Back to Journals » Journal of Inflammation Research » Volume 19
Comparative Analysis of Ferroptosis- and Mitochondria-Related Genes in Ischemic Stroke via Bioinformatics Analysis and Experimental Validation
Authors Ma K, Jiang Y, Yang Y, Rao T, Zhan Y ![]() , Yin Z, Dan Y, Xu S, Yang S
, Yin Z, Dan Y, Xu S, Yang S
Received 3 March 2026
Accepted for publication 20 June 2026
Published 10 July 2026 Volume 2026:19 605715
DOI https://doi.org/10.2147/JIR.S605715
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Professor Ning Quan
Ke Ma,1,* Yijing Jiang,2,3,* Yihan Yang,1 Ting Rao,1,2 Ying Zhan,1 Zihan Yin,1 Yuqin Dan,1 Sihan Xu,1 Shanli Yang1,2
1College of Rehabilitation Medicine, Fujian University of Traditional Chinese Medicine, Fuzhou, People’s Republic of China; 2The Affiliated Rehabilitation Hospital of Fujian University of Traditional Chinese Medicine, Fuzhou, People’s Republic of China; 3Fujian Key Laboratory of Rehabilitation Technology, Fuzhou, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Shanli Yang, Email [email protected]
Background: Ischemic stroke (IS) is a major cause of death and long-term disability worldwide. Multiple complex biological processes contribute to IS-related neuronal death, among which oxidative stress plays a central role in disease progression. Increasing evidence suggests that oxidative stress–induced neuronal injury is closely associated with ferroptosis and mitochondrial dysfunction, both of which contribute to excessive reactive oxygen species accumulation, lipid peroxidation, and impaired cellular energy metabolism during cerebral ischemia. However, their relative contributions and associated molecular signatures in IS have not been systematically compared. Therefore, this study aimed to identify ferroptosis- and mitochondria-associated genes involved in oxidative stress and neuronal injury in IS and to explore their potential as therapeutic targets for ischemic brain injury.
Methods: Two public microarray datasets (GSE22255 and GSE58294) were integrated and analyzed using weighted gene co-expression network analysis (WGCNA) to identify IS-associated gene modules. Ferroptosis-related genes from FerrDb and mitochondria-associated genes from MitoCarta3.0 were intersected with key modules to screen candidate genes. Protein–protein interaction analysis and CytoHubba were applied to identify hub genes. Logistic regression models were constructed to compare the diagnostic performance of ferroptosis- and mitochondria-related gene signatures. Functional enrichment analyses were conducted using Gene Ontology and KEGG. Key genes were further validated in a rat middle cerebral artery occlusion/reperfusion (MCAO/R) model treated with the ferroptosis inhibitor Ferrostatin-1 (Fer-1).
Results: Nine ferroptosis-related hub genes and nine mitochondria-related hub genes were identified. The ferroptosis-based diagnostic model showed significantly higher discriminatory power than the mitochondrial model (AUC = 0.949 vs. 0.829). Among these genes, GSK3B, IDH1, and PRDX1 exhibited the most prominent differential expression and were selected as core genes. In vivo experiments demonstrated that Fer-1 markedly reduced infarct volume, improved neurological function, attenuated oxidative stress, and restored the ACSL4/GPX4/TFR1 signaling axis. Fer-1 also reversed MCAO-induced dysregulation of GSK3B phosphorylation, IDH1, and PRDX1 expression, indicating effective suppression of ferroptosis.
Conclusion: Ferroptosis-related gene signatures outperform mitochondria-associated genes in the diagnosis of ischemic stroke. GSK3B, IDH1, and PRDX1 represent key molecular regulators linking oxidative stress to ferroptotic neuronal injury and may serve as promising biomarkers and therapeutic targets for ischemic stroke.
Keywords: ischemic stroke, ferroptosis, oxidative stress, GSK3B, IDH1, PRDX1, bioinformatics analysis
Introduction
Stroke is one of the leading causes of long-term disability and mortality worldwide, among which ischemic stroke (IS) accounts for more than 71% of all stroke cases.1 IS is primarily caused by a sudden interruption of cerebral blood flow, resulting in severe ischemic injury and subsequent permanent neurological dysfunction, which markedly reduces patients quality of life.2 Increasing evidence has demonstrated that multiple complex biological processes are involved in IS-related neuronal death, including oxidative stress, neuroinflammation, excitotoxicity, and various forms of regulated cell death.3 Among these pathological mechanisms, oxidative stress has been recognized as a central contributor to IS progression. In particular, ferroptosis and mitochondrial dysfunction have emerged as two critical and closely interconnected factors contributing to neuronal injury during IS.
Ferroptosis is a regulated form of cell death driven by iron-dependent lipid peroxidation and excessive reactive oxygen species (ROS) accumulation. Increasing evidence indicates that ferroptosis plays an essential role in the pathological progression of IS and significantly influences neurological outcomes.4 During cerebral ischemia/reperfusion (CIR), excessive ROS are rapidly generated, which not only cause secondary injury to the cerebral vasculature but also disrupt neuronal networks, thereby aggravating ischemic damage.5,6 Iron overload further amplifies ROS production through Fenton reactions, promotes lipid peroxidation, and ultimately drives ferroptotic neuronal death, forming a vicious cycle of iron accumulation, oxidative stress, and membrane damage.7,8 Experimental evidence from middle cerebral artery occlusion (MCAO) models has shown that pharmacological inhibition of ferroptosis significantly attenuates neuronal injury and oxidative stress, partly through modulation of the xCT/GPX4 signaling axis.9 Notably, neurons are particularly vulnerable to ferroptosis because of their high content of polyunsaturated fatty acids and limited antioxidant capacity. Collectively, these findings suggest that ferroptosis-related genes may play important regulatory roles in IS progression and could serve as potential diagnostic biomarkers and therapeutic targets.
Mitochondrial dysfunction is another crucial pathological event involved in IS. Following cerebral ischemia, interruption of blood supply results in oxygen and glucose deprivation, which severely impairs mitochondrial respiratory function and ATP production.10 As the primary intracellular source of reactive oxygen species (ROS), dysfunctional mitochondria further exacerbate oxidative stress, while excessive oxidative stress in turn damages mitochondrial structure and regulatory mechanisms, ultimately forming a self-amplifying cycle of mitochondrial injury and ROS accumulation.11 Acute cerebral ischemia has been reported to induce mitochondrial membrane damage, neuronal membrane rupture, blood–brain barrier disruption, and eventual neuronal death.11,12 Increasing evidence suggests that maintaining mitochondrial structural integrity and functional homeostasis is essential for neuronal survival and neurological recovery after IS.13 Recent studies have demonstrated that compounds such as Salidroside and Hesperidin can alleviate ischemic neuronal injury by attenuating mitochondrial calcium overload and suppressing oxidative stress.11 In addition, astrocytic low-density lipoprotein receptor-related protein 1 (LRP1) has been shown to promote mitochondrial transfer to injured neurons through inhibition of the CD44/RhoA signaling pathway, thereby mitigating ischemic brain injury. Collectively, these findings indicate that mitochondria-related genes are closely associated with energy metabolism, oxidative homeostasis, and neuronal survival during IS progression.
Importantly, accumulating evidence indicates that ferroptosis and mitochondrial dysfunction are not independent pathological events but are tightly interconnected during IS.14 Mitochondria are major intracellular sources of ROS and key regulators of iron metabolism, thereby directly participating in the initiation and execution of ferroptosis.15 Conversely, ferroptosis lipid peroxidation can severely impair mitochondrial membrane integrity and bioenergetic function, further amplifying oxidative damage and neuronal injury.16 Given that ferroptosis-related signatures mainly reflect iron-dependent lipid peroxidation, whereas mitochondria-related signatures are more closely associated with cellular energy metabolism and oxidative homeostasis, these two molecular patterns may exhibit distinct diagnostic characteristics in IS. However, despite growing recognition of their critical roles, most previous studies have focused on ferroptosis or mitochondrial dysfunction individually, while systematic comparisons between ferroptosis-related genes and mitochondria-associated genes in IS remain limited. In particular, the relative diagnostic value and predictive performance of these two molecular signatures have not yet been fully elucidated.
Given the complex and multifactorial nature of IS, single biomarkers are often insufficient to achieve accurate and stable diagnosis, highlighting the necessity of constructing multi-gene diagnostic models. Therefore, the present study aimed to: (1) identify key ferroptosis- and mitochondria-related differentially expressed genes in IS; (2) construct and validate a multi-gene diagnostic model using logistic regression analysis; and (3) explore the associated signaling pathways and verify the expression of core diagnostic genes in animal models. Collectively, these findings are expected to provide new insights into the molecular mechanisms underlying IS and facilitate the identification of potential diagnostic biomarkers and therapeutic targets.
Methods
Data Preparation and Processing
Two microarray datasets related to ischemic stroke (IS), GSE22255 and GSE58294, were obtained from the Gene Expression Omnibus (GEO) database (http://www.ncbi.nlm.nih.gov/geo/). Both datasets were generated using the GPL570 platform. GSE22255 contains peripheral blood samples from 20 IS patients and 20 healthy controls, collected between 2010 and 2019. GSE58294 includes peripheral blood samples from 69 IS patients and 23 healthy controls, collected between 2014 and 2019. In the present study, we analyzed the expression data from these datasets. Batch effects between GSE22255 and GSE58294 were corrected using R version 4.4.1 to enable integrated downstream analyses.
Weighted Gene Co-Expression Network Analysis (WGCNA)
WGCNA was conducted to identify key co-expression modules associated with IS. First, sample clustering was performed to remove potential outlier samples and ensure analytical reliability. An appropriate soft-thresholding power (β) was then chosen to approximate a scale-free topology. A weighted adjacency matrix was constructed based on pairwise correlation coefficients, and subsequently transformed into a topological overlap matrix (TOM). Gene modules were identified using the hybrid dynamic tree-cutting algorithm, with the minimum module size set to 150 genes and the MEDissThres parameter set to 0.2 to merge similar modules. Module–trait correlations were then assessed to determine the association between each gene module and clinical traits (IS and control). Genes within the module showing the strongest association with IS were defined as key module genes for further analysis.
Identification of Hub Genes
FerrDb is the first dedicated database for ferroptosis regulators and ferroptosis–disease associations; therefore, ferroptosis-related human genes were downloaded from FerrDb V2 for subsequent analyses. MitoCarta3.0, a curated inventory of human and mouse genes with strong experimental evidence for mitochondrial localization, was used to extract mitochondria-associated human genes. Overlap analyses were then performed between IS-related module genes and ferroptosis-related genes, as well as between IS-related module genes and mitochondria-related genes. The intersecting genes were visualized using Venn diagrams.
To further explore the interactions among the overlapping genes, the STRING database (https://string-db.org/) was used to construct a protein–protein interaction (PPI) network, which was subsequently visualized in Cytoscape v3.10.3. The CytoHubba plugin was applied to calculate node degree and assess the topological importance of each gene. The top 10 genes ranked by degree were identified as hub genes.
Construction and Validation of the Logistic Regression Model
The expression levels of the 10 hub genes were visualized using violin plots, and genes with statistically significant differences between IS patients and controls (P < 0.05) were selected for constructing the logistic regression model. A logistic regression classifier was then established to distinguish IS patients from healthy controls. The predictive performance of the model was assessed using receiver operating characteristic (ROC) curve analysis in R version 4.4.1.
Among the hub genes, those with significant associations (P < 0.05) were incorporated into a nomogram to further estimate the probability of IS occurrence. The expression patterns of the selected hub genes were presented using violin plots to illustrate their differential expression between groups.
Functional Enrichment Analysis
Functional enrichment analyses were conducted based on the three Gene Ontology (GO) categories: biological process (BP), cellular component (CC), and molecular function (MF). In addition, Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analysis was performed to explore the involvement of the selected genes in biological pathways, diseases, and chemical–drug interactions. All enrichment analyses were carried out using the clusterProfiler package in R, enabling the systematic identification of biological functions and pathways associated with the gene set.
Animals and Ethics
Specific pathogen-free (SPF) grade male Sprague–Dawley (SD) rats (Slaughter, Shanghai; Production License No. SCXK [Shanghai] 2023–0004), 2 months of age and weighing 280±30 g, were used in this study. All procedures were approved by the Experimental Animal Centre of Fujian University of Traditional Chinese Medicine (License No. SYXK [Min] 2023–0004). Rats were housed five per cage under controlled environmental conditions (temperature 21–25°C, relative humidity 40–60%, noise <60 dB) with free access to food and water and maintained on a 12h light/dark cycle. Animals were acclimated for one week prior to experimentation.
All animal experiments were approved by the Experimental Animal Management Committee of Fujian University of Traditional Chinese Medicine and conducted in strict accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals. For all surgical procedures, anesthesia was induced and maintained with inhaled isoflurane. Adequate anesthesia was confirmed by the absence of a pedal withdrawal reflex. Post-operative care included maintaining body temperature until full recovery from anesthesia. Humane endpoints were established to minimize animal distress. Animals were monitored daily for signs of severe weight loss (>20% body weight), dehydration, labored breathing, or markedly abnormal behavior (eg, unresponsiveness or immobility). At the end of the experiments, or immediately upon meeting any of these pre-defined humane endpoints, animals were humanely euthanized. Euthanasia was performed by an overdose of inhaled isoflurane followed by cervical dislocation as a secondary physical method, in accordance with the AVMA Guidelines for the Euthanasia of Animals (2020 edition).
A total of 36 rats were randomly assigned to three groups (n = 12 per group): a sham-operated group, an MCAO group, and a Ferrostatin-1 (Fer-1) treatment group. Ferrostatin-1 (Cat No. CM00719, Proteintech), a potent ferroptosis inhibitor that prevents lipid peroxidation,4 was dissolved in 10% NaCl and administered via intraperitoneal injection at a dose of 5 mg/kg/day.17 Fer-1 treatment was initiated 24 h before MCAO/R surgery and continued once daily for seven consecutive days. During the experimental period, all rats had free access to food and water.
Group Allocation and Blinding
Animals were randomly assigned to experimental groups using a random number table. Group allocation was performed by a technician who was not involved in any subsequent parts of the study. During the experiment, the surgeons performing the procedures were aware of the group assignments due to the nature of the surgical model. However, the outcome assessors and the statistician responsible for data analysis were kept blinded to group allocation until the final analysis was completed.
MCAO/R Model
MCAO/R surgery was performed in both the model and Fer-1 treatment groups, following a 12-hour fasting period. Rats were placed in the supine position and anesthetized with 3% isoflurane in a gas mixture containing 30% O2 and 67% N2. A midline cervical incision was made, and blunt dissection was carefully performed to expose the common carotid artery (CCA). The internal carotid artery (ICA), external carotid artery (ECA), and CCA were then isolated. The ECA was ligated to interrupt its blood supply. Subsequently, the CCA was temporarily ligated with a slipknot, and an arterial clip was placed on the ICA. A small incision was made in the CCA using ophthalmic scissors, and a monofilament (spigot) was gently advanced 18–20 mm from the insertion point to occlude the middle cerebral artery. After confirming correct placement, the ligature was tightened, the filament was secured and disinfected, and the skin incision was sutured. After 1.5 hours of occlusion, the filament was withdrawn to allow reperfusion, which continued for 24 hours. In the sham-operated group, only anesthesia and vascular isolation were performed without filament insertion or ischemia induction.
Neurological Deficit Score
After 24 hours of reperfusion, the extent of the neurological deficit was scored using the Zea Longa method. The following points were assigned: no symptoms of neurological deficit = 0 points; inability to fully extend the contralateral forelimb = 1 point; turning to the hemiplegic side when walking = 2 points; tilting to the hemiplegic side when walking = 3 points; inability to walk spontaneously and impaired consciousness = 4 points; death = 5 points. Inclusion criteria: 1) a score of 1 to 3, and 2) no subarachnoid hemorrhage. Rats that did not meet the inclusion criteria were excluded from the experiment.
MRI
At 24 h and 7 days after MCAO/R, localization images and T2-weighted imaging (T2WI) were acquired using a 7.0 T small-animal MRI scanner. T2WI parameters were as follows: TR = 4200 ms, TE = 35 ms, flip angle = 90°, and slice thickness = 1.6 mm. After image acquisition, the raw MRI data were processed for subsequent analysis. The infarct regions and the whole brain were manually delineated using ITK-SNAP software. Infarct volume was calculated using the following formula: infarct volume (%) = (number of infarct voxels / total brain voxel count) × 100%.
Oxidative Stress Level
The oxidative stress biomarkers malondialdehyde (MDA; Nanjing Jiancheng), superoxide dismutase (SOD; Beyotime Biotechnology), and glutathione (GSH; Nanjing Jiancheng) were measured using commercially available assay kits. On ice, ipsilateral ischemic cortical tissues were carefully isolated 7 days after Fer-1 administration and used to prepare a 10% ischemic brain tissue homogenate. The homogenates were then centrifuged, and the supernatants were collected for subsequent analysis. Protein concentration in each sample was determined using a BCA assay kit and recorded accordingly. The same supernatant samples were subsequently used to prepare control and experimental wells according to the manufacturers’ protocols for each assay. All procedures were performed strictly in accordance with the kit instructions. Finally, absorbance was measured using a microplate reader, and the concentrations of MDA, SOD, and GSH were calculated based on the corresponding formulas provided with the kits.
Western Blot
Ipsilateral ischemic cortical tissues were rapidly lysed on ice using RIPA buffer (Boster, Wuhan, China) to extract total proteins. Protein concentrations were determined using a BCA assay kit (Boster, Wuhan, China). Equal amounts of protein were separated by SDS–PAGE (Boster, Wuhan, China), transferred onto PVDF membranes, and subsequently blocked. The membranes were washed with TBST (Boster, Wuhan, China) and incubated with the following primary antibodies at 4 °C overnight: ACSL4/FACL4 Polyclonal antibody (Proteintech, Cat No. 22401-1-AP, 1:10,000), GPX4 Monoclonal antibody (Proteintech, Cat No. 67763-1-Ig, 1:2000), Anti-CD71/TFRC Antibody (Boster, Cat No PB9233, 1:1000), GSK3B Recombinant antibody (Proteintech, Cat No. 82061-1-RR, 1:10,000), IDH1 Polyclonal antibody (Proteintech, Cat No. 12332-1-AP, 1:6000), PRDX1 Polyclonal antibody (Proteintech, Cat No. 15816-1-AP, 1:10,000), Phospho-GSK3B (Ser9) Polyclonal antibody (Proteintech, Cat No. 67558-1-Ig, 1:6000), and GAPDH Monoclonal antibody (Proteintech, Cat No. 60004-1-Ig, 1:20,000) After washing with TBST, membranes were incubated with the corresponding HRP-conjugated secondary antibodies: Goat Anti-Mouse IgG (H+L) (Proteintech, Cat No. SA00001-1, 1:5000) or Goat Anti-Rabbit IgG (H+L) (Proteintech, Cat No. SA00001-2, 1:5000) for 1 h at room temperature. Protein bands were visualized using a chemiluminescent developer solution (Proteintech, Wuhan, China), and band intensities were quantified using ImageJ software.
Rotarod Test
The rotarod test was performed on days 1 and 7 after MCAO/R to evaluate motor endurance and resistance to fatigue. Each rat was initially placed on a stationary rod, and the test commenced once the rod began rotating. Rats that remained on the rod for 1 min at a constant speed proceeded to the accelerating phase, during which the rotation speed increased from 4 m/min to 40 m/min over a 5-min period. The latency to fall was recorded for each trial. Each rat underwent three trials with a 20-min rest interval between trials.
Statistical Analysis
To reduce methodological bias, group allocation was independently randomized by three investigators, and all outcome assessments were performed by a single investigator to ensure consistency in measurement. Statistical analyses for wet lab experiments were carried out using IBM SPSS version 27.0 (SPSS Inc., Chicago, USA). Data are reported as mean ± standard deviation (SD). The normality of each dataset was evaluated using the Shapiro–Wilk test. When data conformed to a normal distribution, comparisons among three groups were conducted using one-way ANOVA, followed by LSD post hoc testing for pairwise contrasts. For datasets that did not meet normal distribution criteria, comparisons between two groups were performed using the Mann–Whitney U-test. Statistical significance was set at P < 0.05. Statistical analyses for bioinformatic data were performed using R version 4.4.1, as detailed in the corresponding Methods sections for each analysis.
ARRIVE Statement
This study was designed, conducted, and reported in accordance with the ARRIVE 2.0 guidelines, including adherence to the Essential 10.
Result
WGCNA and Identification of Core Modules
Weighted gene co-expression network analysis (WGCNA) was performed, and a scale-free network was constructed using a soft-thresholding power of 17 (R2 = 0.9) (Figure 1A). Module eigengenes, representing the principal component of each modules gene expression profile, were subsequently calculated and clustered based on their correlations. A total of seven distinct modules were identified, each assigned a unique color. The correlations between module eigengenes and the clinical phenotype were then evaluated. Among these, the yellow module showed the strongest association with IS. Therefore, the 1252 genes contained in the yellow module were selected for further analysis (Figure 1B).
 |
Figure 1 (A) Analysis of scale-free topology index (top) and mean connectivity (bottom) across different soft-thresholding powers (β). (B) Heatmap showing the correlations between module eigengenes and clinical phenotypes, with the yellow module exhibiting the strongest association with IS. |
Identification of Hub Genes
A total of 409 human ferroptosis-related genes were retrieved from the FerrDb V2 database, including 190 drivers, 216 suppressors, and 3 markers. In addition, MitoCarta3.0 listed 19,274 mitochondria-associated genes. To determine the overlap between IS-related module genes identified by WGCNA and ferroptosis-related genes from FerrDb, a Venn diagram analysis was performed, revealing 24 overlapping genes (13 drivers and 11 suppressors) (Figure 2A). Similarly, overlapping WGCNA-derived IS-related genes with mitochondria-associated genes yielded 1034 shared genes, as shown by the Venn diagram (Figure 2B).
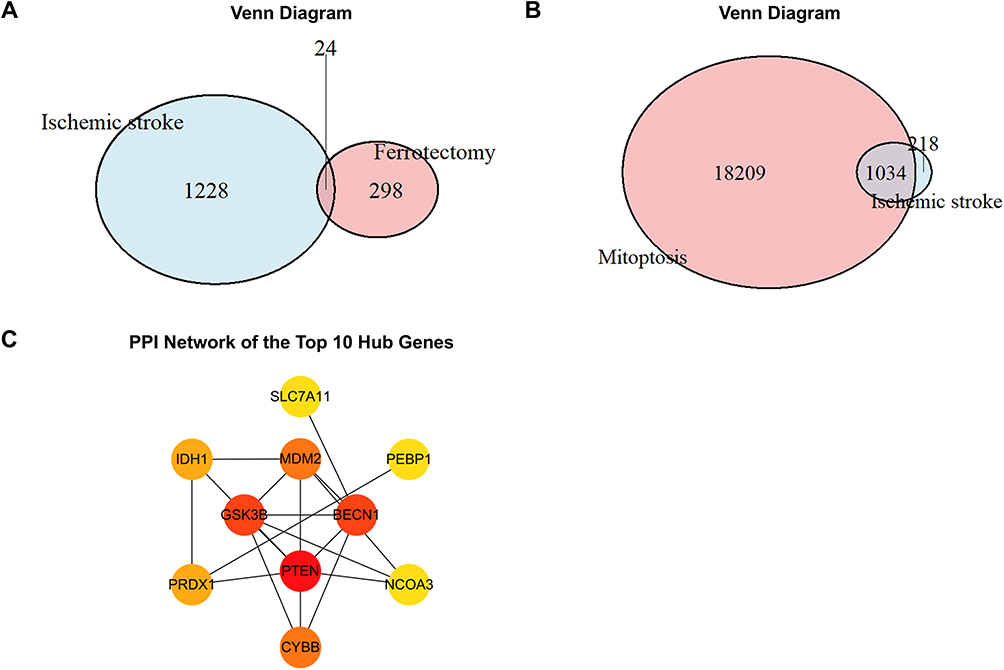 |
Figure 2 (A) Venn diagram showing the overlap between IS-related module genes and ferroptosis-related genes. (B) Venn diagram showing the overlap between IS-related module genes and mitochondria-related genes. (C) PPI network of the top 10 hub genes identified among the ferroptosis-related overlapping genes. |
Protein–protein interaction (PPI) networks of the 24 ferroptosis-related overlapping genes were constructed using the STRING database and visualized in Cytoscape v3.10.3. Based on node degree calculated using the CytoHubba plugin, PTEN, BECN1, GSK3B, MDM2, CYBB, PRDX1, IDH1, SLC7A11, PEBP1, and NCOA3 were identified as the top 10 hub genes (Figure 2C). Using the same analytical procedure for the set of 1034 mitochondria-associated overlapping genes, RPS3, RPS6, RPS28, RPS19, RPS20, RPS2, RPS5, RPL3, RPLP0, and RPL13A were identified as hub genes.
Construction and Validation of the Logistic Regression Models
Among the ferroptosis-related hub genes, PTEN, BECN1, GSK3B, MDM2, CYBB, PRDX1, IDH1, SLC7A11, and PEBP1 were screened and identified as significant predictors for distinguishing IS patients from controls (P < 0.05). A logistic regression model constructed using these genes demonstrated strong discriminatory ability, with an area under the ROC curve (AUC) of 0.949 (Figure 3A). A corresponding nomogram was established to further estimate the probability of IS occurrence (Figure 3C). Violin plots depicting gene expression levels revealed that GSK3B, IDH1, and PRDX1 showed particularly notable differences between the two groups (Figure 3D).
 |
Figure 3 (A) ROC curve evaluating the performance of the ferroptosis-related logistic regression model, with an AUC of 0.949. (B) ROC curve evaluating the performance of the mitochondria-related logistic regression model, with an AUC of 0.829. (C) Nomogram for predicting the occurrence of IS. Ferroptosis-related hub genes—including PTEN, BECN1, GSK3B, MDM2, CYBB, PRDX1, IDH1, SLC7A11, and PEBP1 (P < 0.05)—were incorporated into the model. (D) Violin plots showing the expression levels of the hub genes. |
Using a similar approach, a logistic regression model was built based on the mitochondrial hub genes RPS3, RPS6, RPS28, RPS19, RPS20, RPS2, RPS5, RPL3, and RPL13A. These genes also significantly differentiated IS patients from controls (P < 0.05). The model yielded an AUC of 0.829 (Figure 3B), suggesting that ferroptosis-related gene signatures exhibited higher predictive specificity for IS than mitochondrial dysfunction. The ROC curves and corresponding nomograms for both models are presented in the figure.
Functional Enrichment Analysis
GO enrichment analysis was conducted on the 24 ferroptosis-related module genes across the biological process (BP), cellular component (CC), and molecular function (MF) categories (Figure 4A). The significantly enriched GO-BP terms were primarily associated with stress responses, including response to oxidative stress and chemical stress. GO-CC analysis indicated that the overlapping genes were significantly enriched in the tertiary granule. The GO-MF terms were mainly related to ubiquitin-like protein ligase binding, ubiquitin protein ligase binding, and rRNA binding.
 |
Figure 4 (A) Gene Ontology (GO) functional enrichment analysis of ferroptosis-related module genes. (B) Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analysis of ferroptosis-related module genes. |
KEGG pathway analysis revealed that the overlapping genes were predominantly enriched in pathways related to prostate cancer and ferroptosis, with the ferroptosis pathway being particularly prominent (Figure 4B). These results suggest that the overlapping genes may play critical roles in the pathophysiological mechanisms investigated in this study and warrant further in-depth exploration.
Neuroprotective Effects of Fer-1 in a Rat Model of Ischemic Stroke
To verify the successful establishment of the ischemic stroke model, all rats underwent T2WI and neurological assessments on postoperative day 1. The results confirmed pronounced cerebral ischemic lesions accompanied by clear neurological deficits (Figure 5A and C). After 7 days of treatment, Fer-1 significantly improved neurological function, as reflected by reduced Zea–Longa neurological deficit scores compared with the MCAO group (Figure 5C). After 7 days, Fer-1 treatment markedly reduced infarct volume and showed a trend toward improved rotarod performance (Figure 5B and D).
 |
Figure 5 (A) Representative T2-weighted MRI images from the Sham, MCAO, and Fer-1 groups on postoperative days 1 and 7. (B) Quantification of infarct volume changes (difference between Day 1 and Day 7), showing that Fer-1 treatment significantly attenuated infarct progression compared with the MCAO group (n=8). **** p < 0.0001; ns, not significant. (C) Neurological deficits assessed by the Zea–Longa score on Days 1 and 7 across different groups. **** p < 0.0001; ns, not significant. (D) Motor performance in the rotarod test measured at 1 day (1d) and 7 days (7d) after surgery (n=8). MCAO group compared to Fer-1 group: ** p < 0.01. |
Fer-1 Mitigates Oxidative Stress Following MCAO Rats
To evaluate the antioxidant effects of Fer-1, we assessed three oxidative stress biomarkers (MDA, GSH, and SOD) as well as overall antioxidant capacity. Following MCAO, antioxidant capacity markedly decreased; however, treatment with Fer-1 effectively reversed this decline. Compared with the model group, Fer-1–treated rats exhibited significantly lower MDA levels and significantly higher GSH and SOD levels (Figure 6A–C). These results indicate that Fer-1 strongly mitigates MCAO-induced oxidative stress.
 |
Figure 6 (A) Levels of MDA in the brains of MCAO rats (n = 3). (B) Levels of GSH in the brains of MCAO rats (n = 3). (C) Levels of SOD in the brains of MCAO rats (n = 3).* p < 0.05; ** p < 0.01. |
Fer-1 Reduces Ferritin Deposition and MCAO/R Injury via the ACSL4/GPX4/TfR1 Axis
To confirm the occurrence of ferroptosis following cerebral I/R injury, we measured cortical iron content and the expression of ferroptosis-related markers (TfR1, ACSL4, and GPX4) after MCAO/R in rats. MCAO/R resulted in increased cortical iron accumulation, up-regulation of TfR1 and ACSL4, and down-regulation of GPX4, collectively indicating the presence of ferroptosis. Pharmacological inhibition with Fer-1 reversed these alterations: TfR1 and ACSL4 levels were markedly reduced, whereas GPX4 expression was restored (Figure 7A–D). These findings suggest that Fer-1 suppresses neuronal ferroptosis by modulating TfR1, ACSL4, and GPX4 expression, thereby alleviating cerebral I/R injury.
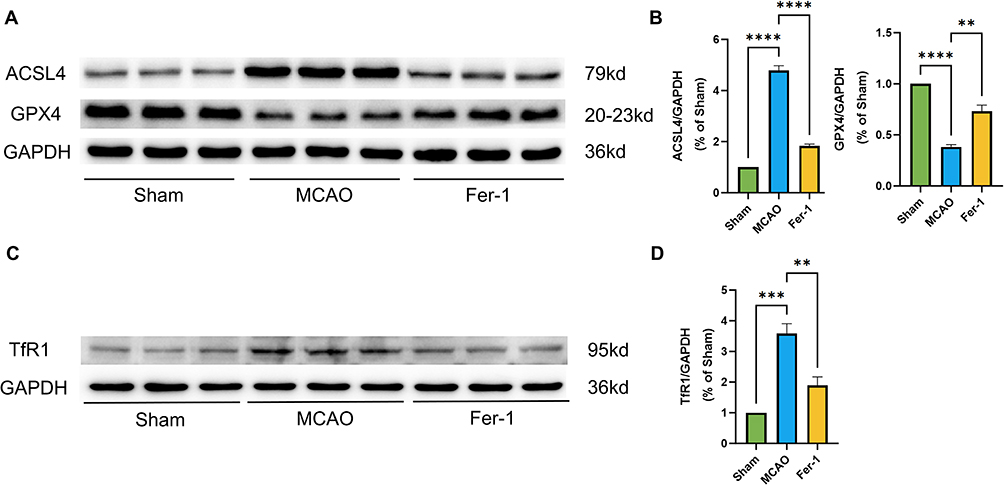 |
Figure 7 (A–D) Representative Western blot bands and quantitative analysis of ACSL4, GPX4, and TfR1 protein levels in MCAO rats following Fer-1 treatment (n = 3). ** p < 0.01; **** p < 0.0001. |
PRDX1, IDH1, and GSK3β Confer Neuroprotection Against MCAO/R Injury Through Ferroptosis Inhibition
To further verify whether the identified key proteins were associated with the ferroptosis pathway, we examined the expression of PRDX1, IDH1, GSK3B, and its phosphorylated form p-GSK3B (Ser9). MCAO markedly decreased PRDX1 levels while significantly increasing IDH1 expression. Although total GSK3B levels remained unchanged, the inhibitory phosphorylation site p-GSK3B (Ser9) was substantially reduced following MCAO. These molecular changes are consistent with characteristic ferroptosis-related alterations, including depletion of the antioxidant protein PRDX1, stress-induced upregulation of IDH1, and enhanced GSK3B activity (reflected by reduced p-GSK3B). In contrast, Fer-1 treatment significantly reversed these abnormalities, increasing PRDX1 levels, decreasing IDH1 expression, and restoring p-GSK3B (Ser9) (Figure 8A–D). Collectively, these findings suggest that Fer-1 mitigates MCAO-induced ferroptosis by enhancing antioxidant defenses, modulating metabolic stress responses, and suppressing aberrant GSK3B activation.
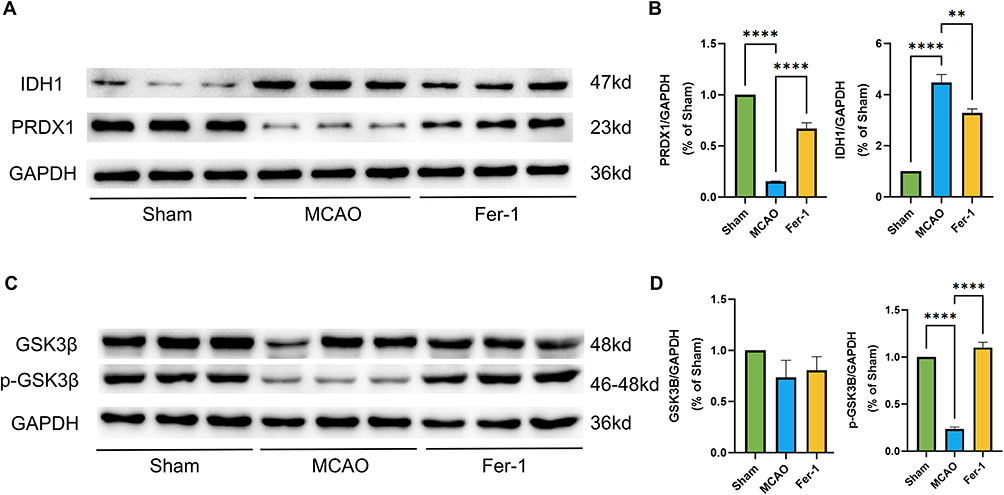 |
Figure 8 (A–D) Representative Western blot bands and quantitative analysis of IDH1, PRDX1, and p-GSK3β/GSK3β protein levels in MCAO rats following Fer-1 treatment (n = 3). ** p < 0.01; **** p < 0.0001. |
Discussion
In this study, we integrated bioinformatics analysis with in vivo experimental validation to identify ferroptosis- and mitochondria-associated genes involved in ischemic stroke (IS) and to clarify their biological relevance. Through WGCNA and PPI network analyses, we screened hub genes associated with IS. Logistic regression further demonstrated that the ferroptosis-related gene signature exhibited higher predictive specificity for IS than the mitochondrial dysfunction–related gene set. Based on these analyses, GSK3B, IDH1, and PRDX1 emerged as the most significant IS-associated genes. Subsequent animal experiments confirmed their functional involvement in oxidative stress and ferroptosis during ischemia–reperfusion injury. Collectively, our findings highlight the central role of ferroptosis in IS pathophysiology and indicate that GSK3B, IDH1, and PRDX1 may serve as promising biomarkers and potential therapeutic targets.
Although both ferroptosis and mitochondrial dysfunction contribute to neuronal injury, our comparative modeling revealed that ferroptosis-related genes exhibited substantially stronger discriminatory power than mitochondrial genes (AUC: 0.949 vs. 0.829). This observation aligns with accumulating evidence suggesting that lipid peroxidation and iron-dependent oxidative stress occur rapidly and extensively in IS, preceding overt mitochondrial structural collapse.14 Given the tight interplay between ferroptosis, lipid peroxidation, and ROS accumulation, increased mitochondrial iron content may serve as an important upstream trigger that drives ferroptotic cell death. Previous studies indicate that mitochondrial ROS accumulation acts as a key late-stage amplifier of ferroptosis, further supporting the involvement of mitochondrial dysfunction in its execution.18 In our study, the significant enrichment of module genes in oxidative stress-related pathways provides additional mechanistic support for this interpretation. Interestingly, KEGG analysis also identified enrichment in prostate cancer-related pathways. This finding does not indicate a direct association between prostate cancer and IS, but rather reflects shared molecular mechanisms, as many cancer-related pathways are closely involved in oxidative stress, apoptosis and inflammation, which are also critical processes in ischemic brain injury.19,20 In contrast, the ferroptosis pathway showed a more direct biological relevance to IS pathology and was therefore a major focus of this study. Collectively, these findings suggest that ferroptosis is not merely a downstream consequence of ischemic injury but functions as a primary driver of neuronal death during the progression of IS. By highlighting the predominance of ferroptosis signaling over mitochondrial impairment in predictive modeling and pathway enrichment, our results emphasize the pivotal role of ferroptosis in the pathophysiology of ischemic stroke.
In vivo experiments demonstrated that Fer-1 effectively reduced infarct volume and improved neurological deficit scores, indicating partial preservation of neurological function. Although Fer-1 also showed a trend toward attenuating motor impairment in the rotarod test, the improvement did not reach statistical significance. This may reflect the multifactorial nature of ischemic stroke pathology. While ferroptosis contributes importantly to neuronal injury, additional mechanisms including excitotoxicity, inflammation, apoptosis, and mitochondrial dysfunction also participate in post-ischemic neurological impairment. Therefore, inhibition of ferroptosis alone may be insufficient to fully restore complex motor coordination within the relatively short observation period. In addition, functional recovery after stroke often requires longer-term neuroplasticity and neural circuit remodeling, which may not be fully achieved within 7 days. Biochemically, Fer-1 restored antioxidant capacity by increasing SOD and GSH levels while reducing MDA accumulation, findings consistent with its established ability to inhibit lipid peroxidation and stabilize membrane integrity.21 Unlike conventional antioxidants, Fer-1 specifically blocks iron-dependent peroxidation of polyunsaturated phospholipids, thereby disrupting a key amplification loop of ferroptosis.
Mechanistic evidence further supports the critical involvement of ferroptosis in neuronal injury following ischemic stroke. Transferrin receptor 1 (TFR1) mediates cellular uptake of transferrin-bound Fe3⁺, which is subsequently reduced to Fe2⁺ and transported into the cytosol by divalent metal transporter 1 (DMT1). Upregulation of TFR1 and DMT1 promotes intracellular iron accumulation—a hallmark of ferroptosis—and inhibition of TFR1 has been shown to markedly reduce iron uptake.22 Excess Fe2⁺accelerates Fenton chemistry, generating large amounts of reactive oxygen species (ROS) and driving lipid peroxidation. Ischemia–reperfusion further exacerbates ROS production, disrupting redox homeostasis and amplifying oxidative damage. GPX4, a central suppressor of ferroptosis, detoxifies lipid hydroperoxides using GSH as a cofactor; thus, either GSH depletion or GPX4 loss greatly enhances sensitivity to ferroptosis cell death.23,24 GPX4 downregulation has been consistently observed in acute ischemic stroke and is associated with increased neuronal vulnerability.25 SOD contributes additional antioxidant protection by removing superoxide radicals, whereas elevated levels of malondialdehyde (MDA)—a terminal product of lipid peroxidation—reflect the extent of oxidative injury during ischemia. ACSL4 is another essential determinant of ferroptosis, catalyzing the esterification of long-chain polyunsaturated fatty acids (PUFAs), particularly arachidonic acid (AA) and adrenic acid (AdA), thereby increasing membrane susceptibility to lipid peroxidation.26 Notably, both ACSL4 and TFR1 expression, as well as iron accumulation, remain elevated during the post-stroke recovery period. Guo et al reported that TFR1 levels and iron content continue to exceed baseline even 28 days after stroke, suggesting prolonged ferroptotic pressure within ischemic lesions.27 Within this mechanistic framework, our results demonstrate that Fer-1 effectively reverses MCAO-induced upregulation of TFR1 and ACSL4 while restoring GPX4 expression.
Our findings suggest that GSK3B, IDH1, and PRDX1 participate in complementary but interconnected pathways regulating oxidative stress and ferroptosis after ischemic stroke. Specifically, enhanced GSK3B activity may amplify oxidative and inflammatory signaling, PRDX1 depletion weakens endogenous ROS-scavenging capacity, whereas dysregulated IDH1 reflects impaired NADPH-dependent redox homeostasis. Together, these alterations promote excessive lipid peroxidation and ferroptotic neuronal injury during cerebral ischemia/reperfusion. This is consistent with growing evidence identifying ferroptosis as a critical driver of ischemic brain injury.28 PRDX1, a cytosolic antioxidant enzyme, detoxifies peroxide species and maintains redox homeostasis in the post-ischemic brain. Reduced PRDX1 expression compromises endogenous peroxide-scavenging capacity, promotes ROS accumulation, disrupts microglial function, and enlarges infarct size in MCAO/R models.29 Conversely, PRDX1 preservation mitigates oxidative injury and inflammation, supporting tissue salvage and functional recovery. The pronounced reduction of PRDX1 observed in our study suggests increased neuronal susceptibility to lipid peroxidation—the biochemical hallmark of ferroptosis. This aligns with previous work demonstrating that PRDX1 protects neural tissue against ischemia-induced oxidative injury, whereas its loss exacerbates infarct expansion and neuronal death (1). IDH1 is a metabolic enzyme that contributes reducing equivalents to the cytosolic NADPH pool, linking central carbon metabolism to lipid synthesis and antioxidant defenses. Acute ischemic stress often induces compensatory increases in IDH1 as cells attempt to enhance NADPH-dependent redox buffering.30 However, persistent or maladaptive IDH1 upregulation may indicate metabolic stress and heightened susceptibility to ROS-driven lipid peroxidation. The elevated IDH1 levels observed after MCAO likely reflect this compensatory response; yet excessive or prolonged activation has been associated with increased vulnerability to ferroptosis, consistent with previous reports that disrupted redox metabolism facilitates ferroptotic cell death in stroke.31 Reduced phosphorylation of GSK3B at Ser9 indicates enhanced kinase activity, a pathological state known to amplify oxidative and inflammatory signaling, impair mitochondrial function, and worsen ischemic outcomes.32 Restoration of Ser9 phosphorylation is widely recognized as neuroprotective, supporting mitochondrial integrity and reducing cell death, consistent with earlier studies implicating aberrant GSK3B activation in ischemic neuronal injury.33 Although some studies were not conducted in ischemic stroke models, they indicate that p-GSK3B (Ser9) acts as a core activator in the GSK3β/Nrf2/GPX4 anti-ferroptosis pathway, further supporting the potential mechanistic role of GSK3B in regulating ferroptotic neuronal injury.34 Importantly, Ferrostatin-1 treatment reversed these pathological changes—restoring PRDX1 expression, normalizing IDH1 levels, and increasing p-GSK3B (Ser9). This coordinated correction strongly suggests that these three proteins function not merely as downstream indicators but as active mediators of ferroptosis injury.
Notably, the expression patterns of GSK3B, IDH1, and PRDX1 observed in our rat MCAO model closely mirrored the expression trends identified in the clinical GEO datasets. This consistency between in vivo validation and bioinformatic prediction strengthens the robustness of our findings and further confirms that these three genes represent conserved, clinically relevant regulators of ferroptosis in ischemic stroke. Their concordant alterations across experimental and clinical contexts underscore their mechanistic significance and reinforce their potential as prognostic biomarkers and therapeutic targets.
Collectively, the coordinated dysregulation of GSK3B, IDH1, and PRDX1 highlights their central roles in ferroptosis-mediated neuronal injury after stroke, consistent with previous studies emphasizing ferroptosis-related pathways as key determinants of stroke severity and functional recovery.28,35
Clinical Implications
These findings underscore the therapeutic potential of targeting oxidative stress and ferroptosis in ischemic stroke. The coordinated dysregulation of GSK3B, IDH1, and PRDX1 suggests that ferroptosis plays an active role in driving neuronal loss and functional impairment. Pharmacological inhibition with Fer-1 effectively reverses these molecular alterations, reinforcing ferroptosis as a promising target for neuroprotection. Moreover, these proteins may serve as biomarkers reflecting oxidative stress status or therapeutic responsiveness, providing opportunities for early intervention and personalized treatment strategies.
Limitations
Despite the novel insights provided by this study, several limitations should be acknowledged. First, the in vivo experiments were conducted using a rat MCAO model, which may not fully recapitulate the complexity and heterogeneity of human ischemic stroke, and external clinical validation was not performed. Second, differences in gene set size between ferroptosis- and mitochondria-associated genes, as well as the lack of adjustment for potential clinical confounding variables in the GEO datasets, may have influenced the comparative bioinformatics results. Third, although GSK3B, IDH1, and PRDX1 were identified as ferroptosis-associated genes closely related to oxidative stress and neuronal injury in stroke, the molecular analyses were primarily based on expression changes, and direct functional validation, such as gene knockdown or overexpression experiments, was not performed. In addition, the upstream regulatory mechanisms of these genes and their potential interactions with other programmed cell death pathways were not investigated. Fourth, the GEO datasets used for bioinformatics analysis had relatively small sample sizes, which may limit the robustness and generalizability of the logistic regression models. Finally, long-term neurological outcomes and potential adverse effects associated with ferroptosis inhibition were not evaluated. Therefore, future studies involving larger clinical cohorts, external validation, and more comprehensive mechanistic investigations are needed to further clarify the specific roles of these genes in ferroptosis-related ischemic injury and to support their translational potential.
Conclusion
This study identified GSK3B, IDH1, and PRDX1 as ferroptosis-associated genes closely related to oxidative stress and neuronal injury during ischemic stroke progression. Pharmacological inhibition of ferroptosis with Fer-1 attenuated ischemia/reperfusion-induced brain injury, alleviated oxidative stress, and modulated the expression of ACSL4, GPX4, and TFR1. Collectively, these findings further support the involvement of GSK3B, IDH1, and PRDX1 in ferroptosis-related pathological processes during ischemic stroke and suggest that these ferroptosis-associated pathways may represent potential targets for neuroprotective intervention.
Data Sharing Statement
The original contributions presented in the study are included in the article, further inquiries can be directed to the corresponding authors.
Ethics Statement
All animal experiments were approved by the Experimental Animal Management Committee of Fujian University of Traditional Chinese Medicine (Approval No. 82474611) and were conducted in strict accordance with local laws and regulations, institutional guidelines, and the National Institutes of Health Guide for the Care and Use of Laboratory Animals.
Author Contributions
The listed authors contributed to this paper as follows: Ke Ma and Yijing Jiang (co–first authors) contributed equally to this work and were responsible for Conceptualization, Data curation, Formal analysis, Methodology, Software, Validation, Writing-original draft, and Writing-review & editing. Shanli Yang (corresponding author) contributed to Conceptualization, Methodology, Funding acquisition, Supervision, Writing-original draft, and Writing -review & editing. Yihan Yang contributed to Formal analysis, Writing-original draft, and Writing-review & editing. Ting Rao contributed to Software, Resources, Writing-original draft, and Writing-review & editing. Ying Zhan and Zihan Yin contributed to Investigation, Formal analysis, Writing-original draft, and Writing-review & editing. Yuqin Dan and Sihan Xu contributed to Data curation, Resources, Writing-original draft, and Writing-review & editing. All authors gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
This work was supported by the National Natural Science Foundation of China (No. 82474611), the Major Scientific Research Project of the Fujian Provincial Health Commission (No. 2024ZD01007), and the Joint Fund Project for Scientific and Technological Innovation of the Fujian Provincial Department of Science and Technology (No. 2025Y9603).
Disclosure
The authors declare they have no competing interests.
References
1. Tuo QAPL. Ferroptosis in ischemic stroke: animal models and mechanisms. Zool Res. 2024;6(45):1235–16.
2. Kim S, Lee W, Jo H, et al. The antioxidant enzyme Peroxiredoxin-1 controls stroke-associated microglia against acute ischemic stroke. Redox Biol. 2022;54:102347. doi:10.1016/j.redox.2022.102347
3. Liu T, Li X, Zhou X, et al. PI3K/AKT signaling and neuroprotection in ischemic stroke: molecular mechanisms and therapeutic perspectives. Neural Regen Res. 2025;20(10):2758–2775. doi:10.4103/NRR.NRR-D-24-00568
4. You R, Sun B, Luo J, Shao N, Si W. Exploring the role of Mitoferrin-1 in Ferroptosis and mitochondrial damage in acute ischemic stroke. Int Immunopharmacol. 2025;155:114625. doi:10.1016/j.intimp.2025.114625
5. Hong T, Zhao T, He W, et al. Exosomal circBBS2 inhibits ferroptosis by targeting miR-494 to activate SLC7A11 signaling in ischemic stroke. FASEB J. 2023;37(9):e23152. doi:10.1096/fj.202300317RRR
6. Jung YS, Lee SEW, Park JH, Seo HB, Choi BT, Shin HK. Electroacupuncture preconditioning reduces ROS generation with NOX4 down-regulation and ameliorates blood-brain barrier disruption after ischemic stroke. J Biomed Sci. 2016;23(1). doi:10.1186/s12929-016-0249-0
7. Li Y, Wang Y, Yang W, et al. ROS-responsive exogenous functional mitochondria can rescue neural cells post-ischemic stroke. Front Cell Dev Biol. 2023;11:1207748.
8. Liu D, Yang S, Yu S. Interactions between ferroptosis and oxidative stress in ischemic stroke. Antioxidants. 2024;13(11):1329. doi:10.3390/antiox13111329
9. Ye Y, Xie X, Bi Y, et al. Nrf2 alleviates acute ischemic stroke induced ferroptosis via regulating xCT/GPX4 pathway. Free Radical Bio Med. 2025;231:153–162. doi:10.1016/j.freeradbiomed.2025.02.040
10. Sharma R, Malviya R, Srivastava S, Ahmad I, Rab SO, Uniyal P. Targeted Treatment Strategies for Mitochondria Dysfunction: correlation with Neurological Disorders. Curr Drug Targets. 2024;25(10):683–699. doi:10.2174/0113894501303824240604103732
11. Zhang C, Lan X, Wang Q, et al. Decoding ischemic stroke: perspectives on the endoplasmic reticulum, mitochondria, and their crosstalk. Redox Biol. 2025;82:103622. doi:10.1016/j.redox.2025.103622
12. Zhang Y, Zhang H, Zhao F, et al. Mitochondrial-targeted and ROS-responsive nanocarrier via nose-to-brain pathway for ischemic stroke treatment. Acta Pharm Sin B. 2023;13(12):5107–5120. doi:10.1016/j.apsb.2023.06.011
13. Liu L, Chen D, Zhou Z, et al. Traditional Chinese medicine in treating ischemic stroke by modulating mitochondria: a comprehensive overview of experimental studies. Front Pharmacol. 2023;14:1138128.
14. Wang P, Cui Y, Ren Q, et al. Mitochondrial ferritin attenuates cerebral ischaemia/reperfusion injury by inhibiting ferroptosis. Cell Death Dis. 2021;12(5):447.
15. She R, Liu D, Liao J, Wang G, Ge J, Mei Z. Mitochondrial dysfunctions induce PANoptosis and ferroptosis in cerebral ischemia/reperfusion injury: from pathology to therapeutic potential. Front Cell Neurosci. 2023;17:1191629.
16. Yang XC, Jin YJ, Ning R, et al. Electroacupuncture attenuates ferroptosis by promoting Nrf2 nuclear translocation and activating Nrf2/SLC7A11/GPX4 pathway in ischemic stroke. Chin Med. 2025;20(1). doi:10.1186/s13020-024-01047-0
17. Xiang J, Li L, Li J, Mo X, Chen W, Hu Y. Protective mechanism of wenyang fuyuan prescription on nerve injury in rats with cerebral ischemia-reperfusion injury based on ferroptosis. Tradit Chin Drug Res Clin Pharmacol. 2023;1649–1657.
18. Song X, Hao X, Zhu BT. Role of mitochondrial reactive oxygen species in chemically-induced ferroptosis. Free Radical Bio Med. 2024;223:473–492. doi:10.1016/j.freeradbiomed.2024.07.006
19. Beyaztas H, Ersoz C, Ozkan BN, et al. The role of oxidative stress and inflammation biomarkers in pre- and postoperative monitoring of prostate cancer patients. Free Radical Res. 2024;58(2):98–106. doi:10.1080/10715762.2024.2320381
20. Mei W, Wei M, Tang C, et al. BCAT2 binding to PCBP1 regulates the PI3K/AKT signaling pathway to inhibit autophagy-related apoptosis and ferroptosis in prostate cancer. Cell Death Dis. 2025;16(1). doi:10.1038/s41419-025-07559-3
21. Scarpellini C, Klejborowska G, Lanthier C, Hassannia B, Berghe TV, Augustyns K. Beyond ferrostatin-1: a comprehensive review of ferroptosis inhibitors. Trends Pharmacol Sci. 2023;44(12):902–916. doi:10.1016/j.tips.2023.08.012
22. Stockwell BR, Angeli JPF, Bayir H, et al. Ferroptosis: a regulated cell death nexus linking metabolism, redox biology, and disease. Cell. 2017;171(2):273–285. doi:10.1016/j.cell.2017.09.021
23. Dodson M, Castro-Portuguez R, Zhang DD. NRF2 plays a critical role in mitigating lipid peroxidation and ferroptosis. Redox Biol. 2019;23:101107. doi:10.1016/j.redox.2019.101107
24. Ursini F, Maiorino M. Lipid peroxidation and ferroptosis: the role of GSH and GPx4. Free Radical Bio Med. 2020;152:175–185. doi:10.1016/j.freeradbiomed.2020.02.027
25. Alim I, Caulfield JT, Chen Y, et al. Selenium drives a transcriptional adaptive program to block ferroptosis and treat stroke. Cell. 2019;177(5):1262–1279. doi:10.1016/j.cell.2019.03.032
26. Cui Y, Zhang Y, Zhao X, et al. ACSL4 exacerbates ischemic stroke by promoting ferroptosis-induced brain injury and neuroinflammation. Brain Behav Immun. 2021;93:312–321. doi:10.1016/j.bbi.2021.01.003
27. Guo X, Jin X, Han K, et al. Iron promotes neurological function recovery in mice with ischemic stroke through endogenous repair mechanisms. Free Radical Bio Med. 2022;182:59–72. doi:10.1016/j.freeradbiomed.2022.02.017
28. Bu ZQ, Yu HY, Wang J, et al. Emerging role of ferroptosis in the pathogenesis of ischemic stroke: a new therapeutic target? Asn Neuro. 2021;13:175909142110375. doi:10.1177/17590914211037505
29. Liu Q, Zhang Y. PRDX1 enhances cerebral ischemia-reperfusion injury through activation of TLR4-regulated inflammation and apoptosis. Biochem Bioph Res Co. 2019;519(3):453–461. doi:10.1016/j.bbrc.2019.08.077
30. He Q, Chen J, Xie Z, Chen Z. Wild-type isocitrate dehydrogenase-dependent oxidative decarboxylation and reductive carboxylation in cancer and their clinical significance. Cancers. 2022;14(23):5779. doi:10.3390/cancers14235779
31. Jiang X, Stockwell BR, Conrad M. Ferroptosis: mechanisms, biology and role in disease. Nat Rev Mol Cell Bio. 2021;22(4):266–282. doi:10.1038/s41580-020-00324-8
32. Guo XH, Pang L, Gao CY, Meng FL, Jin W. Lyoniresinol attenuates cerebral ischemic stroke injury in MCAO rat based on oxidative stress suppression via regulation of Akt/GSK-3β/Nrf2 signaling. Biomed Pharmacother. 2023;167:115543. doi:10.1016/j.biopha.2023.115543
33. Xu B, Xu J, Cai N, et al. Roflumilast prevents ischemic stroke-induced neuronal damage by restricting GSK3β-mediated oxidative stress and IRE1α/TRAF2/JNK pathway. Free Radical Bio Med. 2021;163:281–296. doi:10.1016/j.freeradbiomed.2020.12.018
34. Wu ZH, Han LJ, Zhang JX, et al. Kaempferide alleviates glucocorticoid-induced osteoporosis by targeting ferroptosis through the GSK3β/Nrf-2/GPX4 pathway. Int Immunopharmacol. 2026;179:116601. doi:10.1016/j.intimp.2026.116601
35. Yeh SJ, Chen CH, Lin YH, et al. Association of ferroptosis with severity and outcomes in acute ischemic stroke patients undergoing endovascular thrombectomy: a case-control study. Mol Neurobiol. 2023;60(10):5902–5914. doi:10.1007/s12035-023-03448-y
 © 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
Recommended articles
The Emerging Role of Ferroptosis in Various Chronic Liver Diseases: Opportunity or Challenge
Zhu L, Luo S, Zhu Y, Tang S, Li C, Jin X, Wu F, Jiang H, Wu L, Xu Y
Journal of Inflammation Research 2023, 16:381-389
Published Date: 31 January 2023
Variation of Ferroptosis-Related Markers in HaCaT Cell Photoaging Models Induced by UVB
Zhang PC, Hong Y, Zong SQ, Chen L, Zhang C, Tian DZ, Ke D, Tian LM
Clinical, Cosmetic and Investigational Dermatology 2023, 16:3147-3155
Published Date: 1 November 2023
Ciprofol Ameliorates Myocardial Ischemia/Reperfusion Injury by Inhibiting Ferroptosis Through Upregulating HIF-1α

Ding J, Wang BY, Yang YF, Kuai LY, Wan JJ, Zhang M, Xia HY, Wang Y, Zheng Z, Meng XW, Peng K, Ji FH
Drug Design, Development and Therapy 2024, 18:6115-6132
Published Date: 18 December 2024
Enteric-Coated Aspirin Induces Small Intestinal Injury via the Nrf2/Gpx4 Pathway: A Promising Model for Chronic Enteropathy

Zhang M, Xia S, Feng L, Han X, Zhang Y, Huang Y, Liu Y, Zhao K, Guan J, Tian D, Liao J, Yu Y
Drug Design, Development and Therapy 2025, 19:891-910
Published Date: 11 February 2025
Vaccarin Ameliorates Renal Fibrosis by Inhibiting Ferroptosis via Nrf2/SLC7A11/GPX4 Signaling Pathway
Cui M, Xu Q, Duan L, Lu J, Hu J
Drug Design, Development and Therapy 2025, 19:1609-1626
Published Date: 6 March 2025