")
Back to Journals » Cancer Management and Research » Volume 10
Combined use of CDK4/6 and mTOR inhibitors induce synergistic growth arrest of diffuse intrinsic pontine glioma cells via mutual downregulation of mTORC1 activity
Authors Asby DJ , Killick-Cole CL, Boulter LJ, Singleton WGB, Asby CA, Wyatt MJ, Barua NU, Bienemann AS, Gill SS
Received 6 March 2018
Accepted for publication 5 May 2018
Published 12 September 2018 Volume 2018:10 Pages 3483—3500
DOI https://doi.org/10.2147/CMAR.S167095
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Leylah Drusbosky
Daniel J Asby,1 Clare L Killick-Cole,1 Lisa J Boulter,1 William GB Singleton,1,2 Claire A Asby,3 Marcella J Wyatt,1 Neil U Barua,1,2 Alison S Bienemann,1 Steven S Gill1,2
1Functional Neurosurgery Research Group, Translational Health Sciences, Bristol Medical School, University of Bristol, Bristol, UK; 2Department of Neurosurgery, North Bristol NHS Trust, Southmead Hospital, Bristol, UK; 3Department of Neurology, North Bristol NHS Trust, Southmead Hospital, Bristol, UK
Background: Diffuse intrinsic pontine glioma (DIPG) is a lethal type of pediatric brain tumor that is resistant to conventional chemotherapies. Palbociclib is a putative novel DIPG treatment that restricts the proliferation of rapidly dividing cancer cells via selective inhibition of cyclin-dependent kinase (CDK) 4 and CDK6. However, implementing palbociclib as a monotherapy for DIPG is unfeasible, as CDK4/6 inhibitor resistance is commonplace and palbociclib does not readily cross the blood–brain barrier (BBB) or persist in the central nervous system. To inhibit the growth of DIPG cells, we aimed to use palbociclib in combination with the rapamycin analog temsirolimus, which is known to ameliorate resistance to CDK4/6 inhibitors and inhibit BBB efflux.
Materials and methods: We tested palbociclib and temsirolimus in three patient-derived DIPG cell lines. The expression profiles of key proteins in the CDK4/6 and mammalian target of rapamycin (mTOR) signaling pathways were assessed, respectively, to determine feasibility against DIPG. Moreover, we investigated effects on cell viability and examined in vivo drug toxicity.
Results: Immunoblot analyses revealed palbociclib and temsirolimus inhibited CDK4/6 and mTOR signaling through canonical perturbation of phosphorylation of the retinoblastoma (RB) and mTOR proteins, respectively; however, we observed noncanonical downregulation of mTOR by palbociclib. We demonstrated that palbociclib and temsirolimus inhibited cell proliferation in all three DIPG cell lines, acting synergistically in combination to further restrict cell growth. Flow cytometric analyses revealed both drugs caused G1 cell cycle arrest, and clonogenic assays showed irreversible effects on cell proliferation. Palbociclib did not elicit neurotoxicity in primary cultures of normal rat hippocampi or when infused into rat brains.
Conclusion: These data illustrate the in vitro antiproliferative effects of CDK4/6 and mTOR inhibitors in DIPG cells. Direct infusion of palbociclib into the brain, in combination with systemic delivery of temsirolimus, represents a promising new approach to developing a much-needed treatment for DIPG.
Keywords: palbociclib, temsirolimus, brain tumor, DIPG, retinoblastoma protein, cyclin-dependent kinase
Introduction
Diffuse intrinsic pontine glioma (DIPG) is a lethal, high-grade, pediatric glioma that accounts for up to 85% of all brainstem gliomas.1 Approximately 100–150 new cases of DIPG in children are recorded each year in the USA.2 The genetic basis of DIPG is complex, and several genetic and epigenetic alterations are associated with the disease. However, ~80% of DIPG tumors exhibit the missense mutation lysine 27 to methionine (K27M) in the genes that encode histones H3.1 and H3.3 (H3.1K27M and H3.3K27M, respectively).3 DIPG is difficult to treat due to the heterogeneity generated from the various mutations associated with the disease, as well as its sensitive location in the brainstem. Fractionated radiation is the conventional treatment but has no long-term benefit, with the 2-year survival rate currently below 20%.1 Furthermore, chemotherapies have proved ineffective and new treatments are urgently required. The results of combinatorial approaches utilizing synergistic targeted therapies have been encouraging.3
Palbociclib (also known as PD-0332991) is a selective inhibitor of cyclin-dependent kinases (CDK) 4 and 6.4 Both CDK4 and 6 form functionally identical heterodimeric complexes with cyclin D1 (cycD1), cycD2, or cycD3 that phosphorylate and inactivate retinoblastoma (RB) protein.5 Inactivation of RB relieves negative regulation of the E2F transcription factor, which facilitates progression through the G1/S transition in the cell cycle, thus permitting DNA synthesis and cell proliferation. Palbociclib inhibits the kinase activity of CDK4/6, thereby preventing phosphorylation of RB and cell cycle progression. This leads to cell cycle arrest in the G1 phase of the cell cycle. Thus, treatment with palbociclib results in cells accumulating in G1 and inhibition of cell proliferation. However, malignancies can develop resistance to CDK4/6 inhibitors,6 putatively via cycD1 expression flux, with downregulation of the mammalian target of rapamycin (mTOR) being one approach to abrogate this.4
Temsirolimus (previously known as CCI-779) is an ester analog, or rapalog, of the mTOR inhibitor rapamycin, which possesses increased aqueous solubility and improved pharmacokinetics compared to its parent molecule.7 Temsirolimus is anti-tumorigenic in vitro through targeted inhibition of the mTORC1 complex and was approved for intravenous systemic delivery for the treatment of renal cell carcinoma in 2007.8 The effectiveness of temsirolimus as a single agent therapy against other types of cancer has been limited, though it has shown some capacity to treat glioblastoma10 and has been shown to readily cross the blood-brain barrier (BBB).11 Temsirolimus combination therapies are currently being trialed for treatment of central nervous system (CNS) tumors, where it has been found to be well tolerated by patients and appears amenable to combination with other targeted therapies.12
We used three cell lines encompassing both archetypal DIPG histone mutations (H3.1K27M found in SU-DIPG IV cells and H3.3K27M found in SF7761 and SF8628 cells) to demonstrate that, by selectively disrupting the CDK4/6-cycD1-RB and mTOR signaling pathways, sustained inhibition of DIPG cell proliferation can be achieved through synergistic cytostatically driven effects on cell growth, increasing the therapeutic potential of these two candidate DIPG therapeutics.
Materials and methods
Reagents were purchased from ThermoFisher Scientific (Loughborough, Leicestershire, UK) unless otherwise stated. Palbociclib and temsirolimus were purchased from Selleckchem (Houston, TX, USA). Drug synergy analysis, based on cell viability assays, compared single-agent treatments to equivalent combination treatments, as previously described.13 Statistical analyses were conducted using GraphPad Prism 6 and MiniTab 17. All values are expressed as mean of triplicate determinations ± SEM. Values of p < 0.05 were considered as statistically significant.
Cell culture and cell treatments
Patient-derived SF7761 and SF8628 cell lines were isolated from DIPG tumor tissue acquired by the University of California San Francisco (UCSF) Tissue Bank. SU-DIPG IV cells were isolated from a DIPG patient at Stanford University. All procedures were conducted with Institutional Review Board approval. SF7761 and SF8628 cells were obtained from Nalin Gupta (UCSF) and SU-DIPG IV from Michelle Monje (Stanford University) via material transfer agreements. Cells were authenticated by short tandem repeat (STR) profiling (Public Health England, London, UK). Cells were used within ten passages from thawing and confirmed to be mycoplasma free (in-house testing). SF7761 and SF8628 culture has been described previously.13 SU-DIPG IV cells were grown in tumor stem media: Dulbecco’s modified Eagle medium / Ham’s F-12 (DMEM/F12) and Neurobasal-A medium [1:1 ratio], with B27 neural cell culture supplement (2%), human basic fibroblast growth factor (hFGF-basic; 20 ng/ml; Peprotech, London, UK), mouse epidermal growth factor (mEGF; 20 ng/ml; Peprotech), human platelet-derived growth factor AA (hPDGF-AA; 10 ng/ml; Generon, Maidenhead, UK), hPDGF-BB (10 ng/ml; Generon) and heparin (2 mg/ml, StemCell Technologies, Grenoble, France). Cells were seeded 16 hours prior to treatment in all instances and maintained at 5% CO2 and 37°C. Cells were treated with drugs for 24 hours unless stated otherwise. Serially diluted stock solutions of palbociclib and temsirolimus were reconstituted in artificial cerebrospinal fluid (Torbay Pharmaceuticals, Paignton, Devon, UK) and dimethyl sulfoxide, respectively.
Protein immunoblotting
Immunoblotting was done as previously described.14 Nitrocellulose membranes were probed overnight at 4°C with the following antibodies at their respective dilutions. From Cell Signaling Technology (Danvers, MA, USA): RB (4H1) #9309 1:1,000; phospho-RB (Ser807/811) #8516 1:1,000; CDK6 (D4S8S) #13331 1:500; cyclD1 (92G2) #2978 1:500; mTOR #2972 1:1,000; phospho-mTOR (Ser2448) #2971 1:1,000; S6 kinase (49D7) #2708 1:1,000; phospho-S6 kinase (Thr389, 108D2) #9234 1:1,000; 4EBP1 (53H11) #9644 1:2,000; phospho-4EBP1 (Thr37/46) #9459 1:500; Rictor (53A4) #2114 1:1,000; phospho-rictor (Thr1135, D30A3) #3806 1:1,000; anti-rabbit HRP #7074 1:2,000; and anti-mouse HRP #7076 1:1,000. In addition to p16INK4A #MAB4133 (Millipore, Watford, Hertfordshire, UK) and CDK4 #559677 (Becton Dickinson, Wokingham, Berkshire, UK). Densitometry values are the mean of triplicate determinations, and representative blots are shown.
Cell viability assays
Cell viability and cytotoxicity were measured with a two-color fluorescent-based live/dead kit (Life Technologies, Paisley, UK) utilizing calcein acetoxymethyl (calcein-AM) and ethidium homodimer-1 (ethD-1). Cells were seeded in 96-well plates and
incubated for 16 hours prior to drug treatments. Cells were dosed with various concentrations of each drug, as described in the Results section below. Cells were then washed with PBS and incubated with 2 μM calcein-AM and 4 μM ethD-1 for 30 minutes. Fluorescence was measured on a microplate reader (FLUOstar Omega; BMG Labtech, Aylesbury, Buckinghamshire, UK) and normalized against appropriate controls.
Flow cytometry
Cells were seeded on 12-well plates and dosed with palbociclib, temsirolimus, or both, as described. SF7761, SF8628, and SU-DIPG IV cells were seeded at 3.5 × 105, 0.4 × 105, and 1 × 105 cells per well, respectively, for experiments requiring 24-hour drug treatments, and at 1 × 105, 0.2 × 105, and 0.4 × 105 cells per well, respectively, for 72-hour experiments. Cells were then detached from the growing surface using Accutase solution and collected. For cell viability analysis, calcein-AM and ethD-1 were diluted in PBS at 1:10,000 and 1:250 concentrations, respectively, prior to their addition to cell suspensions at a 1:1 ratio. For cell cycle analyses, DRAQ5 (eBioscience, San Diego, CA, USA) was diluted 1:200 in PBS and added to cell suspensions at a 1:1 ratio.
Clonogenic assays
SU-DIPG IV cells were seeded at 800 cells per 6-cm dish, 16 hours prior to treatments described in this article. Cells were processed as previously described.14 At the end of the experiment, cells were fixed by the addition of methanol/acetic acid (3:1) for 5 minutes, then stained with 0.5% (v/v) crystal violet solution (diluted in methanol) for 10 minutes. Colonies were counted using the colony area plugin for ImageJ (version 1.46r; National Institutes of Health, USA).
In vitro neurotoxicity assessment via fluorescent immunocytochemistry
Experiments were conducted on rat E18 hippocampal cultures grown on poly-d-lysine-coated glass coverslips after cell extraction had been carried out using previously described protocols.15 Palbociclib toxicity was assessed after 24 hours of treatment using immunocytochemistry assays conducted on untreated and palbociclib-treated primary hippocampal cells as described previously.13
In vivo neurotoxicity assessment via fluorescent immunohistochemistry
Targeted delivery of palbociclib to the brain striatum of Wistar rats was achieved using a custom cannula system and rate-controlled microinfusion pump, as previously described.15 The animals used in this study were handled according to the protocols approved by the ethical committee of University of Bristol, and all the protocols were undertaken in accordance with the UK Animal Scientific Procedures Act 1986. Immunohistochemical analysis of tissue sections was conducted as previously described.15 Briefly, animals were transcardially perfused with 4% paraformaldehyde and the brains were removed; these were then fixed using 4% paraformaldehyde. Rat brains were then cut into 35-μm thick sections at −20°C and mounted on microscope slides. Anti-NeuN (1:100; Millipore, Burlington, MA, USA) and anti-GFAP (1:300; Millipore) primary antibodies were used together with standard immunofluorescent protocols to identify any neuronal disruption and gliosis, respectively.
Results
Combination treatment with palbociclib and temsirolimus inhibit both the CDK4/6-cycD1-RB and mTORC1 signaling pathways in DIPG cells
Barton et al has previously shown that palbociclib is effective at inhibiting the CDK4/6-cycD1-RB signaling pathway at submicromolar concentrations in genetically engineered murine DIPG cells.16 Furthermore, analogs of temsirolimus, such as everolimus, have been shown to be effective at inhibiting cell viability at low-micromolar concentrations in non-DIPG brain malignancies, such as glioblastoma.17,18 Thus, in order to establish whether palbociclib and temsirolimus could be clinically relevant to DIPG, we initially examined their effects on the expression of key proteins in the CDK4/6-cycD1-RB and mTOR signaling pathways in three ex vivo human DIPG cell lines, at concentrations known to elicit strong on-target effects (0.5 and 10 µM, respectively).
Heterodimeric CDK4/6–cycD complexes phosphorylate and, consequently, inhibit RB activity. Therefore, hypophosphorylated RB is an indicator of inhibition of CDK4/6 and release of the block on RB activity. We found that palbociclib effectively inhibited CDK4/6 after 24 hours, eliciting marked decreases in the phosphorylation of RB in all three cell lines (Figure 1), and in a dose-dependent manner across a range of doses in SF7761 (Figure 2). As palbociclib is thought to have maximal activity in situations where RB is intact and p16INK4A is lacking, we wished to clarify that p16INK4A expression was absent in our cells (although deletion of CDKN2A and a lack of p16INK4A protein expression has been shown previously in SF7761,19 SF862820, and SU-DIPG IV20). As our cell lines all lacked p16INK4A expression (Figure S1), we suggest that, in DIPG cells, palbociclib acts as a proxy for the p16INK4A-mediated inhibition of CDK4/6 and the consequential attenuation of cell cycle progression, which is normally found in noncancerous cells.4 Furthermore, as the expression of CDK4, CDK6, and cycD1 were largely unaffected by palbociclib treatment, it appears more likely that the observed impact of palbociclib on cell proliferation resulted from its archetypal effects on CDK4/6 inhibition alone, rather than any off-target effects or nonspecific hypertoxicity affecting the CDK4/6-cycD-RB pathway.
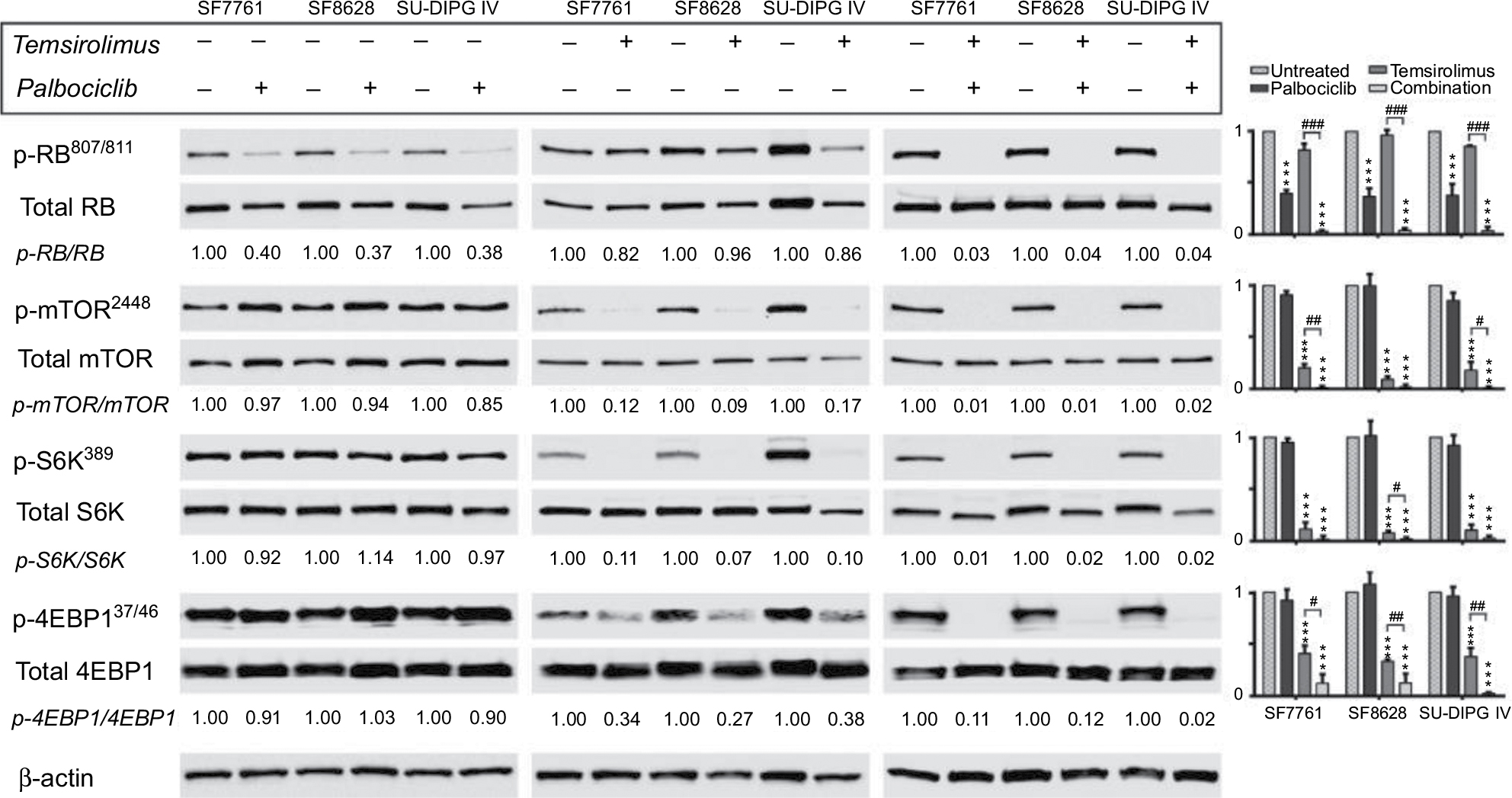 | Figure 1 CDK4/6 and mTOR inhibitors cross-regulate, leading to enhanced inhibition of the respective pathways when used in combination. Notes: SF7761, SF8628, and SU-DIPG IV cells were treated with vehicle, 0.5 µM palbociclib, 10 µM temsirolimus, or coadministered 0.5 µM palbociclib with 10 µM temsirolimus for 24 hours prior to total protein extraction and immunoblot analyses. Quantitative densitometry values are shown below each blot. Densitometry graphs compare single-agent treatments and combination treatment to vehicle-treated controls (right). β-actin was used as a loading control. Data are the mean ± SEM of triplicate determinations in all instances. *p < 0.05; **p < 0.01; ***p < 0.001 (compared to respective vehicle treatment). #p < 0.05; ##p < 0.01; ###p < 0.001 (compared to respective temsirolimus single-agent treatment). Abbreviations: RB, retinoblastoma protein; CDK, cyclin-dependent kinase; mTOR, mechanistic target of rapamycin protein; S6K, p70 ribosomal protein S6 kinase; 4EBP1, eukaryotic translation initiation factor 4E-binding protein 1; rictor, rapamycin-insensitive companion of mammalian target of rapamycin; p-, phosphorylated form of protein. |
 | Figure 2 Coadministration of palbociclib with temsirolimus yields beneficial enhancement of CDK4/6 and mTOR inhibition in DIPG cells. Notes: SF7761 cells were treated with a range of doses of palbociclib, temsirolimus or temsirolimus coadministered with 0.5 µM palbociclib for 24 hours prior to total protein extraction. Immunoblot analyses showed palbociclib and temsirolimus effectively inhibited CDK4/6-cycD1-RB and mTOR signaling pathways in DIPG cells, respectively, while each drug also exhibits inherent attributes to disrupt the other’s target protein, leading to an ostensible synergistic outcome when used in combination. Quantitative densitometry values are shown below each blot. Densitometry graphs compare single-agent temsirolimus treatment and temsirolimus given in combination with 0.5 µM palbociclib (right). The dotted line emphasizes 0.5 µM palbociclib single-agent results versus combination effects. β-actin was used as a loading control. Data are the mean ± SEM of triplicate determinations in all cases. #p < 0.05; ##p < 0.01; ###p < 0.001 (compared to respective temsirolimus single-agent treatment). Abbreviations: RB, retinoblastoma protein; CDK, cyclin-dependent kinase; mTOR, mechanistic target of rapamycin protein; S6K, p70 ribosomal protein S6 kinase; 4EBP1, eukaryotic translation initiation factor 4E-binding protein 1; rictor, rapamycin-insensitive companion of mammalian target of rapamycin; p-, phosphorylated form of protein; PD, palbociclib; TM, temsirolimus. |
In contrast, temsirolimus had a limited effect on RB activation in the cell lines tested (Figure 1), and only demonstrated an effect on RB at the highest doses in SF7761 (ie, 10–40 µM; Figure 2). Concurrent use of both drugs potentiated inhibition of CDK4/6 by palbociclib and enhanced RB activation, indicative of antiproliferative effects. Furthermore, immunoblotting revealed that the combination treatment produced a conspicuous decrease in CDK expression, which was not readily observable following single-agent treatments. Moreover, this may contribute to inhibition of cell proliferation.
Temsirolimus is a rapamycin analog and, thus, a specific inhibitor of mTOR. It binds with the FK506-binding protein 12 (FKBP12), which then directly binds mTORC1 and obscures the correct alignment of substrates to its catalytic cleft, thus preferentially inhibiting phosphorylation of the mTOR substrate p70 ribosomal protein S6 kinase (S6K) over 4E-binding protein 1 (4EBP1).21 In all three cell lines, temsirolimus elicited a significant decrease in mTORC1 activity, indicated by decreased phosphorylation of mTOR and its downstream targets S6K and 4EBP1 (Figure 1). This effect was dose-dependent in SF7761 and no phosphorylated mTOR protein was detectable following treatment with 20 µM temsirolimus (Figure 2). Moreover, dose-dependent reduction of cycD1 resulted from temsirolimus treatment, which showed some correlation with reduced mTOR activity in the lower dose range (fold change for cycD1 and p-mTOR/mTOR, respectively, at: 0.2 µM = 0.65 and 0.48; 2 µM = 0.38 and 0.21; 10 µM = 0.33 and 0.08; 20 µM = 0.17 and 0.01; and 40 µM = 0.07 and 0.01 µM). This reflects the role of mTORC1 as a positive regulator of the translation of cycD1 protein,22 indicating on-target effects of temsirolimus in DIPG cells.
Furthermore, palbociclib potentiated inhibition of mTORC1 by temsirolimus, with the addition of 0.5 µM palbociclib bringing about additional significant downregulation of mTOR, S6K, and 4EBP1 phosphorylation. Palbociclib’s augmentation of the effectiveness of temsirolimus on mTOR pathway inhibition shows the hallmarks of being more than just an additive effect, because when palbociclib was used as a single drug at this low dose it had an unremarkable effect on mTOR, S6K, or 4EBP1. With single-agent temsirolimus eliciting substantial reductions in mTOR activity, palbociclib’s potentiation of the direct effect of temsirolimus on mTOR was difficult to quantify, with increased effects on mTOR slightly observable in only one cell line (SF7761; Figure 2). However, increased effects on mTOR following combination treatment were observed indirectly via increased hypophosphorylation of 4EBP1 in all three cell lines (Figure 1). For example, in SF7761, using 0.02 µM temsirolimus in combination with 0.5 µM palbociclib led to a fold change of 0.32 in mTOR phosphorylation, whereas the same dose of temsirolimus only elicited a fold change of 0.93 for the single-agent treatment (Figure 2). Likewise, S6K and 4EBP1 showed significantly increased hypophosphorylation in SF7761 across a range of doses following combination treatment compared with temsirolimus alone (eg, p-S6k/S6K: fold change at 0.02 µM temsirolimus = 0.90 and 0.01 for temsirolimus and temsirolimus + palbociclib, respectively; and p-4EBP/4EBP: fold change at 0.02 µM temsirolimus = 0.97 and 0.53 for temsirolimus and temsirolimus + palbociclib, respectively).
Concurrent use of temsirolimus with palbociclib also resulted in increased activation of RB, despite the equivalent single-agent doses having minimal effects on RB phosphorylation (Figures 1 and 2). Specifically, in SF7761 cells, 0.5 µM palbociclib and 0.02 µM temsirolimus only elicited a fold change in RB phosphorylation of 0.5 and 0.93, respectively. In combination, these doses caused a fold change of 0.06. Furthermore, combination treatment produced a conspicuous decrease in CDK expression, which was not readily observed following single-agent treatments (Figure 2). Finally, use of both drugs together promoted hypophosphorylation of the mTORC2-associated rictor protein (Figure 2), signifying rictor activation and enhanced mTORC2 activity. As S6K is a negative regulator of rictor,23 it is unsurprising that the increased inhibition of S6K observed following combination treatment then stimulated rictor activation. However, this is an undesirable side effect of an anti-tumorigenic treatment because increased mTORC2 activity signifies increased cell growth and proliferation. Nevertheless, considering the increased antiproliferative effects of the drugs in combination observed in our other assays (see below), we propose that the concurrent use of both drugs establishes a cellular environment not conducive to cell expansion and, thus, mTORC2 activation is ineffectual.
CDK4/6 and mTOR inhibition decrease viability in DIPG cells without triggering extensive cytotoxicity
After we had established that palbociclib and temsirolimus had significant on-target effects on DIPG cells, we attempted to establish whether these molecular effects translated to significant inhibition of DIPG cell growth. We first measured changes in cell viability in our three DIPG cell lines treated with a range of concentrations of either palbociclib or temsirolimus (200 nM–200 µM). Following 24-hour treatment with palbociclib, cell viability was found to decrease in a dose-dependent manner in all three cell lines (Figure 3A). A highly significant reduction in viability was reached with 15 µM palbociclib in all cell lines. However, significant cytotoxicity was only evident in cells at 25 µM palbociclib (Figure 3B). This implies a cytostatic mode of action at IC50 doses of palbociclib (Table 1 and Figure S2). Similarly, temsirolimus provoked a dose-dependent loss in cell viability in all three cell lines (Figure 3C) and showed cytostatic traits. Specifically, temsirolimus elicited significant loss of viability in SF7761 and SU-DIPG IV following treatment with 10 µM temsirolimus, whereas only causing substantial toxicity at 25 µM (Figure 3D); this is higher than the calculated IC50 values of 20.9 and 16.8 µM, for SF7761 and SU-DIPG IV, respectively (Table 1 and Figure S2). In SF8628, no significant cytotoxicity following temsirolimus treatment was observed at any of the concentrations tested (Figure 3D).
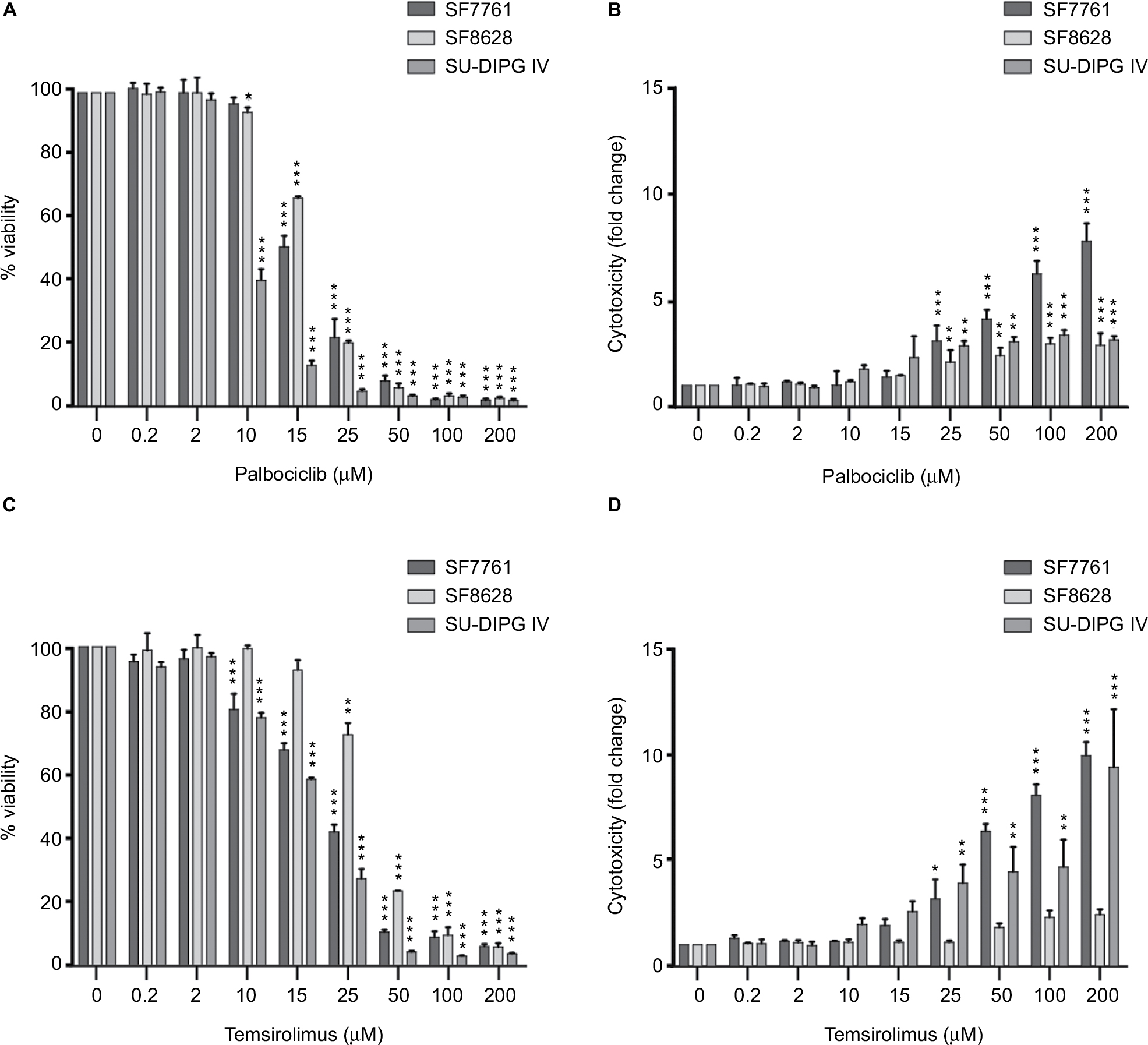 | Figure 3 Palbociclib and temsirolimus treatments as monotherapies reduce cell viability in DIPG. Notes: (A) calcein-AM cell viability assays were conducted on SF7761, SF8628, and SU-DIPG-IV DIPG cell lines treated with increasing concentrations of palbociclib for 24 hours. (B) drug cytotoxicity was measured following 24 hours treatment with palbociclib, using ethidium homodimer-1 (ethD-1). (C) cell viability assays were conducted on SF7761, SF8628, and SU-DIPG IV cell lines treated with increasing concentrations of temsirolimus for 24 hours. (D) drug cytotoxicity was measured following 24-hour treatment with temsirolimus, using ethD-1. Data are the mean ± SEM of triplicate determinations. *p < 0.05, **p < 0.01, and ***p < 0.001 (compared with the respective vehicle treatment). |
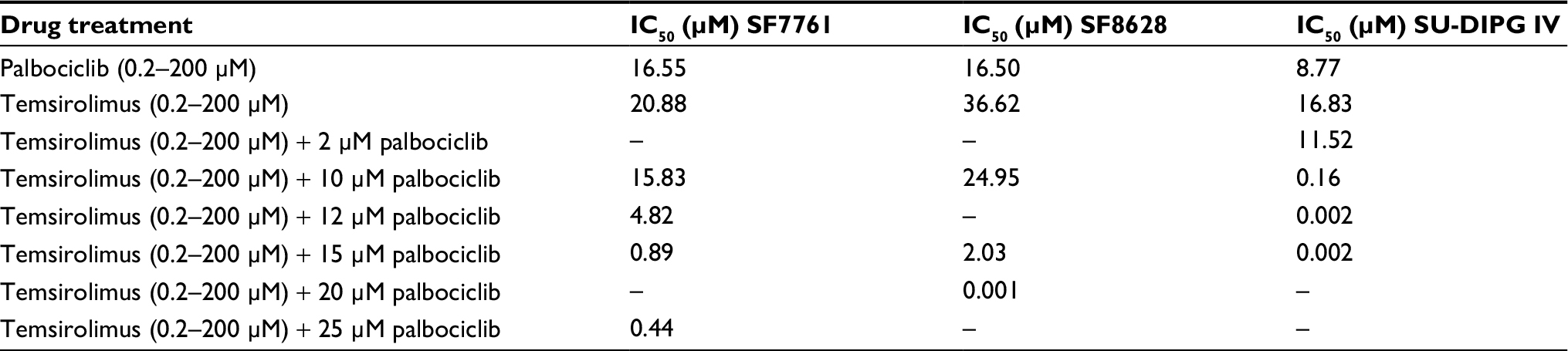 | Table 1 IC50 values for 24-hour single-agent and combination treatments in SF7761, SF8628, and SU-DIPG IV cells |
Palbociclib and temsirolimus act synergistically to restrict the growth of DIPG cells
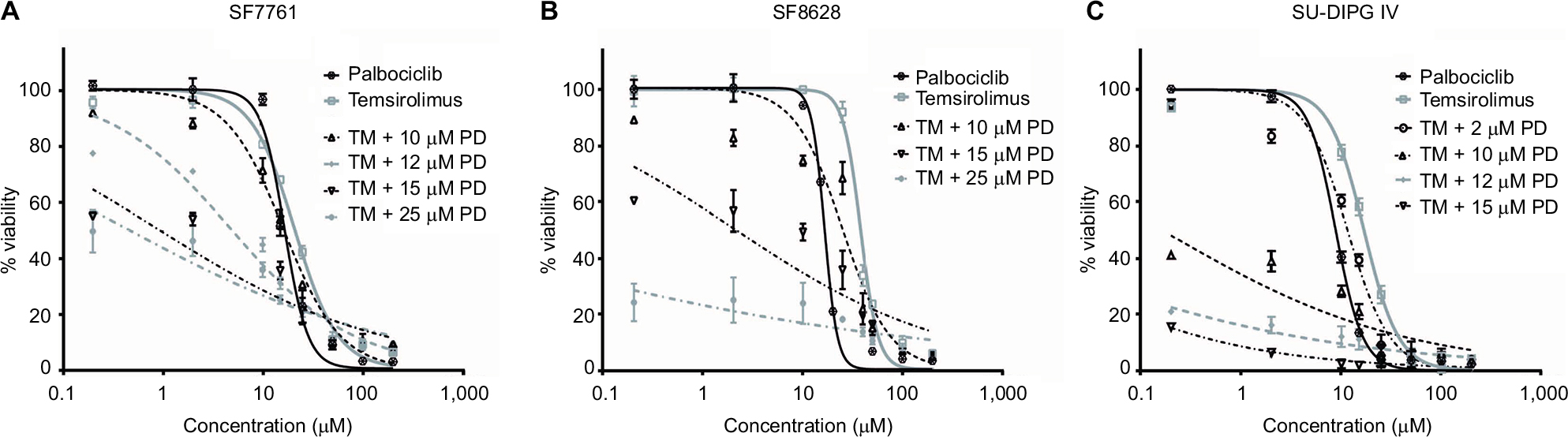
It has been shown previously in other malignancies that overcoming CDK4/6 inhibitor resistance and increasing the therapeutic potency of CDK4/6 inhibitors can be achieved by combining this class of therapeutic with other pathway-selective agents.24 As palbociclib and temsirolimus appeared effective at inhibiting DIPG cell growth as monotherapies, we next assessed the effect of the drugs in combination on cell viability. To achieve this, we treated cells with a variable concentration of temsirolimus (200 nM–200 µM) in combination with a single fixed dose of palbociclib for 24 hours (0, 2, 10, 12, 15, or 25 µM). Combination treatments demonstrated that the two drugs worked effectively in combination, and increasing the concentration of the fixed dose of palbociclib triggered greater reductions in cell viability (Figure 4). Quantitative assessment of synergy generated combination indices that were indicative of additive or synergistic effects. For example, in SF7761 and SF8628 cells, a 15 µM fixed dose of palbociclib yielded highly synergistic combinations with various doses of temsirolimus, exclusively producing combination index values <1, with most ranging between 0.3 and 0.7, consistent with “true synergism”.25 In experiments involving SU-DIPG IV, a lower dose of 10 µM palbociclib was sufficient to bring about strong synergistic effects on cell viability.
 | Figure 4 Coadministration of palbociclib and temsirolimus decreases cell viability synergistically. Notes: SF7761 (A), SF8628 (B), and SU-DIPG IV (C) cells were treated with single-agent temsirolimus at increasing concentrations (0.2–200 μM) or in the same concentration range combined with a single fixed dose of palbociclib (2, 10, 12, 15, or 25 μM). Cell viability was assessed using calcein-AM staining. A combination index score was assigned to each of the different combinations and is shown on the right. Data are the mean ± SEM of triplicate determinations. |
A more detailed examination of cell viability using flow cytometry (Figure 5) supported our initial findings, whereas also indicating that although palbociclib caused only limited cell death at lower doses, cell numbers generally decreased steadily to below the percentage of live cells in all instances (Figure 5, left panels). Doses of palbociclib above 25 µM appeared to be highly toxic, causing considerable cell death, consistent with our initial cytotoxicity assays (Figure 3B). Thus, palbociclib doses of this magnitude appeared fundamentally cytotoxic and were excluded from the experiments that were conducted subsequently. Cytostatic-driven reductions in cell viability were more evident in cells treated with temsirolimus. In SF7761 and SF8628, temsirolimus produced negligible cell death at all concentrations up to 50 µM, whereas cell numbers consistently decreased in a dose-dependent manner relative to untreated cells (Figure 5, center panels). Coadministration of a low fixed dose of palbociclib (either 15 µM for SF7761 and SF8628, or 10 µM for SU-DIPG IV cells), combined with varying concentrations of temsirolimus potentiated the effects on cells (Figure 5, right panels), provoking a significant decrease in the number of cells, together with cell viability (particularly in SF7761 and SU-DIPG IV) – implying a shift from a cytostatic to cytotoxic mode of action, indicative of increased anti-tumorigenic properties. These doses of palbociclib were chosen as they were close to the drug’s IC50 and elicited strong synergism with temsirolimus (established in the above-described previous experiments), whereas they were largely sublethal in the single-drug flow cytometric analyses.
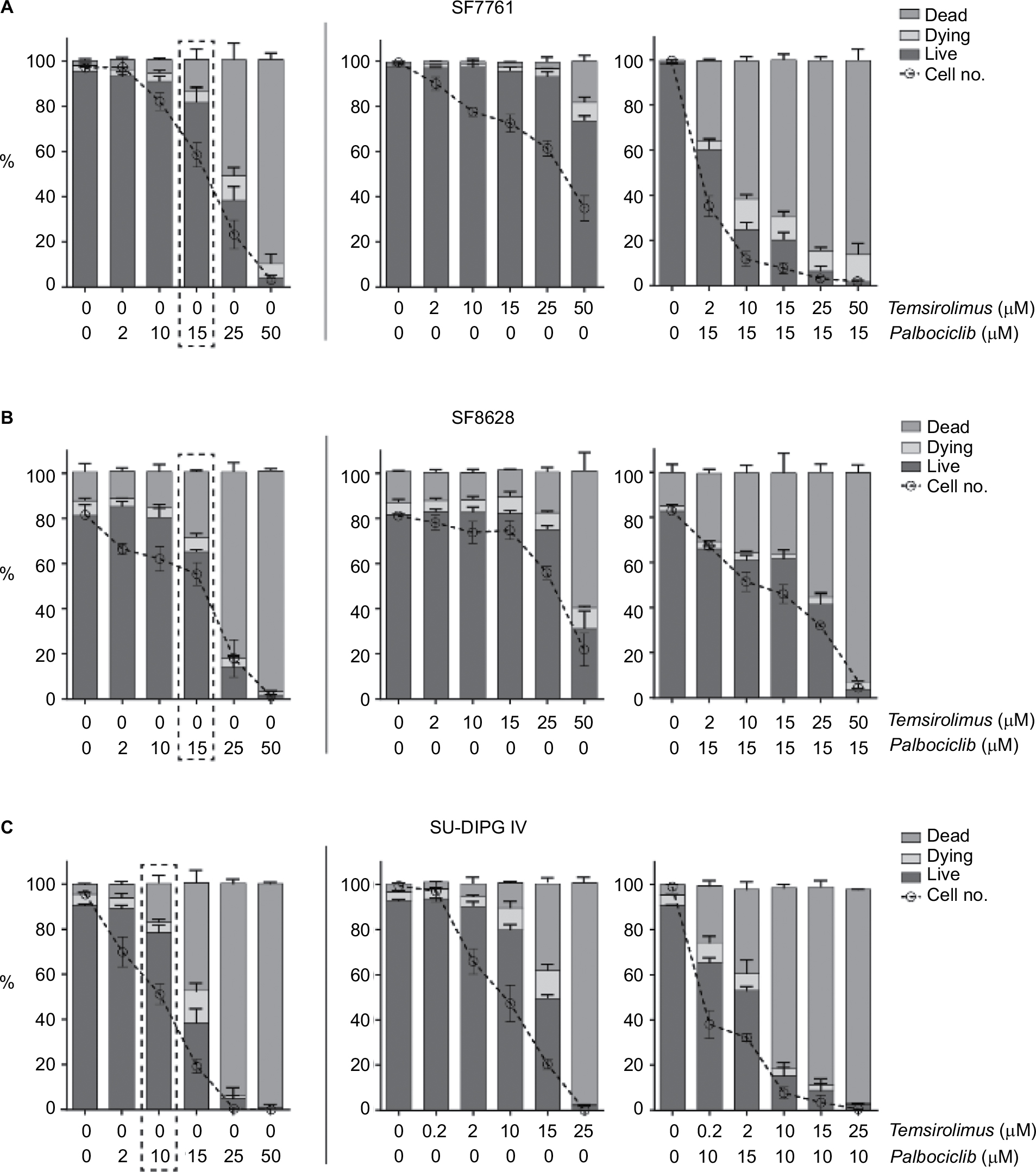 | Figure 5 Coadministration of palbociclib and temsirolimus decreases cell viability principally through restricting cell proliferation. Notes: Bivariate flow cytometric analyses of cell viability in SF7761 (A), SF8628 (B) and SU-DIPG IV (C) cells. Cells were treated with palbociclib (0–50 mM), temsirolimus (0–50 mM) or both (temsirolimus 0–50 mM, with a fixed dose of either 10 mM or 15 mM palbociclib, dependent on cell line sensitivity). Analysis of “% live cells” (calcein-AM positive), “% dead cells” (ethD-1 positive) and “% dying cells” (double positive) was performed. The dotted line box in each of the panels on the left-hand side emphasizes the dose of palbociclib used for the combinatorial analyses. Data are the mean ± SEM of triplicate determinations. |
Combined CDK4/6 and mTOR inhibition cytostatically inhibit proliferation and trigger G0–G1 cell cycle arrest
Having observed that the combination of palbociclib and temsirolimus potentiated each drug’s cytostatic effects, we next investigated whether these effects continued beyond the initial 24-hour period. Low, sublethal doses of each drug were selected on the basis of previous data, as well as cell-line-specific drug sensitivity and some preliminary experiments that examined the effects of the drugs over time (data not shown). These criteria led to us utilizing a dose of 2 µM palbociclib for all three cell lines, as this drug concentration had no effect on cell death in any of the three cell lines, and only a very limited effect on cell proliferation in SF8628 and SU-DIPG IV. A concentration of 10 µM temsirolimus was utilized for SF7761 and SF8628, whereas a lower dose of 0.2 µM temsirolimus was used for SU-DIPG IV, as this cell line was more sensitive than the other cell lines to temsirolimus and showed significant loss of viability at 10 µM in the previous flow cytometric analyses. Over a 72-hour period the viability of control cells remained high (Figure 6A–C, left). Similarly, treatment with either palbociclib or temsirolimus did not impact cell viability. However, cells treated with palbociclib or temsirolimus clearly demonstrated diminished proliferation, which intensified over time (Figure 6A–C, center and right, respectively).
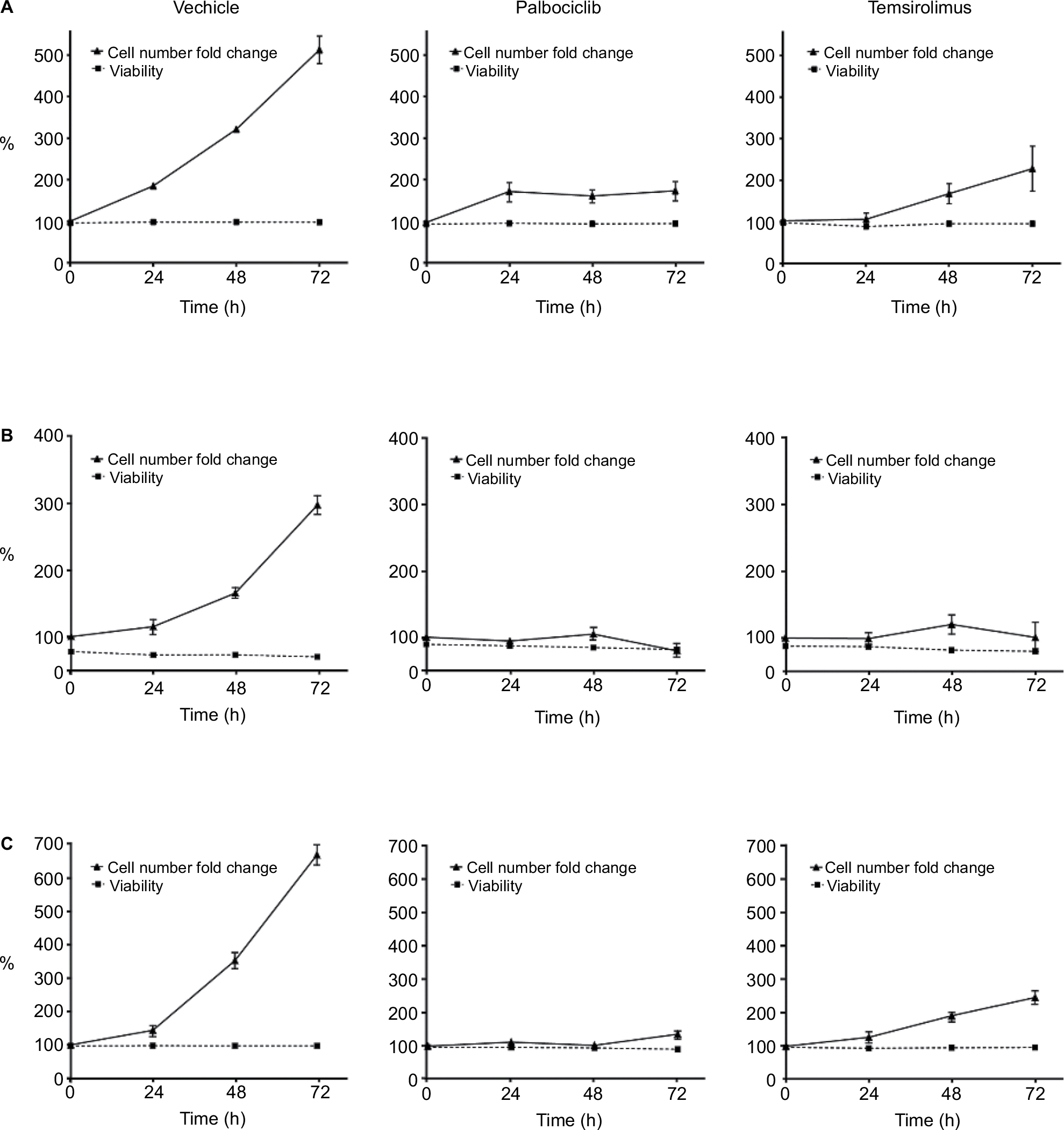 | Figure 6 Palbociclib and temsirolimus inhibit the growth of DIPG cells cytostatically. Notes: SF7761 (A), SF8628 (B), and SU-DIPG IV (C) cells were treated with vehicle, palbociclib (2 µM), or temsirolimus (10 µM for SF7761 and SF8628; 0.2 µM for SU-DIPG IV) for 0–72 hours. Cells were then counted and stained with calcein-AM to deduce % viability by using flow cytometry. Data are the mean ± SEM of triplicate determinations in all cases. |
Effects on cell proliferation were investigated further via cell cycle analyses. In SF7761 cells, treatment with palbociclib and temsirolimus for 24 hours caused a marked increase in the percentage of cells in the G0–G1 phase of the cell cycle, consistent with CDK4/6 and mTOR inhibition, respectively (Figure 7A). G0–G1 arrest persisted over 72 hours in all three cell lines tested, with SU-DIPG IV demonstrating the greatest amount of cell cycle arrest (Figure 7B, 7C, and S3). To further assess the antiproliferative effects of the drugs, we conducted clonogenic assays using SU-DIPG IV cells that, unlike SF7761 and SF8628, exhibit strong intrinsic clonogenicity. We found palbociclib and temsirolimus inhibited colony formation time dependently as single agents, and concurrent treatment with both drugs potentiated the effect of temsirolimus at all three time points (Figure 7D and S4). The clonogenicity of SU-DIPG IV showed very high sensitivity to palbociclib in our preliminary combination experiments and, thus, a dose of 0.5 µM palbociclib (in line with our initial immunoblotting assays) was used in combination with temsirolimus, which was sufficient to considerably augment the ability of temsirolimus to reduce colony numbers. Taken together, these data suggest the drugs provided sustained antiproliferative effects via cell cycle arrest.
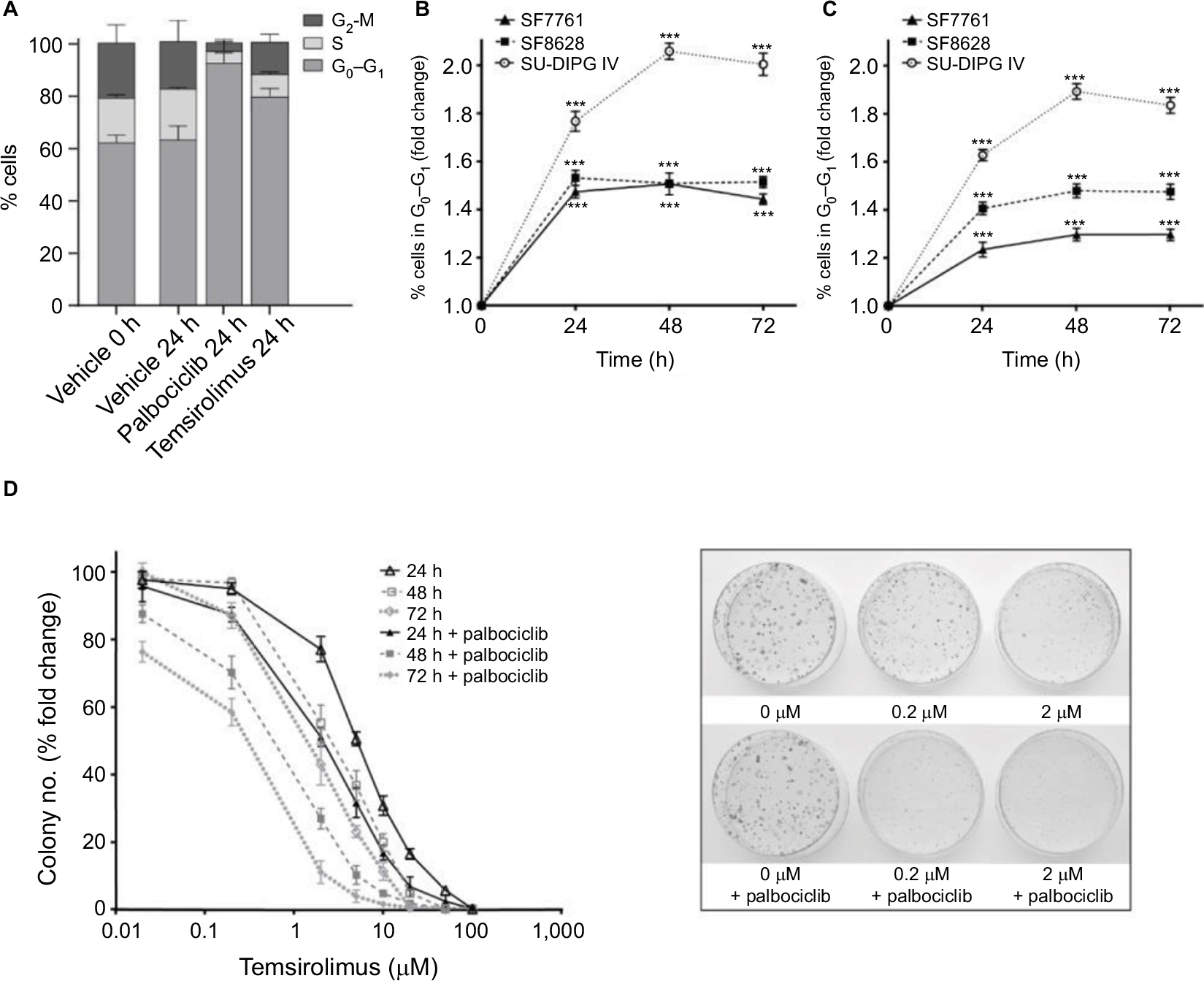 | Figure 7 Palbociclib and temsirolimus treatments lead to cell cycle arrest. Notes: (A) DRAQ5 fluorescent dye was used to conduct flow cytometric cell cycle analysis on SF7761 cells treated with vehicle, 2 µM palbociclib, or 10 µM temsirolimus for 24 hours. (B) DRAQ5 cell cycle analysis of SF7761, SF8628, and SU-DIPG IV cells treated with 2 µM palbociclib demonstrated a significant increase in the percentage of cells in the G0–G1 phase over time. (C) DRAQ5 cell cycle analysis of SF7761 and SF8628 cells treated with 10 µM temsirolimus, and SU-DIPG IV cells treated with 0.2 µM temsirolimus, demonstrated a significant increase in the percentage of cells in the G0–G1 phase over time. (D) SU-DIPG IV cells were treated with temsirolimus, or the combination of temsirolimus and 0.5 µM palbociclib for 24–72 hours, and colonies were counted after 14 days. Representative plates following 72 hours treatment are shown (right). Data are the mean ± SEM of triplicate determinations in all cases. *p < 0.05, **p < 0.01, and ***p < 0.001 (compared with respective vehicle treatment). |
Palbociclib does not cause significant neurotoxicity in normal brain tissue
It has previously been demonstrated that the lipophilic nature of temsirolimus enables it to readily cross the BBB.11 In contrast, palbociclib does not readily cross the BBB.26 To address this issue, we propose it would be prudent to deliver palbociclib directly into the brain of DIPG patients to circumvent the BBB, whereas temsirolimus could be infused intravenously, which does not pose any known toxicity issues.12 With this in mind, we examined the effect of palbociclib on normal brain tissue. To achieve this, we initially exposed normal neurons and glial cells within primary rat hippocampal cultures to 2 µM palbociclib and observed no discernable toxic effects, relative to the general morphology and numbers of cells seen in vehicle-treated cultures (Figure 8). We went on to investigate the toxicity of palbociclib in vivo by carrying out direct infusions of either 100 or 400 µM palbociclib into the striatum of normal rat brains. Analyses of the brain tissue showed no significant neurotoxicity caused by palbociclib, compared with control animals (Figure 9).
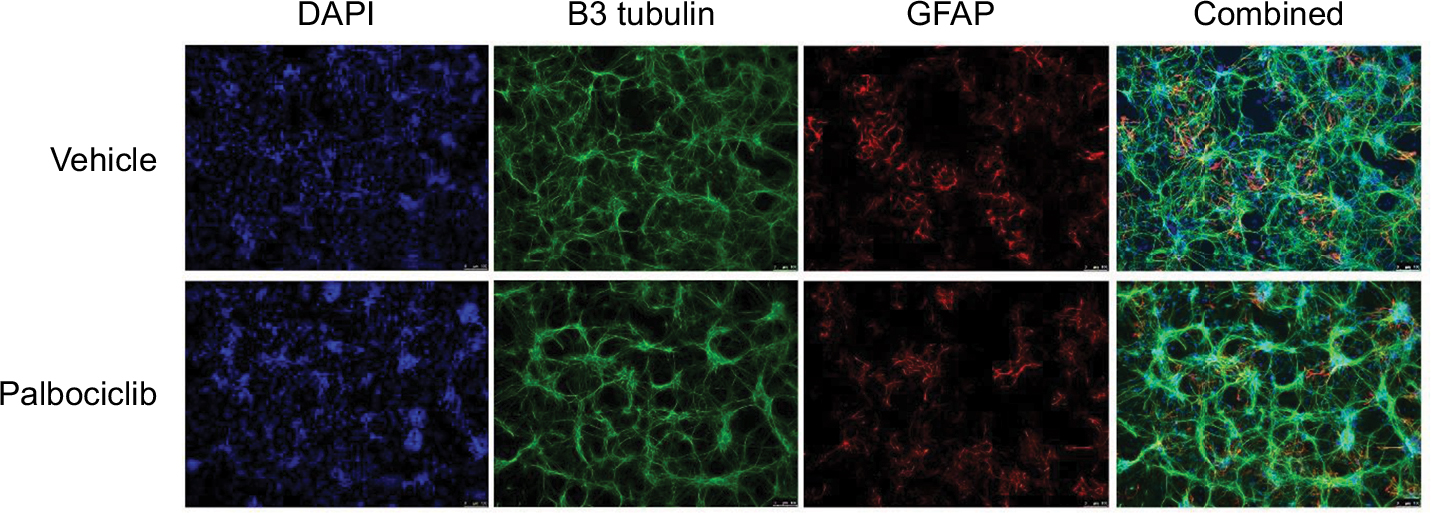 | Figure 8 Treatment of rat hippocampal primary cultures with palbociclib did not elicit cytotoxicity. Notes: Primary hippocampal culture treated with vehicle solution or 2 µM palbociclib for 24 hours. Intact neuronal networks and normal glial cell morphology was observed in cultures treated with palbociclib, suggesting no significant toxicity. Cell nuclei were stained with DAPI (blue); neurons were stained with B3 tubulin antibody (green); glial cells were stained with the GFAP antibody (red/orange). Abbreviations: DAPI, 4′,6-diamidino-2-phenylindole; B3 tubulin, tubulin, beta III isoform; GFAP, glial fibrillary acidic protein. |
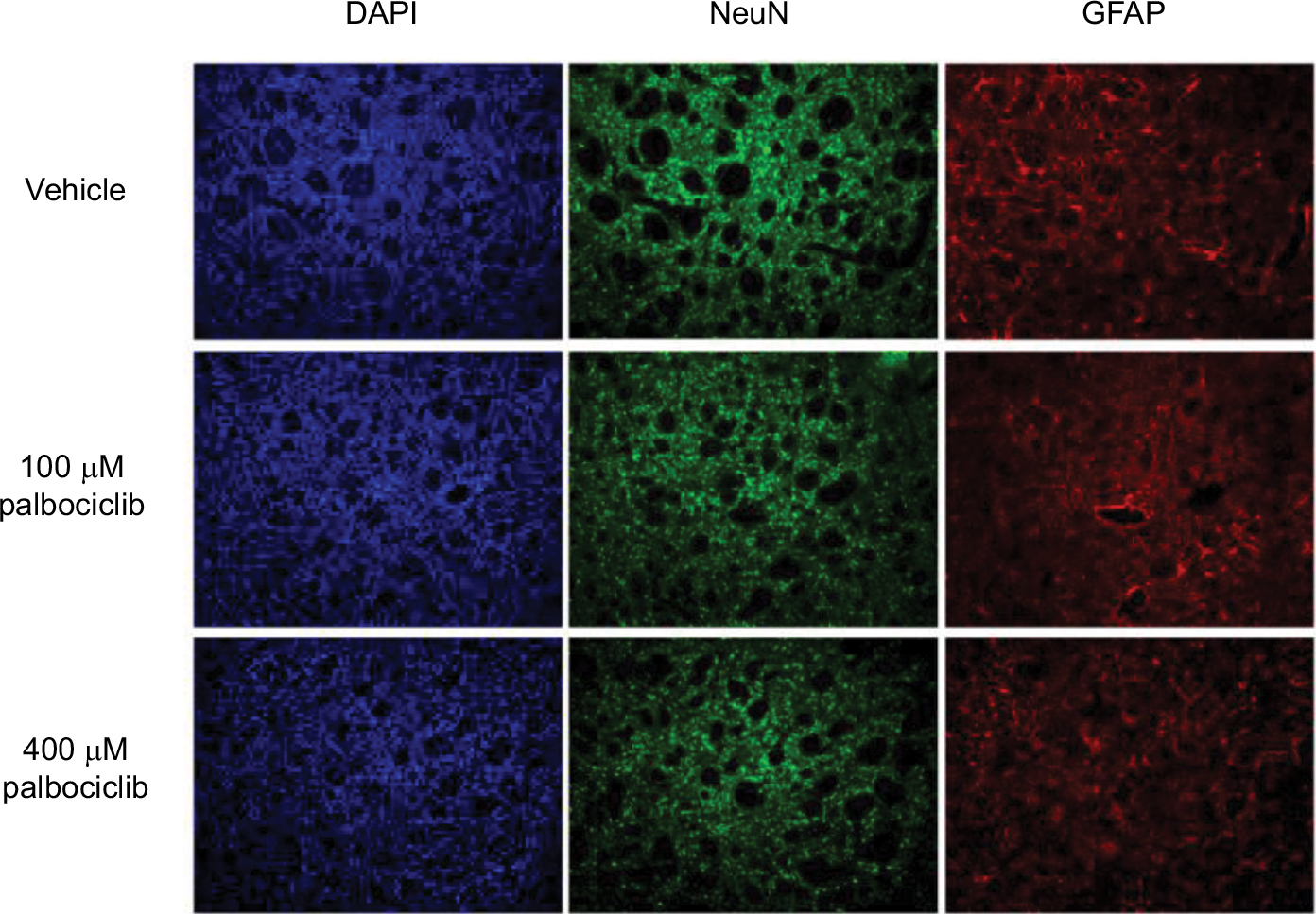 | Figure 9 Direct delivery of palbociclib into rat brain did not elicit discernible toxicity. Notes: Palbociclib was infused into the striatum of rat brains at a rate of 1 µL/min (5 µL total) at either 100 or 400 µM. Tissue was harvested after 48 hours and analyzed using immunofluorescence staining. Tissue morphology appeared normal in rats dosed with palbociclib, suggesting no significant toxicity. Cell nuclei were stained with DAPI (blue); neurons were stained with NeuN antibody (green); glial cells were stained with GFAP antibody (red). Abbreviations: DAPI, 4′,6-diamidino-2-phenylindole; NeuN, neuronal nuclear protein; GFAP, glial fibrillary acidic protein. |
Discussion
DIPG is a highly heterogeneous disease characterized by lethal tumors that differ in cellular origin and pathogenesis. In part, this heterogeneity is derived from the genetic basis of the disease and two archetypal causative mutations.27,28 In the present study, we have shown that palbociclib and temsirolimus can reduce DIPG cell proliferation in vitro in three different cell lines encompassing the two histone H3 mutations most frequently associated with DIPG (H3.3K27M represented by SF7761 and SF8628; and H3.1K27M in SU-DIPG IV). On the whole, we found that palbociclib and temsirolimus were more efficacious in SU-DIPG IV cells, suggesting that DIPG tumors harboring the H3.1K27M mutation may be more suited to the proposed drug combination. Although SF7761 and SF8628 cells both harbor the H3.3K27M mutation, SF7761 was generally more sensitive to drug treatments. This variation in the two H3.3K27M cell lines likely relates to other genotypic, and phenotypic, differences that exist between these cells. Crucially, the SF7761 cell line has been modified to express telomerase, whereas SF8628 is unmodified and has been established directly from DIPG biopsy. Consequently, SF7761 cells can potentially survive indefinitely in vitro, whereas SF8628 cells senesce over time. Furthermore, SF7761 cells have a shorter doubling time compared to SF8628 (Figure 6), are typically much smaller cells, and are cultured in a different culture media formulation. Furthermore, these cell lines have been shown to have distinctive endogenous genetic profiles beyond the shared H3 histone mutation. For example, expression of the nuclear kinase WEE1 is much higher in SF8628 compared with SF7761.29 Asymmetrical expression of this important cell cycle regulator could conceivably contribute to the dissimilar response to our proposed novel drug combination, which specifically targets cell cycle arrest. Irrespective of these differences, the combination of palbociclib and temsirolimus brought about strong growth inhibition in both SF7761 and SF8628.
Many novel DIPG therapeutics have been proposed in recent years, but none have been successfully implemented.30,31 Single-agent rapalog treatments for CNS tumors, including DIPG, have had limited success.32–36 However, temsirolimus has been well tolerated in pediatric patients.12 The situation is similar for CDK4/6 inhibitors, where use of these drugs as robust single-agent treatments for different cancers has proven unconvincing.32,38–40 Nevertheless palbociclib has been well tolerated, increased survival in a DIPG mouse model,16 and can elicit anti-tumor immunological effects.41 Both rapalogs and CDK4/6 inhibitors have shown tolerability in the CNS, but have produced mixed results for treating CNS tumors.3,7,12,23
Palbociclib has strong potential utility for treating DIPG, because RB disruption is uncommon in DIPG and cells typically lack functional p16INK4A – key prerequisites for palbociclib efficacy. Furthermore, p16INK4A has been shown to be repressed by aberrant histone H3.3K27M expression, which is a common mutation in DIPG.26,42 In addition, rapalogs have intrinsic potential for treating DIPG and can be used to overcome the characteristic hyperactivated PI3K/AKT/mTOR signaling exhibited in DIPG cells.37 For example, the platelet-derived growth factor receptor (PDGFR) is commonly overexpressed in DIPG and drives cancer cell proliferation via the PI3K/AKT/mTOR signaling pathway. Combining rapalogs with CDK4/6 inhibitors has proven effective previously4,18 and, in accord with other groups, we found that palbociclib potentiates the effects of temsirolimus and may facilitate circumvention of its downstream repression by AKT. We propose palbociclib negates the intrinsic CDK4–cycD1 complex inhibition of tuberous sclerosis complex 2,41 thereby increasing mTORC1 inhibition and sensitization to temsirolimus. Furthermore, the decreased cycD1 and CDK4/6 expression, along with augmented hypophosphorylation of RB observed following our combination treatments, would serve to decrease mTOR activity further within this paradigm.
In addition to palbociclib potentially supplementing the effects of temsirolimus, the latter may potentiate the former. Documented undesirable effects, such as attenuation of its own anti-mitogenic effects through increased cellular metabolism, that have curtailed interest in palbociclib as a cancer treatment can be abrogated by mTOR inhibition.24 This was observed in our experiments via enhanced RB activation. We hypothesize this may arise through the capacity of temsirolimus to block global promitogenic cellular effects caused by palbociclib (as described earlier), in addition to a blockade of cycD1 expression, which is positively regulated by mTOR and would, therefore, be restricted by temsirolimus. With a cycD1 deficit, there would be a propensity for further restriction of CDK4/6–cycD1 complex formation and, thus, decreased RB phosphorylation. Interestingly, if CDK4/6–cycD1 complexes form part of the mTOR regulation machinery in DIPG cells, and palbociclib and temsirolimus synergize to drive a significant deficit of cycD1, then ultimately, a negative feedback loop would ensue whereby decreased cycD1 expression intensifies mTOR inhibition, and mTOR inhibition decreases cycD1 expression. Thus, palbociclib and temsirolimus potentially elicit an anti-mitogenic positive feedback loop when used concurrently, with palbociclib inducing rapalog sensitization, and temsirolimus inhibiting the negative side effects of palbociclib. We propose that pathway cross-regulation and mutual potentiation of the inhibition of target proteins drives increased antiproliferative effects and the observed shift toward cytotoxicity in cancer cells.
Despite advocating that palbociclib potentiates the inhibitory effects of temsirolimus on mTOR and facilitates circumvention of AKT-driven restoration of mTORC1 activity, we observed that concurrent use of palbociclib and temsirolimus increased activation of the mTORC2-associated rictor protein. This did not, however, appear to affect cell viability. This may, in part, be due to the relatively short 24-hour exposure time for most of our drug treatments, chosen principally to mimic patient drug infusion times, but which also illustrated rapid drug efficacy. Furthermore, where longer treatment times were used (ie, Figures 6 and 7), promitogenic mTORC2 effects did not appear significant and cell proliferation continued to decline. We are unable to fully rationalize this discrepancy and intend to undertake further investigation but suggest temsirolimus may directly inhibit mTORC2 in DIPG cells. Rapalogs have been found to have an inhibitory effect on mTORC2 in some cancer cells, dependent on high expression of FKBP12.43 Pertinently, FKBP12 is significantly overexpressed in childhood astrocytomas.44
Conclusion
Taken together, our results support the notion that the best clinical potential for developing an efficacious DIPG therapeutic will stem from concurrent inhibition of multiple oncogenic pathways. Our data suggest palbociclib and temsirolimus synergistically reduce cell viability in SF7761, SF8628, and SU-DIPG IV cells primarily through increased cytostatic effects that drive down cell proliferation, with some cytotoxicity likely stemming from the loss of metabolic compensatory pathways. As this project is principally an in vitro study, further investigation is required to translate these findings to the clinical setting. Frequently, treatment failures occur because agents cannot cross the BBB due to efflux transporters. The ability of palbociclib to sufficiently enter the brain remains a concern,26 but mTOR inhibitors have been shown to ameliorate palbociclib efflux and facilitate increased drug concentrations in the CNS.18,45 Nevertheless, our results demonstrate that simultaneous inhibition of the CDK4/6-cycD1-RB and PI3K-AKT-mTOR pathways is a feasible approach to DIPG treatment.
Acknowledgments
The authors thank Abbie’s Army DIPG brain tumor research charity for their generous financial support. The authors also thank Dr Nalin Gupta for the SF7761 and SF8628 cell lines, Dr Michelle Monje for the SU-DIPG IV cell line, and Dr Alex Hoose for general advice and assistance. This study was funded by Abbie’s Army DIPG brain tumor research charity (to NU Barua and AS Bienemann), the Functional Neurosurgery Research Fund held by Southmead Hospital Charities (SS Gill), the Gatsby foundation (to WGB Singleton), and joint funding from the Medical Research Council and the Brain Tumour Charity (grant no. MR/N00130/1 to WGB Singleton).
Disclosure
SS Gill is a consultant for Renishaw PLC. NU Barua is a consultant for Renishaw PLC. The authors report no other conflicts of interest in this work.
References
Freeman CR, Farmer JP. Pediatric brain stem gliomas: a review. Int J Radiat Oncol Biol Phys. 1998;40(2):265–271. | ||
Smith MA, Freidlin B, Ries LA, Simon R. Trends in reported incidence of primary malignant brain tumors in children in the United States. J Natl Cancer Inst. 1998;90(17):1269–1277. | ||
Long W, Yi Y, Chen S, Cao Q, Zhao W, Liu Q. Potential new therapies for pediatric diffuse intrinsic pontine glioma. Front Pharmacol. 2017;8:495. | ||
Heilmann AM, Perera RM, Ecker V, et al. CDK4/6 and IGF1 receptor inhibitors synergize to suppress the growth of p16INK4A-deficient pancreatic cancers. Cancer Res. 2014;74(14):3947–3958. | ||
Malumbres M, Barbacid M. Mammalian cyclin-dependent kinases. Trends Biochem Sci. 2005;30(11):630–641. | ||
Herrera-Abreu MT, Palafox M, Asghar U, et al. Early adaptation and acquired resistance to CDK4/6 inhibition in estrogen receptor-positive breast cancer. Cancer Res. 2016;76(8):2301–2313. | ||
Hidalgo M, Rowinsky EK. The rapamycin-sensitive signal transduction pathway as a target for cancer therapy. Oncogene. 2000;19(56):6680–6686. | ||
Kwitkowski VE, Prowell TM, Ibrahim A, et al. FDA approval summary: temsirolimus as treatment for advanced renal cell carcinoma. Oncologist. 2010;15(4):428–435. | ||
Margolin K, Longmate J, Baratta T, et al. CCI-779 in metastatic melanoma: a phase II trial of the California Cancer Consortium. Cancer. 2005;104(5):1045–1048. | ||
Galanis E, Buckner JC, Maurer MJ, et al; North Central Cancer Treatment Group. Phase II trial of temsirolimus (CCI-779) in recurrent glioblastoma multiforme: a North Central Cancer Treatment Group Study. J Clin Oncol. 2005;23(23):5294–5304. | ||
Zhao H, Cui K, Nie F, et al. The effect of mTOR inhibition alone or combined with MEK inhibitors on brain metastasis: an in vivo analysis in triple-negative breast cancer models. Breast Cancer Res Treat. 2012;131(2):425–436. | ||
Becher OJ, Gilheeney SW, Khakoo Y, et al. A phase I study of perifosine with temsirolimus for recurrent pediatric solid tumors. Pediatr Blood Cancer. 2017;64(7). | ||
Killick-Cole CL, Singleton WGB, Bienemann AS, et al. Repurposing the anti-epileptic drug sodium valproate as an adjuvant treatment for diffuse intrinsic pontine glioma. PLoS One. 2017;12(5):e0176855. | ||
Asby DJ, Cuda F, Beyaert M, Houghton FD, Cagampang FR, Tavassoli A. AMPK activation via modulation of de novo purine biosynthesis with an inhibitor of ATIC homodimerization. Chem Biol. 2015;22(7):838–848. | ||
Arshad A, Yang B, Bienemann AS, et al. Convection-enhanced delivery of carboplatin PLGA nanoparticles for the treatment of glioblastoma. PLoS One. 2015;10(7):e0132266. | ||
Barton KL, Misuraca K, Cordero F, et al. PD-0332991, a CDK4/6 inhibitor, significantly prolongs survival in a genetically engineered mouse model of brainstem glioma. PLoS One. 2013;8(10):e77639. | ||
Berenguer-Daizé C, Astorgues-Xerri L, Odore E, et al. OTX015 (MK-8628), a novel BET inhibitor, displays in vitro and in vivo antitumor effects alone and in combination with conventional therapies in glioblastoma models. Int J Cancer. 2016;139(9):2047–2055. | ||
Olmez I, Brenneman B, Xiao A, et al. Combined CDK4/6 and mTOR inhibition is synergistic against glioblastoma via multiple mechanisms. Clin Cancer Res. 2017;23(22):6958–6968. | ||
Aoki Y, Hashizume R, Ozawa T, et al. An experimental xenograft mouse model of diffuse pontine glioma designed for therapeutic testing. J Neurooncol. 2012;108(1):29–35. | ||
Piunti A, Hashizume R, Morgan MA, et al. Therapeutic targeting of polycomb and BET bromodomain proteins in diffuse intrinsic pontine gliomas. Nat Med. 2017;23(4):493–500. | ||
Weichhart T, Hengstschläger M, Linke M. Regulation of innate immune cell function by mTOR. Nat Rev Immunol. 2015;15(10):599–614. | ||
Averous J, Fonseca BD, Proud CG. Regulation of cyclin D1 expression by mTORC1 signaling requires eukaryotic initiation factor 4E-binding protein 1. Oncogene. 2008;27(8):1106–1113. | ||
Jhanwar-Uniyal M, Gillick JL, Neil J, Tobias M, Thwing ZE, Murali R. Distinct signaling mechanisms of mTORC1 and mTORC2 in glioblastoma multiforme: a tale of two complexes. Adv Biol Regul. 2015;57:64–74. | ||
Franco J, Witkiewicz AK, Knudsen ES. CDK4/6 inhibitors have potent activity in combination with pathway selective therapeutic agents in models of pancreatic cancer. Oncotarget. 2014;5(15):6512–6525. | ||
Chou TC. Theoretical basis, experimental design, and computerized simulation of synergism and antagonism in drug combination studies. Pharmacol Rev. 2006;58(3):621–681. | ||
de Gooijer MC, Zhang P, Thota N, et al. P-glycoprotein and breast cancer resistance protein restrict the brain penetration of the CDK4/6 inhibitor palbociclib. Invest New Drugs. 2015;33(5):1012–1019. | ||
Hashizume R, Andor N, Ihara Y, et al. Pharmacologic inhibition of histone demethylation as a therapy for pediatric brainstem glioma. Nat Med. 2014;20(12):1394–1396. | ||
Monje M, Mitra SS, Freret ME, et al. Hedgehog-responsive candidate cell of origin for diffuse intrinsic pontine glioma. Proc Natl Acad Sci U S A. 2011;108(11):4453–4458. | ||
Mueller S, Hashizume R, Yang X, et al. Targeting Wee1 for the treatment of pediatric high-grade gliomas. Neuro Oncol. 2014;16(3):352–360. | ||
Frazier JL, Lee J, Thomale UW, Noggle JC, Cohen KJ, Jallo GI. Treatment of diffuse intrinsic brainstem gliomas: failed approaches and future strategies. J Neurosurg Pediatr. 2009;3(4):259–269. | ||
Johung TB, Monje M. Diffuse intrinsic pontine glioma: new pathophysiological insights and emerging therapeutic targets. Curr Neuropharmacol. 2017;15(1):88–97. | ||
Grasso CS, Tang Y, Truffaux N, et al. Functionally defined therapeutic targets in diffuse intrinsic pontine glioma. Nat Med. 2015;21(7):827. | ||
Lombardi G, Pambuku A, Bellu L, et al. Effectiveness of antiangiogenic drugs in glioblastoma patients: a systematic review and meta-analysis of randomized clinical trials. Crit Rev Oncol Hematol. 2017;111:94–102. | ||
Moreno-Smith M, Lakoma A, Chen Z, et al. p53 nongenotoxic activation and mTORC1 inhibition lead to effective combination for neuroblastoma therapy. Clin Cancer Res. 2017;23(21):6629–6639. | ||
Geoerger B, Kieran MW, Grupp S, et al. Phase II trial of temsirolimus in children with high-grade glioma, neuroblastoma and rhabdomyosarcoma. Eur J Cancer. 2012;48(2):253–262. | ||
City of Hope Medical Center. A feasibility, dose-escalation study using intracerebral microdialysis to assess the neuropharmacodynamics of temsirolimus in patients with primary or metastatic brain tumors. Available from: http://www.clinicaltrials.gov/ct2/show/NCT00784914. NLM identifier: NCT00784914. Accessed January 12, 2018. | ||
Puget S, Philippe C, Bax DA, et al. Mesenchymal transition and PDGFRA amplification/mutation are key distinct oncogenic events in pediatric diffuse intrinsic pontine gliomas. PLoS One. 2012;7(2):e30313. | ||
Schröder LB, McDonald KL. CDK4/6 inhibitor PD0332991 in glioblastoma treatment: does it have a uture? Front Oncol. 2015;5:259. | ||
Pfizer. A study of PD 0332991 in patients with recurrent Rb positive glioblastoma (PD0332991). Available from: https://www.clinicaltrials.gov/ct2/show/NCT01227434. NLM identifier: NCT01227434. Accessed January 15, 2018. | ||
Pediatric Brain Tumor Consortium. Palbociclib isethionate in treating younger patients with recurrent, progressive, or refractory central nervous system tumors. Available from: https://www.clinicaltrials.gov/ct2/show/NCT02255461. NLM identifier: NCT02255461. Accessed January 16, 2018. | ||
Goel S, Wang Q, Watt AC, et al. Overcoming therapeutic resistance in HER2-positive breast cancers with CDK4/6 inhibitors. Cancer Cell. 2016;29(3):255–269. | ||
Cordero FJ, Huang Z, Grenier C, et al. Histone H3.3K27M represses p16 to accelerate gliomagenesis in a murine model of DIPG. Mol Cancer Res. 2017;15(9):1243–1254. | ||
Schreiber KH, Ortiz D, Academia EC, Anies AC, Liao CY, Kennedy BK. Rapamycin-mediated mTORC2 inhibition is determined by the relative expression of FK506-binding proteins. Aging Cell. 2015;14(2):265–273. | ||
Khatua S, Peterson KM, Brown KM, et al. Overexpression of the EGFR/FKBP12/HIF-2alpha pathway identified in childhood astrocytomas by angiogenesis gene profiling. Cancer Res. 2003;63(8):1865–1870. | ||
Minocha M, Khurana V, Qin B, Pal D, Mitra AK. Co-administration strategy to enhance brain accumulation of vandetanib by modulating P-glycoprotein (P-gp/Abcb1) and breast cancer resistance protein (Bcrp1/Abcg2) mediated efflux with m-TOR inhibitors. Int J Pharm. 2012;434(1–2):306–314. |
Supplementary materials
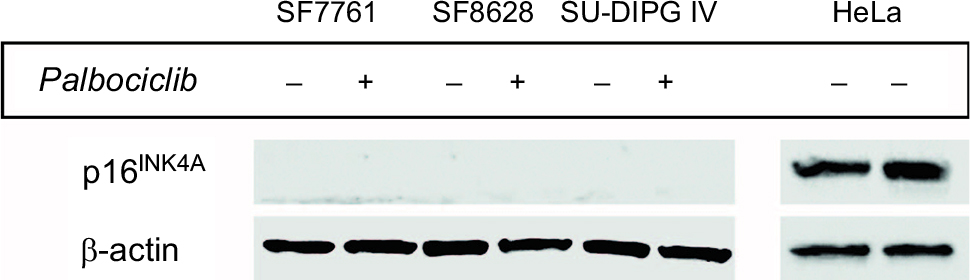 | Figure S1 DIPG cells, ostensibly, do not express p16INK4A. Notes: Immunoblot using p16INK4A-specific antibody against protein from SF7761, SF8628, and SU-DIPG IV cells treated with vehicle (−) or 10 µM palbociclib (+) for 24 hours. HeLa cell protein was used as a positive control for p16INK4A expression. β-actin was used as a loading control. Representative immunoblot of three determinations. Abbreviation: p16INK4A, cyclin-dependent kinase inhibitor 2A. |
 | Figure S2 Logarithmic dose-response curves for palbociclib and temsirolimus single-agent and combination treatments. Notes: (A) SF7761, (B) SF8628, and (C) SU-DIPG IV cells were treated for 24 hours with increasing concentrations of single-agent palbociclib (0.2–200 µM), single-agent temsirolimus (0.2–200 µM) or temsirolimus (0.2–200 µM) combined with a single fixed dose of palbociclib (2, 10, 12, 15 or 25 µM). Cell viability was assessed using calcein-AM staining and an IC50 modeled in each instance. Data are the mean ± SEM of triplicate determinations. Abbreviations: PD, palbociclib; TM, temsirolimus. |
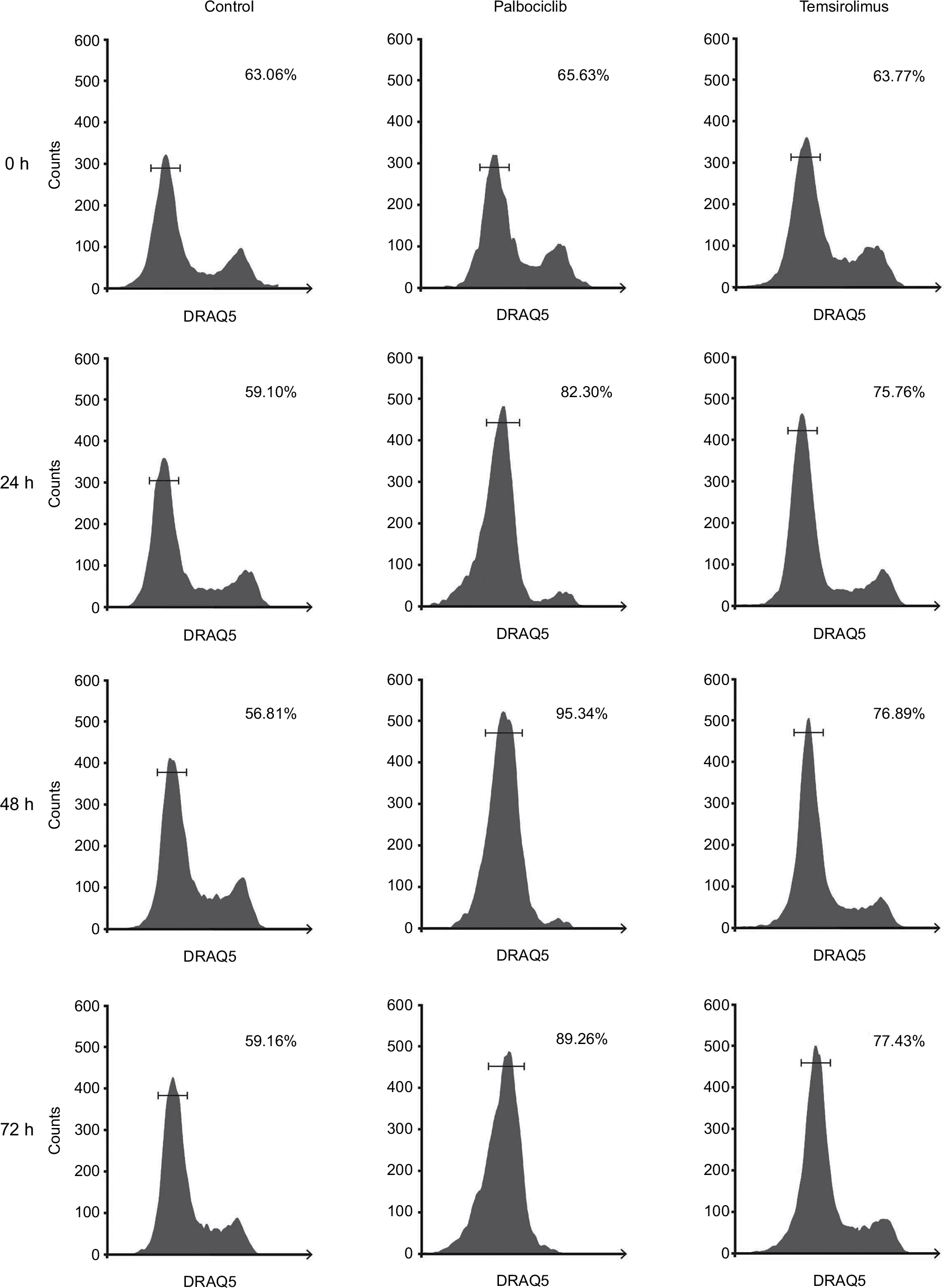 | Figure S3 Representative cell cycle analysis histograms illustrating G1-S arrest in DIPG cells in response to palbociclib and temsirolimus treatment compared to control cells. Notes: SF7761 cells were treated with vehicle, 2 µM palbociclib or 10 µM temsirolimus for 0, 24, 48, or 72 hours, as shown. DRAQ5 fluorescent dye was used to conduct flow cytometric cell cycle analysis on cells following treatment. G1 peak (left), G2 peak (right), and S-phase cells (transitional central area) are shown in all instances. Percentage value (top right) indicates the percentage of total cells in G1 phase. Each panel is a representative histogram of three determinations. |
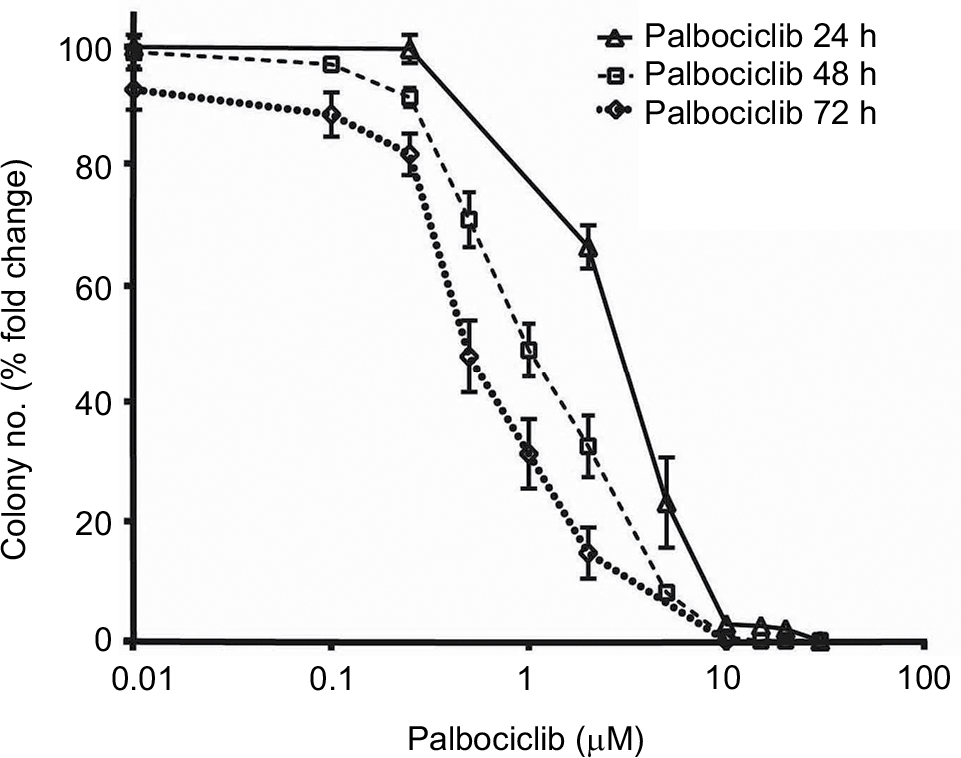 | Figure S4 Palbociclib dose-dependently reduces clonogenicity in DIPG cells. Notes: SU-DIPG IV cells were treated with different concentrations of palbociclib for 24–72 hours, and colonies were counted after 14 days. Data are the mean ± SEM of triplicate determinations. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.