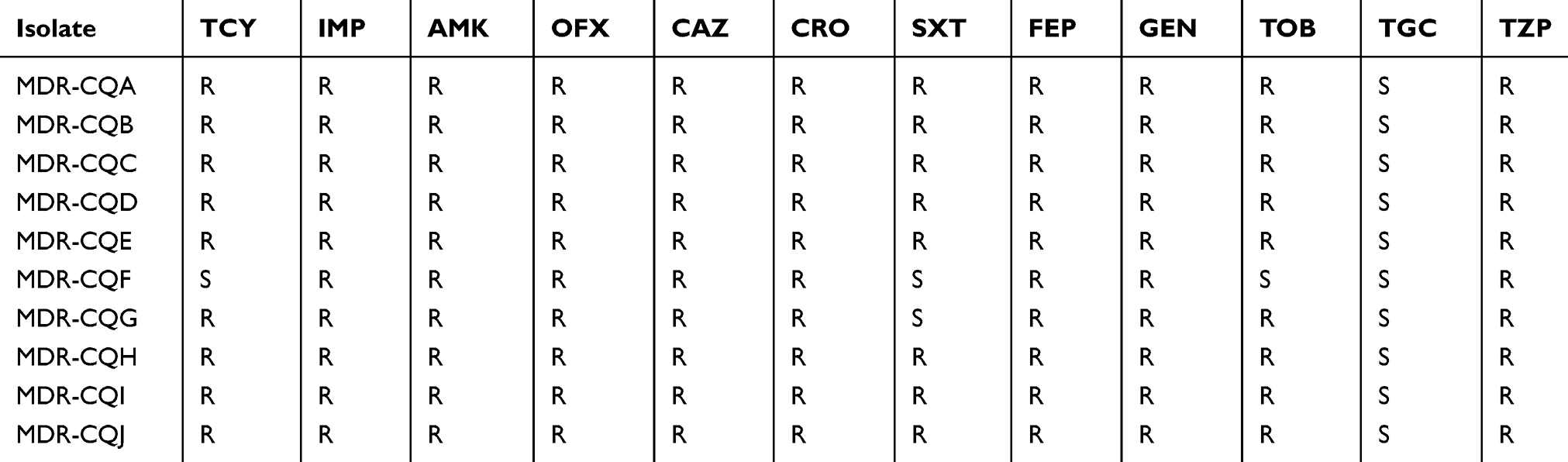 |
Table 2 Antimicrobial susceptibility of 10 Acinetobacter baumannii isolates |
Whole-genome resequencing and comparison with MDR-CQ
Detailed quality control statistics for the WGRS results for the 10 A. baumannii isolates are shown in Table S1. The total G + C% content was between 39.05% and 39.43%, and the average sequencing depth for each strain was between 273.95 and 428.99. The percentage of mapped reads for each strain was between 92.93% and 99.27%, and there was between 96.16% and 100% coverage of the reference sequence for a minimum depth of 20X; this high homology with the reference sequence confirmed that each isolate was A. baumannii. A total of 12,354 SNPs and 42 InDels (Tables S2 and S3) were detected by whole-genome resequencing, and multiple structural variations and copy number variations were found (Tables S4 and Tables S5). An overview of the WGRS results for the ten isolates, visualized using Circos, is shown in Figure 1.
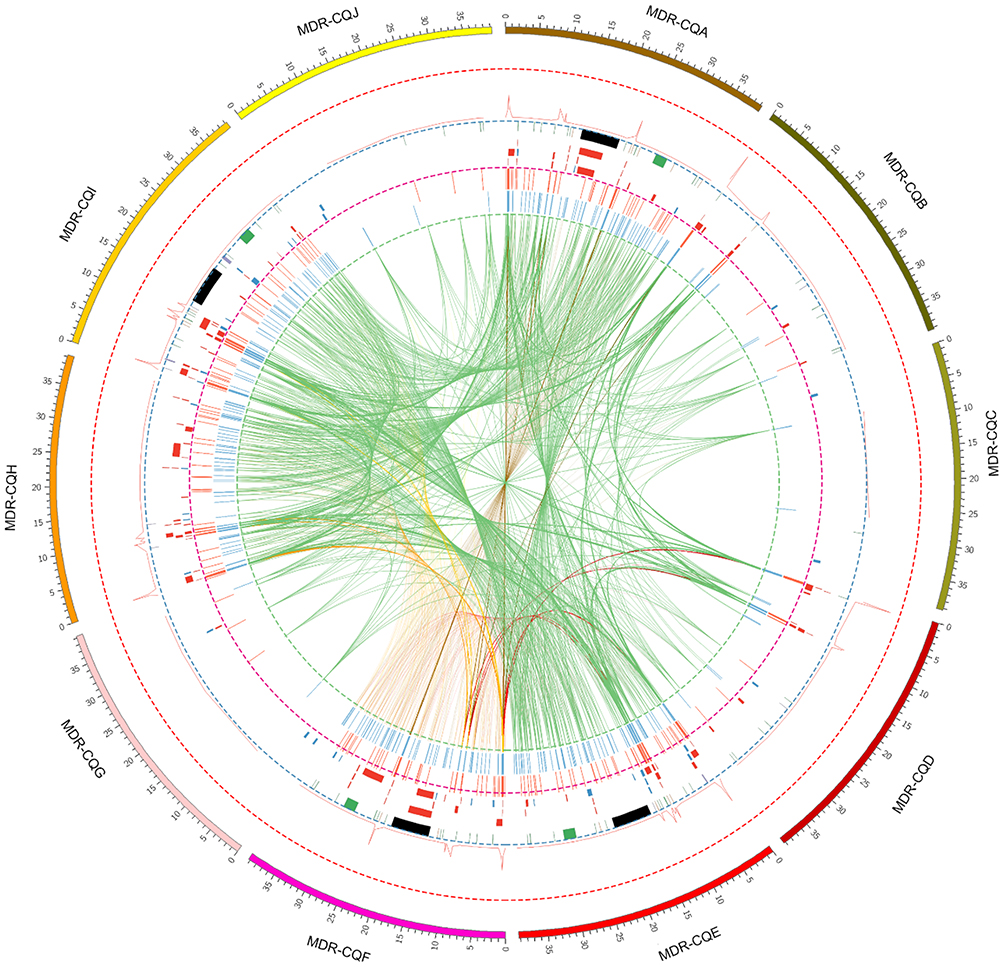 |
Figure 1 Chord diagram of whole-genome resequencing results. Notes: The outermost part is the genome length of ten isolates, and the outermost circle is the coordinate axis, where each square on the scale represents 10,000 bases—used to display the mutation information that occurred at the corresponding location. The center line in the figure shows the correlation between different chromosomal regions, which is used to show that SNPs sites are repeated in these 10 strains. In the histogram, the synonymous and non-synonymous mutations of the SNPs site in the strain are respectively indicated, blue indicates a non-synonymous mutation, and red indicates a synonymous mutation. Red indicates the variation of the deletion type in the single copy number variation, and blue indicates the variation of the duplication type in the single copy number variation. The figure also contains the detected structural variations, such as DEL, INV, INS, ITX, CTX, and other types. Among them, green indicates the DEL type variation; orange indicates the INS type, purple indicates the ITX type, black indicates INV, and gray indicates CTX type variation. Furthermore, we also calculated the density of SNPs, using the form of a line graph to represent the number of SNPs per 10,000 bases. Abbreviations: SNP, Single nucleotide polymorphism; SV, Structural variation; CNV, Copy number variations; DEL, Deletion; INV, Inversion; INS, Insertion; ITX, Intra-chromosomal translocation; CTX, Inter-chromosomal translocation. |
Multilocus sequence typing and phylogenetic grouping
A total of three STs were identified (Table 1): ST208 (MDR-CQA, MDR-CQE, MDR-CQF, MDR-CQH, and MDR-CQI), ST369 (MDR-CQB and MDR-CQD) and ST195 (MDR-CQC, MDR-CQG, MDR-CQJ, and reference strain MDR-CQ). ST208 was the most prevalent, occurring mainly in two hospitals, the Second Affiliated Hospital of Chongqing Medical University and Chongqing People’s Hospital. ST369 was only identified at the Second Affiliated Hospital of Chongqing Medical University. The two strains from the Chongqing No.5 People’s Hospital belonged to ST195. All three STs belonged to clonal complex CC92.18–20
The 10 A. baumannii isolates were classified into four phylogenetic groups (Figure 2), namely, a, b, c, and d. Strain ATCC-17978 was found in early studies and was susceptible to a variety of multiple antibiotics; Strain AYE was isolated from France in 2001 and was a multidrug-resistant strain.21 In group a, MDR-CQA had high similarity with MDR-CQF. In group b, MDR-TJ and MDR-ZJ06, located in the middle of the phylogenetic tree, were isolated from the eastern coastal areas of China, Tianjin and Zhejiang Province, respectively. MDR-CQB and MDR-CQD originated from MDR-TJ and MDR-ZJ06, indicating that these two strains may be transmitted from the eastern coast of China. In group c, MDR-CQG, MDR-CQJ and MDR-CQC originated from MDR-CQ. In group d, MDR-CQH, MDR-CQE, and MDR-CQI also had high similarity, independent of the above three groups, suggesting that A. baumannii has a rich origin resulting in substantial diversity.
 |
Figure 2 Phylogenetic tree of 10 A. baumannii and other reference genomes. Notes: The sequences of the other nine A. baumannii (AB0057, ACICU, AB5075-UW, AYE, MDR-TJ, MDR-ZJ06, and ATCC17978) were used as external roots. |
Differences in drug resistance genes compared to three typical chinese A. baumannii strains
A total of 33 antibiotic-related genes with high mutation rates were obtained following removal of genes with similarity less than 80% and coverage less than 70%. The number of occurrences of these drug resistance genes is shown in Table 3.
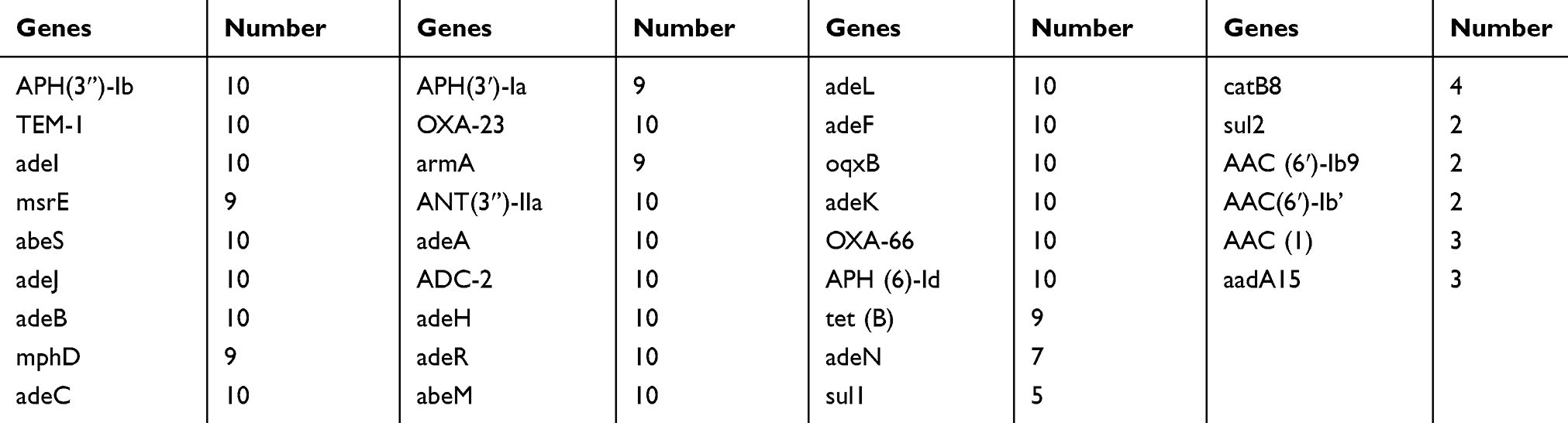 |
Table 3 Statistics on drug resistance genes of ten Acinetobacter baumannii isolates |
It can be seen from the table that a total of 19 drug resistance genes, namely, APH(3’’)-Ib, TEM-1, adeA, adeB, adeC, OXA-23, ANT(3’’)-IIa, ADC-2, adeF, adeH, adeJ, adeK, adeL, abeM, adeR, abeS, oqxB, OXA-66, and APH(6)-Id, were present in all 10 multidrug-resistant A. baumannii; eleven of the 19 were efflux pump family genes (adeABC, adeRS, etc.). The genes APH(3’’)-Ib, APH(6)-Id, APH(3ʹ)-Ia, ANT(3’’)-IIa, AAC(1) and aadA15 are correlated with aminoglycoside resistance,22–24 TEM-1 and ADC-2 are associated with β-lactam resistance,25,26 and OXA-23 and OXA-66 are associated with carbapenem resistance.27
In addition, the sulfonamide resistance genes sul1 and sul2 were present in 5 strains and 2 strains, respectively,28 the quinolone resistance genes AAC(6ʹ)-Ib9 and AAC(6ʹ)-Ib’ were found in only 2 strains,29 and the chloramphenicol resistance gene catB8 was present in 4 strains.30 With the exception of MDR-CQF, the other 9 strains each contained the tetracycline resistance-related gene tet (B),31 the macrolide resistance-related gene msrE,32 and the aminoglycoside resistance-related gene armA.33
To obtain additional genetic resistance information, the ten multidrug-resistant A. baumannii isolates were compared with multidrug-resistant reference strains for differences in InDels and SNPs. We analyzed the InDel mutations associated with drug resistance genes after comparison with three reference strains, and the details of the InDel variation are shown in Table S6. In terms of the InDel differences, the isolates in this study were not substantially different from HRAB-85, MDR-TJ, and MDR-ZJ06, as the only InDel difference was in the class I integron AacA4 (Table 4). Compared with HRAB-85, only MDR-CQC had a deletion of 19 bases (AGCAGCAACGATGTTACGC) in the coding region of 1,367,637. Compared with MDR-ZJ06, only MDR-CQE and MDR-CQI harbored a deletion of the sequence AGCAGCAACGATGTTACGC in the coding region of 1,367,637. Thus, 19-bp mutations in AacA4 were associated with significant changes in aminoglycoside resistance. There were no InDel differences identified for MDR-TJ.
 |
Table 4 InDels changes closely related to antibiotic resistance of Acinetobacter baumannii |
Finally, we analyzed the SNP variation associated with drug resistance genes following comparison with the three reference strains, and the SNP variation details are shown in Table S7. Unlike for the InDels, the SNP differences in comparison with HRAB-85, MDR-TJ and MDR-ZJ06 were quite abundant (Table 5). Nonsynonymous mutations occurred in the LysR-family gene, the AacA4-family gene, the ANT (3’’)-II-family gene, the AdeS gene, the ADC-82 gene and the QacE-family gene. In contrast, stop-loss mutations occurred in the MarC-family gene.
 |
Table 5 SNP changes closely related to antibiotic resistance of Acinetobacter baumannii |
Among these, the LysR transcriptional regulator family is a class of transcriptional regulatory proteins ubiquitous in prokaryotes.34 There was one mutation found in the highly conserved LysR regulator of MDR-CQC strains. Specifically, there was a C to T mutation at position 343 in the coding region, and the corresponding protein harbors a H115Y mutation, which may increase the resistance to carbapenems.
There were a total of 49 gene variants of ANT(3’’)-II found among the 10 A. baumannii isolates. ANT(3’’)-II is a new subclass of aminoglycoside nucleotide transferases; the ANT(3’’)-II gene frequently undergoes horizontal gene transfer in A. baumannii, and the ANT(3’’)-II protein is resistant to aminoglycosides.35 MDR-CQG contained only four nonsynonymous mutations, whereas the other nine strains had five. The five nonsynonymous mutations resulted in the variants A262T, P206S, M205K, T135A, K20E, and MDR-CQG lacked the K20E variation.
AdeS/AdeR is a two-component regulatory system located upstream of the efflux pump AdeABC.36 Members of the Ade ABC efflux pump family are currently the main determinants of antibiotic resistance. Due to their active efflux activities, their expression has extensive effects on antibiotic resistance, including against aminoglycosides, trimethoprim, and quinolones.37 In MDR-CQJ, the AdeS gene harbored a C to T mutation at position 281 in the coding region, corresponding to amino acid change A94V, which resulted in increased expression and downstream regulation. Enhanced expression of the AdeABC pump results in increased efflux, thereby generating resistance to multiple drugs.38
ADC-82 is a recently discovered allelic variant that is resistant to a wide spectrum of cephalosporins.39 In MDR-CQD, there was a T to C point mutation at position 731 in the ADC-82 gene, corresponding to amino acid change L244P, which might have resulted in increased expression.
The disinfectant resistance gene QacE confers resistance to quaternary ammonium salt disinfectants.40 A mutation in this gene was found in each of the ten A. baumannii isolates. Specifically, the base at position 263 in the coding region was mutated from C to T, corresponding to amino acid change T88I, which might also lead to increased expression.
The MarC gene encodes a member of the RND family of multidrug efflux pumps.41 A mutation in this gene was found in MDR-CQB, MDR-CQC, MDR-CQD, MDR-CQG, and MDR-CQJ. Specifically, there was an A to G stop-loss mutation at base position 615 in the coding region, allowing downstream genes to be continually overexpressed.
Discussion
Comparative genomics based on high-throughput sequencing technology can yield more comprehensive and accurate targeted microbial genome information than traditional methods. In this study, whole-genome sequences of 10 multidrug-resistant A. baumannii isolated from Chongqing were studied. The sequence of a representative A. baumannii strain commonly studied by scholars was used as a reference to assess differences in drug susceptibility and drug-resistance gene content to obtain a better understanding of A. baumannii multidrug resistance in China.
According to the results of the resequencing analysis, we can see that the MDR-CQC, MDR-CQG, and MDR-CQJ strains have less variation relative to the MDR-CQ reference genome, as the number of SNPs, InDels, and SV variants was far less than that in other strains. The SNPs in the MDR-CQG strain occurred at the same sites as in other strains. Although there was a unique mutation site in the MDR-CQC and MDR-CQJ strains, this site was a nonsynonymous mutation and may not have had a significant effect on the functions of the strains. Combined with the phylogenetic tree, these data also support the conclusion that MDR-CQC, MDR-CQG, and MDR-CQJ are closely related to the reference strain, MDR-CQ.
The MDR-CQA, MDR-CQE, MDR-CQF, and MDR-CQI strains had substantial chromosomal structural variations, especially chromosomal inversions. At the same time, we also found that the strain with a large number of SNP sites had a higher SNP density across the first 1,000 bps of the genome. In contrast, the density of SNPs remained at a lower level within the first 1,000 bp in the other strains.
MLST results showed that ST208, one of the most common types in China, accounted for the most (5 of 10), which is one of the most common types in China. MDR-CQB and MDR-CQD may be transmitted from the eastern coast of China. The strains clustered into four groups, and the gap between each group was obvious, which indicates that A. baumannii strains in Chongqing had a rich origin resulting in substantial diversity.
We used HRAB-85, MDR-TJ, and MDR-ZJ06 as reference sequences to analyze differences in drug resistance genes. HARB-85 was isolated at the No.307 Hospital of Beijing Liberation Army in China in 2017.42 MDR-TJ was isolated at the Second Affiliated Hospital of Tianjin Medical University in 2012.43 MDR-ZJ06 was isolated at the First Affiliated Hospital of Zhejiang University in 2011.44 All three are multidrug-resistant strains for which genome-wide sequencing has been used to systematically analyze drug resistance genes. The three strains are highly valuable as references for analyzing the domestic migration of A. baumannii resistance genes.
In this study, a total of 34 drug resistance genes were found in 10 multidrug-resistant A. baumannii strains and included 11 efflux pump genes belonging to several families (adeABC, adeRS, etc.) in each strain.45 Class I integrants [aac(6)-Ib, catB8 and aadA1] were also found.46 Nonsynonymous point mutations occurred in the regulatory gene of the adeABC efflux pump system, adeS, causing the system to be overexpressed and leading to efflux-mediated drug resistance against aminoglycosides, quinolones, β-lactam, macrolides, tetracyclines, and other drugs, which is consistent with the research of Meredith et al3. It can be seen that multidrug efflux pumps play a key role in the ten multidrug-resistant A. baumannii isolates in this study because this class accounts for the largest proportion of the identified drug resistance genes. By comparing the differences in drug resistance genes and drug sensitivity to those of MDR-CQF, we can infer that the tet(B) and armA genes encode resistance to tetracycline and tobramycin, respectively.
Compared with the three reference strains from the eastern coastal region, the isolates in this study harbored InDel differences only in the class I integron AacA4, differences characterized by a 19-bp deletion that confers resistance to aminoglycosides.47 In terms of the differences in SNPs, nonsynonymous variations occurred in the LysR-family gene, the AacA4-family gene, the ANT (3’’)-II-family gene, the AdeS gene, the ADC-82 gene, the QacE-family gene, and the MarC-family gene. It can be inferred that the A. baumannii from Chongqing have different variants of drug resistance genes than do strains from the eastern coastal areas of China, and most of the variants are the result of single-site mutations. The ANT (3’’)-II gene, which was found in each A. baumannii isolate, had the most mutations; this is an natural resistance gene and may function as a streptomycin/spectinomycin resistance gene in nature. Furthermore, the ANT (3’’)-II gene represented the main difference in drug resistance genes among A. baumannii examined in this study. The variants associated with changes in amino acids, such as A262T, P206S, M205K, T135A, and K20E, may lay the foundation for the next step in antibiotic development.
In contrast to previous studies, this work was the first to use resequencing technology to study the differences in drug resistance genes of A. baumannii in China. Furthermore, we used three typical Chinese A. baumannii strains as reference strains to study the differences in InDels and SNPs within the drug resistance genes, an approach that was relatively rare in the previous studies. This study has some limitations. For example, only ten clinical isolates were used, which may have affected genetic differentiation analysis and the study of cross-regional spread. Likewise, the resistant reference strains did not include worldwide epidemic strains, and only the differences in drug resistance genes from China were examined. Last, we did not conduct any experiments to confirm that the genetic diversity of InDels and SNPs was associated with antibiotic resistance or to verify the potential relationship between amino acid substitution and increased gene expression. Further investigation of the function of the various genes identified in this study is needed.
Conclusion
The situation with A. baumannii resistance in Chongqing is very serious. However, the overall evolution tends to be stable. In addition, the multidrug-resistant A. baumannii in China are relatively independent from foreign strains, forming an autonomous system, and there are cases of cross-regional migration. The main mechanism of multidrug-resistance in these strains involves the presence of genes associated with efflux pumps. In most of these genes, there are nonsynonymous SNPs that may be involved in drug resistance. Here, we briefly reported the genomic variation among A. baumannii from Chongqing, China. These variants and their association with antibiotic resistance mechanisms may provide a reference for the development of new antibiotics.
Ethics approval
The study was carried out in accordance with the approved guidelines of the Ethics Committee of Chongqing People’s Hospital with written informed consent from all subjects.
Acknowledgments
The authors should like to acknowledge and thank all microbiologists and technical staff for isolating the A. baumannii strain. The authors also thank Shanghai Personal Biotechnology Company for providing bioinformatics analysis of these isolates. This work was financially supported by the National Natural Science Foundation of China (Grant No. 81572089), Medical Research Project of Chongqing Health Commission (Grant No. 2015MSXM111) and Key Research and Development Projects of Science and Technology Department of Sichuan Province (Grant No. 2019YFS0319). This manuscript was revised by Jake George James and the grammatical errors have been corrected.
Disclosure
Zuoyi Jian is an employee of Novogene Biotechnology Co, Ltd. The authors report no other conflicts of interest in this work.
References
1. Mirnejad R, Heidary M, Bahramian A, Goudarzi M, Pournajaf A. Evaluation of Polymyxin B susceptibility profile and detection of drug resistance genes among Acinetobacter Baumannii Clinical Isolates in Tehran, Iran during 2015–2016. Mediterr J Hematol Infect Dis. 2018;10(1):e2018044. doi:10.4084/mjhid.2018.044
2. Loman NJ, Constantinidou C, Chan JZ, et al. High-throughput bacterial genome sequencing: an embarrassment of choice, a world of opportunity. Nature Rev Microbiol. 2012;10(9):599–606. doi:10.1038/nrmicro2850
3. Wright MS, Iovleva A, Jacobs MR, Bonomo RA, Adams MD. Genome dynamics of multidrug-resistant Acinetobacter baumannii during infection and treatment. Genome Med. 2016;8(1):26. doi:10.1186/s13073-016-0279-y
4. Mardis ER. Next-generation DNA sequencing methods. Annu Rev Genomics Hum Genet. 2008;9:387–402. doi:10.1146/annurev.genom.9.081307.164359
5. Shah MW, Yasir M, Farman M, et al. Antimicrobial susceptibility and molecular characterization of clinical strains of Acinetobacter baumannii in Western Saudi Arabia. Microb Drug Resist. 2019. doi:10.1089/mdr.2019.0018
6. Ju F, Zhang T. 16S rRNA gene high-throughput sequencing data mining of microbial diversity and interactions. Appl Microbiol Biotechnol. 2015;99(10):4119–4129. doi:10.1007/s00253-015-6536-y
7. Thi Khanh Nhu N, Riordan DW, Do Hoang Nhu T, et al. The induction and identification of novel Colistin resistance mutations in Acinetobacter baumannii and their implications. Sci Rep. 2016;6:28291. doi:10.1038/srep28291
8. Snitkin ES, Zelazny AM, Montero CI, et al. Genome-wide recombination drives diversification of epidemic strains of Acinetobacter baumannii. Pro Natl Acad Sci USA. 2011;108(33):13758–13763. doi:10.1073/pnas.1104404108
9. Post V, White PA, Hall RM. Evolution of AbaR-type genomic resistance islands in multiply antibiotic-resistant Acinetobacter baumannii. J Antimicrob Chemother. 2010;65(6):1162–1170. doi:10.1093/jac/dkq095
10. Jiang M, Liu L, Ma Y, et al. Molecular epidemiology of multi-drug resistant Acinetobacter baumannii Isolated in Shandong, China. Front Microbiol. 2016;7:1687. doi:10.3389/fmicb.2016.01687
11. Zarrilli R, Pournaras S, Giannouli M, Tsakris A. Global evolution of multidrug-resistant Acinetobacter baumannii clonal lineages. Int J Antimicrob Agents. 2013;41(1):11–19. doi:10.1016/j.ijantimicag.2012.09.008
12. Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25(14):1754–1760. doi:10.1093/bioinformatics/btp324
13. Matsuda F, Tsugawa H, Fukusaki E. Method for assessing the statistical significance of mass spectral similarities using basic local alignment search tool statistics. Anal Chem. 2013;85(17):8291–8297. doi:10.1021/ac401564v
14. Krzywinski M, Schein J, Birol I, et al. Circos: an information aesthetic for comparative genomics. Genome Res. 2009;19(9):1639–1645. doi:10.1101/gr.092759.109
15. Yu J, Sun Z, Liu W, et al. Multilocus sequence typing of Streptococcus thermophilus from naturally fermented dairy foods in China and Mongolia. BMC Microbiol. 2015;15:236. doi:10.1186/s12866-015-0551-0
16. McArthur AG, Waglechner N, Nizam F, et al. The comprehensive antibiotic resistance database. Antimicrob Agents Chemother. 2013;57(7):3348–3357. doi:10.1128/AAC.00419-13
17. Lee CR, Lee JH, Park M, et al. Biology of Acinetobacter baumannii: pathogenesis, antibiotic resistance mechanisms, and prospective treatment options. Front Cell Infect Microbiol. 2017;7:55. doi:10.3389/fcimb.2017.00517
18. Runnegar N, Sidjabat H, Goh HM, Nimmo GR, Schembri MA, Paterson DL. Molecular epidemiology of multidrug-resistant Acinetobacter baumannii in a single institution over a 10-year period. J Clin Microbiol. 2010;48(11):4051–4056. doi:10.1128/JCM.01208-10
19. Wang X, Qiao F, Yu R, Gao Y, Zong Z. Clonal diversity of Acinetobacter baumannii clinical isolates revealed by a snapshot study. BMC Microbiol. 2013;13:234. doi:10.1186/1471-2180-13-234
20. Deng M, Zhu MH, Li JJ, et al. Molecular epidemiology and mechanisms of tigecycline resistance in clinical isolates of Acinetobacter baumannii from a Chinese university hospital. Antimicrob Agents Chemother. 2014;58(1):297–303. doi:10.1128/AAC.01727-13
21. Vallenet D, Nordmann P, Barbe V, et al. Comparative analysis of Acinetobacters: three genomes for three lifestyles. PLoS One. 2008;3(3):e1805. doi:10.1371/journal.pone.0001805
22. Lin MF, Yang CM, Lin CH, Huang ML, Tu CC, Liou ML. Clinical features and molecular epidemiology of multidrug-resistant Acinetobacter calcoaceticus-A baumannii complex in a regional teaching hospital in Taiwan. Am J Infect Control. 2009;37(9):e1–e3. doi:10.1016/j.ajic.2009.03.008
23. Ou HY, Kuang SN, He X, et al. Complete genome sequence of hypervirulent and outbreak-associated Acinetobacter baumannii strain LAC-4: epidemiology, resistance genetic determinants and potential virulence factors. Sci Rep. 2015;5:8643. doi:10.1038/srep08643
24. Valentine SC, Contreras D, Tan S, Real LJ, Chu S, Xu HH. Phenotypic and molecular characterization of Acinetobacter baumannii clinical isolates from nosocomial outbreaks in Los Angeles County, California. J Clin Microbiol. 2008;46(8):2499–2507. doi:10.1128/JCM.00367-08
25. Krizova L, Poirel L, Nordmann P, Nemec A. TEM-1 beta-lactamase as a source of resistance to sulbactam in clinical strains of Acinetobacter baumannii. J Antimicrob Chemother. 2013;68(12):2786–2791. doi:10.1093/jac/dkt275
26. Villalon P, Valdezate S, Medina-Pascual MJ, Carrasco G, Vindel A, Saez-Nieto JA. Epidemiology of the Acinetobacter-derived cephalosporinase, carbapenem-hydrolysing oxacillinase and metallo-beta-lactamase genes, and of common insertion sequences, in epidemic clones of Acinetobacter baumannii from Spain. J Antimicrob Chemother. 2013;68(3):550–553. doi:10.1093/jac/dks448
27. Nigro SJ, Hall RM. Antibiotic resistance islands in A320 (RUH134), the reference strain for Acinetobacter baumannii global clone 2. J Antimicrob Chemother. 2012;67(2):335–338. doi:10.1093/jac/dkr447
28. Soltani B, Heidari H, Ebrahim-Saraie HS, Hadi N, Mardaneh J, Motamedifar M. Molecular characteristics of multiple and extensive drug-resistant Acinetobacter baumannii isolates obtained from hospitalized patients in Southwestern Iran. Le Infezioni Med. 2018;26(1):67–76.
29. Gholami M, Haghshenas M, Moshiri M, et al. Frequency of 16S rRNA methylase and aminoglycoside-modifying enzyme genes among clinical isolates of Acinetobacter baumannii in Iran. Iran J Pathol. 2017;12(4):329–338.
30. Zhu Y, Yi Y, Liu F, et al. Distribution and molecular profiling of class 1 integrons in MDR Acinetobacter baumannii isolates and whole genome-based analysis of antibiotic resistance mechanisms in a representative strain. Microbiol Res. 2014;169(11):811–816. doi:10.1016/j.micres.2014.04.002
31. Vilacoba E, Almuzara M, Gulone L, et al. Widespread dispersion of the resistance element tet(B):: ISCR2in XDR Acinetobacter baumannii isolates. Epidemiol Infect. 2016;144(7):1574–1578. doi:10.1017/S0950268815002897
32. Blackwell GA, Hall RM. The tet39 determinant and the msrE-mphE genes in Acinetobacter plasmids are each part of discrete modules flanked by inversely oriented pdif (XerC-XerD) sites. Antimicrob Agents Chemother. 2017;61(8). doi:10.1128/AAC.00780-17
33. Blackwell GA, Holt KE, Bentley SD, Hsu LY, Hall RM. Variants of AbGRI3 carrying the armA gene in extensively antibiotic-resistant Acinetobacter baumannii from Singapore. J Antimicrob Chemother. 2017;72(4):1031–1039. doi:10.1093/jac/dkw542
34. Yoon EJ, Courvalin P, Grillot-Courvalin C. RND-type efflux pumps in multidrug-resistant clinical isolates of Acinetobacter baumannii: major role for AdeABC overexpression and AdeRS mutations. Antimicrob Agents Chemother. 2013;57(7):2989–2995. doi:10.1128/AAC.02556-12
35. Zhang G, Leclercq SO, Tian J, et al. A new subclass of intrinsic aminoglycoside nucleotidyltransferases, ANT(3”)-II, is horizontally transferred among Acinetobacter spp. by homologous recombination. PLoS Genet. 2017;13(2):e1006602. doi:10.1371/journal.pgen.1006602
36. Rumbo C, Gato E, Lopez M, et al. Contribution of efflux pumps, porins, and beta-lactamases to multidrug resistance in clinical isolates of Acinetobacter baumannii. Antimicrob Agents Chemother. 2013;57(11):5247–5257. doi:10.1128/AAC.00730-13
37. Adams FG, Stroeher UH, Hassan KA, Marri S, Brown MH. Resistance to pentamidine is mediated by AdeAB, regulated by AdeRS, and influenced by growth conditions in Acinetobacter baumannii ATCC 17978. PLoS One. 2018;13(5):e0197412. doi:10.1371/journal.pone.0197412
38. Lari AR, Ardebili A, Hashemi A. AdeR-AdeS mutations & overexpression of the AdeABC efflux system in ciprofloxacin-resistant Acinetobacter baumannii clinical isolates. Indian J Med Res. 2018;147(4):413–421. doi:10.4103/ijmr.IJMR_1375_16
39. Saranathan R, Kumari R, Kalaivani R, et al. Detection of ISAba1 in association with a novel allelic variant of the beta-lactamase ADC-82 and class D beta-lactamase genes mediating carbapenem resistance among the clinical isolates of MDR A. baumannii. J Med Microbiol. 2017;66(2):103–111. doi:10.1099/jmm.0.000395
40. Babaei M, Sulong A, Hamat R, Nordin S, Neela V. Extremely high prevalence of antiseptic resistant quaternary ammonium compound E gene among clinical isolates of multiple drug resistant Acinetobacter baumannii in Malaysia. Ann Clin Microbiol Antimicrob. 2015;14:11. doi:10.1186/s12941-015-0113-1
41. McDermott PF, McMurry LM, Podglajen I, et al. The marC gene of Escherichia coli is not involved in multiple antibiotic resistance. Antimicrob Agents Chemother. 2008;52(1):382–383. doi:10.1128/AAC.00930-07
42. Li P, Huang Y, Yu L, et al. Isolation and whole-genome sequence analysis of the imipenem heteroresistant Acinetobacter baumannii clinical isolate HRAB-85. Int J Infect Dis. 2017;62:94–101. doi:10.1016/j.ijid.2017.07.005
43. Huang H, Yang ZL, Wu XM, et al. Complete genome sequence of Acinetobacter baumannii MDR-TJ and insights into its mechanism of antibiotic resistance. J Antimicrob Chemother. 2012;67(12):2825–2832. doi:10.1093/jac/dks327
44. Zhou H, Zhang T, Yu D, et al. Genomic analysis of the multidrug-resistant Acinetobacter baumannii strain MDR-ZJ06 widely spread in China. Antimicrob Agents Chemother. 2011;55(10):4506–4512. doi:10.1128/AAC.01134-10
45. Ruzin A, Keeney D, Bradford PA. AdeABC multidrug efflux pump is associated with decreased susceptibility to tigecycline in Acinetobacter calcoaceticus-Acinetobacter baumannii complex. J Antimicrob Chemother. 2007;59(5):1001–1004. doi:10.1093/jac/dkm058
46. Lee Y, D’Souza R, Yong D, Lee K. Prediction of putative resistance islands in a carbapenem-resistant Acinetobacter baumannii global clone 2 clinical isolate. Ann Lab Med. 2016;36(4):320–324. doi:10.3343/alm.2016.36.4.320
47. Azizi O, Shakibaie MR, Badmasti F, et al. Class 1 integrons in non-clonal multidrug-resistant Acinetobacter baumannii from Iran, description of the new blaIMP-55 allele in In1243. J Med Microbiol. 2016;65(9):928–936. doi:10.1099/jmm.0.000315
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.