")
Back to Journals » International Journal of Chronic Obstructive Pulmonary Disease » Volume 16
Combined Pulmonary Fibrosis and Emphysema (CPFE) Clinical Features and Management
Authors Hage R , Gautschi F, Steinack C, Schuurmans MM
Received 23 October 2020
Accepted for publication 30 December 2020
Published 28 January 2021 Volume 2021:16 Pages 167—177
DOI https://doi.org/10.2147/COPD.S286360
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Richard Russell
René Hage,1,2 Fiorenza Gautschi,1,2 Carolin Steinack,1,2 Macé M Schuurmans1,2
1University Hospital Zurich, Division of Pulmonology, Zurich, Switzerland; 2University of Zurich, Faculty of Medicine, Zurich, Switzerland
Correspondence: René Hage
University Hospital Zurich, Division of Pulmonology, Raemistrasse 100, Zurich 8091, Switzerland
Tel +41 44 2559111
Email [email protected]
Background: Combined pulmonary fibrosis and emphysema (CPFE) is an underrecognized syndrome characterized by chronic, progressive disease with a dismal prognosis. Frequent co-morbidities with a higher incidence than in idiopathic pulmonary fibrosis or emphysema alone are pulmonary hypertension (WHO group 3) in 47– 90% of the patients and lung cancer in 46.8% of the patients.
Objective: Review current evidence and knowledge concerning diagnosis, risk factors, disease evolution and treatment options of CPFE.
Methods: We searched studies reporting CPFE in original papers, observational studies, case reports, and meta-analyses published between 1990 and August 2020, in the PubMed, Embase, Cochrane Library, Wiley Online Library databases and Google Scholar using the search terms [CPFE], [pulmonary fibrosis] OR [IPF] AND [emphysema]. Bibliographies of retrieved articles were searched as well. Further inclusion criteria were publications in English, French, German and Italian, with reference to humans. In vitro data and animal data were not considered unless they were mentioned in studies reporting predominantly human data.
Results: Between May 1, 1990, and September 1, 2020, we found 16 studies on CPFE from the online sources and bibliographies. A total of 890 patients are described in the literature. Although male/female ratio was not reported in all studies, the large majority of patients were male (at least 78%), most of them were current or former heavy smokers.
Conclusion: CPFE is a syndrome presenting with dyspnea on exertion followed by disruptive cough and recurrent exacerbations. The disease may progress rapidly, be aggravated by pulmonary hypertension WHO group 3 and is associated with an increased risk of lung cancer. Smoking and male sex are important risk factors. There is a need for more research on CPFE especially relating to etiology, influence of genetics, treatment and prevention options. Antifibrotic therapy might be an interesting treatment option for these patients.
Keywords: COPD, pulmonary hypertension, autoimmune hypothesis
Introduction
Respiratory physicians are aware of a relatively new interstitial syndrome which is linked to heavy current or former smoking called Combined Pulmonary Fibrosis and Emphysema (CPFE). The disease was first described by Cottin in 2005. It has since been increasingly recognized as a separate clinical entity and is characterized by progressive worsening of respiratory symptoms, decline of lung function and a high mortality. Currently, no specific drug treatment is available, so treatment focuses on both the emphysema component and the fibrosis component. Major symptoms include dyspnea on exertion (functional class III or IV of the New York Heart Association)1 in almost all patients and a chronic productive or non-productive cough.2 There is a strong male predominance and the majority of patients are current or former heavy smokers. Most patients have abnormal auscultation findings with predominantly bilateral crackles in the lower lung zones, while some patients can have wheezing and diminished breath sounds.2 Other possible signs and symptoms consist of finger clubbing, hypoxemia and chest pain.2
Radiologically, computed tomography (CT) scans of the chest show both fibrosis predominantly in the lower zones and emphysema predominantly in the upper zones.2 The pulmonary function tests show a relatively preserved lung function with a low diffusing capacity of the lung (DLCO). Chronic respiratory failure, acute exacerbations, pulmonary hypertension (PH) WHO group 3 and lung carcinoma are the major causes of mortality. Lung transplantation is sometimes considered when all medical options have been attempted.
The co-existence of emphysema and fibrosis was first reported in dogs who were subjected to experimental cigarette smoking.3 In autopsies of human lungs, fibrotic changes with emphysema had already been described by Auerbach,4 but the first report of the combination of emphysema and fibrosis was documented in 1990, when a case series of 8 patients was described.5 It was Cottin who coined the term CPFE in 2005 and defined it as a CT-defined syndrome of combined pulmonary fibrosis and emphysema characterized by subnormal spirometry, severe impairment of gas exchange, high prevalence of pulmonary hypertension and poor survival.2
The exact incidence of CPFE is unknown, and some patients previously diagnosed as idiopathic pulmonary fibrosis (IPF) could in fact have a CPFE. In a series of 110 patients initially diagnosed as IPF, 28% of the patients were reevaluated and then classified as CPFE.6
In this article, we summarize the clinical, lung functional, radiological and pathological characteristics of CPFE, as described in the literature. We also propose a flow chart as a guide for clinicians.
Methods
We searched studies reporting CPFE in original papers, observational studies, case reports, and meta-analyses published from 1990 to the end of August 2020, in the PubMed, Embase, Cochrane Library, Wiley Online Library databases and Google Scholar using the search terms [CPFE], [pulmonary fibrosis] OR [IPF] AND [emphysema]. Further inclusion criteria were publications in English, French, German and Italian with reference to humans. Bibliographies were also searched for references missed by the search strategy.
Results
Between May 1, 1990, and September 1, 2020, we found 16 studies on CPFE using our search strategy. The studies are summarized in Table 1.
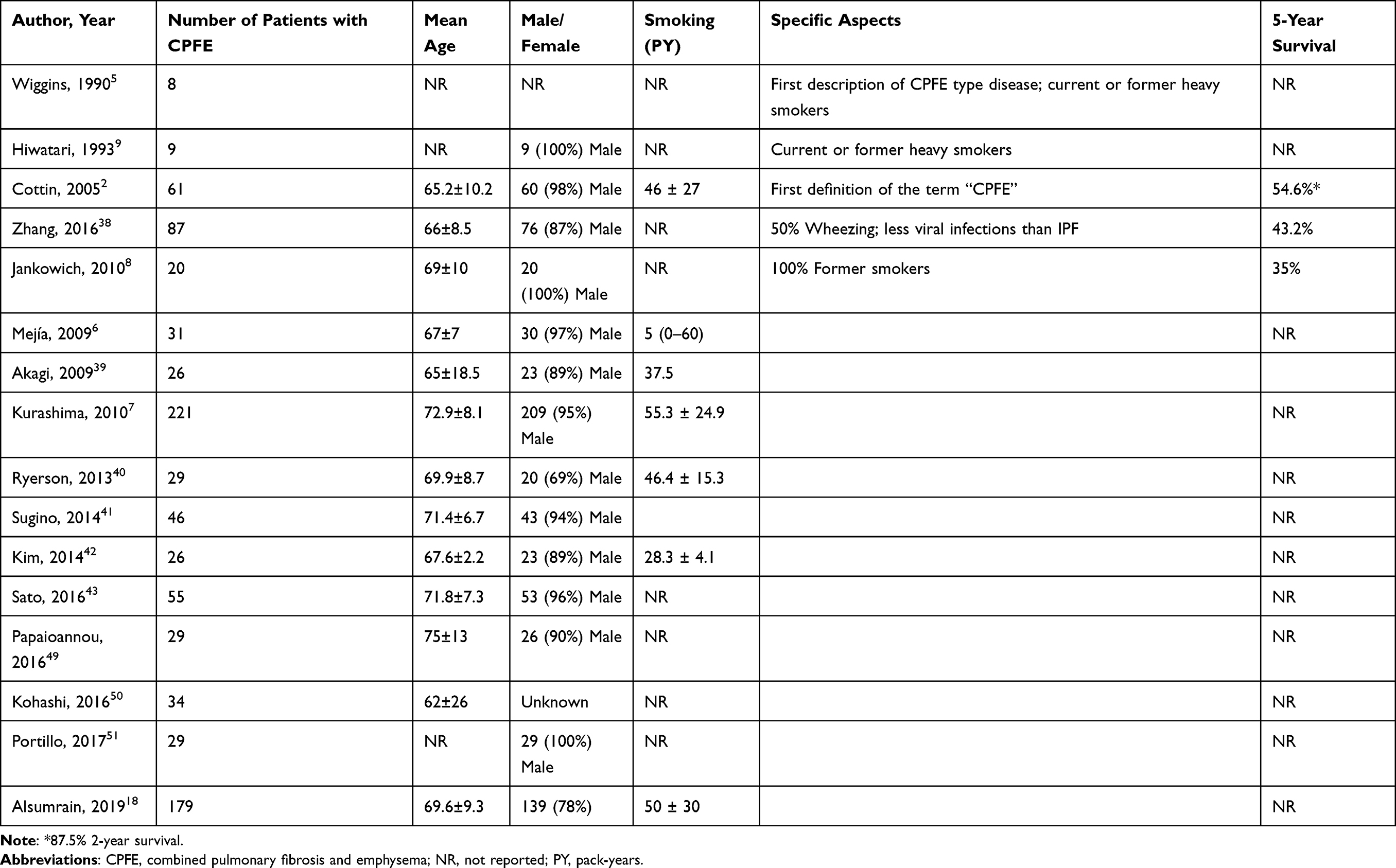 |
Table 1 Studies on Patients with Combined Pulmonary Fibrosis and Emphysema (CPFE) |
A total of 890 patients have been described in literature. Although male/female ratio has not been reported in all studies, in studies who reported the male/female ratio, the large majority (85.4%) of patients were male (760/890 patients). Most patients were current or former smokers, although several studies did not report smoking history or smoking status, as is shown in Table 1. The number of pack years in the CPFE patients has only been reported in 43.8% of the studies (7 of 16), ranging from 56 to 55.37 pack years. Some studies reported a 5-year survival rate of 35%8 to 54.6%,2 although these data could only be found in 18.8% (3/16) of the included studies.
The largest series has been reported by Kurashima et al,7 who analyzed 660 patients with a usual interstitial pneumonia (UIP) pattern on HRCT, in whom CPFE was found in 221/660 patients (33.5%). Although this study did not report the 5-year survival rates, it reported a better survival in CPFE compared to patients with a UIP pattern without emphysema (median survival 8.5 and 7.5 years, respectively).
Discussion
Due to the fairly recent description of CPFE less than two decades ago, the published evidence on its pathogenesis, epidemiology, clinical features and treatment options are still fairly limited. Some features are discussed here in detail:
Pathogenesis: Four Different Theories
The pathogenesis and possible pathophysiologic mechanisms leading to CPFE are still largely unknown. Different theories have been described in the medical literature. Most are not mutually exclusive. A number of pathways may lead to this diagnosis:
CPFE Starts with Fibrosis, Subsequently Resulting in Emphysema
One theory is that the fibrosis, with predominance in the basal lung parts, exerts traction on the upper parts of the lung, resulting in the development of emphysema.2 However, emphysema most of the time precedes the fibrotic changes, which would question the appropriateness of this theory.9
CPFE is Due to Gastroesophageal Reflux (GER) Promoted by Smoking Behavior
Gastroesophageal reflux (GER) has been identified to be associated with interstitial lung diseases and is therefore considered a risk factor for the development of lung fibrosis. Smoking can increase GER and thus be responsible for development of the emphysema and indirectly (via increased GER) for the fibrotic lung changes that develop over time. This relationship is controversial10 and can be confounded by other effects of smoking on the lung tissue.
The mechanism behind this could also be due to a sequence of events that leads first to emphysema and then additionally to fibrotic changes triggered by late-onset increased gastropharyngeal reflux. In certain individuals who are susceptible to tobacco smoke, a symptomatic smoking-related emphysema might develop. As part of standard care, a smoking cessation intervention is performed, which leads not only to smoking abstinence but also frequently to a relevant weight gain. The weight gain itself promotes increased gastropharyngeal reflux (with micro-aspirations) and thus may trigger development of fibrotic changes in the lungs. This hypothesis is compatible with the frequently observed temporal sequence of development of emphysema prior to the fibrotic changes and also with the frequently observed history (in our cohort) of strong weight gain after smoking cessation. So far, no published case series or cohort studies have systematically documented this sequence of events and the possible role of reflux and micro-aspirations in CPFE patients.
CPFE as an Autoimmune Phenomenon
A third hypothesis in a subgroup of CPFE patients could be an autoimmune phenomenon.11 One multicenter study investigated 40 patients with CPFE and 60 patients with IPF. A statistically significant number of CPFE patients with elevated serum ANA with or without positive p-ANCA titers were observed compared with patients with IPF without emphysema.11 Patients with CPFE and positive autoimmune markers showed improved survival compared to patients with a negative autoimmune profile.11 Moreover, a massive infiltration of clusters of CD20+ B cells forming lymphoid follicles within the fibrotic lung in CPFE patients with positive serum immunologic profile compared to patients with negative profile was noted and positively correlated with improved survival.11
Cottin et al reported a relationship between patients with connective tissue disease (CTD) and CPFE.12 These CTD patients had rheumatoid arthritis (RA, n=18), systemic sclerosis (SSc, n=10), mixed or overlap CTD (n=4), or other CTDs (n=2). In this study patients with combined CTD and CPFE were significantly younger than a historical control group of patients with idiopathic CPFE and were more frequently female.12 In addition, patients with CTD and CPFE had higher lung volumes, lower diffusing capacity, higher pulmonary pressures, and were more frequently male than those with CTD and lung fibrosis without emphysema.12
CPFE in Development Pathways Based on Genetic Factors
A genetic component may contribute to the development of CPFE. These studies are complex and so far cannot explain CPFE development in all patients. Collum et al demonstrated that both adenosine and its receptor ADORA2B are elevated in chronic lung diseases.13 Activation of ADORA2B leads to elevated levels of hyaluronan synthases (HAS) and thus higher concentration of hyaluronan. Hyaluronan is a glycosaminoglycan that contributes to chronic lung injury,13 suggesting that ADORA2B and hyaluronan contribute to CPFE.
Another study found an association between ABCA3 mutations and CPFE in a 41-year-old nonsmoking male presenting with dyspnea on mild exertion.14 The ABCA3 gene is involved in surfactant metabolism. Recessive loss-of-function mutations in ABCA3 present as lethal surfactant deficiency in the newborn, whereas other recessive mutations in ABCA3 can result in interstitial lung disease in older children.14
Some animal experiments are also of interest. One study in transgenic mice showed overexpression of tumor necrosis factor (TNF-) α, driven by the surfactant protein C promoter, inducing pathological changes consistent with CPFE.15 Another study showed overexpression of Interleukin (IL-)13 and transforming growth factor (TGF-) β1 in transgenic mice, inducing CPFE.14 The severity of CPFE development, induced by IL-13 and TGF-β1 might result from a disbalance of apoptosis, proteolysis and fibrosis.16
Lung Function
The fibrotic (restrictive disorder) and emphysematous (obstructive disorder) components in CPFE have counterbalancing effects in lung function measurements leading to a combined disorder pattern with a relatively normal lung function. The latter aspect may explain why some of these patients are diagnosed fairly late, since spirometry is often non-diagnostic. Preservation of lung function can therefore be misleading, underestimating the CPFE severity, or even lead to a delayed or missed diagnosis.
The isolated emphysematous component normally shows a low FEV1/FVC ratio. This is caused by the loss of elastic recoil and the high lung compliance, leading to expiratory airway collapse and hyperinflation. The hyperinflation (air trapping) explains the frequently observed large RV and/or increased RV/TLC ratio. In CPFE, due to the coexisting fibrosis and emphysema, the restrictive component can counterbalance the obstructive component, resulting in a (near) normal FEV1/FVC ratio and a (near) normal RV and/or RV/TLC ratio.
An isolated markedly impaired diffusing capacity for carbon monoxide (DLCO) in pulmonary function testing can be an important clue to the diagnosis of CPFE.
As a result of the progressive functional loss of alveoli, thereby decreasing the surface area for gas exchange, there is a progressively severe reduction in the DLCO. The fibrotic component of CPFE leads to a decreased pulmonary capillary volume, and both fibrosis and emphysema lead to ventilation/perfusion abnormalities, resulting in a decreased DLCO. Differential diagnosis of decreased DLCO should include an increased carboxyhemoglobin (smoking), anemia and pulmonary vascular disease. The chances of diagnosing CPFE are best when body plethysmography and diffusion capacity are performed and subsequently computed tomography (CT) of the chest is requested.
Radiology
Typical radiological findings of CPFE in chest CT images are emphysema in the upper parts of the lungs, fibrosis in the lower parts of the lung17 and honeycombing.18 The study of Cottin, who first defined CPFE, also additionally found traction bronchiectasis in 42 patients (69%), ground-glass opacities in 40 (66%) and bullae in 33 (54%).2
Emphysema can be centrilobular, paraseptal or (most frequently) mixed.18 Large relatively thick-walled cysts may be present in addition to the pulmonary emphysema,17 sometimes the cysts appear within the fibrotic areas. Fibrosis can have different patterns, such as (consistent or possible) usual interstitial pneumonia (UIP), non-specific interstitial pneumonia (NSIP-) like, chronic hypersensitivity pneumonitis (cHP-) like, or unclassifiable. The clinical interstitial lung diseases (ILD) causing CPFE may be IPF, unclassifiable or classifiable (Figure 1). A large retrospective cohort study, including 179 CPFE patients showed that the largest underlying clinical ILD was ‘unclassifiable’ (n=79, 44%), followed by UIP/IPF (n=58, 32%) and classifiable ILD in 23%, most of them being connective tissue disease (CTD-) related ILD (45%).18
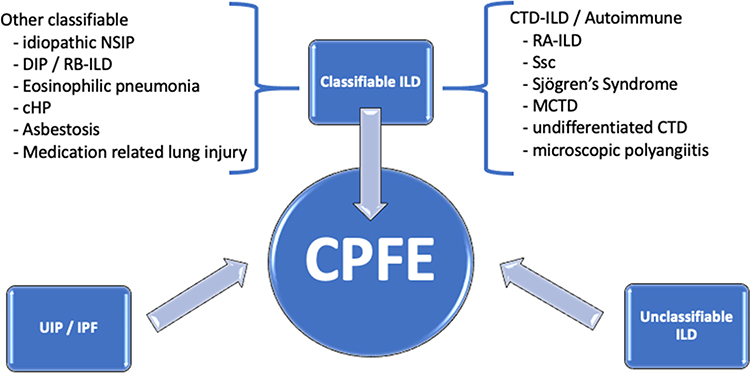 |
Figure 1 Clinicoradiological interstitial lung diseases in CPFE. Abbreviations: cHP, chronic hypersensitivity pneumonitis; CPFE, combined pulmonary fibrosis and emphysema; CTD-ILD, connective tissue disease interstitial lung disease; DIP, desquamative interstitial pneumonia; MCTD, mixed connective tissue disease; NSIP, non-specific interstitial pneumonia; RA-ILD, rheumatoid arthritis–associated interstitial lung disease; RB-ILD, respiratory bronchiolitis interstitial lung disease; Ssc, systemic sclerosis. |
In the case of pulmonary hypertension, characteristical CT findings are dilatation of the central pulmonary arteries, enlargement of the right-sided heart chambers, reduced number of peripheral pulmonary artery branches, and mosaic attenuation of the lung parenchyma.17,19
Histopathology
Pathological features in CPFE have been studied by Inomata et al, who retrospectively reviewed the clinical charts and examined chest CT images and pathological findings of an autopsy series of 22 CPFE patients.20 Histopathological characteristics were the UIP pattern, and thick-walled cystic lesions (TWCLs). These TWCLs showed emphysematous destruction and enlargement of membranous and respiratory bronchioles with fibrosis.20 These TWCLs, with a prevalence of both radiological and pathological changes in 72.7% of CPFE patients, were not observed in patients with IPF or emphysema alone.20 The cystic lesions were always larger than the cysts of honeycombing.20
TWCLs should be considered as an important pathological (and radiological) feature of CPFE.20
Bronchoalveolar Lavage (BAL)
In CPFE, the BAL fluid shows the same characteristics as in IPF.6 In a study examining 27 patients with CPFE, the BAL leucocyte count was 240 ± 200 × 106/L, with a differential cell count including macrophages 76% ± 24 (range 10–90), neutrophils 10% ± 19 (2–73), eosinophils 2% ± 10 (0–43), and lymphocytes 5% ± 9 (0–43).2 These cell counts are all non-specific, as in IPF.
Predictors of Survival or Outcome
Different longitudinal measurements such as lung function (FEV1, FVC, DLCO), symptoms and radiological studies (HRCT scans), are used to classify disease severity. However, none of these parameters are fully satisfactory in predicting prognosis in CPFE patients. Wells et al tried to develop a more reliable predictor of prognosis in IPF patients, by using the composite physiologic index (CPI).21 This index was derived to represent the extent of fibrosis in IPF patients, with adjustment for the emphysema component. In IPF patients, extent of IPF on CT was calculated by a formula incorporating multiple components of pulmonary function: extent of disease on CT = 91.0 – (0.65 × percent predicted DLCO) – (0.53 × percent predicted FVC) + (0.34 × percent predicted FEV1). In IPF patients, CPI and longitudinal changes in DLCO were more predictive than FVC and FEV1.22 However, CPI is not helpful in predicting prognosis in CPFE patients: Schmidt et al demonstrated that longitudinal change in FEV1 was most predictive of mortality in CPFE patients, whereas a significant increase (ie by at least five points over 6 or 12 months) in CPI was the best predictor in patients with IPF without emphysema.22
The clinicoradiological patterns of the fibrotic disease of CPFE are also relevant in the estimation of the prognosis. The study of Alsumrain et al showed that the overall mortality for the study period (11 years) was greatest in those with CPFE with UIP/IPF pattern, compared to other classifiable and unclassifiable ILD patterns (69% vs 45% vs 38%, respectively, P = 0.016).18
Clinically, prognosis of CPFE is predominantly determined by three complications: pulmonary hypertension, acute exacerbations (AE) and lung cancer.
The likelihood of developing pulmonary hypertension in CPFE, defined by a mean pulmonary arterial pressure (mPAP) of ≥25 mmHg at rest, is higher than in patients with isolated idiopathic pulmonary fibrosis.23 In the pathogenesis of CPFE not only pulmonary hypertension WHO group 3 (as a result of an underlying lung disease) but also idiopathic pulmonary arterial hypertension (IPAH) might play a role24 and probably differs from the vasculopathy seen in patients with chronic obstructive pulmonary disease (COPD). Vasculopathy in COPD has mainly been observed in small arteries and arterioles.24 In CPFE, vasculopathy is broad and heterogeneous throughout the lungs, in arteries/arterioles, veins/venules and capillaries,24 with the predominant vasculopathy in the arteries/arterioles.
Demonstration of pulmonary hypertension by right-heart catheterization in CPFE was associated with a 1-year survival rate of only 60%.25 In a retrospective study of 61 patients with CPFE, pulmonary hypertension was present in 47% of the patients, with a survival of 87.5% after 2 years and 54.6% after 5 years and a median survival of 6.1 years.2 It was concluded that the presence of pulmonary hypertension at diagnosis was a critical determinant of prognosis. This is in accordance with the findings of Mejía et al.26 As mentioned before, the worse prognosis of CPFE is predominantly caused by the pulmonary hypertension.25 However, this is in contrast to the statement of Sakai, who believes that the prognosis of CPFE is better than that of IPF/UIP, despite a high prevalence of lung cancer reported in cases with CPFE.17
In a study of 38 lung transplant recipients, who underwent a lung transplantation due to fibrotic end-stage lung disease, there were 21 patients with IPF and 8 with CPFE.26 In this study, they found significant pulmonary arterial vasculopathy which did not correlate with the clinical severity of the underlying lung disease and did not correlate with the presence and/or severity of PH as measured by right-heart catheterization.26 In 21%, there were plexiform lesions, which are traditionally a hallmark of WHO group 1 PH, termed PAH.26 The investigators suggested that the observed vasculopathy might be derived from severe and prolonged hypoxia sharing biochemical mechanisms similar to those seen with WHO group 1 PAH. They concluded that these findings indicate that advanced pulmonary arteriopathy is common and may develop in a heterogeneous regional pattern in advanced lung disease prior to the clinical detection of PH.26
The second major complication in CPFE is the acute exacerbation (AE-CPFE). AE can be attributed to the emphysematous component (AE-CPFE, COPD-type), or fibrotic component of CPFE (AE-CPFE, IPF-type). Treatment differs depending on the predominant underlying type of exacerbation.27
The first type of AE-CPFE, the COPD-type can be defined as per GOLD guidelines published in 2018, specifically, the severe category of AE; an acute worsening of respiratory symptoms that requires hospitalization.27 Clinically it is characterized by airflow obstruction and acute bronchospasm, and radiologically it may show airway wall thickening, mucous impaction, atelectasis, consolidations and mediastinal adenopathy.27 Standard care comprises inhalation of bronchodilators and systemic corticosteroids. In case of clear signs of bacterial infection, additionally, antibiotics are often given. This is usually recommended when two of the three Anthonisen criteria are present.
The AE-CPFE, IPF-type can be defined by the clinical and radiological criteria described by Collard et al in analogy for acute exacerbations of IPF.28 These clinical criteria are an acute, clinically significant, respiratory deterioration, characterized by evidence of new, widespread alveolar abnormality, leading to acute hypoxic respiratory failure. The radiological criteria are a change in baseline imaging including presence of ground-glass opacities, interlobular septal thickening or new consolidations on a background pattern consistent with a UIP, that is not fully explained by cardiac failure or fluid overload. Standard care comprises high dose systemic corticosteroids and/or antibiotic therapy. One study showed that patients who experienced AE-CPFE, IPF-type had significantly higher morbidity, requiring more often invasive mechanical ventilation and ECMO as compared to patients with AE-CPFE, COPD-type.27
Although the exact frequency is unknown, AE of CPFE is rare, compared to AE of idiopathic pulmonary fibrosis (IPF). A Japanese single-center study reported, 21 patients with AE of CPFE and 41 patients with AE of IPF revealing that the survival time after AE for patients with CPFE was longer than that for patients with IPF.29 In this study, the 30- and 90-day survival rates for patients in the AE-CPFE group were 95.2% and 85.7%, respectively; these values were significantly higher than those (61.0% and 43.9%) for patients in the AE-IPF group.29
The third major complication in patients with CPFE is lung cancer. In CPFE, the risk to develop lung cancer is high, and prognosis is much worse compared to non-CPFE patients. Kitaguchi showed lung cancer in 46.8% of CPFE patients.30 The predominant histological type (42.3%) is the squamous cell carcinoma (SqCC), followed by adenocarcinoma (34.4%).31 Compared with lung cancer population with an otherwise normal lung, the OR to develop SqCC in CPFE is 9.06 (95% CI, 6.08–13.5).31 The median survival for CPFE patients with lung cancer (19.5 months) is significantly shorter than in non-CPFE (53.1 months).31
In a review by Koo et al, 620 patients in nine original articles were assessed.31 Most patients were elderly (mean age 70.4 years) and heavy smokers (mean 53.5 packyears).31 The males were remarkably predominant (92.6%).31 Since there is a significantly elevated risk of lung cancer in CPFE patients, radiological evaluation should be considered on a regular basis.8
Lung cancer profoundly influences the prognosis of CPFE patients, as their lung function may be too poor to allow surgery or chemotherapy.30 In addition, radiotherapy could further aggravate fibrosis. However, Li et al conducted a meta-analysis of 13 studies in patients with CPFE and lung cancer, and assumed that chemotherapy, best supportive care and radiotherapy might benefit lung cancer patients with CPFE. The validity of these outcomes was stated to be questionable because of confounding factors. Only three studies were included in the subgroup analysis.32 The authors suggested that more studies on selecting optimal treatment modalities for lung cancer patients with CPFE should be conducted to define the most effective and safe treatment strategy.32
In an autopsy study including 22 CPFE patients, comparing these with findings with 8 idiopathic pulmonary fibrosis (IPF) patients and with 17 emphysema-only patients, it was shown that there was a higher number of patients with lung cancer in the CPFE and emphysema-only group, compared to the IPF group.20 The authors suggested that emphysema leads to a higher lung cancer risk than IPF.20 They also observed that lung cancer tended to develop close to areas of dense fibrosis with architectural distortion in the CPFE patients, particularly in the 8 (42.1%) patients with fibrotic lesions and the 4 (21%) patients with thick-walled cystic lesions (TWCLs).20 Thus, TWCLs, which are lesions that combine fibrosing interstitial pneumonia and emphysema, could also be considered a source of lung cancer development.20
Diagnostic steps and management are summarized in Figure 2.
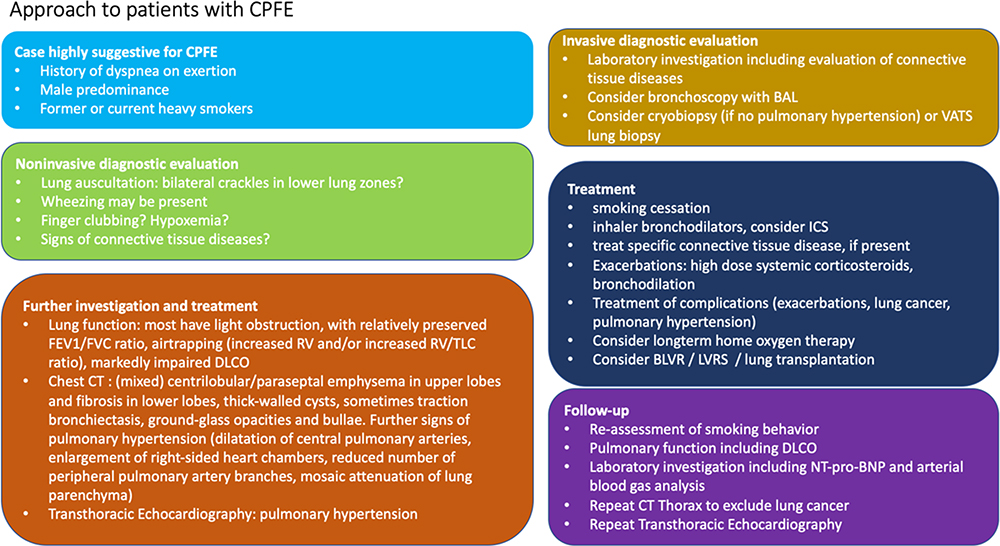 |
Figure 2 Diagnosis and management of patients with interstitial lung diseases in CPFE. Abbreviations: BAL, bronchoalveolar lavage; BLVR, bronchoscopic lung volume reduction; CT, computed tomography; DLCO, diffusing capacity of the lung; FEV1, forced expiratory volume in 1 second; FVC, forced vital capacity; ICS, inhaled corticosteroids; LVRS, lung volume reduction surgery; NT-pro-BNP, N-terminal prohormone brain natriuretic peptide; RV, residual volume; TLC, total lung capacity. |
Diagnosis of Underlying Interstitial Lung Disease
Diagnosing interstitial lung disease can be a challenge. With regard to the IPF component in CPFE, the presence of a definite UIP pattern in the HRCT scan is sufficient for the diagnosis of IPF.33 In the case of a probable UIP or indefinite pattern, a surgical lung biopsy for an accurate diagnosis can be considered, depending on the multidisciplinary discussion by the clinician, radiologist and pathologist. However, in these patients, the risks of a surgical lung biopsy may be excessive and depends on age, severity of the disease, and comorbidity.34 Although not standard of care, cryobiopsy could be considered as an alternative to thoracic surgery.35 Importantly, before cryobiopsy is considered, pulmonary hypertension should be evaluated, in order to decrease the risk of a massive pulmonary hemorrhage complication.35 However, to our knowledge, clear guidelines concerning pulmonary hypertension in cryobiopsy do not exist.
Treatment
Considering treatment options, smoking cessation is certainly indicated for both components of CPFE. For those that have not stopped smoking yet professional counselling in addition to smoking cessation medication is standard care. For the obstructive lung disease or emphysema component inhaled bronchodilators may be effective, especially long-acting compounds which are associated with better medication adherence than short-acting compounds. Combined inhaled corticosteroids (ICS)/long-acting beta2-agonists (LABA) have been studied in 45 patients with CPFE.36 It showed that ICS/LABA therapy could improve the lung function in patients with CPFE and reduce exacerbation frequency and severity of disease during the acute episode.36
In CPFE with underlying connective tissue disease, a disease-specific treatment should be given. Case reports describing these treatments are limited.37 Case reports on patients with CPFE show that they could possibly improve with lung volume reduction (LVR) in case of advanced emphysema, even without plethysmographic evidence of severe hyperinflation.38 If LVR is not possible and after extensive exclusion of (lung) cancer, patients should be evaluated early for lung transplantation as the ultimate treatment modality of CPFE.
Outlook: Antifibrotic Drugs?
Treatment with antifibrotic drugs, such as pirfenidone and nintedanib, may be effective in CPFE but further trials are awaited.
What can we learn from antifibrotic treatment in IPF? In the Assessment of Pirfenidone to Confirm Efficacy and Safety in Idiopathic Pulmonary Fibrosis (ASCEND) study, treatment with pirfenidone for 52 weeks was shown to be a potentially effective oral antifibrotic drug. In this randomized, double-blind, placebo-controlled trial it significantly reduced disease progression and improved progression-free survival in IPF patients.39 However, in this trial, patients with FEV1/FVC ratio <0.8 after administration of bronchodilator at screening were excluded. Therefore, we have no data on patients with an emphysema component, or on patients with CPFE.
On the other hand, in the INPULSIS trials, the therapeutic effect of nintedanib was not influenced by emphysema.40 It also showed a reduction in the decline in FVC after 52 weeks in IPF patients in two randomized, double-blind Phase 3 trials (INPULSIS-1 and INPULSIS-2).41 Moreover, nintedanib has been shown to decrease the annual rate of decline in the FVC not only in IPF patients but also in patients with other (non-UIP-like) fibrotic patterns.42 Treatment with nintedanib showed a clinically relevant slower FVC decrease (−154.2 mL per year), which was only slightly lower than that in patients with a UIP-like fibrotic pattern.42 However, this change in physiological outcomes was not accompanied by meaningful changes in measures of quality of life.42 The question remains, if slowing down the FVC decrease will be relevant to CPFE disease progression, since FVC is not the best longitudinal lung function parameter, as discussed above. Nevertheless, data for antifibrotic therapy as a therapeutic option for non-IPF fibrosing lung diseases are promising, although to the best of our knowledge these are currently not available for CPFE. If antifibrotics become available for CPFE, it will be important to discuss the optimal timing of initiation of these drugs. Furthermore, clinicians need answers concerning questions regarding combining or replacing existing immunosuppressive drugs with antifibrotics in CPFE patients with connective tissue diseases. Further research is needed to identify parameters that allow a more accurate prediction of CPFE disease progression, to then evaluate early pharmacological and/or interventional treatment resulting in the preservation of lung function and reduction of mortality. The role of pulmonary function tests for following response to therapy is not yet clearly defined for CPFE patients since FVC may be subnormal or falsely normal due the combined ventilatory defect and therefore using it as a treatment response parameter may be inadequate. The same holds true for DLCO, which reflects the combined impact of emphysema and fibrosis. Patients with more extensive emphysema and less fibrosis can have the same level of DLCO impairment as patients with less emphysema and more fibrosis, whereas the prognosis is likely different for the two conditions. Also in patients with both progressive emphysema and progressive fibrosis, DLCO can remain unchanged compared to the previous values obtained when both pathologies were milder.43
Conclusion
CPFE is a syndrome presenting with dyspnea on exertion followed by disruptive cough and recurrent exacerbations. The disease may progress rapidly, be aggravated by pulmonary hypertension WHO group 3 and is associated with an increased risk of lung cancer. Smoking and male sex are important risk factors. The exact pathogenesis is unknown. Pulmonary function tests show a subnormal or normal FEV1/FVC ratio with a markedly impaired DLCO. The main chest CT findings are emphysema (in the upper zone of the lung) with fibrosis (in the lower zone). Histopathology shows UIP, emphysema and TWCLs. The BAL findings are non-specific. Frequent complications in CPFE are pulmonary hypertension, acute exacerbations and lung cancer, all contributing to a poor prognosis. Currently, there is no specific treatment, and smoking cessation is the most important intervention. Some patients might benefit from ICS/LABA treatment. Data on antifibrotics in CPFE are currently not available, and studies with CPFE patients are difficult as interpretation of lung function parameters can be challenging in the follow-up of these patients.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Portillo K, Morera J. Combined pulmonary fibrosis and emphysema syndrome: A new phenotype within the spectrum of smoking-related interstitial lung disease. Pulm Med. 2012. 2012. doi:10.1155/2012/867870
2. Cottin V, Nunes H, Brillet PY, et al. Combined pulmonary fibrosis and emphysema: A distinct underrecognised entity. Eur Respir J. 2005;26(4):586–593. doi:10.1183/09031936.05.00021005
3. Frasca JM, Auerbach O, Parks VR, Jamieson JD. Electron microscopic observations on pulmonary fibrosis and emphysema in smoking dogs’. Experimental Molecular Pathol. 1971;16:108–125. doi:10.1016/0014-4800(71)90022-0
4. Auerbach O, Garfinkel L, Cuykr Hammond E. Relation of smoking and age to findings in lung parenchyma: a microscopic study. Chest. 1974;65(1):29–35. doi:10.1378/chest.65.1.29
5. Wiggins J, Strickland B, Turner-Warwick M. Combined cryptogenic fibrosing alveolitis and emphysema: the value of high resolution computed tomography in assessment. Respir Med. 1990;84(5):365–369. doi:10.1016/S0954-6111(08)80070-4
6. Mejía M, Carrillo G, Rojas-Serrano J, et al. Idiopathic pulmonary fibrosis and emphysema: decreased survival associated with severe pulmonary arterial hypertension. Chest. 2009;136(1):10–15. doi:10.1378/chest.08-2306
7. Kurashima K, Takayanagi N, Tsuchiya N, et al. The effect of emphysema on lung function and survival in patients with idiopathic pulmonary fibrosis. Respirology. 2010;15(5):843–848. doi:10.1111/j.1440-1843.2010.01778.x
8. Jankowich MD, Rounds S. Combined pulmonary fibrosis and emphysema alters physiology but has similar mortality to pulmonary fibrosis without emphysema. Lung. 2010;188(5):365–373. doi:10.1007/s00408-010-9251-6
9. Hiwatari N, Shimura S, Takishima T. Pulmonary emphysema followed by pulmonary fibrosis of undetermined cause. Respiration. 1993;60(6):354–358. doi:10.1159/000196235
10. Bédard Méthot D, Leblanc É, Lacasse Y. Meta-analysis of gastroesophageal reflux disease and idiopathic pulmonary fibrosis. Chest. 2019;155(1):33–43. doi:10.1016/j.chest.2018.07.038
11. Tzouvelekis A, Zacharis G, Oikonomou A, et al. Increased incidence of autoimmune markers in patients with combined pulmonary fibrosis and emphysema. BMC Pulm Med. 2013;13:1. doi:10.1186/1471-2466-13-31
12. Cottin V, Nunes H, Mouthon L, et al. Combined pulmonary fibrosis and emphysema syndrome in connective tissue disease. Arthritis Rheum. 2011;63(1):295–304. doi:10.1002/art.30077
13. Collum SD, Molina JG, Hanmandlu A, et al. Adenosine and hyaluronan promote lung fibrosis and pulmonary hypertension in combined pulmonary fibrosis and emphysema. DMM. 2019;12:5. doi:10.1242/dmm.038711
14. Epaud R, Delestrain C, Louha M, Simon S, Fanen P, Tazi A. Combined pulmonary fibrosis and emphysema syndrome associated with ABCA3 mutations. Eur Respir J. 2014;43(2):638–641. doi:10.1183/09031936.00145213
15. Lundblad LKA, Thompson-Figueroa J, Leclair T, et al. Tumor necrosis factor-α overexpression in lung disease: A single cause behind a complex phenotype. Am J Respir Crit Care Med. 2005;171(12):1363–1370. doi:10.1164/rccm.200410-1349OC
16. Fulkerson PC, Fischetti CA, Hassman LM, Nikolaidis NM, Rothenberg ME. Persistent effects induced by IL-13 in the lung. Am J Respir Cell Mol Biol. 2006;35(3):337–346. doi:10.1165/rcmb.2005-0474OC
17. Sakai F, Tominaga J, Kaga A, et al. Imaging diagnosis of interstitial pneumonia with emphysema (combined pulmonary fibrosis and emphysema). Pulm Med. 2012;2012:1–9. doi:10.1155/2012/816541;.
18. Alsumrain M, de Giacomi F, Nasim F, et al. Combined pulmonary fibrosis and emphysema as a clinicoradiologic entity: characterization of presenting lung fibrosis and implications for survival. Respir Med. 2019;146:106–112. doi:10.1016/j.rmed.2018.12.003
19. Devaraj A, Wells AU, Meister MG, Corte TJ, Hansell DM. The effect of diffuse pulmonary fibrosis on the reliability of CT signs of pulmonary hypertension. Radiology. 2008;249(3):1042–1049. doi:10.1148/radiol.2492080269
20. Inomata M, Ikushima S, Awano N, et al. An autopsy study of combined pulmonary fibrosis and emphysema: correlations among clinical, radiological, and pathological features. BMC Pulm Med. 2014;14:1. doi:10.1186/1471-2466-14-104
21. Wells AU, Desai SR, Rubens MB, et al. Idiopathic pulmonary fibrosis: A composite physiologic index derived from disease extent observed by computed tomography. Am J Respir Crit Care Med. 2003;167(7):962–969. doi:10.1164/rccm.2111053
22. Schmidt SL, Nambiar AM, Tayob N, et al. Pulmonary function measures predict mortality differently in IPF versus combined pulmonary fibrosis and emphysema. Eur Respir J. 2011;38(1):176–183. doi:10.1183/09031936.00114010
23. Heathcote KL, Cockcroft DW, Fladeland DA, Fenton ME. Normal expiratory flow rate and lung volumes in patients with combined emphysema and interstitial lung disease: a case series and literature review case presentation. Can Respir J. 2011;18(5):e73–76. doi:10.1155/2011/354325.
24. Sato T, Tsujino I, Tanino M, Ohira H, Nishimura M. Broad and heterogeneous vasculopathy in pulmonary fibrosis and emphysema with pulmonary hypertension. Respirol Case Rep. 2013;1(1):10–13. doi:10.1002/rcr2.7
25. Cottin V, le Pavec J, Prévot G, et al. Pulmonary hypertension in patients with combined pulmonary fibrosis and emphysema syndrome. Eur Respir J. 2010;35(1):105–111. doi:10.1183/09031936.00038709
26. Dotan Y, Stewart J, Gangemi A, et al. Pulmonary vasculopathy in explanted lungs from patients with interstitial lung disease undergoing lung transplantation. BMJ Open Respir Res. 2020;7:1. doi:10.1136/bmjresp-2019-000532
27. Zantah M, Dotan Y, Dass C, Zhao H, Marchetti N, Criner GJ. Acute exacerbations of COPD versus IPF in patients with combined pulmonary fibrosis and emphysema. Respir Res. 2020;21:1. doi:10.1186/s12931-020-01432-x
28. Collard HR, Ryerson CJ, Corte TJ, et al. Acute exacerbation of idiopathic pulmonary fibrosis an international working group report. Am J Respir Crit Care Med. 2016;194(3):265–275. doi:10.1164/rccm.201604-0801CI
29. Ikuyama Y, Ushiki A, Kosaka M, et al. Prognosis of patients with acute exacerbation of combined pulmonary fibrosis and emphysema: A retrospective single-centre study. BMC Pulm Med. 2020;20:1. doi:10.1186/s12890-020-01185-9
30. Kitaguchi Y, Fujimoto K, Hanaoka M, Kawakami S, Honda T, Kubo K. Clinical characteristics of combined pulmonary fibrosis and emphysema. Respirology. 2010;15(2):265–271. doi:10.1111/j.1440-1843.2009.01676.x
31. Koo HJ, Do KH, Lee JB, Alblushi S, Lee SM. Lung cancer in combined pulmonary fibrosis and emphysema: A systematic review and meta-analysis. PLoS One. 2016;11:9. doi:10.1371/journal.pone.0161437
32. Li C, Wu W, Chen N, et al. Clinical characteristics and outcomes of lung cancer patients with combined pulmonary fibrosis and emphysema: A systematic review and meta-analysis of 13 studies. J Thorac Dis. 2017;9(12):5322–5334. doi:10.21037/jtd.2017.12.72
33. Raghu G, Remy-Jardin M, Myers JL, et al. Diagnosis of idiopathic pulmonary fibrosis. an official ATS/ERS/JRS/ALAT clinical practice guideline. American J Respir Critical Care Med. 2018;198:e44–e68. doi:10.1164/rccm.201807-1255ST
34. Wells AU. “Any fool can make a rule and any fool will mind it.”. BMC Med. 2016;14:1. doi:10.1186/s12916-016-0562-1
35. Lentz RJ, Taylor TM, Kropski JA, et al. Utility of flexible bronchoscopic cryobiopsy for diagnosis of diffuse parenchymal lung diseases. J Bronchology Interv Pulmonol. 2018;25(2):88–96. doi:10.1097/LBR.0000000000000401
36. Dong F, Zhang Y, Chi F, et al. Clinical efficacy and safety of ICS/LABA in patients with combined idiopathic pulmonary fibrosis and emphysema. Int J Clin Exp Med. 2015;8(6):8617–8625.
37. Sogkas G, Hirsch S, Olsson KM, et al. Lung involvement in primary sjögren’s syndrome—an under-diagnosed entity. Front Med. 2020;7:7. doi:10.3389/fmed.2020.00332
38. Straub G, Caviezel C, Frauenfelder T, Bloch KE, Franzen D. Successful lung volume reduction surgery in combined pulmonary emphysema and fibrosis without body-plethysmographic hyperinflation-a case report. J Thorac Dis. 2018;10:S2830–S2834. doi:10.21037/jtd.2018.06.60
39. King TE, Bradford WZ, Castro-Bernardini S, et al. A phase 3 trial of pirfenidone in patients with idiopathic pulmonary fibrosis. New England J Med. 2014;370(22):2083–2092. doi:10.1056/nejmoa1402582
40. Cottin V, Azuma A, Raghu G, et al. Therapeutic effects of nintedanib are not influenced by emphysema in the Inpulsis trials. Eur Respir J. 2019;53:4. doi:10.1183/13993003.01655-2018
41. Richeldi L, Du Bois RM, Raghu G, et al. Efficacy and safety of nintedanib in idiopathic pulmonary fibrosis. New England J Med. 2014;370(22):2071–2082. doi:10.1056/nejmoa1402584
42. Flaherty KR, Wells AU, Cottin V, et al. Nintedanib in progressive fibrosing interstitial lung diseases. New England J Med. 2019;381(18):1718–1727. doi:10.1056/NEJMoa1908681
43. Cottin V, Cordier JF, Wells AU. Centrilobular emphysema combined with pulmonary fibrosis results in improved survival: A response. Fibrogenesis Tissue Repair. 2011;4:1. doi:10.1186/1755-1536-4-16
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.