Back to Journals » Vascular Health and Risk Management » Volume 18
Comparative Assessment of Echocardiographic Patterns Among Chronic Myeloid Leukemia Patients on Tyrosine Kinase Inhibitor and Healthy Controls
Authors Bamgboje AO, Durosinmi MA ![]() , Mene-Afejuku TO, Fagbayimu MO
, Mene-Afejuku TO, Fagbayimu MO ![]() , Fajobi O
, Fajobi O ![]() , Balogun MO
, Balogun MO ![]()
Received 22 November 2021
Accepted for publication 27 January 2022
Published 15 February 2022 Volume 2022:18 Pages 27—42
DOI https://doi.org/10.2147/VHRM.S348744
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Konstantinos Tziomalos
Abayomi O Bamgboje,1 Muheez A Durosinmi,2 Tuoyo O Mene-Afejuku,3 Micheal O Fagbayimu,4 Olusola Fajobi,5 Michael O Balogun6
1Department of Internal Medicine, SCL Healthcare St Vincent’s/Holy Rosary Hospital, Miles city, Montana, 59101, USA; 2Department of Hematology and Immunology Obafemi Awolowo University Teaching Hospitals Complex Ile-Ife, Osun state, Nigeria; 3Department of Cardiology Tower Health System, Reading Hospital, West Reading, Pennsylvania, USA; 4Department of Surgery Obafemi Awolowo University Teaching Hospitals Complex Ile-Ife, Osun State, Nigeria; 5Department of Community Medicine Awolowo University Teaching Hospitals Complex Ile-Ife, Osun State, Nigeria; 6Department of Internal Medicine Cardiology Unit Obafemi Awolowo University Teaching Hospitals Complex Ile-Ife, Osun State, Nigeria
Correspondence: Abayomi O Bamgboje, Department of Internal Medicine, 1233 North 30th Street, Billings, MT, 59101, USA, Tel +1 9293189246, Email [email protected]
Purpose: Chronic myeloid leukemia (CML) is one of the common hematological malignancies in Nigeria. Cardiac abnormalities are associated with CML irrespective of treatment with tyrosine kinase inhibitors such as imatinib, which is available gratis in Nigeria.
Objective: To assess the prevalence and patterns of cardiac dysfunction among patients with CML irrespective of treatment with imatinib using transthoracic echocardiography, and 12-lead surface electrocardiography.
Patients and Methods: CML patients without Imatinib, CML patients with imatinib, and apparently healthy (age- and sex-matched) controls were 70 each in the study. Various echocardiographic parameters were measured and data obtained were analyzed, and the level of significance was taken as p < 0.05.
Results: Of 70 CML patients with imatinib, 54.3% were men and 45.7% were women, while the CML group without imatinib had 62.9% men and 37.1% women, non-CML control had 54.3% men and 45.7% women. The average hematocrit was significantly lower in the CML group without Imatinib compared with the other groups (p< 0.001). And, 12.9% and 17.1% of CML groups with and without imatinib had LVH, respectively, and none of the non-CML controls had LVH (P< 0.041). Impaired left ventricular relaxation in 25.71% and 28.57% of CML patients with and without imatinib respectively but only 8.57% of the non-CML control had impaired left ventricular relaxation (p=0.236). Mitral valve regurgitation was the most frequent valvular abnormality across the groups. Pulmonary hypertension in 17.4% and 20% of CML patients with and without imatinib, respectively, but none of the non-CML controls had pulmonary hypertension (p< 0.001). Pericardial effusion in 32.86% and 45.71% of CML patients with and without imatinib, respectively, but none of the non-CML controls had pericardial effusion (p< 0.001). There was no significant difference in the QTC interval across the three groups.
Conclusion: Cardiac abnormalities are present in CML patients with or without Imatinib treatment, with significant prevalence than what is seen in the non-CML control group.
Keywords: chronic myeloid leukemia, CML, tyrosine kinase inhibitor-imatinib, cardiac dysfunction
Introduction
Chronic myeloid leukemia (CML) is a myeloproliferative neoplasm characterized by clonal expansion of hematopoietic cells. In more than 95% of the cases, these cells carry the Philadelphia chromosome or BCR-ABL1 chimeric gene.1 Symptom onset in patients with CML can be insidious and may be attributed to splenomegaly, anemia, hypermetabolism, and hearing impairment; however, some patients are diagnosed incidentally.2,3
Historically, chemotherapy and interferon-alpha have been the standard of treatment for CML, as well as stem cell transplantation. Tyrosine kinase inhibitors (TKIs) have been recommended as first-line treatment for most patients with CML since 2001.4,5
The first-generation TKI imatinib has shown substantial survival benefits compared to historical CML treatments.6 A randomized trial that compared imatinib with interferon-alpha plus cytarabine in the chronic phase of CML demonstrated the superiority of imatinib in all standard indicators of the disease within a median follow-up of 19 months.7
Cardiac abnormalities, including coronary arterial thrombosis, myocardial infarction, pulmonary hypertension (PHT), asymptomatic pericardial effusion, cardiac tamponade, intractable cardiac failure due to intraventricular thrombosis, and valvular heart diseases, have been associated with CML.8,9 Additionally, cardiac abnormalities may also be associated with the use of imatinib. Given the impact of cardiovascular abnormalities in the general population, cardiac abnormalities may contribute to the morbidity and mortality observed in patients with CML.
Previous studies have shown that CML affects blood vessels, cardiac structures, and cardiac function. Studies have also shown that TKIs used in the treatment of CML cause perturbations in cardiac structure and function. However, these studies are not recent and were not conducted in Nigeria.9
Generally, there are few studies on the echocardiographic patterns of CML patients being treated with TKIs. This study may also be the first in Nigeria to assess the echocardiographic pattern, including the structure and function of the heart, in TKI-treated CML patients.
Objective
- To determine the prevalence of cardiac abnormalities using echocardiography, in CML patients attending a hematology outpatient clinic at OAUTHC, Ile-Ife, Nigeria, and treated with or without imatinib.
- To compare the prevalence of left ventricular systolic and diastolic dysfunction, valvular abnormalities, pericardial abnormalities, and PHT among CML patients, CML patients on imatinib attending a hematology outpatient clinic at OAUTHC, Ile-Ife, Nigeria, and an apparently healthy control population.
Materials and Methods
This is a Comparative cross-sectional study in which 70 imatinib-treated CML patients, 70 imatinib-naive CML patients, and 70 age and sex-matched apparently healthy volunteers were recruited consecutively.
This study was reviewed and approved by the Ethics and Research committee of the Obafemi Awolowo University Teaching Hospitals Complex (OAUTHC), Ile-Ife and all participants gave written consent prior to enrolling into the study. The study conformed to the principles outlined in the Declaration of Helsinki, on the ethical principles for medical research involving human subjects.
Inclusion Criteria
- Subjects aged 18 years and above who consented to the study.
- Patients diagnosed with CML at the hematology outpatient clinic, OAUTHC, Ile-Ife, Nigeria, according to standard WHO clinical, hematological, and cytogenetic criteria,10 and just before starting imatinib.
- Patients diagnosed with CML at the hematology outpatient clinic, OAUTHC, Ile-Ife, Nigeria, according to standard WHO clinical, hematological and cytogenetic criteria,10 and who had been taking 400 mg imatinib for at least 3 months.
- Controls: Healthy subjects aged 18 years and above who gave informed consent to participate in the study and were age- and sex-matched with the CML groups.
Exclusion Criteria
- Patients with CML and a premorbid history of heart disease, diabetes, hypertension, or chronic kidney disease.
- Patients who were not willing to participate in the study.
- Patients with a history of allogeneic hematopoietic stem cell transplant.
- CML patients with a history of cigarette smoking.
- Patients who were on other drugs with known cardiotoxicity.
- Patients with significant alcohol consumption.
The patients were interviewed and examined in detail. CML diagnosis was based on blood counts, presence of leukocytosis, and differential counts (basophilia and immature granulocytes from the metamyelocyte to the myeloblast). CML diagnoses were confirmed in bone marrow aspirate by measuring the Philadelphia (Ph) chromosome (22q−), which is a result of the balanced translocation t (9;22) (q34; q11), and/or by measuring a BCR-ABL1 rearrangement in peripheral blood. The presence of normal blood counts was considered non-CML in the control population.
Echocardiography was performed according to standard procedure using a General Electric (GE) medical system Vivid 7-dimension ultrasound instrument with a P5s MHz transducer. Complete 2D echocardiographic examinations were performed according to the recommendations of the American Society of Echocardiography (ASE)11 taking leading-edge-to-leading-edge recordings.12
Left ventricular mass was derived using the Devereux modified American Society of Echocardiography (ASE) formula, as follows:13

where 1.04 = specific gravity of the myocardium
0.8 = correction factor
LVIDd = left ventricular internal diameter in diastole (cm)
PWTd = left ventricular posterior wall thickness in diastole (cm)
IVSTd = interventricular septal thickness in diastole (cm)
The LV mass index was obtained by dividing the LV mass by the body surface area. The upper limits for the LV mass index were 134 and 110 g/m2 in men and women, respectively.14,15
Relative wall thickness (RWT) was calculated using the formula:

A RWT value <0.45 is indicative of normal left ventricular geometry or eccentric hypertrophy while ≥0.45 indicates concentric left ventricular hypertrophy or remodeling for both men and women.15
Left ventricular systolic function was evaluated using the ejection fraction:16 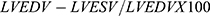 , where normal ≥50%.
, where normal ≥50%.
Left ventricular diastolic function was evaluated using transmitral peak early (E) and late (A) diastolic velocities, the ratio of E to A, and E-wave deceleration time (EDT).17,18
Left ventricular global myocardial performance was assessed using the TEI index calculated by the summation of isovolumic relaxation time and isovolumic contraction time divided by the left ventricular ejection time: 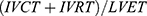 19,20
19,20
PHT was defined as a mean pulmonary arterial pressure (mPAP) of ≥25 mmHg and21,22 was calculated from PASP using the Chemla formula as follows: 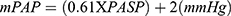 .22,23 Continuous-wave Doppler echocardiography was used to measure the maximum velocity of the tricuspid regurgitant jet (v) in the apical 4-chamber view. The placement of the cursor was guided by color Doppler, which showed the extent of maximum regurgitation across the tricuspid valve. Having obtained the maximum velocity of the tricuspid regurgitant jet (v), the trans-tricuspid pressure gradient was calculated using the modified Bernoulli equation (4v2). Right ventricular systolic pressure (RVSP) was estimated by adding the trans-tricuspid pressure gradient to right atrial pressure (RAP). RVSP was equivalent to PASP after ruling out pulmonary valve stenosis.24 RAP was estimated using the diameter and collapse of the inferior vena cava (IVC) during spontaneous respiration. In this method, an IVC diameter ≤2.1 cm that collapses >50% with a sniff suggests a normal RAP of 3 mmHg (range 0–5 mm Hg), whereas an IVC diameter >2.1 cm that collapses <50% with a sniff indicates a high RA pressure of 15 mmHg (range 10–20 mmHg). In indeterminate cases in which the IVC diameter and collapse did not fit this definition, an intermediate value of 8 mmHg (range, 5–10 mm Hg) was used.25 Mild PHT was defined by an mPAP of 25.0–34.9 mmHg, moderate PHT by an mPAP of 35.0–44.9 mmHg, and severe PHT by an mPAP ≥45.0 mmHg.26
.22,23 Continuous-wave Doppler echocardiography was used to measure the maximum velocity of the tricuspid regurgitant jet (v) in the apical 4-chamber view. The placement of the cursor was guided by color Doppler, which showed the extent of maximum regurgitation across the tricuspid valve. Having obtained the maximum velocity of the tricuspid regurgitant jet (v), the trans-tricuspid pressure gradient was calculated using the modified Bernoulli equation (4v2). Right ventricular systolic pressure (RVSP) was estimated by adding the trans-tricuspid pressure gradient to right atrial pressure (RAP). RVSP was equivalent to PASP after ruling out pulmonary valve stenosis.24 RAP was estimated using the diameter and collapse of the inferior vena cava (IVC) during spontaneous respiration. In this method, an IVC diameter ≤2.1 cm that collapses >50% with a sniff suggests a normal RAP of 3 mmHg (range 0–5 mm Hg), whereas an IVC diameter >2.1 cm that collapses <50% with a sniff indicates a high RA pressure of 15 mmHg (range 10–20 mmHg). In indeterminate cases in which the IVC diameter and collapse did not fit this definition, an intermediate value of 8 mmHg (range, 5–10 mm Hg) was used.25 Mild PHT was defined by an mPAP of 25.0–34.9 mmHg, moderate PHT by an mPAP of 35.0–44.9 mmHg, and severe PHT by an mPAP ≥45.0 mmHg.26
In the absence of detectable tricuspid regurgitant signal, difficult visualization of the tricuspid regurgitant jet, or the presence of pulmonic stenosis, we used the right ventricular acceleration time (AT) and the ratio of acceleration time to ejection time 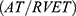 from the pulmonary ejection flow jet obtained by continuous-wave Doppler.
from the pulmonary ejection flow jet obtained by continuous-wave Doppler.
PTH was assessed based on the following:
OR
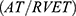 ratio of <0.30. This method included patients without a detectable tricuspid regurgitant velocity.
ratio of <0.30. This method included patients without a detectable tricuspid regurgitant velocity.Echocardiographic evaluation of the valve morphology and valvular abnormalities was performed using color-coded Doppler imaging. In addition, conventional resting 12-lead surface ECG was performed. Baseline clinical and laboratory parameters such as body mass index (BMI) and hematocrit were estimated.
Statistics
Data were analyzed using the IBM SPSS software (version 20.0; IBM Corporation, Armonk, NY, USA). Categorical variables are expressed as proportions and percentages, whereas continuous variables are expressed as means ± standard deviation or as ranges. Bivariate analysis was performed using the chi-square and Fisher’s exact tests for discrete variables. The t-test was used to compare cardiac function between patients with CML treated with imatinib and healthy controls. ANOVA was used to compare cardiac function among CML patients, CML patients on imatinib, and apparently healthy controls, and the Bonferroni post hoc test was used to determine the significance between groups. A p-value ˂0.05 was considered significant.
Results
Demographic and Physical Characteristics and Laboratory and Echocardiographic Parameters of the Study Subjects
A total of 210 subjects completed the study. This included 70 imatinib-treated CML subjects, 70 imatinib-naïve CML subjects, and 70 apparently healthy subjects without CML. The demographics, physical characteristics, and laboratory parameters of the study population are summarized in Table 1. Imatinib-treated CML patients and imatinib-naive CML patients were matched for age and sex with healthy non-CML controls. CML patients on imatinib included 38 male and 32 female patients, CML patients that were imatinib-naïve included 44 male and 26 female patients, and the non-CML control patients included 38 male and 32 female patients.
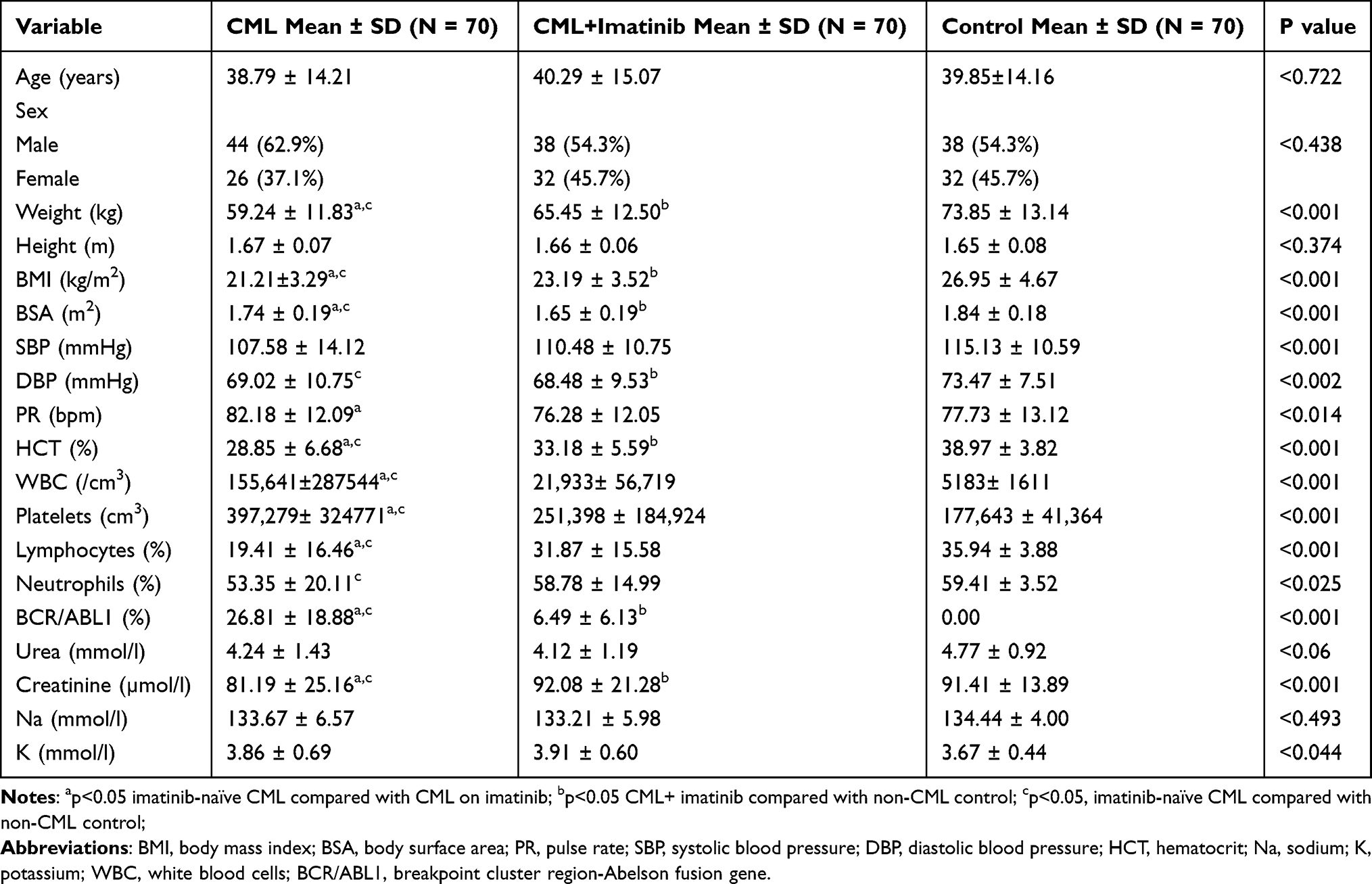 |
Table 1 Demographic and Physical Characteristics and Laboratory Parameters of Study Subjects |
There were no statistically significant differences in gender across the groups (p<0.438). Age ranges in the imatinib-treated CML, imatinib-naïve CML, and non-CML control groups were 18–79 years, 18–82 years, and 18–75 years, respectively. There were significant differences in weight, body mass index, and body surface area among the three groups. However, height was not significantly different among the three groups. The pulse rate and systolic and diastolic blood pressures were also significantly different among the three groups.
As shown in Table 1, CML patients not treated with imatinib had significantly lower hematocrit than CML patients treated with imatinib. The white blood cell and platelet counts were significantly higher in the imatinib-native CML group compared to the imatinib-treated CML group. There was also a significant difference between the hematocrit of imatinib-treated CML patients and non-CML controls: the hematocrit of CML patients treated with imatinib was significantly lower than non-CML controls; however, CML patients treated with imatinib had a significantly higher white blood cell count and platelet count than non-CML controls. The percentages of lymphocytes and neutrophils were significantly lower in the imatinib-naive CML group compared to the imatinib-treated CML group, which had significantly lower percentages of lymphocytes and neutrophils compared to the non-CML controls.
The BCR/ABL1 ratio was significantly different between the two CML groups. The mean BCR/ABL1 ratio in the imatinib-naive CML group was 26.81±18.88, while the mean BCR/ABL1 ratio in the imatinib-treated CML group was 6.49 ± 6.13. The BCR/ABL1 ratio was significantly higher in the imatinib-naive CML group than in CML patients who had been treated with imatinib for at least 3 months. The BCR/ABL1 ratio was also significantly higher in the imatinib-treated CML group than in the non-CML control group.
From the two-dimensional and M-mode echocardiographic parameters highlighted in Table 2, there was a statistically significant increase in pericardial effusion rates in imatinib-naïve CML patients compared to the imatinib-treated CML group (p<0.0001). Of the imatinib-treated CML patients, 32.9% had pericardial effusion, while 45.7% of the imatinib-naive CML patients showed evidence of pericardial effusion. The imatinib-treated CML group had a higher AOD of 2.92 ± 0.31 compared to the other two groups; however, the difference between the CML groups with or without imatinib was not significant. The mean left ventricular posterior wall thickness (LVPWT) was significantly higher in the CML group without imatinib and in non-CML controls than that in the imatinib-treated CML group (p<0.026).
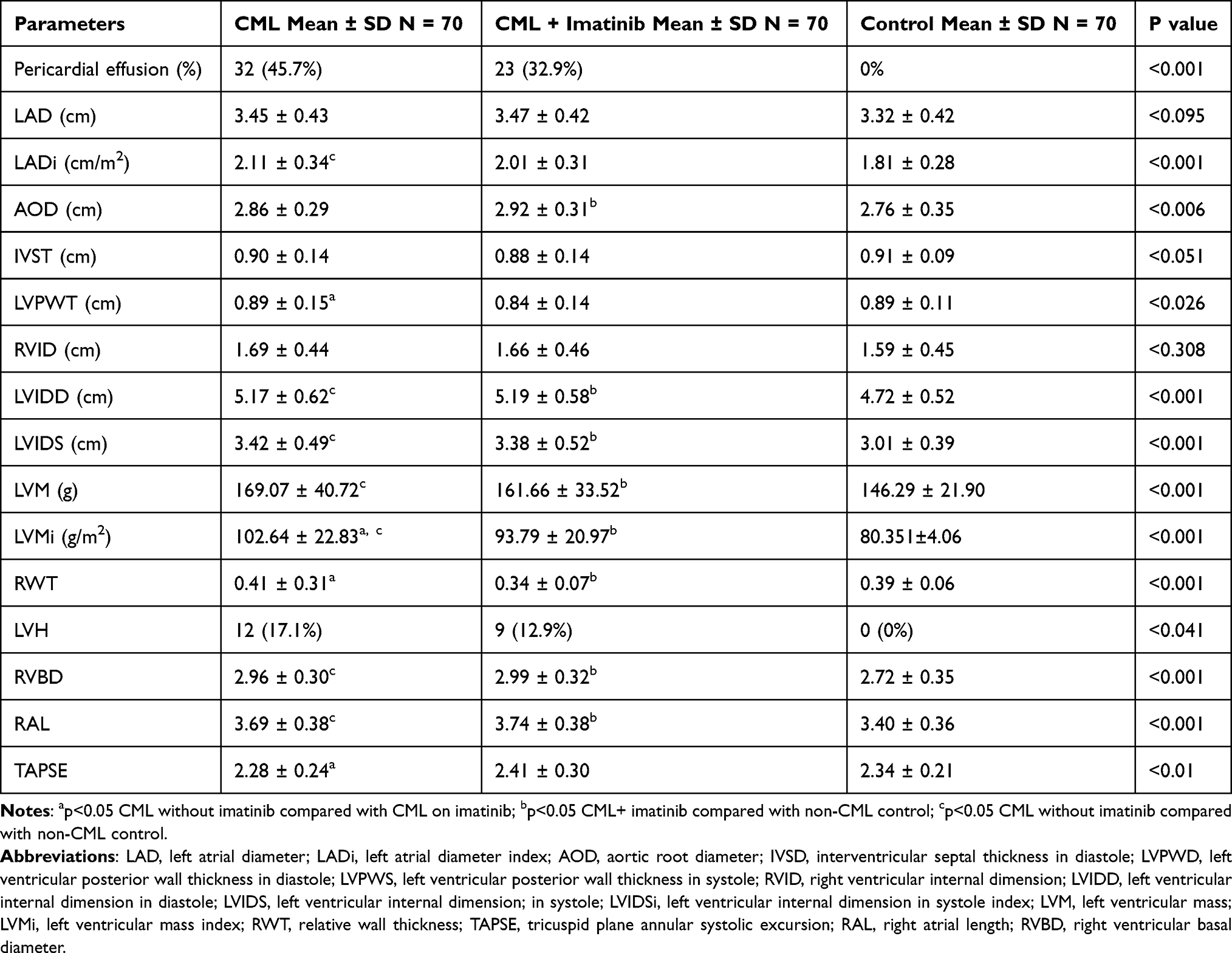 |
Table 2 Two-Dimensional and M-Mode Echocardiographic Parameters of the Study Population |
The mean left ventricular internal dimension in systole (LVIDS) and left ventricular mass (LVM) were significantly higher in the CML groups with or without imatinib than in the non-CML group. Left ventricular hypertrophy (LVH) was observed in 17.1% of imatinib-treated CML patients, while 12.9% of imatinib-naïve CML patients had LVH. In Table 3 the mean stroke volume was higher in the imatinib-treated CML group compared with the other two groups and was significantly higher than the control group (86.20 ± 20.54 mL vs 70.76 ± 17.47 mL; p<0.001). The mean left ventricular ejection fraction was higher in the control group (66.79 ± 4.38%) than the other groups and was significantly higher than in the imatinib-naïve CML group (64.26 ± 6.16%) (p<0.029). There was no statistically significant difference in the mean left ventricular fractional shortening across the three groups (p<0.131). None of the three groups had LVEF <50%.
 |
Table 3 Left Ventricular Systolic Function in the Study Population |
Table 4 summarizes the Doppler echocardiographic findings in the study population. There were no significant differences among the groups for MV E velocity, MV A velocity, and MV E/A ratio (p=0.051, 0.707, and 0.237, respectively); and the deceleration time was longer in imatinib-naïve CML patients and imatinib-treated CML patients compared with the non-CML control group (p<0.004). The isovolumic contraction time was also significantly longer in the imatinib-treated CML group and the imatinib-naïve CML group than in the non-CML control group (p<0.001). There was no significant difference in the isovolumic relaxation time between imatinib-naïve CML patients and imatinib-treated CML patients; however, the isovolumic relaxation time was significantly higher in non-CML controls than in the other two groups (p<0.001). The difference in the TEI across the three groups was not statistically significant (p<0.175). The TV E, TV A velocities, and TV E/A ratios were not significantly different across the groups. There was no significant difference in the e’ and MV E/e’ across the three groups; however, the means of the a’ and s’ velocities were significantly higher in the imatinib-naive CML group than in the control group, and the imatinib-treated CML group had higher mean velocities than the control group. The mean e’/a’ ratio was significantly lower in the imatinib-naïve CML group than in the other groups.
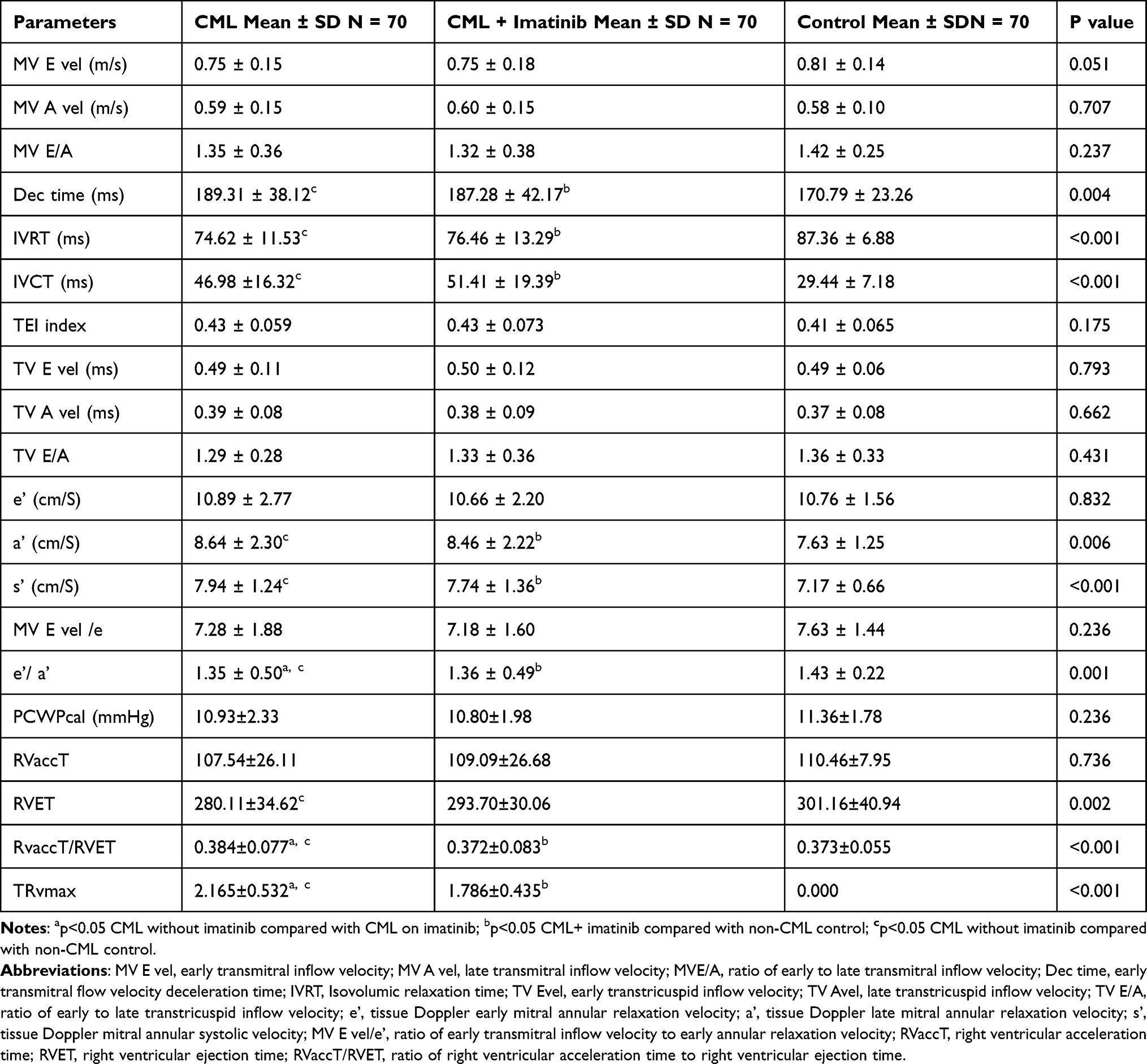 |
Table 4 Doppler Echocardiographic Characteristics of the Study Population |
Twenty (28.57%) imatinib-naive CML patients had diastolic dysfunction, 18 (25.71%) imatinib-treated CML patients had diastolic dysfunction, only 6 (8.57%) of the non-CML controls had diastolic dysfunction (p=0.001). Mitral regurgitation was commoner in the CML group without imatinib, 40%, compared with the other groups. The CML group on imatinib had a higher 34.3% frequency of mitral regurgitation compared with 12.9% in the non-CML control group (P<0.001). Tricuspid regurgitation was more prevalent in the CML group without imatinib, 35.7% compared with 25.7% in the CML group with imatinib and 0% in the non-CML control groups (P<0.001). The frequency of aortic regurgitation was 5.7% in the CML groups with or without imatinib but 0% in the non-CML control. There was no significant difference in AR across the three groups (P=0.125).
PHT in the Study Population
Figure 1 summarizes the prevalence of PHT in the study subjects. The prevalence of PHT was 2.9% in the imatinib-naïve CML group, and none of the other groups had PHT. However, when PHT was defined by RV acceleration time/(RVET) <0.3 ms and RV acceleration time <100 ms, the frequency of PHT was 20% in the imatinib-naïve CML group, which was significantly higher than the control group (p<0.001). All the CML patients without imatinib who had a mean pulmonary artery pressure >25 mmHg also had PHT as defined by RV acceleration time/(RVET) <0.3 ms and RV acceleration time <100 ms. The frequency of PHT was 17.14% in the CML group treated with imatinib, which was significantly higher than that in the non-CML control group.
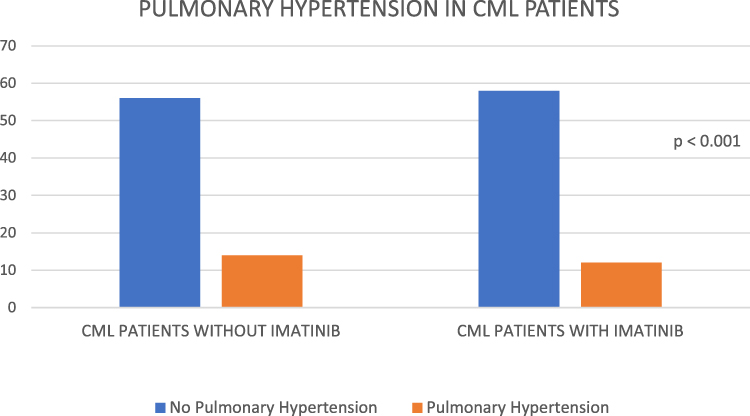 |
Figure 1 Pulmonary hypertension using right ventricular acceleration time/right ventricular ejection time <0.3 ms, and right ventricular acceleration time <100 ms. |
ECG Abnormalities in the Study Subjects
As shown in Table 5, imatinib-treated patients had a higher incidence of sinus bradycardia, premature atrial complexes, ST-segment depression, or elevation and T-wave abnormalities. However, this difference was not statistically significant. The incidence of first-degree AV block was higher in the imatinib-naïve CML group.
 |
Table 5 ECG Abnormalities in Study Subjects |
As shown in Table 6, there was no significant difference in PR intervals across the groups. The imatinib-treated CML group had a significantly longer QRS duration than the non-CML control group. The mean QT and QTC intervals across the groups were not significantly different. The mean P-wave duration was longer in the non-CML control group than in the other groups.
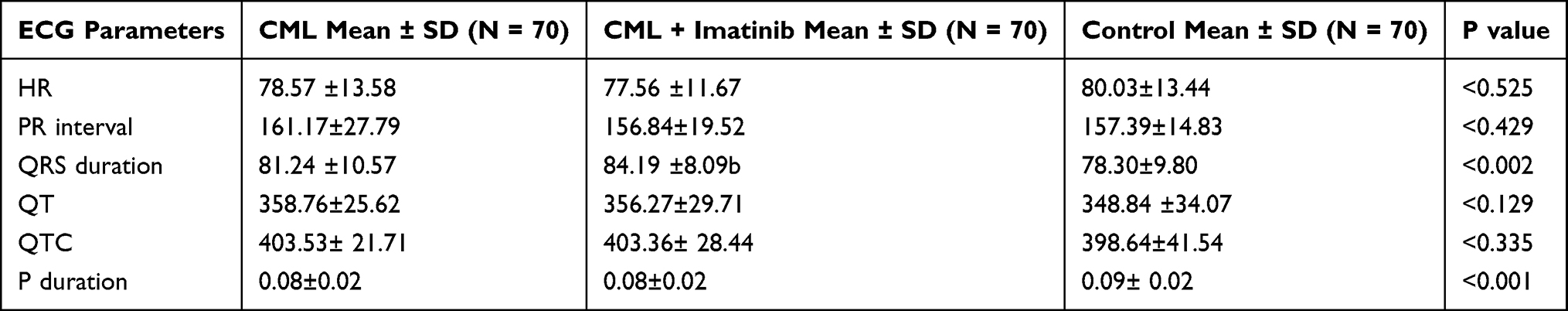 |
Table 6 ECG Parameters in the Study Subjects |
Discussion
Clinical and Demographic Characteristics of Participants
This study included 70 imatinib-treated CML patients, 70 imatinib-naïve CML patients, and 70 apparently healthy age- and sex-matched non-CML patients. In this study, more men than women had CML, similar to observations reported in other African studies.27–30
The mean age of CML patients with or without imatinib was 39.78 ± 14.8 years, which compares favorably with the study of Omoti et al31 who reported a mean age of 38.8 years. Louw et al32 reported a mean age of 42 years, and Mendizabal et al33 reported a mean age of 39.5 years. The age range observed in this study was 18–79 and 18–82 years for the CML groups with and without imatinib, respectively, which is similar to previous Nigerian studies.28,29 A double age peak for the disease was seen in this study, similar to previous studies.27 This study corroborates previous studies’29,34,35 assertion that CML occurs in younger age groups in Africans, since the median age of CML patients was 36 years, while the modal age is 30 years.
Body Weight and BMI
There was a significant difference in body weight and BMI between the imatinib-naïve CML group, the imatinib-treated CML group, and the apparently healthy controls. There was also a statistically significant difference in weight and BMI between the imatinib-naïve and imatinib-treated CML groups. In addition, the weight and BMI of the imatinib-naïve CML group were significantly lower than those of the imatinib-treated CML group.
In the absence of treatment failure, most patients with CML show clinical improvement and attain cytological and hematological responses within 3 months of imatinib treatment.36 Improvement in oral intake may account for the significantly higher mean weight and BMI in the imatinib-treated CML group than in the imatinib-naive CML group. This observation is somewhat similar to the findings of Breccia et al,37 who reported that 50% of CML patients treated with TKIs had a normal BMI. In addition, the release of cachexin and other cytokines can lead to weight loss in CML groups.
Laboratory Parameters Among the Study Population
There was a significant difference in hematocrit (HCT) levels among the study subjects. Imatinib-naïve CML patients were more anemic than imatinib-treated CML patients. However, non-CML controls had normal hematocrit. This finding is similar to those of other studies.9,29 Anemia can be explained by the fact that CML patients have poor oral intake, and therefore often suffer from nutritional anemia, reflecting either iron deficiency anemia or megaloblastic anemia. In addition, patients with CML may also have anemia due to chronic disease. Although most patients with CML on imatinib attain hematological and cytogenetic remission after 3–6 months of treatment with attendant improvement in hematological parameters,36 it is possible that imatinib may cause myelosuppression.38
White blood cell counts in the CML group were significantly higher than those in the non-CML control group. This finding is similar to those observed in previous Nigerian studies.29 The white blood cell count was significantly higher in the imatinib-naïve CML group than in the imatinib-treated CML group. This is likely due to hematological remission, which is rapidly attained in CML patients treated with imatinib. The mean percentages of lymphocytes and neutrophils were significantly lower in the CML group without imatinib than in the other groups.
Platelet count was also higher in the imatinib-naïve CML group as compared to other groups, which corroborates several previous studies.29 The BCR/ABL1 ratio was significantly higher in the imatinib-naïve CML group than in the CML group who had been on imatinib for at least 3 months. This likely reflects a reduction in the BCR-ABL 1 transcript level to ≤10%, which usually occurs after 3 months of treatment.36
A case of acute renal failure was reported by Pou et al39 in an imatinib-treated CML patient; however, none of the patients in our study had any clinical or biochemical evidence of acute renal failure. Previous studies have shown normal renal function in patients with CML with or without imatinib.40
Cardiovascular Parameters
Diastolic blood pressure was lower in the imatinib-treated CML group than in the imatinib-naive CML group, similar to results obtained by Ribeiro et al.40 Systolic blood pressure was lower in the imatinib-naive CML group than in the imatinib-treated CML group.
Anemia is one of the causes of hyperdynamic circulation, and the higher levels of anemia in the CML group, and particularly in the imatinib-naïve CML group, may account for the significantly higher stroke index, cardiac output, and cardiac index in the imatinib-naïve CML group.41 The mean cardiac index in the imatinib-naïve CML group was higher than normal. Weiskopf et al42 observed that a lower hemoglobin concentration corresponds with a higher heart rate, stroke volume index, and cardiac index. They also noted a 58% reduction in the systemic vascular resistance index.
Pericardial Effusion
Pericardial effusion was observed in 45.7% of the imatinib-naïve CML patients, 32.9% of the imatinib-treated CML patients, and none of the non-CML controls. Previously, Cassis et al43 reported a case of massive hemopericardium as the initial presentation of CML. Quintás-Cardama et al44 found that 29% of the patients with concurrent pleural effusion had pericardial effusion. Huang et al45 reported a case of cardiac tamponade as the initial presentation of a blast crisis in CML. According to Hammoud et al,46 pericardial effusion in patients with malignancy can be malignant pericardial effusion, radiation-induced pericarditis, drug-induced pericarditis, or idiopathic pericarditis. Huang et al45 provided a clearer description of the mechanisms through which pericardial effusion develops in patients with CML, including leukemic cell infiltration, extramedullary hematopoiesis, infection, and bleeding. More patients in our study had an echocardiographic diagnosis of pericardial effusion compared with the findings of Kadikoylu et al.9 This may be explained by the late presentation of CML in Nigeria. According to Krauth et al,47 serosal inflammation develops more commonly in patients with advanced disease. In addition, PHT is associated with the development of pericardial effusion according to Bossone et al,48 where the attributed pericardial effusion in PHT patients to impaired venous and lymphatic drainage secondary to elevated right atrial pressure. Some CML patients in this study had PHT, which might have contributed to the frequency of pericardial effusion in this study.
Echocardiographic Parameters
This study found significantly higher left atrial diameter index (LADi) in the imatinib-naïve CML group than in the control group. However, no significant differences were observed between the CML groups. The LAD in this study was not significantly different across the groups. The LAD in the imatinib-naïve CML, imatinib-treated CML, and non-CML control groups was 3.45 ± 0.43, 3.47 ± 0.42 and 3.32 ± 0.42, respectively. This is similar to a previous study that found a LAD of 3.5 ± 0.6 and 3.5 ± 0.5 in the imatinib-naïve CML and non-CML control groups, respectively.9 In addition, Ribeiro et al40 found a median LAD of 3.6 in CML patients treated with or without imatinib. The AOD was significantly higher in the CML group treated with imatinib than in the non-CML control group. There was no significant difference in the AOD between the two CML groups. The AOD in this study in the CML group without imatinib was 2.86 ± 0.29, which compares favorably with the 3.10 ± 0.3 found by Kadikoylu et al.9 The LVIDD, LVIDS, LVM, RVBD, and RAL values were significantly higher in the CML groups with or without imatinib than in the non-CML controls (P<0.001). The difference in the aforementioned cardiac dimensions between the two CML groups was not statistically significant. The LVIDD for the CML group without imatinib and non-CML controls in this study was 5.17 ± 0.62 and 4.72 ± 0.52, respectively, Kadikoylu et al9 found LVIDD to be 5.0 ± 0.4 and 4.9 ± 0.5 in the CML group without imatinib and non-CML controls respectively. In this study, LVM and LVMi were significantly higher in the CML groups with or without imatinib than in the non-CML controls.
LVH detected by echocardiography was observed in 17.1% of the CML group without imatinib and 12.9% of the CML group with imatinib. None of the non-CML controls showed left ventricular hypertrophy. LVH has been linked to multiple factors leading to cardiac growth and differentiation in chronically anemic patients.41,49 According to Kerkela et al, LVH is an adaptive response to a long-term increase in volume or pressure overload.50 An alteration in left ventricular wall stress is the primary signal for LVH, which develops in a pattern specific to the inciting signal (pressure or volume overload). Volume overload is a result of chronic anemia in CML patients and primarily results in the addition of new sarcomeres in series and parallel. This pattern results in a wall thickness sufficient to counterbalance the increased radius (eccentric hypertrophy).41,51 This suggests that the relative wall thickness remains within the normal range, as observed in this study. The RWT was significantly higher in the imatinib-naïve CML group than in the other two groups, and was also higher in the control group than in the imatinib-treated CML group. However, none of the subjects in the three groups had a relative wall thickness >0.45.
The RVIDD in this study was not significantly different between groups. The imatinib-naïve CML group had a mean RVIDD of 1.69 ± 0.44. In comparison, Ribeiro et al40 reported a median value of 1.8. Therefore, our data corroborate Ribeiro et al’s finding that imatinib has no direct effect on the right ventricular internal dimension.
Tricuspid plane annular systolic excursion (TAPSE) was lower in the imatinib-naïve CML group than in the imatinib-treated CML group. All the studied subjects had normal TAPSE, indicating that right ventricular systolic function was preserved in CML patients with or without imatinib.
Doppler Findings
This study showed no significant differences in mitral A velocity, E velocity, or E/A ratio across the groups. This compares favorably with the study by Kadikoylu et al,9 which reported an MV E velocity of 0.75 ± 0.15, an A velocity of 0.59 ± 0.15 and an E/A ratio of 1.35 ± 0.36 in imatinib-treated CML patients, and an E velocity 0.81 ± 0.14, and A velocity of 0.58 ± 0.1, and E/A ratio of 1.42 ± 0.25 in non-CML controls. In this study the mitral E velocity was 0.75 ± 0.15, A velocity was 0.59 ± 0.15 and E/A ratio was 1.35 ± 0.36 in the imatinib-naïve CML group. The mitral deceleration time was longer in imatinib-naïve CML patients than in imatinib-treated CML patients; however, this difference was not statistically significant. This finding is consistent with that of Ribeiro et al.40 Given that the hematocrit was significantly lower and the heart rate was significantly higher in the imatinib-naïve CML than in the other groups, the higher degree of anemia might have led to the tachycardia observed in this group. This could then have led to ischemia, increased ventricular remodeling, increased left ventricular stiffness, and a longer mean deceleration time.
The E/e ratio in this study was not significantly different across the groups (p=0.236). The E/e’ ratios were 7.18 ± 1.60 and 7.28 ± 1.88 in the CML groups with and without imatinib, respectively. Ribeiro et al40 found E/e’ ratios of 7.1 and 7.4 in the CML groups with and without imatinib, respectively (p=0.34).
Cardiac Function: Systolic Function
This study found no significant difference in the mean ejection fraction between CML groups with or without imatinib. The ejection fractions in the CML groups with and without imatinib were 64.77 ± 6.89 and 64.26 ± 6.16, respectively. This is similar to the median ejection fraction of 68 and 69 in CML patients with and without imatinib, respectively, reported by Ribeiro et al.40 Kadikoylu et al9 reported an ejection fraction of 66 ±7 in CML patients without imatinib and 67 ± 5 in non-CML controls. None of the CML patients with or without imatinib had an LVEF <50%. However, Kerkela et al52 reported a lower ejection fraction of 56 ± 7% in 10 imatinib-naïve patients with CML. These patients developed symptoms of heart failure, and ejection fraction was reduced to 25 ± 8% [(P < 0.001) versus pre-treatment ejection fraction] after a mean of 7.2 ± 5.4 months on imatinib. Kerkela et al52 also performed myocardial biopsies on two individuals with no history of coronary artery disease. Transmission electron micrographs of the hearts showed prominent membrane whorls in the myocytes of the two patients. The aforementioned pathology, though nonspecific, has been reported to be a characteristic of toxin-induced myopathies.53 In addition, the Kerkela et al52 study has been criticized by Atallah et al,54 since only 10 imatinib-treated patients who had developed congestive heart failure were included. In addition, the study by Kerkela et al52 did not assess the frequency of this adverse event or its associated potential risk factors. Tiribelli et al55 argued that imatinib is potentially cardiotoxic at concentrations that are higher than those used in current clinical practice and that Kerkela et al’s52 observed pathological changes in cardiac myocytes do not necessarily translate to clinically significant cardiotoxicity. Consequently, heart failure is a rare event in imatinib-treated CML patients, as observed in several studies.56–58 A prospective study of CML patients who took imatinib for a mean duration of 3.4 years was performed by Estabragh et al,59 in which the authors reported no evidence of myocardial deterioration either at baseline or after 12 months of imatinib treatment. They further concluded that imatinib cardiotoxicity was not an important clinical consideration for patients with CML or their advisors.
Cardiac Function: Diastolic Function
In this study, 25.71% of the imatinib-treated CML group and 28.57% of the imatinib-naïve CML group had impaired left ventricular relaxation, while only 8.57% of the non-CML controls had diastolic dysfunction. These results are similar to those of Kadikoylu et al.9
Diastolic dysfunction is present in 25% of the CML population, according to Kadikoylu et al,9 whereas Hassanain et al60 found diastolic dysfunction in 6.25% of the studied CML population. Considering that the mean duration of symptoms at presentation in our patients was 15.77 ± 17.36 months, it is likely that many of our CML patients presented late. This might explain the more severe reduction in mean hematocrit observed in this group, which may have led to the tachycardia with subsequent ischemia, increased ventricular remodeling, and increased left ventricular stiffness.
Valvular Abnormalities
In this study, the most prevalent valvular abnormality was mitral regurgitation (MR). MR was significantly higher in the imatinib-naïve CML group than in the non-CML control group. The prevalence of mitral regurgitation in the CML group with and without imatinib and in non-CML controls was 40%, 34.3%, and 12.9%, respectively. These values are similar to those reported by Kadikoylu et al,9 where MR prevalence was 50% in CML patients without imatinib and 13% in non-CML controls. Hassan et al60 reported MR in 31.25% of CML subjects, and 6.7% of the control subjects. The prevalence of valvular regurgitation was not significantly different between CML groups with and without imatinib. Tricuspid regurgitation was seen in 35.7% of CML patients without imatinib, 25.7% of CML patients with imatinib, but in none of the controls. In comparison, Kadikoylu et al9 found that the frequency of tricuspid regurgitation was 38% in imatinib-naïve CML patients. In addition, we observed aortic regurgitation in 5.7% of CML patients with or without imatinib, and in none of the non-CML controls. This is similar to the prevalence reported by Hassan et al,60 which was 6.25% in CML subjects. There was no significant difference in the frequency of aortic regurgitation between the CML groups. The prevalence of aortic regurgitation in our study was similar to, although lower than, the prevalence reported by Kadikoylu et al.9 Valvular abnormalities were observed in 63% of the patients with myeloproliferative disorders, studied by Reisner et al.61
In myeloproliferative disorders, clonal involvement of megakaryocytopoiesis results in elevated levels of platelet-specific proteins, increased thromboxane generation, and expression of activation-dependent epitopes on the platelet surface. This then leads to increased microthrombi formation and deposition on the heart valves, which can cause valvular damage.62
Reisner et al61 noted that valvular damage and regurgitation may result from high pressure, turbulence, and jet effects that are prominent in the left heart in a hyperdynamic state caused by anemia, a condition that is more prominent in CML patients without imatinib.
In addition, anemia is an independent predictor of moderate or severe functional MR in non-ischemic DCM patients, according to Tigen et al.63
PHT
In this study, PHT was observed in 20% of the imatinib-naïve CML patients and 17.14% of the imatinib-treated CML patients. None of the non-CML controls had PHT. The frequency of PHT was significantly higher in the CML groups with or without imatinib than in the non-CML control group. Similarly, Kadikoylu et al9 reported a 25% frequency of PHT in imatinib-naïve CML patients. In another study, PHT was observed in 13.3% of the patients with myeloproliferative disorders.61
In this study, the prevalence of PHT was lower in the imatinib-treated CML group than in the imatinib-naïve CML group.
12-Lead Surface Electrocardiographic Parameters
In this study, the mean heart rates were not significantly different between the groups. PR interval, QT interval, and corrected QTC were not significantly different between the groups. The PR interval was prolonged in three of the CML patients without imatinib, putting the frequency of first-degree AV block in this group at 4.29%. Neither the imatinib-treated CML group nor the non-CML control had a prolonged PR interval. The mean QTC interval seen in this study was 403.53 ± 21.71, 403.36 ± 28.44 and 398.64 ± 41.54 in the CML group without imatinib, with imatinib, and the non-CML control, respectively. This is similar to data from Ribeiro et al40 who reported a QTC interval of 410 ± 22 and 410 ± 22 in the CML groups with and without imatinib. There was no significant difference in the QTC interval. None of the participants in this study had a prolonged QTC interval. Studies have linked prolonged QTC intervals with the use of nilotinib, a more potent derivative of imatinib.64,65
The average QRS durations found in this study were 84.19 ± 8.09, 81.24 ± 10.57 and 78.30 ± 9.80 for the CML groups with imatinib, without imatinib, and the non-CML control, respectively. The CML group treated with imatinib had a significantly longer mean QRS duration than the non-CML group (p=0.002). There was no significant difference in QRS duration in this study. The average QRS duration in this study was similar to that reported by Ribeiro et al.40
Sinus bradycardia was observed in 2.9% and 4.3% of CML patients with and without imatinib, respectively, and none of the non-CML controls had sinus bradycardia. The prevalence of sinus tachycardia in this study may be a result of anemia in the CML groups. There were no incidences of atrial fibrillation or flutter in the study subjects. This finding also agrees with those of Ribeiro et al40 The frequency of atrial fibrillation according to Kadikoylu et al9 in CML patients was 13%. The mean age of the CML group studied by Kadikoylu et al9 was higher than that of our study subjects, which might account for the higher incidence of atrial fibrillation. By contrast, Ribeiro et al40 reported the frequency of ventricular premature complexes as 4% and 2% in CML patients with and without imatinib, respectively. However, in our study, the patients had atrial premature complexes, rather than ventricular premature complexes. There was no significant difference in the frequency of electrocardiographically diagnosed left ventricular hypertrophy in the CML groups with or without imatinib in this study, which is similar to Ribeiro et al.40 Finally, in our study, T-wave abnormalities were seen in 5.71% of imatinib-naïve CML and 12.86% of CML patients on imatinib, similar to the findings of Ribeiro et al.40
Conclusions
Systolic dysfunction was not observed in imatinib-naïve or imatinib-treated CML patients or non-CML controls. The incidence of diastolic dysfunction was higher in the CML group than in the non-CML group. Pericardial effusion was observed in the CML group, but none of the non-CML controls had pericardial effusion. Valvular regurgitation was more common in the CML group than in the non-CML control group, and the most prevalent valvular abnormality in the CML group was mitral regurgitation.
Due to the lack of resources and time constraint global longitudinal strain, troponin I and a follow-up study could have been performed in these groups of patients. This would have helped detect the development of more subtle cardiac abnormalities that may be attributed to imatinib.
Abbreviations
ACC, American College of Cardiology; AHA, American Heart Association; ASE, American Society of Echocardiography; AV BLOCK, atrioventricular block; BCR-ABL gene, breakpoint cluster region-Abelson fusion gene; BP, blood pressure; BMI, body mass index; BSA, body surface area; CML, chronic myeloid leukaemia; DBP, diastolic blood pressure; HIV, human immunodeficiency virus.; HR, heart rate; LV, left ventricle; LVH, left ventricular hypertrophy; LVM, left ventricular mass; LVMI, left ventricular mass index; MAP, mean arterial blood pressure; MPAP, mean pulmonary arterial pressure; PASP, pulmonary artery systolic pressure; PAC, premature atrial complexes; PCWP, pulmonary capillary wedge pressure; Ph+, Philadelphia chromosome positive; PVC, premature ventricular contraction; RAP, right atrial pressure; RVSP, right ventricular systolic pressure; RAL, right atrial length; RVBD, right ventricular basal diameter; RWT, relative wall thickness; TAPSE, tricuspid annular plane systolic excursion; TKi, tyrosine kinase inhibitor; SBP, systolic blood pressure; SVT, supraventricular tachycardia; VT, ventricular tachycardia; WHO, World Health Organization.
Funding
There is no funding to report.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Frazer R, Irvine AE, McMullin MF. Chronic myeloid leukaemia in the 21st century. Ulster Med J. 2007;76(1):8–17.
2. Omunakwe HE, Nwauche CA, Kaladada KI. Chronic Myeloid Leukemia: clinical and laboratory features at presentation to a referral hospital in Southern Nigeria. Blood. 2013;122(21):5174. doi:10.1182/blood.V122.21.5174.5174
3. Sanyaolu AA, Yemisi BA, Muheez AD, Akeem OL. Otological diseases in patients with chronic Myeloid Leukemia. J Leukemia. 2014;2(128):1–3.
4. O’Brien S, Berman E, Moore JO, et al. NCCN Task Force report: tyrosine kinase inhibitor therapy selection in the management of patients with chronic myelogenous leukemia. J Natl Compr Cancer Netw. 2011;9(Suppl 2):
5. Russo D, Martinelli G, Malagola M, et al. Effects and outcome of a policy of intermittent imatinib treatment in elderly patients with chronic myeloid leukemia. Blood. 2013;121(26):5138–5144. doi:10.1182/blood-2013-01-480194
6. Lang K, McGarry LJ, Huang H, Dorer D, Kaufman E, Knopf K. Mortality and vascular events among elderly patients with chronic myeloid leukemia: a retrospective analysis of linked SEER-medicare data. Clin Lymphoma Myeloma Leuk. 2016;16(5):275–285.e271. doi:10.1016/j.clml.2016.01.006
7. O’Brien SG, Guilhot F, Larson RA, et al. Imatinib compared with interferon and low-dose cytarabine for newly diagnosed chronic-phase chronic myeloid leukemia. New Eng J Med. 2003;348(11):994–1004. doi:10.1056/NEJMoa022457
8. Saif MW, Khan U, Greenberg BR. Cardiovascular manifestations of myeloproliferative disorders: a review of the literature. Hosp Phy. 1999;23:43–54.
9. Kadikoylu G, Onbasili A, Tekten T, Barutca S, Bolaman Z. Functional and morphological cardiac changes in myeloproliferative disorders (clinical study). Int J Cardiol. 2004;97(2):213–220. doi:10.1016/j.ijcard.2003.08.013
10. Vardiman JW, Harris NL, Brunning RD. The World Health Organization (WHO) classification of the myeloid neoplasms. Blood. 2002;100(7):2292–2302. doi:10.1182/blood-2002-04-1199
11. Henry WL, DeMaria A, Gramiak R, et al. Report of the American Society of Echocardiography Committee on nomenclature and standards in two-dimensional Echocardiography. Circulation. 1980;62(2):212–217. doi:10.1161/01.CIR.62.2.212
12. Sahn DJ, DeMaria A, Kisslo J, Weyman A. Recommendations regarding quantitation in M-mode echocardiography: results of a survey of echocardiographic measurements. Circulation. 1978;58(6):1072–1083. doi:10.1161/01.CIR.58.6.1072
13. Devereux RB, Alonso DR, Lutas EM, et al. Echocardiographic assessment of left ventricular hypertrophy: comparison to necropsy findings. Am J Cardiol. 1986;57(6):450–458. doi:10.1016/0002-9149(86)90771-X
14. Ajayi OE, Ajayi EA, Akintomide OA, et al. Ambulatory blood pressure profile and left ventricular geometry in Nigerian hypertensives. J Cardiovasc Dis Res. 2011;2(3):164–171. doi:10.4103/0975-3583.85263
15. Levy D, Savage DD, Garrison RJ, Anderson KM, Kannel WB, Castelli WP. Echocardiographic criteria for left ventricular hypertrophy: the Framingham Heart Study. Am J Cardiol. 1987;59(9):956–960. doi:10.1016/0002-9149(87)91133-7
16. Diaz RA, Obasohan A, Oakley CM. Prediction of outcome in dilated cardiomyopathy. Br Heart J. 1987;58(4):393–399. doi:10.1136/hrt.58.4.393
17. Wiegers SE. Diastolic echo parameters: meaningless numbers or crucial information? Circ Cardiovasc Imaging. 2011;4(5):460–462. doi:10.1161/CIRCIMAGING.111.968131
18. Bukachi F, Waldenström A, Mörner S, Lindqvist P, Henein MY, Kazzam E. Pulmonary venous flow reversal and its relationship to atrial mechanical function in normal subjects – Umeå General Population Heart Study. Eur Heart J Cardiovasc Imaging. 2005;6(2):107–116.
19. Akintunde AA. The clinical value of the Tei index among Nigerians with hypertensive heart failure: correlation with other conventional indices: cardiovascular topics. Cardiovasc J Afr. 2012;23(1):40–43. doi:10.5830/CVJA-2011-032
20. Tei C, Ling LH, Hodge DO, et al. New index of combined systolic and diastolic myocardial performance: a simple and reproducible measure of cardiac function–a study in normals and dilated cardiomyopathy. J Cardiol. 1995;26(6):357–366.
21. Galiè N, Hoeper MM, Humbert M, et al. Guidelines for the diagnosis and treatment of pulmonary hypertension. Eur Heart J. 2009;30(20):2493–2537. doi:10.1093/eurheartj/ehp297
22. Chemla D, Castelain V, Humbert M, et al. New formula for predicting mean pulmonary artery pressure using systolic pulmonary artery pressure. CHEST J. 2004;126(4):1313–1317. doi:10.1378/chest.126.4.1313
23. Sciomer S, Magrì D, Badagliacca R. Non-invasive assessment of pulmonary hypertension: Doppler–echocardiography. Pulm Pharmacol Ther. 2007;20(2):135–140. doi:10.1016/j.pupt.2006.03.008
24. Rudski LG, Lai WW, Afilalo J, et al. Guidelines for the echocardiographic assessment of the right heart in adults: a report from the American Society of Echocardiography: endorsed by the European Association of Echocardiography, a registered branch of the European Society of Cardiology, and the Canadian Society of Echocardiography. J Am Soc Echocardiogr. 2010;23(7):685–713. doi:10.1016/j.echo.2010.05.010
25. McLaughlin VV, Archer SL, Badesch DB, et al. ACCF/AHA 2009 expert consensus document on pulmonary hypertension: a report of the American College of Cardiology Foundation Task Force on expert consensus documents and the American Heart Association developed in collaboration with the American College of Chest Physicians; American Thoracic Society, Inc.; and the Pulmonary Hypertension Association. J Am Coll Cardiol. 2009;53(17):1573–1619. doi:10.1016/j.jacc.2009.01.004
26. Karaye KM, Saidu H, Bala MS, Yahaya IA. Prevalence, clinical characteristics and outcome of pulmonary hypertension among admitted heart failure patients. Ann Afr Med. 2013;12(4):197–204. doi:10.4103/1596-3519.122685
27. Fleming AF. Epidemiology of the leukaemias in Africa. Leuk Res. 1979;3(2):51–59. doi:10.1016/0145-2126(79)90062-6
28. Okany CC, Akinyanju OO. Chronic leukaemia: an African experience. Med Oncol Tumor Pharmacother. 1989;6(3):189–194. doi:10.1007/BF02985189
29. Boma PO, Durosinmi MA, Adediran IA, Akinola NO, Salawu L. Clinical and prognostic features of Nigerians with chronic myeloid leukemia. Niger Postgrad Med J. 2006;13(1):47–52.
30. Williams O, Bamgboye E. Estimation of incidence of human leukaemia subtypes in an urban African population. Oncology. 1983;40(6):381–386. doi:10.1159/000225769
31. Omoti CE, Imiere EO. Trends in the pattern of leukaemia incidence in a tertiary health center in Nigeria: 1990–2004. J Biomed Sci. 2006;5(2):44–94.
32. Louw VJ. Chronic myeloid leukaemia in South Africa. Hematology. 2012;17(sup1):S75–S78. doi:10.1179/102453312X13336169155817
33. Mendizabal AM, Garcia-Gonzalez P, Levine PH. Regional variations in age at diagnosis and overall survival among patients with chronic myeloid leukemia from low and middle income countries. Cancer Epidemiol. 2013;37(3):247–254. doi:10.1016/j.canep.2013.01.002
34. Oyekunle AA, Bolarinwa RA, Oyelese AT, Salawu L, Durosinmi MA. Determinants of overall and progression-free survival of Nigerian patients with Philadelphia-positive chronic Myeloid Leukemia. Adv Hematol. 2015;2015:1–5. doi:10.1155/2015/908708
35. Durosinmi MA, Faluyi JO, Oyekunle AA, et al. The use of Imatinib mesylate in Nigerians with chronic myeloid leukemia. Cell Ther Transplant. 2008;1(2):58–62.
36. Baccarani M, Deininger MW, Rosti G, et al. European LeukemiaNet recommendations for the management of chronic myeloid leukemia: 2013. Blood. 2013;122(6):872–884. doi:10.1182/blood-2013-05-501569
37. Breccia M, Colafigli G, Molica M, Alimena G. Cardiovascular risk assessments in chronic myeloid leukemia allow identification of patients at high risk of cardiovascular events during treatment with nilotinib. Am J Hematol. 2015;90(5):E100–101. doi:10.1002/ajh.23976
38. Sneed TB, Kantarjian HM, Talpaz M, et al. The significance of myelosuppression during therapy with imatinib mesylate in patients with chronic myelogenous leukemia in chronic phase. Cancer. 2004;100(1):116–121. doi:10.1002/cncr.11863
39. Pou M, Saval N, Vera M, et al. Acute renal failure secondary to imatinib mesylate treatment in chronic myeloid leukemia. Leuk Lymphoma. 2003;44(7):1239–1241. doi:10.1080/1042819031000079140
40. Ribeiro AL, Marcolino MS, Bittencourt HN, et al. An evaluation of the cardiotoxicity of imatinib mesylate. Leuk Res. 2008;32(12):1809–1814. doi:10.1016/j.leukres.2008.03.020
41. Metivier F, Marchais SJ, Guerin AP, Pannier B, London GM. Pathophysiology of anaemia: focus on the heart and blood vessels. Nephrol Dial Transplant. 2000;15(suppl 3):14–18. doi:10.1093/oxfordjournals.ndt.a027970
42. Weiskopf RB, Viele MK, Feiner J, et al. Human cardiovascular and metabolic response to acute, severe isovolemic anemia. JAMA. 1998;279(3):217–221. doi:10.1001/jama.279.3.217
43. Cassis N, Porterfield J, Rogers JS, Shah S, Storey E. Massive hemopericardium as the initial manifestation of chronic myelogenous leukemia. Arch Intern Med. 1982;142(12):2193–2194. doi:10.1001/archinte.1982.00340250159025
44. Quintás-Cardama A, Kantarjian H, O’Brien S, et al. Pleural effusion in patients with chronic myelogenous leukemia treated with dasatinib after imatinib failure. J Clin Oncol. 2007;25(25):3908–3914. doi:10.1200/JCO.2007.12.0329
45. Huang C-T, Yu S-H, Chen Y-H, Lin S-F. Cardiac tamponade as a symptom of the blast crisis of chronic myeloid leukemia. Kaohsiung J Med Sci. 2016;32(3):160–161. doi:10.1016/j.kjms.2016.02.007
46. Hammoud D, Shaheen M, Nassar R. Imatinib (Gleevec) induced worsening of pericardial effusion in CML. Firat Tip Dergisi. 2011;16(2):100–101.
47. Krauth MT, Herndlhofer S, Schmook MT, Mitterbauer-Hohendanner G, Schlogl E, Valent P. Extensive pleural and pericardial effusion in chronic myeloid leukemia during treatment with dasatinib at 100 mg or 50 mg daily. Haematologica. 2011;96(1):163–166. doi:10.3324/haematol.2010.030494
48. Bossone E, D’Andrea A, D’Alto M, et al. Echocardiography in pulmonary arterial hypertension: from diagnosis to prognosis. J Am Soc Echocardiogr. 2013;26(1):1–14. doi:10.1016/j.echo.2012.10.009
49. Rossi M, Carillo S. Pathogenesis of cardiac hypertrophy in iron deficiency anaemia: the role of noradrenaline. Br J Exp Pathol. 1982;63(3):269.
50. Kerkela R, Force T. Recent insights into cardiac hypertrophy and left ventricular remodeling. Curr Heart Fail Rep. 2006;3(1):14–18. doi:10.1007/s11897-006-0026-6
51. O’Riordan E, Foley RN. Effects of anaemia on cardiovascular status. Nephrol Dial Transplant. 2000;15(suppl_3):19–22. doi:10.1093/oxfordjournals.ndt.a027971
52. Kerkela R, Grazette L, Yacobi R, et al. Cardiotoxicity of the cancer therapeutic agent imatinib mesylate. Nat Med. 2006;12(8):908–916. doi:10.1038/nm1446
53. Khan MA. Effects of myotoxins on skeletal muscle fibers. Prog Neurobiol. 1995;46(5):541–560. doi:10.1016/0301-0082(95)00015-N
54. Atallah E, Durand J-B, Kantarjian H, Cortes J. Congestive heart failure is a rare event in patients receiving imatinib therapy. Blood. 2007;110(4):1233–1237. doi:10.1182/blood-2007-01-070144
55. Tiribelli M, Medeot M. Cardiotoxicity of imatinib: at the heart of the problem. Leuk Res. 2011;35(1):36–37. doi:10.1016/j.leukres.2010.09.021
56. Baccarani M, Cilloni D, Rondoni M, et al. The efficacy of imatinib mesylate in patients with FIP1L1-PDGFRα-positive hypereosinophilic syndrome. Results of a multicenter prospective study. Haematologica. 2007;92(9):1173–1179. doi:10.3324/haematol.11420
57. Hochhaus A, O’brien S, Guilhot F, et al. Six-year follow-up of patients receiving imatinib for the first-line treatment of chronic myeloid leukemia. Leukemia. 2009;23(6):1054–1061. doi:10.1038/leu.2009.38
58. Breccia M, Cannella L, Frustaci A, Stefanizzi C, Levi A, Alimena G. Cardiac events in imatinib mesylate-treated chronic myeloid leukemia patients: a single institution experience. Leuk Res. 2008;32(5):835–836. doi:10.1016/j.leukres.2007.08.016
59. Estabragh ZR, Knight K, Watmough SJ, et al. A prospective evaluation of cardiac function in patients with chronic myeloid leukaemia treated with imatinib. Leuk Res. 2011;35(1):49–51. doi:10.1016/j.leukres.2010.08.020
60. Hassan HH, Alawad AS, Murad NS. Echocardiographic evaluation of cardiac involvement in myeloproliferative disorders. Med J Babylon. 2008;5(3–4):556–574.
61. Reisner SA, Rinkevich D, Markiewicz W, Tatarsky I, Brenner B. Cardiac involvement in patients with myeloproliferative disorders. Am J Med. 1992;93(5):498–504. doi:10.1016/0002-9343(92)90576-W
62. Leone G, Sica S, Chiusolo P, Teofili L, De Stefano V. Blood cells diseases and thrombosis. Haematologica. 2001;86(12):1236–1244.
63. Tigen K, Karaahmet T, Kirma C, et al. The association of functional mitral regurgitation and anemia in patients with non-ischemic dilated cardiomyopathy. Cardiol J. 2010;17(3):274–280.
64. Nicolini FE, Turkina A, Shen ZX, et al. Expanding Nilotinib access in clinical trials (ENACT). Cancer. 2012;118(1):118–126. doi:10.1002/cncr.26249
65. Haouala A, Widmer N, Duchosal MA, Montemurro M, Buclin T, Decosterd LA. Drug interactions with the tyrosine kinase inhibitors imatinib, dasatinib, and nilotinib. Blood. 2010;117(8):e75–87. doi:10.1182/blood-2010-07-294330
 © 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.