")
Back to Journals » International Journal of Nanomedicine » Volume 18
Combined Donepezil with Astaxanthin via Nanostructured Lipid Carriers Effective Delivery to Brain for Alzheimer’s Disease in Rat Model
Authors Shehata MK , Ismail AA, Kamel MA
Received 8 May 2023
Accepted for publication 19 July 2023
Published 27 July 2023 Volume 2023:18 Pages 4193—4227
DOI https://doi.org/10.2147/IJN.S417928
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Farooq A. Shiekh
Mustafa K Shehata,1 Assem A Ismail,1 Maher A Kamel2
1Department of Pharmaceutics, Faculty of Pharmacy, Alexandria University, Alexandria, Egypt; 2Department of Biochemistry, Medical Research Institute, Alexandria University, Alexandria, Egypt
Correspondence: Mustafa K Shehata, Department of Pharmaceutics, Faculty of Pharmacy, Alexandria University, Khartoum Square, Azzarita, Alexandria, 21521, Egypt, Tel +20 1114740302, Fax +002/23/4871668, Email [email protected]; [email protected]
Introduction: Donepezil (DPL), a specific acetylcholinesterase inhibitor, is used as a first-line treatment to improve cognitive deficits in Alzheimer’s disease (AD) and it might have a disease modifying effect. Astaxanthin (AST) is a natural potent antioxidant with neuroprotective, anti-amyloidogenic, anti-apoptotic, and anti-inflammatory effects. This study aimed to prepare nanostructured lipid carriers (NLCs) co-loaded with donepezil and astaxanthin (DPL/AST–NLCs) and evaluate their in vivo efficacy in an AD-like rat model 30 days after daily intranasal administration.
Methods: DPL/AST–NLCs were prepared using a hot high-shear homogenization technique, in vitro examined for their physicochemical parameters and in vivo evaluated. AD induction in rats was performed by aluminum chloride. The cortex and hippocampus were isolated from the brain of rats for biochemical testing and histopathological examination.
Results: DPL/AST–NLCs showed z-average diameter 149.9 ± 3.21 nm, polydispersity index 0.224 ± 0.017, zeta potential – 33.7 ± 4.71 mV, entrapment efficiency 81.25 ± 1.98% (donepezil) and 93.85 ± 1.75% (astaxanthin), in vitro sustained release of both donepezil and astaxanthin for 24 h, spherical morphology by transmission electron microscopy, and they were stable at 4– 8 ± 2°C for six months. Differential scanning calorimetry revealed that donepezil and astaxanthin were molecularly dispersed in the NLC matrix in an amorphous state. The DPL/AST–NLC-treated rats showed significantly lower levels of nuclear factor-kappa B, malondialdehyde, β-site amyloid precursor protein cleaving enzyme-1, caspase-3, amyloid beta (Aβ1‑42), and acetylcholinesterase, and significantly higher levels of glutathione and acetylcholine in the cortex and hippocampus than the AD-like untreated rats and that treated with donepezil–NLCs. DPL/AST–NLCs showed significantly higher anti-amyloidogenic, antioxidant, anti-acetylcholinesterase, anti-inflammatory, and anti-apoptotic effects, resulting in significant improvement in the cortical and hippocampal histopathology.
Conclusion: Nose-to-brain delivery of DPL/AST–NLCs is a promising strategy for the management of AD.
Keywords: Alzheimer’s disease, donepezil, astaxanthin, intranasal delivery, nanostructured lipid carriers
Introduction
Alzheimer’s disease (AD) is a devastating neurodegenerative disorder that is characterized by severe dementia, progressive memory loss, and cognitive impairment. AD represents a challenge to healthcare systems as the number of people with AD is steadily increasing worldwide, and it tends to have various comorbidities and medical requirements for long-term care. The commonly prescribed medications for AD are only symptomatic that are used to improve the cognitive symptoms of AD, and they do not stop the progressive pathology of the disease. Despite numerous clinical trials conducted, no approved disease-modifying treatment for AD is currently available.1,2 Based on the evidence, AD is a multifactorial disorder as different mechanisms, such as oxidative stress, neuronal inflammation, accumulation of amyloid-beta (Aβ) plaques, tau pathology, and glutamate excitotoxicity are involved in the pathophysiology of AD, which together lead to neuronal and synaptic loss, and atrophy in some areas of the brain such as the cortex and hippocampus, responsible for cognitive and behavioral processes.3
 |
Figure 1 Particle size distribution curve of the optimized nanostructured lipid carriers with donepezil and astaxanthin (DPL/AST–NLCs). |
 |
Figure 2 Transmission electron microscopy (TEM) photograph of the optimized nanostructured lipid carriers with donepezil and astaxanthin (DPL/AST–NLCs) (25,000X magnification). |
 |
Figure 3 Differential scanning calorimetry (DSC) thermograms of (a) glyceryl palmitostearate, (b) astaxanthin (AST), (c) donepezil (DPL), and (d) nanostructured lipid carriers with donepezil and astaxanthin (DPL/AST–NLCs). |
 |
Figure 4 The in vitro release profiles of donepezil (DPL) and astaxanthin (AST) from the optimized nanostructured lipid carriers with donepezil and astaxanthin (DPL/AST–NLCs) and solution of free drugs (DPL/AST–Solution) for 24 h using dialysis method in phosphate buffer (pH 7.4) at 37 ± 0.5°C. Data are presented as mean ± SD (standard deviation), n = 3. |
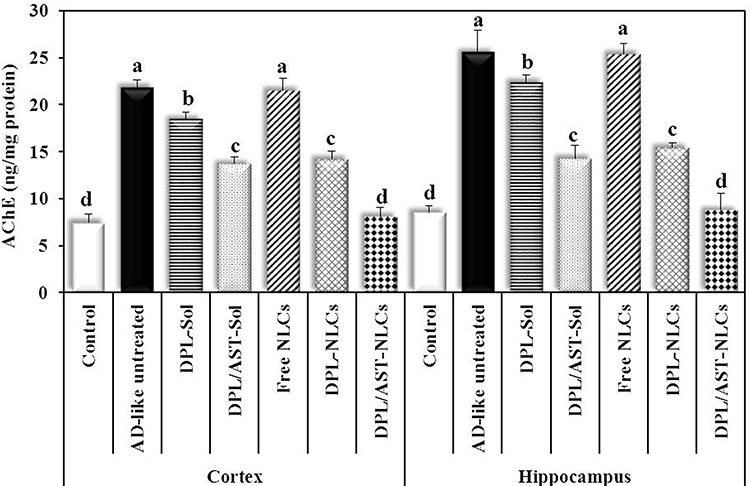 |
Figure 5 The cortical and hippocampal levels of acetylcholinesterase (AChE) in different groups. Data are presented as mean ± standard deviation, n = 6. In the same tissue (the cortex or hippocampus), means with any common letter are not significantly different, while means with completely different letters (from a - d) are significantly different at p < 0.05. Abbreviations: DPL, donepezil; AST, astaxanthin; AD-like, Alzheimer’s disease-like; NLCs, nanostructured lipid carriers; Sol, solution. |
 |
Figure 6 The cortical and hippocampal levels of acetylcholine (ACh) in different groups. Data are presented as mean ± standard deviation, n = 6. In the same tissue (the cortex or hippocampus), means with any common letter are not significantly different, while means with completely different letters (from a - d) are significantly different at p < 0.05. Abbreviations: DPL, donepezil; AST, astaxanthin; AD-like, Alzheimer’s disease-like; NLCs, nanostructured lipid carriers; Sol, solution. |
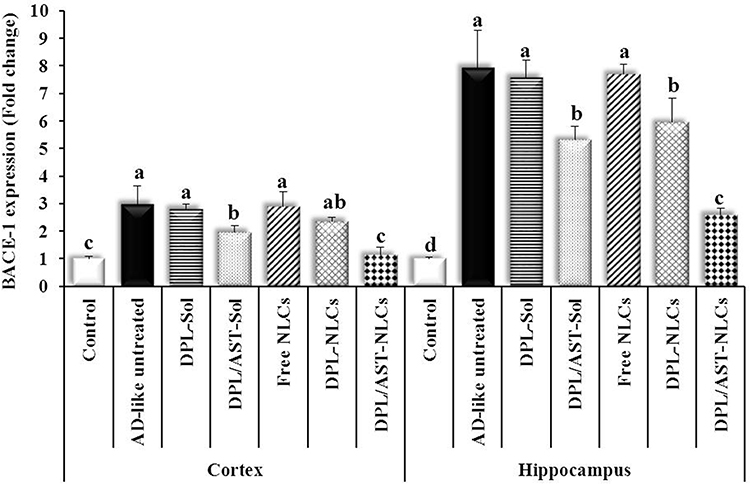 |
Figure 7 The cortical and hippocampal expression of β-site amyloid precursor protein cleaving enzyme-1 (BACE-1) in different groups. Data are presented as mean ± standard deviation, n = 6. In the same tissue (the cortex or hippocampus), means with any common letter are not significantly different, while means with completely different letters (from a - d) are significantly different at p < 0.05. Abbreviations: DPL, donepezil; AST, astaxanthin; AD-like, Alzheimer’s disease-like; NLCs, nanostructured lipid carriers; Sol, solution. |
 |
Figure 8 The cortical and hippocampal levels of amyloid beta (Aβ1-42) in different groups. Data are presented as mean ± standard deviation, n = 6. In the same tissue (the cortex or hippocampus), means with any common letter are not significantly different, while means with completely different letters (from a–c) are significantly different at p < 0.05. Abbreviations: DPL, donepezil; AST, astaxanthin; AD-like, Alzheimer’s disease-like; NLCs, nanostructured lipid carriers; Sol, solution. |
 |
Figure 9 The cortical and hippocampal levels of malondialdehyde (MDA) in different groups. Data are presented as mean ± standard deviation, n = 6. In the same tissue (the cortex or hippocampus), means with any common letter are not significantly different, while means with completely different letters (from a–c) are significantly different at p < 0.05. Abbreviations: DPL, donepezil; AST, astaxanthin; AD-like, Alzheimer’s disease-like; NLCs, nanostructured lipid carriers; Sol, solution. |
 |
Figure 10 The cortical and hippocampal levels of reduced glutathione (GSH) in different groups. Data are presented as mean ± standard deviation, n = 6. In the same tissue (the cortex or hippocampus), means with any common letter are not significantly different, while means with completely different letters (from a - c) are significantly different at p < 0.05. Abbreviations: DPL, donepezil; AST, astaxanthin; AD-like, Alzheimer’s disease-like; NLCs, nanostructured lipid carriers; Sol, solution. |
 |
Figure 11 The cortical and hippocampal levels of nuclear factor-kappa B (NF-κB) in different groups. Data are presented as mean ± standard deviation, n = 6. In the same tissue (the cortex or hippocampus), means with any common letter are not significantly different, while means with completely different letters (from a - d) are significantly different at p < 0.05. Abbreviations: DPL, donepezil; AST, astaxanthin; AD-like, Alzheimer’s disease-like; NLCs, nanostructured lipid carriers; Sol, solution. |
 |
Figure 12 The cortical and hippocampal caspase-3 activity in different groups. Data are presented as mean ± standard deviation, n = 6. In the same tissue (the cortex or hippocampus), means with any common letter are not significantly different, while means with completely different letters (from a - d) are significantly different at p < 0.05. Abbreviations: DPL, donepezil; AST, astaxanthin; AD-like, Alzheimer’s disease-like; NLCs, nanostructured lipid carriers; Sol, solution. |
 |
Figure 13 Histology of the cortex stained with hematoxylin and eosin (X400) of (a) control rats, (b) Alzheimer’s disease (AD)–like untreated rats, (c) donepezil-nanostructured lipid carriers (DPL–NLCs) treated rats, (d) donepezil and astaxanthin solution (DPL/AST–Solution) treated rats, and (e) the optimized donepezil and astaxanthin-nanostructured lipid carriers (DPL/AST–NLCs) treated rats. Normal neurons (black arrow), necrotic neurons associated with satellitosis and neuronophagia (red arrow), degenerated neurons (Orange arrow), and glial cells (yellow arrow). |
 |
Figure 14 Histology of the hippocampus stained with hematoxylin and eosin (X400) of (a) control rats, (b) Alzheimer’s disease (AD)–like untreated rats, (c) donepezil-nanostructured lipid carriers (DPL–NLCs) treated rats, (d) donepezil and astaxanthin solution (DPL/AST–Solution) treated rats, and (e) the optimized donepezil and astaxanthin-nanostructured lipid carriers (DPL/AST–NLCs) treated rats. Normal neurons (black arrow), glial cells (yellow arrow), degenerated neurons (red arrow), and pericellular space (Orange arrow). |
Acetylcholine (ACh) is a key neurotransmitter in the brain that is involved in memory, learning, and cognitive functions. According to the cholinergic hypothesis, AD is associated with a reduction in the levels of ACh in the brain due to the increased activity of acetylcholinesterase (AChE) enzyme that is involved in the hydrolysis of ACh, low levels of choline acetyltransferase, which is involved in the synthesis of ACh, and loss of cholinergic neurons, resulting in progressive memory and cognitive deficits.4,5 Thus, a widely studied therapeutic strategy in AD is to improve cerebral cholinergic neurotransmission using AChE inhibitors (AChEIs) that increase ACh concentration in the brain synapses, thereby improving memory and cognitive impairment in AD. AChEIs, such as donepezil, galantamine and rivastigmine, are approved by the Food and Drug Administration (FDA), and they are used as symptomatic treatment for AD.6,7
Donepezil (DPL) is a potent, selective, noncompetitive, and reversible AChEI. Donepezil is a well-known anti-AD agent that increases ACh levels in the synapses, and therefore it provides measurable improvement of cognitive function in AD.7,8 Donepezil is considered a first-line treatment in AD, and it acts primarily as an effective symptomatic therapeutic agent for AD worldwide. Donepezil has high oral bioavailability and a long half-life, and it is commercially available as a tablet (5, 10, and 23 mg) intended for oral administration. The FDA approved donepezil hydrochloride for the treatment of cognitive symptoms in mild, moderate, and severe AD.9,10 Donepezil is used both as monotherapy and in combination therapy with memantine.11 Oral administration of donepezil provides non-targeted delivery, resulting in gastrointestinal adverse events, such as nausea, anorexia, diarrhea, gastric bleeding, muscle convulsions, and sleep disturbance. In addition, donepezil undergoes hepatic first-pass metabolism and it has poor ability to penetrate the blood–brain barrier (BBB) after its oral delivery, due to its hydrophilicity.7,8 In several previous studies, the intranasal (IN) route was used as an alternative non-invasive approach in order to avoid these issues, which allows direct delivery of donepezil to the brain through the trigeminal and olfactory pathways in the nose, avoiding the BBB and hepatic first-pass metabolism and minimized systemic exposure and side effects associated with the oral administration of donepezil.12 In addition, donepezil was incorporated in different nanosystems, like nanoemulsions,13 solid lipid nanoparticles (SLNs),14 liposomes,15 and nanostructured lipid carriers (NLCs),16 in order to overcome the enzymatic degradation and quick removal of donepezil from the nasal cavity and to extend its nasal residence in order to improve the nasomucosal permeability and enhance nose to brain delivery of donepezil for better brain targeting.17 Clinically, early and continuous long-term treatment of AD with donepezil is considered to preserve cognitive function more effectively than delayed treatment. Previously published studies of donepezil have suggested a possible disease-modifying or neuroprotective role of donepezil in AD, highlighting the importance of early diagnosis and treatment initiation in AD.18,19
Astaxanthin (AST) is a potent antioxidant that inhibits the generation of reactive oxygen species and lipid peroxidation, and it improves the outcome of oxidative stress-related diseases including AD.1,20 Several in vitro and in vivo studies have reported the neuroprotective effects of astaxanthin in neurodegenerative disorders owing to its potent antioxidant, anti-inflammatory, anti-amyloidogenic, and anti-apoptotic activities. In addition, astaxanthin can penetrate the BBB and reach the brain, restore cholinergic neurotransmission, and ameliorate behavioral and cognitive deficits. Therefore, astaxanthin could be a potential option for the prevention, reduction of AD progression, and improvement of its prognosis.21,22 Astaxanthin (3,3−-dihydroxy-β,β−-carotene-4,4−-dione) is a natural lipophilic xanthophyll carotenoid produced by various microorganisms and marine animals including algae, bacteria, yeast, shrimp, crustaceans, krill, lobsters, and salmon. The green microalga Haematococcus pluvialis is the natural source of astaxanthin used in the present study. Astaxanthin has different isomeric forms; 3S,3−S-all-trans-AST has higher stability and more powerful antioxidant activity than other isomers.23,24 Astaxanthin is available commercially (4 mg daily) as a dietary supplement approved by the FDA, bringing benefits to public health with no significant adverse effects. Astaxanthin is authorized by the European Food Safety Authority Commission (EFSA) as a food supplement at levels up to 8 mg/day for adults.25,26 In contrast, astaxanthin has poor oral bioavailability due to its high lipophilic nature.27 In previous studies, astaxanthin has been loaded into SLNs28 and NLCs29,30 for better brain targeting via the intranasal route for the management of neurodegenerative disorders, including AD.
Nanostructured lipid carriers (NLCs) are lipid-based nanosystems that have been used to improve the intranasal delivery of drugs to the brain as they improve the nasomucosal permeability, enhance the drug bioavailability in the brain, and provide sustained drug release patterns. Among lipidic nanoparticulate systems, NLCs have sufficient sustained release compared to nanoemulsions, higher stability compared to liposomes, less toxicity than polymeric nanoparticles, and higher drug loading capacity than SLNs. The lipid matrix of NLCs is composed of a blend of solid and liquid lipids with disordered crystalline structure. The presence of liquid lipid in the NLC matrix creates more space for drug accommodation, thereby allowing entrapment of higher amounts of the drug and minimizing drug expulsion during storage compared to SLNs.31,32 Furthermore, NLCs can accommodate both hydrophilic and lipophilic drugs, and dual drug-loaded NLCs have been successfully prepared in previous studies. Sood et al33 developed NLCs co-loaded with hydrophilic donepezil and lipophilic curcumin for the management of AD via the intranasal (IN) route. IN administration of the developed NLCs co-loaded with donepezil and curcumin in a streptozotocin-induced Alzheimer’s model increased drug concentration in the brain than the intravenous route, significantly improved ACh levels, reduced oxidative stress markers, and behavioral tasks showed improved memory and learning compared to pure drugs. Lin et al34 developed NLCs co-loaded with hydrophilic methotrexate and lipophilic calcipotriol as topical therapies for psoriasis. Li et al35 formulated NLCs that co-delivered doxorubicin and lapachone to overcome multidrug resistance during breast cancer therapy. Zhao et al36 formulated NLCs co-loaded with doxorubicin and curcumin to improve the efficacy of chemotherapy in liver cancer. Youssef et al37 developed NLCs co-loaded with natamycin and ciprofloxacin for the treatment of mixed ocular infections.
In previous studies, NLCs have been shown to successfully deliver hydrophilic donepezil or lipophilic astaxanthin individually through the IN route to the brain. Butani16 prepared NLCs loaded with donepezil and incorporated the developed DPL–NLCs in an in situ gel formulation for the treatment of AD. IN treatment of AD-like rats with the developed DPL–NLC gel formulation showed a significant enhancement in cognitive function and higher drug concentration in the brain than the marketed oral donepezil formulation. Gautam et al29 developed NLCs loaded with astaxanthin and incorporated the developed AST–NLCs in an in situ gel formulation to treat Parkinson’s disease. IN treatment of rats with the developed AST–NLC gel formulation showed improved behavior of rats and higher drug bioavailability in the brain than the free astaxanthin gel. In our previous study, we formulated NLCs loaded with astaxanthin, and evaluated the neuroprotective effects of the developed AST–NLCs in a rat model of AD 30 days after daily IN administration. The results showed that the developed AST–NLCs exhibited significantly higher anti-inflammatory, anti-amyloidogenic, anti-apoptotic, anti-acetylcholinesterase, and antioxidant properties.30
The aim of the present study is to develop NLCs co-loaded with donepezil and astaxanthin (DPL/AST–NLCs), in vitro examine their physicochemical parameters and in vivo evaluate their in vivo efficacy in a rat model of AD 30 days after daily intranasal administration for better brain targeting of both drugs (donepezil and astaxanthin) for better management of AD. Combination therapy is based on an acetylcholinesterase inhibitor approach combined with anti-inflammatory, anti-amyloidogenic, anti-apoptotic, and antioxidant approaches. The developed DPL/AST–NLCs were prepared using a hot high-shear homogenization technique, and in vitro examined for their physicochemical parameters, such as zeta potential, z-average diameter, polydispersity index, in vitro drug release, drug loading, entrapment efficiency, stability, morphology study by transmission electron microscopy, and thermal study by differential scanning calorimetry. In addition, the developed DPL/AST–NLCs were administered to the AD-like rats by the intranasal route daily for 30 days, and they were in vivo evaluated for their in vivo efficacy in the AD-like rats 30 days after their daily IN administration using pharmacodynamic studies. Biochemical parameters, such as cholinergic markers (acetylcholine and acetylcholinesterase), oxidative stress markers (glutathione and malondialdehyde), apoptosis markers (caspase-3), amyloidogenic parameters (β-site amyloid precursor protein cleaving enzyme-1 and amyloid beta), and inflammation markers (nuclear factor-kappa B) were estimated in the cortical and hippocampal tissues of rats. In addition, the rat cortical and hippocampal histopathology were observed.
Materials and Methods
Donepezil was a gift from Ranbaxy Research Laboratory (Gurgaon, India). Astaxanthin was purchased from Carbosynth Limited (Berkshire, UK). Precirol® ATO 5 (glyceryl palmitostearate) was a gift from Gattefossé (Saint-Priest, Cedex, France). Oleic acid and Tween® 80 (polysorbate 80) were purchased from Loba Chemie Laboratory Reagents and Fine Chemicals (Mumbai, India). Pluronic® F68 (poloxamer 188) was purchased from BASF SE (Ludwigshafen, Germany). HPLC-grade methanol and acetonitrile were purchased from Sigma-Aldrich (St. Louis, MO, USA). Sodium dihydrogen phosphate and phosphoric acid were purchased from El-Nasr Pharmaceutical Chemicals Co. (Cairo, Egypt).
The Preparation of Nanostructured Lipid Carriers with Donepezil and Astaxanthin
Nanostructured lipid carriers with donepezil and astaxanthin (DPL/AST–NLCs) were prepared by the hot high-shear homogenization technique. Briefly, the lipid phase was prepared by melting 150 mg of glyceryl palmitostearate (solid lipid) at 5○C above its melting point, 50 mg of oleic acid (liquid lipid) was added, and then donepezil (10 mg) and astaxanthin (40 mg) were mixed into the above lipid melt. The hot aqueous surfactant solution, which was prepared by dissolving a mixture of poloxamer 188 and polysorbate 80 (1:1, w/w) in 10 mL of deionized water, was added slowly to the lipid phase under continuous magnetic stirring. The formed coarse pre-emulsion was homogenized at 15,000 rpm for 15 min using high shear homogenizer (IKA T25 digital ultra-TURRAX, Staufen, Germany), and the formed nanoemulsion was cooled down and stored at 4–8 ± 2°C (refrigerator) overnight to allow lipid recrystallization for the formation of DPL/AST–NLCs.38 Formulation parameters such as surfactant concentration, solid-to-liquid lipid ratio, and total lipid-to-total drug ratio were optimized.31
The in vitro Examination of the Developed Nanostructured Lipid Carriers with Donepezil and Astaxanthin
The Determination of z-Average Diameter, Polydispersity Index and Zeta Potential
The z-average diameter (mean particle size PS), polydispersity index (PDI), and zeta potential (ZP) of the prepared DPL/AST–NLCs were determined using a Malvern Zetasizer Nano ZS (Malvern Instruments, Malvern, UK) based on the dynamic light scattering (DLS) technique. The refractive indices of the material (lipid) and the dispersant (water) used for DLS measurements were 1.40 and 1.33, respectively. The samples were diluted with filtered deionized water 10 times (1:10, v/v), and they were measured in triplicate.39
Morphology Study: Transmission Electron Microscopy
The size and morphology of the developed DPL/AST–NLCs were observed by a transmission electron microscopy (TEM, model JEM-100CX, JEOL, Japan). A drop of the diluted sample was stained with a saturated solution of uranyl acetate and dried before microscopic investigation.39
The Determination of Entrapment Efficiency
Percent entrapment efficiency (%EE) of the developed DPL/AST–NLCs was determined by an indirect method that depends on centrifugation of DPL/AST–NLCs (5 mL) at 20,000 rpm for 30 min at 4°C using cooling ultracentrifuge (Sigma Laborzentrifuge GmbH, Osterode, Germany), in order to separate the free drugs (donepezil and astaxanthin) that present in the supernatant from the prepared dual drug-loaded NLCs. For the determination of the amount of free donepezil, the supernatant was analyzed by UV spectrophotometric method (UV–VIS spectrophotometer, UV-160A; Shimadzu, Kyoto, Japan) at λmax of donepezil = 271 nm. For the determination of the amount of free astaxanthin, the supernatant was analyzed by high-performance liquid chromatographic method (HPLC, Agilent Technologies 1200 Infinity Series, USA) at λmax of astaxanthin = 480 nm.40,41 %EE of donepezil or astaxanthin was calculated using the following equation:
Entrapment efficiency (%EE) = (Wtotal − Wfree) × 100 / Wtotal
where Wtotal is the amount of total drug (donepezil or astaxanthin) used in DPL/AST–NLCs, and Wfree is the amount of free drug (donepezil or astaxanthin) in the supernatant.42
Thermal Analysis: Differential Scanning Calorimetry
The physical form of donepezil and astaxanthin in DPL/AST–NLCs was examined by a differential scanning calorimetry DSC (Thermal Analysis Instruments, SDT Q600, USA). Pure glyceryl palmitostearate, donepezil, and astaxanthin were used as controls. The samples (10 mg) were heated from 25°C to 350°C, and the heating rate was 10°C/min.43
The in vitro Release Study
The in vitro release patterns of donepezil and astaxanthin from the prepared DPL/AST–NLCs were determined using the dialysis bag diffusion method (VISKING molecular weight cutoff 12–14 kDa, London, UK). The samples (2 mL of DPL/AST–NLCs) were placed in dialysis bags, and the sealed bags were immersed in phosphate buffer pH 7.4 (100 mL) and then placed in thermostatically controlled shaking water bath (Kottermann, type 3047, Hanigsen, Germany), which was maintained at 100 rpm and 37 ± 0.5°C. Aliquots (5 mL) were withdrawn at different time intervals and replaced with an equivalent volume of fresh medium to maintain sink condition. For the determination of the amount of donepezil and astaxanthin released from DPL/AST–NLCs, the samples were analyzed by UV spectrophotometry at λmax of donepezil = 271 nm and by HPLC at λmax of astaxanthin = 480 nm, respectively. Free donepezil and astaxanthin solution containing the same amounts of donepezil and astaxanthin in the developed DPL/AST–NLCs was used as a control. The in vitro release data of DPL/AST–NLCs were analyzed by different kinetic models.44,45
Stability Study
The prepared DPL/AST–NLCs were subjected to a 6-month stability study at different storage conditions (4–8 ± 2°C and 25 ± 2°C/60 ±5% RH). The samples were periodically analyzed by measuring the z-average diameter, PDI and entrapment efficiencies of donepezil and astaxanthin.46
The in vivo Examination of the Developed Nanostructured Lipid Carriers with Donepezil and Astaxanthin
Animals and Induction of Alzheimer’s Disease
Forty-two male albino rats (three months old and weighing from 150 to 200 gm) were used in the current in vivo study. The animals were treated according to the ethical guidelines of the Institutional Animal Care and Use Committee (IACUC), Alexandria University, Egypt (Approval number: AU062019518251). Induction of AD in rats was performed by oral administration of hydrated aluminum chloride (AlCl36H2O) solution for 6 weeks (daily dose was 75 mg/kg body weight).47 Morris water maze (MWM) test was carried out at the end of the 6 weeks in order to confirm cognitive impairment induced in rats by AlCl3 administration.48
Experimental Design
The rats were divided into seven groups and each group consisted of six rats. Group A was a control (normal) group, and Group B was AD-like untreated group. Treatment groups (from group C to group G) received 20 µL of the tested preparations by the intranasal (IN) route (10 µL in each nostril) daily for 30 days; Group C received DPL–Solution (equivalent to 0.1 mg/Kg body weight of donepezil), Group D received DPL/AST–Solution (equivalent to 0.1 mg/Kg of donepezil and 0.4 mg/Kg of astaxanthin), Group E received free NLCs, Group F received DPL–NLCs (equivalent to 0.1 mg/Kg of donepezil), and Group G received DPL/AST–NLCs (equivalent to 0.1 mg/Kg of donepezil and 0.4 mg/Kg of astaxanthin). MWM test was carried out after the last day of the IN treatment.
The Behavioral Morris Water Maze Test
Briefly, the water-filled pool was divided into 4 quadrants (northwest, northeast, southwest, and southeast), and the escape latency time (the time required for animals to reach the hidden platform) was calculated in the behavioral Morris water maze (MWM) test, which was carried out for 4 days. Probe trial test was carried out to determine the retention memory of rats on the fourth day, and the percentage of the retention time (the time spent in the target quadrant of the pool) was calculated.48
Biochemical Assays
Animals were sacrificed after the end of MWM test, and the brain was removed. The cortical and hippocampal tissues were excised from one hemisphere, cleaned, and homogenized in phosphate-buffered saline (pH 7.4) at a ratio of 1:9. The tissue homogenates were centrifuged at 4 °C, and the supernatants were used for estimation of various biochemical markers.
The Determination of Malondialdehyde
Quantitative measurement of malondialdehyde in the cortical and hippocampal tissues of rats was performed using the colorimetric method described by Draper and Hadley. Briefly, the supernatant of the tissue homogenates was mixed with trichloroacetic acid, and centrifuged. The resulting supernatant was heated with thiobarbituric acid and then cooled, resulting in the formation of pink chromogen that was proportional to the level of malondialdehyde in the tissue sample.49
The Determination of Caspase-3 Activity
Briefly, caspase-3 enzyme cleaved a specific peptide sequence conjugated to a color molecule, p-nitroaniline, in a caspase-3 assay kit (Elabscience, USA), and the released yellow chromophores (p-nitroaniline) was proportional to the caspase-3 activity in the sample.
The Determination of Glutathione
Briefly, reduced glutathione (GSH) was oxidized by 5,5ˋ-dithiobis-(2-nitrobenzoic acid) into oxidized glutathione and 5-thio-2-nitrobenzoic acid that was proportional to the level of GSH in the sample, according to Griffith method.50
The Determination of Acetylcholinesterase, Acetylcholine, Amyloid Beta, and Nuclear Factor-Kappa B
The levels of acetylcholinesterase, acetylcholine, amyloid beta (Aβ1-42), and nuclear factor-kappa B were determined using ELISA reagent kits (Chongqing Biospes Co., Ltd., China) according to the manufacturer’s protocols.
Gene Expression of β-Site Amyloid Precursor Protein Cleaving Enzyme-1
β-site amyloid precursor protein cleaving enzyme-1 (BACE-1) gene expression in the rat cortex and hippocampus was performed by the quantitative real time-polymerase chain reaction using kits (Qiagen, Germany) according to the manufacturer’s protocols. The expression of BACE-1 was quantified relative to the expression of the reference gene (18S rRNA) in the same sample using the 2−ΔΔCt method. The primers used for the determination of rat genes were as follows: 18S rRNA were forward, 5’-GTAACCCGTTGAACCCCATT-3’ and reverse, 5’-AAGCTTATGACCCGCACTT-3’, and BACE-1 were forward, 5’-GCATGATCATTGGTGGTATC-3’ and reverse,5’-CCATCTTGAGATCTTGACCA-3’.51
Histopathological Examination
Briefly, the cortical and hippocampal tissues excised from the other brain hemisphere were stained with hematoxylin for 3 sec, and then stained with eosin for 3 min for histopathological examination.52
Statistical Analysis
Data were analyzed using one-way analysis of variance (ANOVA) followed by Tukey’s multiple comparison post-hoc tests to compare different groups and Pearson’s correlation analysis using the IBM SPSS software package (version 20.0, Armonk, NY, IBM Corp). Data were expressed as mean ± standard deviation (SD). The p value was assumed to be significant at p < 0.05.
Results and Discussion
The Preparation and in vitro Examination of Nanostructured Lipid Carriers with Donepezil and Astaxanthin
DPL/AST–NLCs were successfully prepared by the hot high-shear homogenization technique. Formulation parameters such as surfactant concentration (ranging from 1 to 2.5%w/v), solid-to-liquid lipid ratio (90:10, 75:25, and 60:40, %w/w), and total lipid-to-total drug ratio (ranging from 3:1 to 6:1, w/w) were optimized as they affected the in vitro characteristic parameters of the developed DPL/AST–NLCs (optimization data are not shown).31,38 In the optimized DPL/AST–NLCs, total lipid-to-total drug ratio was 4:1 (w/w), the amount of total lipid used = 200 mg, the amount of donepezil = 10 mg, and that of astaxanthin = 40 mg, solid-to-liquid lipid ratio was 75:25 (%w/w of total lipid amount) (the amount of glyceryl palmitostearate used = 150 mg and that of oleic acid = 50 mg), and surfactant concentration was 1.75%w/v (in 10 mL of deionized water). The optimized DPL/AST–NLCs were in vitro and in vivo evaluated, and their in vitro characteristic parameters are listed in Table 1.
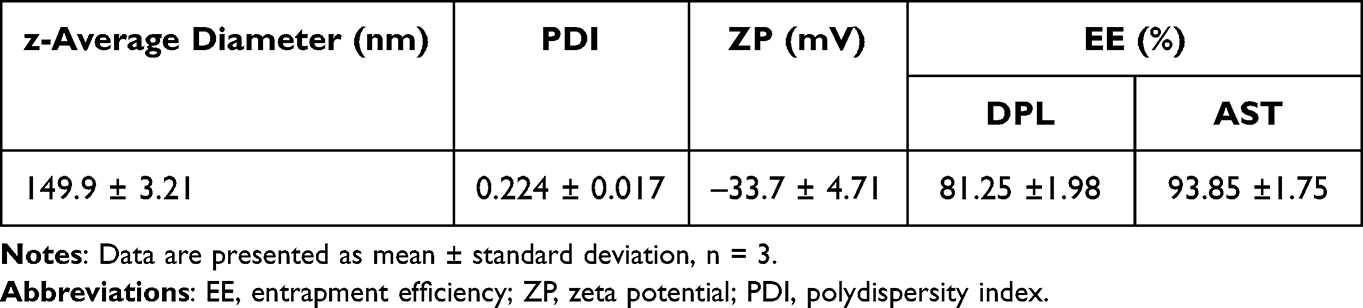 |
Table 1 The in vitro Characteristic Parameters of the Optimized Nanostructured Lipid Carriers with Donepezil and Astaxanthin (DPL/AST–NLCs) |
The Determination of z-Average Diameter, Polydispersity Index and Zeta Potential
The z-average diameter of a nanosystem is a critical parameter because it affects drug permeation via the nasal mucosa, distribution, and drug bioavailability in the brain. The nanosystem with a small mean particle size (PS) showed improved nasal permeability and higher drug bioavailability in the brain.53 As observed in Table 1 and Figure 1, the optimized DPL/AST–NLCs showed z-average diameter = 149.9 ± 3.21 nm (<200 nm), which was found to be optimum for nose-to-brain drug delivery. Polydispersity index (PDI) indicates the distribution of particle size in the nanosystem. The lower value of PDI indicates narrow particle size distribution, while its higher value indicates wide distribution of particle size and particle aggregation may occur.53 The optimized DPL/AST–NLCs had PDI of 0.224 ± 0.017 (<0.3) indicating narrow distribution of particle size and homogeneous nature of the developed formulation. Zeta potential (ZP) represents the electrical charge on the NLC surface and it is a crucial parameter for nanoparticles, as it affects their physical stability. The stability of the formulation increased as its ZP value increased because the particles resisted aggregation. The optimized DPL/AST–NLCs had high ZP value = –33.7 ± 4.71 mV (< –30 mV) that provides sufficient electrostatic repulsion, suggesting good physical stability of the system. The negative charge in DPL/AST–NLCs is due to the presence of free fatty acids in their lipid composition at the surface of the NLC particles. The prepared formulation was better stabilized by the surfactant mixture of poloxamer 188 and polysorbate 80, which was used to increase the stability of the formulation by steric stabilization and prevent agglomeration of the NLC particles.54,55
Morphology Study: Transmission Electron Microscopy
As illustrated from the TEM image in Figure 2, the optimized DPL/AST–NLCs showed spherical particles with no aggregation. The mean PS of DPL/AST–NLCs measured by TEM was found to be in the nanosize range (<200 nm), and it was slightly smaller than that obtained by the DLS method as TEM measurements involved drying of samples.39
The Determination of Entrapment Efficiency
As observed in Table 1, percent entrapment efficiency (%EE) of donepezil and astaxanthin in the optimized DPL/AST–NLCs were 81.25 ±1.98% and 93.85 ±1.75%, respectively. Entrapment efficiency of NLCs mainly depends on the nature of the drug and the lipid in which the drug is encapsulated. NLCs can be loaded with hydrophilic and lipophilic drugs, and they have a higher EE because they have a unique unstructured solid–liquid lipid matrix. The incorporation of liquid lipid (oleic acid) to solid lipid (glyceryl palmitostearate) in the NLC matrix creates a less organized crystal matrix and provides additional space, resulting in entrapment of more drug molecules, higher EE and long-term stability of NLCs owing to minimized drug expulsion during storage. The higher EE of the prepared DPL/AST–NLCs may also be attributed to the small z-average diameter and better stabilization of the formulation by the surfactant mixture.54–56 Butani16 developed NLCs loaded with donepezil alone, with EE of 86.81%. In our previous study, we developed NLCs loaded with astaxanthin alone, with EE = 94.1%.30 In the present study, donepezil and astaxanthin were co-loaded in NLCs with optimum EE of both drugs. The EE of astaxanthin in DPL/AST–NLCs was higher than that of donepezil, as donepezil is a hydrophilic drug, whereas astaxanthin is a lipophilic drug being entrapped in one system.
Thermal Analysis: Differential Scanning Calorimetry
As illustrated from the DSC thermograms in Figure 3, pure donepezil and astaxanthin showed sharp endothermic melting peaks at 214.40°C and 225.92°C, respectively, indicating their crystalline nature. The DSC thermogram of DPL/AST–NLCs showed no endothermic peaks for donepezil or astaxanthin, indicating that both drugs were converted from their crystalline state to an amorphous state in the NLC matrix and may indicate higher solubilization and dispersion of both drugs inside the lipid matrix. Regarding the solid lipid (glyceryl palmitostearate), a reduction in its melting enthalpy value from 176.7 J/g to 112.5 J/g was observed when it was prepared in DPL/AST–NLCs, this may be due to the presence of liquid lipid (oleic acid) in the NLC matrix, resulting in less ordered crystals with decreased enthalpy.30,43
The in vitro Release Study
As observed in Figure 4, the free donepezil and astaxanthin solution (DPL/AST–Solution) showed an immediate release of 79.5% of donepezil and 71.3% of astaxanthin in the first hour, and almost all content of both drugs were found in the release medium after 4 h.57 The in vitro release profiles of donepezil and astaxanthin from the developed DPL/AST–NLCs showed a biphasic release pattern for both drugs, with an initial burst release of 30.5% donepezil and 19.5% astaxanthin after 1 h, which could be ascribed to drugs present on the NLC surface as free moieties or drugs incorporated in the liquid lipid of the NLC matrix, followed by sustained slow release of both drugs for up to 24 h, which could be due to drugs present in the solid lipid.30,54 The biphasic release pattern of donepezil and astaxanthin from the developed DPL/AST–NLCs may indicate that both drugs were evenly distributed in the NLC matrix. The cumulative amount released of astaxanthin from DPL/AST–NLCs at any time was less than that of donepezil, perhaps because of the lipophilicity of astaxanthin.
The in vitro release data of donepezil and astaxanthin from DPL/AST–NLCs were analyzed by Hixon-Crowell, zero-order, Korsmeyer-Peppas, first-order, and Higuchi kinetic models. The correlation coefficient (r2) was determined to detect the best fitted model that describes the release kinetics of donepezil and astaxanthin from DPL/AST–NLCs, and the release exponent “n” value was determined from Korsmeyer-Peppas equation to determine the release mechanism of donepezil and astaxanthin from DPL/AST–NLCs. As observed in Table 2, the best fitted model that describes the release kinetics of both donepezil and astaxanthin from DPL/AST–NLCs was the Korsmeyer-Peppas model, as it had the highest r2 value (r2 for donepezil = 0.9895 and that for astaxanthin = 0.9867), and the release mechanism of both donepezil and astaxanthin from DPL/AST–NLCs was Fickian diffusion (diffusion-controlled release mechanism), because the n value was found to be <0.45 (n for donepezil = 0.344 and that for astaxanthin = 0.429).45,58
 |
Table 2 Models for the in vitro Release Kinetics of Donepezil and Astaxanthin from the Optimized Nanostructured Lipid Carriers with Donepezil and Astaxanthin (DPL/AST–NLCs) |
Stability Study
As illustrated in Table 3, the developed DPL/AST–NLCs were more stable at 4–8 ±2°C compared to 25 ± 2°C/60 ± 5% RH, as there was no significant change in the z-average diameter, PDI and entrapment efficiency of DPL/AST–NLCs at 4–8 ± 2°C up to 6 months, while the z-average diameter and PDI were significantly increased and EE (of both donepezil and astaxanthin) was significantly decreased after storage of DPL/AST–NLCs at 25 ± 2°C/60 ±5% RH for 6 months, which may be due to particle aggregation at higher temperature. Therefore, it was preferred to store DPL/AST–NLCs at 4–8 ± 2°C. The stability of the developed formulation could be due to high electrostatic repulsion, as indicated by its high ZP value, and the use of steric stabilizers.30,55
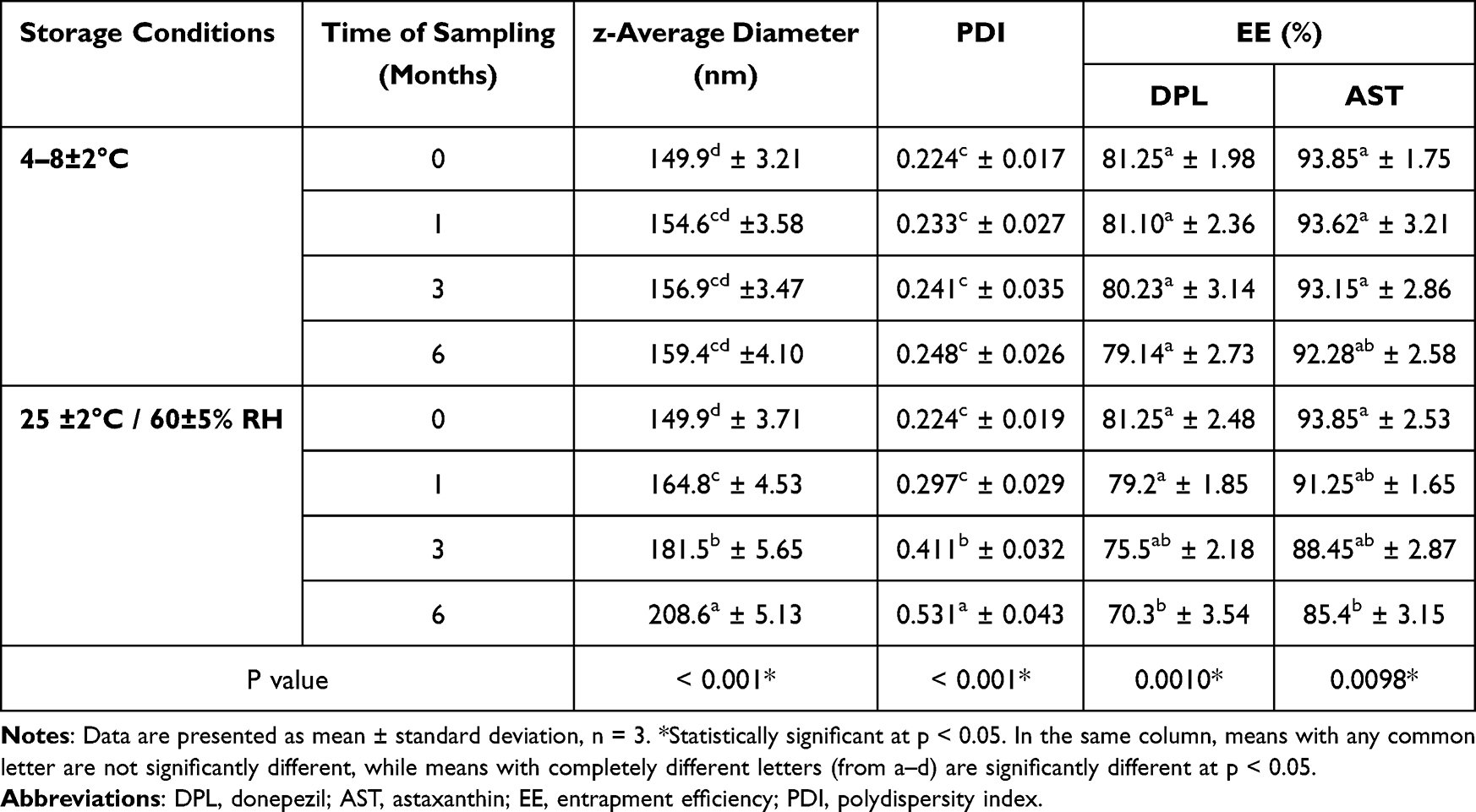 |
Table 3 Stability Parameters of the Optimized Nanostructured Lipid Carriers with Donepezil and Astaxanthin (DPL/AST–NLCs) After Storage for 6 Months at Different Storage Conditions |
The in vivo Examination of the Developed Nanostructured Lipid Carriers with Donepezil and Astaxanthin
The Behavioral Morris Water Maze Test
As observed in Table 4 (after treatment), the escape latency time (the time required for animals to reach the hidden platform) of the AD-like untreated rats was significantly higher in the MWM test during the 4 days (39.29 ± 3.56, 37.27 ± 4.36, 34.67 ± 3.14, 33.84 ± 2.19 sec in days 1,2,3 and 4, respectively) and the percentage of the retention time (the percentage of the time spent in the target quadrant of the pool) in the probe trial test was significantly lower (16.13% ± 2.06) than the control (normal) group (escape latency time; 18.93 ± 2.64, 14.54 ± 2.14, 11.33 ± 1.51, 8.58 ± 1.10 sec in days 1, 2, 3 and 4, respectively, retention time percentage; 32.46% ± 2.59). This clearly indicates cognitive impairment and amnesia induced by AlCl3 administration in rats, which is consistent with previous reports.47 Group treated with donepezil alone in NLCs (DPL–NLCs) demonstrated a significant decline in the escape latency time (29.83 ± 2.64, 25.20 ± 2.37, 21.67 ± 1.51, 18.31 ± 2.37 sec in days 1, 2, 3 and 4, respectively) and a significant increase in the retention time percentage (25.22% ± 1.98) compared to the AD-like untreated group and that treated with DPL–Solution (escape latency time; 36.17 ± 2.04, 32.13 ± 6.38, 28.67 ± 3.72, 27.15 ± 3.83 sec in days 1, 2, 3 and 4, respectively, retention time percentage; 20.09% ± 1.50). Group treated with DPL/AST–Solution (escape latency time; 28.50 ± 2.07, 24.50 ± 1.64, 21.33 ± 1.21, 17.58 ± 1.19 sec in days 1, 2, 3 and 4, respectively, retention time percentage; 25.82% ± 2.43) showed similar results to DPL–NLCs, with no significant difference between them in the MWM and probe trial tests. Group treated with NLCs co-loaded with donepezil and astaxanthin (DPL/AST–NLCs) showed significantly shorter escape latency time (21.50 ± 5.09, 16.67 ± 3.67, 13.33 ± 3.27, 9.34 ± 2.07 sec in days 1, 2, 3 and 4, respectively) and significantly higher retention time percentage (31.36% ± 1.68) compared to the AD-like untreated rats and groups treated with DPL–NLCs, DPL/AST–Solution, or DPL–Solution, and also treatment with DPL/AST–NLCs completely normalized the escape latency time in the MWM test during the 4 days and the retention time percentage in the probe trial test.
 |
Table 4 Morris Water Maze (MWM) Test During 4 Days of Training and Probe Trail Test Before and After Treatment |
Acetylcholine (ACh) is a critical neurotransmitter in the brain for memory, learning, and cognitive functions. In AD, the activity of acetylcholinesterase (AChE) enzyme that breaks down ACh is increased, which is associated with reduced levels of ACh in cholinergic synapses in the brain, resulting in cognitive impairment. AD is also characterized by a loss of neurons, particularly those expressing nicotinic acetylcholine receptors (nAChRs) that play major roles in memory and cognitive functions, thereby leading to a reduction in nAChR numbers and resulting in cognitive impairment..5,59 Donepezil is a potent centrally acting inhibitor of AChE that increases ACh levels in the brain, activates and upregulates nAChRs, thereby enhancing central cholinergic neurotransmission and significantly ameliorating performance and cognitive deficits in AD in several learning and memory tasks.60,61 Cutuli et al62 demonstrated that donepezil administration mitigated working memory dysfunction in rats with cholinergic depletion. Yamada et al63 reported that donepezil prevents spatial working memory deficits induced in rats by amyloid-beta. Riedel et al64 reported that donepezil reversed social memory deficits induced by scopolamine. Su et al65 demonstrated that donepezil prevented spatial memory impairment induced by isoflurane. Butani16 reported that rats treated with DPL–NLC gel (IN) showed a significant enhancement in cognitive function compared to the marketed oral donepezil formulation in a scopolamine-induced amnesia model in the MWM test.
Excessive production and accumulation of amyloid-beta (Aβ) peptides, neuroinflammation, oxidative stress, and apoptosis lead to neurodegeneration, disturbance in cholinergic neurotransmission and synaptic loss, resulting in cognitive impairment in AD.66 Astaxanthin is a natural potent antioxidant with potential neuroprotective properties. Several studies have reported that astaxanthin significantly ameliorated cognitive deficits and enhanced learning and memory in AD-like models, as it has the potential to inhibit oxidative stress, neuroinflammation, Aβ production, and apoptosis, in addition to its AChE inhibitory activity, and it improves cholinergic neurotransmission.67 Che et al68 found that astaxanthin reduced Aβ, phosphorylation of tau, neuroinflammation and oxidative stress and enhanced memory and learning in AD-like mice model. Taksima et al69 reported that astaxanthin reduced the levels of Aβ, malondialdehyde, and superoxide anions, increased glutathione peroxidase activity, increased the retention time and reduced the escape latency time of rats in the MWM test. Rahman et al70 reported that astaxanthin reduced the levels of Aβ, AChE, and inflammatory mediators, increased ACh levels, and reversed cognitive and memory deficits of rats in the MWM test. Al-Amin et al71 reported that astaxanthin reduced spatial memory deficits by reducing oxidative stress. Manabe et al72 reported that astaxanthin accumulated in the cortex and hippocampus of rats and enhanced cognitive function.
The concept of a combination strategy may provide a promising option for the management of AD. Donepezil has been used in combination with other drugs in several studies. Akinyemi et al73 reported that treatment of rats with donepezil in combination with curcumin inhibited AChE activity, decreased malondialdehyde, increased superoxide dismutase and glutathione and improved memory impairment induced in rats by scopolamine. Pattanashetti et al74 indicated that co-administration of donepezil and quercetin significantly decreased brain AChE, Aβ1-42 and lipid peroxidase levels and elevated glutathione levels compared to individual therapy, increased the retention time and decreased transfer latency in cognitive models of MWM, elevated plus maze and passive avoidance test. Keshk et al75 demonstrated that pretreatment with donepezil and losartan increased glutathione-s-transferase, decreased malondialdehyde, and improved memory function better than the single treatment. Cachard et al76 reported that donepezil and prucalopride improved memory deficits in the MWM test in a synergistic way.
In the present study, the AD-like untreated rats showed behavioral changes, memory and cognitive impairment, which may be due to cholinergic dysfunction, neuronal inflammation, degeneration, oxidative stress, and apoptosis induced in rats by AlCl3. This was observed by their significantly higher escape latency time in the MWM test and significantly lower retention time percentage in the probe trial test than the normal group.47 Treatment of rats with DPL–NLCs resulted in a significant decline in the escape latency time and a significant increase in the retention time percentage compared to the AD-like untreated group. Treatment with DPL/AST–NLCs significantly ameliorated AlCl3-induced cognitive impairment compared to DPL–NLCs. Group treated with DPL/AST–NLCs showed significantly higher retention time percentage and significantly lower escape latency time than the DPL–NLC-treated group, and also DPL/AST–NLCs completely normalized the retention time percentage and the escape latency time. Combined treatment with donepezil and astaxanthin in NLCs showed significantly higher antiamnestic effect than donepezil treatment alone in NLCs. The memory and cognitive deficits in the AD-like rats were improved after treatment with the developed DPL/AST-NLCs. This might be due to the improvement of cholinergic neurotransmission through AChE inhibition and reduction of oxidative stress, neuroinflammation, amyloid beta-peptides and apoptosis by donepezil and astaxanthin.
Biochemical Assays
Cholinergic Parameters: Acetylcholinesterase and Acetylcholine
As illustrated in Figures 5 and 6, the AD-like untreated group showed significantly higher levels of acetylcholinesterase (AChE) (21.86 ± 0.79, 25.62 ± 2.34 ng/mg protein) and significantly lower levels of acetylcholine (ACh) (7.58 ± 1.08, 8.38 ± 1.89 pg/mg protein) than the control group (AChE; 7.48 ± 0.87, 8.56 ± 0.72 ng/mg protein, ACh; 23.86 ± 0.82, 35.38 ± 1.13 pg/mg protein) in the cortex and hippocampus, respectively. Group treated with DPL–NLCs showed significantly lower AChE levels (14.20 ± 0.88, 15.45 ± 0.50 ng/mg protein) and significantly higher ACh levels (14.84 ± 1.02, 17.25 ± 1.36 pg/mg protein) than the untreated group and that treated with DPL–Solution (AChE; 18.46 ± 0.73, 22.37 ± 0.79 ng/mg protein, ACh; 10.98 ± 1.23, 11.77 ± 1.72 pg/mg protein) in the cortical and hippocampal tissues. Group treated with DPL/AST–Solution (AChE; 13.70 ± 0.78, 14.29 ± 1.36 ng/mg protein, ACh; 15.32 ± 0.65, 18.73 ± 0.91 pg/mg protein) showed similar results to DPL–NLCs, with no significant difference between them in cholinergic parameters. Group treated with DPL/AST–NLCs showed significantly lower AChE levels (8.13 ± 0.96, 8.76 ± 1.82 ng/mg protein) and significantly higher ACh levels (22.17 ± 1.50, 32.90 ± 1.61 pg/mg protein) than the AD-like untreated rats and groups treated with DPL–NLCs, DPL/AST–Solution, or DPL–Solution in the cortex and hippocampus, and also treatment with DPL/AST–NLCs completely normalized the levels of AChE and ACh in the cortex and hippocampus.
Based on the cholinergic hypothesis of AD, the reduced ACh levels in the brain and deficits in central cholinergic neurotransmission lead to cognitive impairment, which is the main clinical symptom of AD.73 Two enzymes are involved in hydrolysis of ACh, which are butyrylcholinesterase (BChE) and AChE, and the AChE activity is markedly increased in the AD brain.4,5 AChE inhibitors (AChEIs) increase the level of ACh in the brain and therefore they improve learning and memory and attenuate cognition impairment.6 Donepezil is a reversible AChEI that demonstrates high selectivity toward AChE inhibition compared with BChE, and it has a long duration of action.6 Donepezil inhibits AChE, stimulates nicotinic acetylcholine receptors, and prevents cholinergic degeneration, which is associated with enhanced synaptic strengthening that is related to cognitive function improvement and reduced spatial memory impairment.60,61,65
Amyloid-beta (Aβ1-42) peptides cause cholinergic dysfunction and synaptic loss, leading to a decline in ACh levels in the brain.66 In several previous studies, astaxanthin significantly inhibited AChE activity, attenuated the amyloidogenic pathway, and decreased Aβ1-42 levels, which were associated with a significant increase in ACh levels in the brain, resulting in the improvement of memory and cognitive impairment.69,70 Wang et al77 declared that astaxanthin inhibited AChE and prevented substrate binding. Chen et al78 revealed that astaxanthin treatment reversed the loss of choline acetyltransferase, cholinergic fibers, and dendritic spines in the hippocampus, decreased oxidative stress and neuroinflammatory responses, and ameliorated behavioral disorders and memory deficits in AD-like rats infused with ferrous amyloid buthionine. Nai et al79 reported that astaxanthin can modulate cholinergic decline by increasing the nerve growth factor expression, which can decelerate cholinergic neuron degeneration. Astaxanthin can increase the brain-derived neurotrophic factor (BDNF) expression, which is involved in synaptic plasticity, neuronal growth, and dendritic spine genesis that restore the expression of cholinergic fibers.80,81 Damodara et al82 and Wu et al83 reported that astaxanthin increased the BDNF levels, decreased oxidative stress, alleviated brain aging, and modulated spatial learning behavior in rats. In our previous study, the developed AST–NLCs significantly inhibited AChE activity and increased ACh levels in the rat cortex and hippocampus.30
In the present study, AlCl3 administration induced dysfunction in the cholinergic pathway in rats, as indicated by the significantly higher AChE levels and significantly lower ACh levels in the untreated group than the normal group. Treatment of rats with DPL–NLCs resulted in significantly lower AChE levels and significantly higher ACh levels than the AD-like untreated group. Treatment with DPL/AST–NLCs showed significantly higher anti-AChE activity than DPL-NLCs. Group treated with DPL/AST–NLCs showed significantly lower AChE levels and significantly higher ACh levels than the DPL–NLC-treated group and the AD-like untreated rats, and also DPL/AST–NLCs completely normalized the levels of AChE and ACh in the cortex and hippocampus. This may be due to the higher AChE inhibitory potential of both donepezil and astaxanthin, which significantly increased ACh levels, in addition to other neuroprotective effects of astaxanthin that could improve cholinergic neurotransmission.
Amyloidogenic Parameters: β-Site Amyloid Precursor Protein Cleaving Enzyme-1 and Amyloid Beta
As shown in Figures 7 and 8, the AD-like untreated group showed significant upregulation of β-site amyloid precursor protein cleaving enzyme-1 (BACE-1) gene expression (2.98 ± 0.65, 7.95 ± 1.34 fold change) and significantly higher levels of amyloid beta (Aβ1-42) (12.54 ± 1.17, 13.57 ± 0.67 ng/mg protein) than the control group (BACE-1; 1.00 ± 0.08, 1.00 ± 0.06 fold change, Aβ1-42; 2.48 ± 0.40, 2.63 ± 0.63 ng/mg protein) in the cortex and hippocampus, respectively. Group treated with DPL–NLCs showed non-significantly lower cortical BACE-1 expression (2.35 ± 0.15 fold change), significantly lower hippocampal BACE-1 expression (5.94 ± 0.88 fold change) and significantly lower cortical and hippocampal Aβ1-42 levels (7.19 ± 0.82, 9.14 ± 0.53 ng/mg protein) than the AD-like untreated group and that treated with DPL–Solution (BACE-1; 2.81 ± 0.17, 7.58 ± 0.63 fold change, Aβ1-42; 11.35 ± 0.68, 12.79 ± 1.04 ng/mg protein). Group treated with DPL/AST–Solution showed significantly lower cortical and hippocampal BACE-1 expression (1.96 ± 0.25, 5.32 ± 0.49 fold change) and Aβ1-42 levels (6.15 ± 0.49, 8.16 ± 0.42 ng/mg protein) than the untreated group and that treated with DPL–Solution, and there was no significant difference between groups treated with DPL/AST–Solution and DPL–NLCs in amyloidogenic parameters. Group treated with DPL/AST–NLCs showed significantly lower cortical and hippocampal BACE-1 expression (1.14 ± 0.29, 2.60 ± 0.22 fold change) and Aβ1-42 levels (2.62 ± 0.72, 3.90 ± 0.25 ng/mg protein) than the AD-like untreated rats and groups treated with DPL–NLCs, DPL/AST–Solution, or DPL–Solution in the cortex and hippocampus, and also DPL/AST–NLCs completely normalized the cortical BACE-1 expression, and the cortical and hippocampal Aβ1-42 levels.
Amyloid beta (Aβ1-42) peptides are cleavage products derived from amyloid precursor protein (APP). APP is cleaved via two major pathways. In the non-amyloidogenic pathway, APP is processed by α- and γ-secretases into easily removable fragments, while in the amyloidogenic pathway, it is cleaved by β- and γ-secretases into Aβ peptides. BACE-1 is the major β-secretase in the brain and its expression is upregulated in AD, resulting in increased production of Aβ1-42.84,85 Excessive production of Aβ1-42 in the brain produces senile plaques that are involved in AD pathogenesis. Aβ1-42 peptides increase phosphorylation of tau, production of pro-inflammatory cytokines and mitochondrial reactive oxygen species, and neuronal vulnerability to glutamate excitotoxicity, resulting in neuronal cell death. Aβ1-42 binds to nicotinic acetylcholine receptors that causes impairment of cholinergic system, memory loss and cognitive deficits.86,87
Astaxanthin downregulates the amyloidogenic BACE-1 and γ-secretase, and upregulates the non-amyloidogenic α-disintegrin and metalloproteinases (ADAM-10) that form α-secretase, which increases production of soluble APP-α, attenuates Aβ production, and enhances Aβ clearance.70,88 Chen et al78 indicated that astaxanthin reduced Aβ and p-tau. Han et al89 investigated that astaxanthin downregulated β-secretase, reduced Aβ1–42, inflammatory cytokines and oxidative stress, and ameliorated memory loss. Accumulation of Aβ oligomers decreases the expression of type-2 ryanodine receptors (RyR2) that are crucial for synaptic plasticity and increases production of mitochondrial reactive oxygen species (ROS), which leads to synaptotoxicity. Lobos et al90 reported that astaxanthin inhibited downregulation of RyR2 mRNA levels promoted by Aβ oligomers and mitochondrial ROS and protected nerve cells against their harmful effects. Previous studies have demonstrated that insulin resistance in the brain could lead to the accumulation of Aβ and increase the levels of mitochondrial ROS and pro-inflammatory cytokines.91 Rahman et al70 found that astaxanthin attenuated central insulin resistance indicators, reduced the levels of Aβ, AChE, inflammation and oxidative stress. Babalola et al92 reported that astaxanthin enhanced insulin sensitivity and improved Aβ degradation. Huang et al93 reported that astaxanthin upregulated Aβ-degrading enzymes, reduced Aβ levels, and activated mammalian target of rapamycin (mTOR), which is involved in the hippocampal-dependent memory functions. Alghazwi et al94 reported that astaxanthin inhibited Aβ1-42 toxicity and aggregation. In our previous study, the developed AST–NLCs significantly decreased BACE-1 gene expression and Aβ1-42 levels in AD-like rats.30
AChE has been shown to promote the Aβ aggregation. Donepezil reduced Aβ formation and deposition in the brain in previous studies by inhibiting AChE, decreasing β-secretase activity, and activating muscarinic ACh receptors that can activate the non-amyloidogenic processing of APP, suggesting that donepezil may exert a neuroprotective effect and slow disease progression.95 Ye et al96 indicated that donepezil decreased Aβ levels and attenuated Aβ-associated mitochondrial dysfunction in isolated mitochondria and AD-like mice. Dong et al97 reported the protective effects of donepezil against Aβ in AD-like mouse model. Takada-Takatori et al98 reported that donepezil stimulated the expression of sorting nexin protein-33 (SNX-33), which increases the non-amyloidogenic processing of APP and decreased Aβ. Donepezil significantly reduced Aβ-induced neurotoxicity in rat septal neurons.99 Zhang and Gordon100 reported that donepezil reduced the levels of Aβ and tau.
Glycogen synthase kinase-3β (GSK-3β) plays an important role in amyloidogenesis, as it increases BACE-1-mediated cleavage of APP and downregulates α-secretase activity, thereby increasing Aβ production. Interestingly, Aβ prevents inhibitory phosphorylation of GSK-3β and therefore increases its activity that leads to increased β-degradation pathway of APP and Aβ formation.101,102 Aβ1-42 contributes to tau hyperphosphorylation and formation of tau fibrils and neurofibrillary tangles, and tau is involved in Aβ-induced toxicity.103 GSK-3β regulates tau hyperphosphorylation involved in the formation of neurofibrillary tangles, and hyperphosphorylated tau activates GSK-3β.104 GSK-3β overexpression has been observed in AD, resulting in increased BACE-1-mediated cleavage of APP, Aβ production, Aβ-induced neurotoxicity, tau hyperphosphorylation, microglial activation, neuronal death, impaired adult hippocampal neurogenesis and maturation of newborn neurons, cholinergic dysfunction and synaptic loss, resulting in increased cognitive impairment.101,104 Protein phosphatase 2A (PP2A) dephosphorylates tau, and there is a balance between GSK-3β and PP2A actions to control tau phosphorylation. Decreased PP2A activity in AD by Aβ increases tau phosphorylation.105 Consistent with this, inhibition of GSK-3β activity has been shown to reduce Aβ and tau hyperphosphorylation.102 Koh et al106 reported that inhibition of GSK-3β reduced neurotoxicity induced by Aβ. Rahman et al70 and Wen et al107 demonstrated that astaxanthin attenuated GSK-3β activity. Noh et al108,109 reported that donepezil inhibited GSK-3 activity and enhanced PP2A activity. Yoshiyama et al110 reported that donepezil reduced tau and synaptic loss.
In the present study, AlCl3 induced the upregulation of BACE-1 gene expression in rats, which resulted in increased Aβ1-42 levels in the cortex and hippocampus. Treatment of rats with DPL–NLCs significantly decreased the hippocampal BACE-1 expression, and the cortical and hippocampal Aβ1-42 levels compared to the AD-like untreated group. Treatment with DPL/AST–NLCs significantly decreased the cortical and hippocampal BACE-1 expression and Aβ1-42 levels compared to DPL–NLCs and the AD-like untreated rats and completely normalized the cortical BACE-1 expression and the cortical and hippocampal Aβ1-42 levels. Treatment with DPL/AST–NLCs showed significantly higher anti-amyloidogenic activity than DPL–NLCs, which may be due to the anti-amyloidogenic activity of both astaxanthin and donepezil through downregulation of BACE-1 expression that leads to a significant decline in Aβ1-42 levels and may also be mediated by reducing GSK-3 activation.
Oxidative Stress Markers: Malondialdehyde and Reduced Glutathione
As illustrated in Figures 9 and 10, the AD-like untreated group showed significantly higher levels of malondialdehyde (MDA) (6.46 ± 0.57, 0.57 ± 0.07 nmol/g tissue) and significantly lower levels of reduced glutathione (GSH) (2.19 ± 0.33, 2.41 ± 0.23 nmol/mg protein) than the control group (MDA; 2.10 ± 0.21, 0.22 ± 0.04 nmol/g tissue, GSH; 4.60 ± 0.34, 6.64 ± 0.33 nmol/mg protein) in the cortical and hippocampal tissues, respectively. Group treated with DPL–NLCs showed significantly lower cortical and hippocampal MDA levels (4.50 ± 0.37, 0.43 ± 0.03 nmol/g tissue), non-significantly higher cortical GSH level (2.66 ± 0.21 nmol/mg protein) and significantly higher hippocampal GSH level (3.68 ± 0.27 nmol/mg protein) than the AD-like untreated group and that treated with DPL–Solution (MDA; 5.97 ± 0.64, 0.51 ± 0.04 nmol/g tissue, GSH; 2.41 ± 0.21, 2.74 ± 0.25 nmol/mg protein). Group treated with DPL/AST–Solution showed significantly lower MDA levels (3.98 ± 0.48, 0.41 ± 0.03 nmol/g tissue) and significantly higher GSH levels (3.78 ± 0.43, 3.96 ± 0.33 nmol/mg protein) than the untreated group and that treated with DPL–Solution in the cortex and hippocampus. No significant difference was observed between groups treated with DPL/AST–Solution and DPL–NLCs in the cortical and hippocampal MDA levels and the hippocampal GSH levels, whereas the cortical GSH level was significantly higher in DPL/AST–Solution-treated group than the DPL–NLC-treated group. Group treated with DPL/AST–NLCs showed significantly lower MDA levels (2.31 ± 0.44, 0.24 ± 0.02 nmol/g tissue) and significantly higher GSH levels (4.43 ± 0.51, 6.12 ± 0.42 nmol/mg protein) than the AD-like untreated rats and groups treated with DPL–NLCs, DPL/AST–Solution, or DPL–Solution in the cortex and hippocampus, and also treatment with DPL/AST–NLCs completely normalized the cortical and hippocampal MDA and GSH levels.
Oxidative stress refers to an imbalance between the free radical generation and the endogenous antioxidant protective capacity. This balance is disrupted in AD and oxidative stress accumulates. The brain is especially susceptible to the effects of oxidative stress because of its high oxygen demand and abundance of peroxidation-susceptible lipid cells.111,112 Oxidative stress disturbs the mitochondrial dynamics and accelerates mitochondrial dysfunction, resulting in the production of highly reactive oxygen/nitrogen species (ROS/RNS), which cause peroxidation of lipids and proteins, DNA damage, cell death, and increased oxidative stress in the brain.113 Lipid peroxides can accelerate free radical chain reactions, and they are mediators of neuronal inflammation. Extensive oxidative stress in the brain degenerates cholinergic neurons, decreases synaptic activity and cholinergic neurotransmission, increases Aβ accumulation and neuroinflammation, and contributes to reduced dendritic spine density, leading to neurodegeneration and cognitive impairment in AD.114,115
Antioxidants exert obvious neuroprotective effects in AD by alleviating oxidative stress and neuroinflammation in the brain.116 Pena-Bautista et al117 reported that antioxidant supplementation in AD especially in the early stages could prevent disease progression. Carotenoids are used in AD management as they have high antioxidant effect.118 In this regard, astaxanthin is a potent antioxidant carotenoid because of its unique molecular structure that contains a polyene chain (consists of 13 conjugated double bonds) at its center, which is responsible for the strong antioxidant activity of astaxanthin, in addition to hydroxyl and ketonic groups at its periphery that increase the polarity of astaxanthin and enhances its cell membrane penetration capacity, making it a strong antioxidant.119 Astaxanthin inhibits the production of mitochondrial ROS and lipid peroxidation, can maintain the integrity of the cell membrane and has the ability to reverse cellular damage caused by free radicals. These unique chemical properties give astaxanthin powerful antioxidant qualities; astaxanthin shows higher free radical inhibitory capacity than α-, β-carotene and α-tocopherol.120,121
Astaxanthin can suppress oxidative stress by its direct radical scavenging activity, in addition to an indirect mechanism through the activation of nuclear factor erythroid 2-related factor (Nrf2).122,123 Astaxanthin activates the phosphoinositide 3-kinase (PI3K)/protein kinase B (Akt) signaling pathway that facilitates the dissociation of Nrf2 from the chaperone Kelch-like ECH-associated protein 1 (Keap1) and increases its nuclear translocation. Nrf2 activates the antioxidant response element (ARE) pathway and upregulates the expression of Nrf2-regulated antioxidant enzymes, such as glutathione peroxidase-4, catalase, glutathione-S-transferase-α1, superoxide dismutase, and heme oxygenase-1, which can antagonize intracellular oxidative stress. In addition, activation of the PI3K/Akt/Nrf2 pathway by astaxanthin can enhance cell survival, protect neuronal cells from Aβ toxicity, and improve cholinergic neurotransmission, resulting in the improvement of cognitive impairment.124,125 GSK-3β is a downstream target of the PI3K/Akt signaling pathway. GSK-3β increases Nrf2 phosphorylation and degradation via β-transducing repeat-containing protein (β-TrCP).126,127 Astaxanthin activates the PI3K/Akt pathway that inhibits the GSK-3β/β-TrCP pathway and enhances the stability of Nrf2.107,122 Furthermore, astaxanthin is a potential mitochondrial regulator, and it can restore the mitochondrial membrane potential.128 Astaxanthin significantly reduced the mitochondrial ROS and malondialdehyde levels while increasing the endogenous antioxidant activity and glutathione levels.30,129,130
Several studies have shown that donepezil exhibited antioxidant activity. Umukoro et al131 examined the antioxidant effect of donepezil that significantly decreased the levels of MDA and ROS and increased glutathione level in the mouse brain. Atukeren et al132 reported that the antioxidant effect of donepezil in patients with AD was medicated through its AChE inhibitory activity. Munishamappa et al133 studied the in vitro antioxidant effect of donepezil, and they found that the percentage of inhibition of nitric oxide and free radicals by donepezil was increased with an increase in donepezil concentration. Jiang et al134 reported that donepezil attenuated inflammation and oxidative stress in mice. Obafemi et al135 demonstrated that donepezil and metformin combination decreased the levels of MDA and increased superoxide dismutase levels in rats. Keshk et al75 demonstrated that treatment with donepezil and losartan combination decreased MDA levels and increased glutathione-S-transferase levels. Rao et al136 reported that donepezil and resveratrol combination resulted in higher superoxide dismutase activity than resveratrol alone.
MDA is a lipid degradation product, and it is commonly used as a marker for the quantification of lipid peroxidation.115 GSH is used as a marker to measure the endogenous antioxidant activity. An increased GSH content may reduce lipid peroxidation and decrease the MDA levels.137 In the present study, oxidative stress was induced in rats by AlCl3, as indicated by the significantly higher MDA levels and significantly lower GSH levels in the cortex and hippocampus of the AD-like untreated rats than the normal group. Treatment with DPL–NLCs showed significantly lower cortical and hippocampal MDA levels and significantly higher hippocampal GSH levels than the untreated rats. Treatment with DPL/AST–NLCs showed significantly lower MDA levels and significantly higher GSH levels than DPL–NLCs and the AD-like untreated rats in the cortex and hippocampus, and also DPL/AST–NLCs completely normalized the cortical and hippocampal MDA and GSH levels. The significantly higher antioxidant potential of DPL/AST–NLCs compared to DPL–NLCs may be due to the antioxidant effect of both astaxanthin and donepezil as they reduced the oxidative stress, inhibited free radicals and enhanced the antioxidant activity of the brain.
Inflammation Marker: Nuclear Factor-Kappa B
As illustrated in Figure 11, the AD-like untreated rats showed significantly higher cortical and hippocampal levels of nuclear factor-kappa B (NF-κB) (55.78 ± 6.15, 55.36 ± 3.95 pg/mg protein) than the control group (7.93 ± 1.16, 6.36 ± 1.37 pg/mg protein). Group treated with DPL–NLCs showed significantly lower NF-κB levels (33.38 ± 2.02, 32.48 ± 2.63 pg/mg protein) than the AD-like untreated group and that treated with DPL–Solution (47.47 ± 3.50, 51.37 ± 3.30 pg/mg protein) in the cortex and hippocampus. Group treated with DPL/AST–Solution (NF-κB; 29.15 ± 3.33, 30.20 ± 2.31 pg/mg protein) showed similar results to DPL–NLCs, with no significant difference between them in the levels of NF-κB. Group treated with DPL/AST–NLCs showed significantly lower cortical and hippocampal NF-κB levels (11.30 ± 2.90, 14.68 ± 2.52 pg/mg protein) than the AD-like untreated rats and groups treated with DPL–NLCs, DPL/AST–Solution, or DPL–Solution in the cortex and hippocampus, and also treatment with DPL/AST–NLCs completely normalized the cortical NF-κB level.
Abnormal microglial and astrocytic activation induces neuroinflammation in AD.138 Lipopolysaccharides bind to Toll-like receptor-4 (TLR-4) and activate p38-mitogen activated protein kinases (MAPK-p38) and Myeloid differentiation primary response-88/nuclear factor-kappa B/signal transducer and activator of transcription-3 (MyD-88/NF-κB/STAT-3) signaling pathways that increases the expression and activity of cyclooxygenase-2 (COX-2), NADPH oxidase, and inducible nitric oxide synthase (iNOS), and increases the production of ROS, nitric oxide (NO), inflammatory chemokines, and pro-inflammatory cytokines, such as interleukin-1β (IL-1β), interleukin-6 (IL-6), and tumor necrosis factor-alpha (TNF-α), which trigger inflammatory responses and increase oxidative stress and microglial activation, resulting in neurological impairment.139,140 TNF-α increases the expression of iNOS and antagonizes the BDNF stimulatory signals, which causes neuronal excitotoxicity, alteration of synaptic plasticity, and increased dendritic spine damage, resulting in neuronal loss and memory impairment.141,142 Neuroinflammation induces mitochondrial dysfunction that also increases inflammation and oxidative stress.143,144 Like lipopolysaccharides, Aβ has been shown to bind to TLR-4 and activate the MAPK and NF-κB signaling pathways, activate microglia and astrocytes, and increase the levels of pro-inflammatory cytokines.145,146 Regulation of neuroinflammation and microglial activation is a critical target for treating AD.147 NF-κB is a used as a marker of inflammation, it is a prototypical mediator of inflammation and its blocking reduces inflammatory responses.148
Astaxanthin has strong anti-inflammatory action that has been reported in different studies.149,150 Astaxanthin reduces the production of mitochondrial ROS and Aβ, inhibits NF-κB signaling pathway and activation of microglia and astrocytes, and suppresses the levels of iNOS, COX-2, and pro-inflammatory cytokines.151,152 Choi et al153 reported that astaxanthin inhibited the synthesis of NO and inflammatory mediators. Astaxanthin suppressed the activation of NLRP-3 (NOD-, LRR-, and pyrin domain-containing protein-3) inflammasome.154 Astaxanthin can reduce inflammation by activation of the PI3K/AKT/Nrf2 pathway that increases heme oxygenase-1, which catalyzes heme degradation into carbon monoxide that inhibits NF-κB.155,156 Han et al89 reported that astaxanthin reduced neuroinflammation through inactivation of STAT-3. Aβ activates calcineurin that activates nuclear factor of activated T cells (NFAT), which produces inflammation, dendritic spine loss and synaptic plasticity deficits.157,158 Lobos et al90 reported that astaxanthin inhibited the calcineurin/NFAT pathway, neuroinflammation and improved synaptic plasticity deficits. ROS/RNS increase the BBB permeability to peripheral inflammatory cells causing brain inflammation and neuronal injury.159,160 Astaxanthin inhibits peripheral inflammation, protects and maintains the BBB integrity, modulates neuroinflammation and alleviates oxidative stress.149 ROS accumulation during oxidative stress is a crucial trigger for microglial polarization.144 Zhou et al161 reported that astaxanthin suppressed neuroinflammation by inhibiting microglial M1 activation and increased microglial M2 polarization. In our previous study, the developed AST–NLCs significantly suppressed NF-κB levels and reduced neuroinflammation in AD-like rats.30
Donepezil showed anti-inflammatory activity in previous studies. Kim et al162 and Guo et al163 reported that donepezil inhibited microglial activation. Donepezil can inhibit the NF-kB and MAPK pathways and reduce the production of pro-inflammatory mediators.164 Kim et al165 reported that donepezil suppressed inflammation by inhibiting microglia activation, MAPK, NF-κB/STAT-3, NLRP-3 inflammasome, reducing mitochondrial dysfunction, the production of ROS and pro-inflammatory cytokines and improved memory impairment. Haraguchi et al166 indicated that donepezil activated the PI3K/Akt pathway that suppressed the NF-κB activity and Ca2+ elevation induced by TNF-α and decreased inflammation. Donepezil suppressed the production of NO that is involved in Aβ nitration and aggregation.167 There is an evidence of a correlation between the cholinergic and anti-inflammatory pathways.168,169 Donepezil increased the synaptic ACh levels and activated and upregulated α7-nicotinic ACh receptors (α7-nAChRs) that reduced inflammation through the cholinergic anti-inflammatory pathway.170,171 Vagus nerve stimulation releases ACh, leading to dephosphorylation of NF-kB and STAT3 and suppression of the release of pro-inflammatory cytokines.172 De Simone et al173 reported that ACh activated α7-nAChRs, inhibited microglial activation and TNF-α release. Donepezil inhibited the NLRP-3 inflammasome by stimulating α7-nAChRs and the PI3K/AKT pathway and suppressing the NF-κB/STAT-3 pathway related to NLRP-3 inflammasome formation.174,175 Arikawa et al176 demonstrated that donepezil decreased inflammation through inhibition of NF-κB translocation, which was independent of its AChE inhibitory action. GSK-3β increases neuro-inflammation.101 Activation of the PI3K/Akt pathway attenuates GSK-3β and reduces inflammation. Rahman et al70 demonstrated the GSK-3β inhibitory effect of astaxanthin and Noh et al108 reported that donepezil suppressed the GSK-3β activity.
In the present study, neuronal inflammation was induced in rats by AlCl3, as indicated by the significantly higher NF-κB levels in cortex and hippocampus of the AD-like untreated rats than the normal group. Treatment with DPL–NLCs showed significantly lower NF-κB levels than the untreated rats in the cortex and hippocampus. Treatment with DPL/AST–NLCs significantly decreased the NF-κB levels in the cortex and hippocampus compared to DPL–NLCs, and completely normalized the cortical NF-κB level. The significantly higher anti-inflammatory activity of DPL/AST–NLCs compared to DPL–NLCs may be due to the anti-inflammatory action of both astaxanthin and donepezil that mediated through inhibition of NF-κB, increasing ACh levels, and activation of nAChRs.
Apoptosis Marker: Caspase-3
As illustrated in Figure 12, the AD-like untreated rats showed significantly higher cortical and hippocampal caspase-3 activity (18.11 ± 1.15, 19.50 ± 1.24 U/mg protein) than the control group (2.28 ± 0.13, 2.60 ± 0.46 U/mg protein). Group treated with DPL–NLCs showed significantly lower caspase-3 activity (11.78 ± 0.95, 11.86 ± 0.67 U/mg protein) than the untreated group and that treated with DPL–Solution (16.0 ± 0.96, 17.99 ± 1.23 U/mg protein) in the cortex and hippocampus, respectively. Group treated with DPL/AST–Solution (11.06 ± 0.92, 11.58 ± 0.77 U/mg protein) showed similar results to DPL–NLCs, with no significant difference between them in the activity of caspase-3. Group treated with DPL/AST–NLCs showed significantly lower cortical and hippocampal caspase-3 activity (3.75 ± 0.22, 6.55 ± 0.78 U/mg protein) than the AD-like untreated rats and groups treated with DPL–NLCs, DPL/AST–Solution, or DPL–Solution, and also treatment with DPL/AST–NLCs completely normalized the cortical caspase-3 activity.
Accumulation of Aβ, oxidative stress, and neuroinflammatory reactions induce neuronal loss and apoptosis.177 In AD, the anti-apoptotic cytokines, such as Bcl-2 (B-cell lymphoma-2) and Bcl-xL (Bcl-extra large), were decreased, while the pro-apoptotic cytokines, such as Bad (Bcl-2-associated death promoter) and Bax (Bcl-2-associated X), cytochrome-c (Cyt-c) release and the activity of caspase-3 enzyme were increased. Bad and Bax accelerate apoptosis by stimulating the release of Cyt-c from the mitochondria and activating caspases-3/9, whereas Bcl-2 and Bcl-xL inhibit apoptosis by suppressing the release of Cyt-c and decreasing the activation of caspases-3/9. Excessive mitochondrial dysfunction initiates the apoptotic cascade by promoting Bax expression.178,179 GSK-3β stimulates apoptosis by increasing the pro-apoptotic cytokines and decreasing the anti-apoptotic cytokines. Stimulation of the neuroprotective PI3K/Akt pathway inhibits GSK-3β, decreases Bad and Bax, and increases Bcl-2 and Bcl-xL.101
Astaxanthin acts as a shield for cells from inflammatory responses and oxidative stress and reduces neuronal apoptosis due to its ability to stimulate the PI3K/Akt/Nrf2/ARE pathway, inhibit NF-κB and GSK-3β, suppress the production of ROS and pro-inflammatory cytokines, decrease the expression of Bad, Bax, and caspases-3/8/9, inhibit Cyt-c secretion, and increase Bcl-2 level, resulting in suppression of apoptosis and stimulating proliferation of the cells.107,180,181 In our previous study, the developed AST–NLCs significantly reduced caspase-3 activity and suppressed apoptosis in AD-like rats.30
In AD, cholinergic depletion increases the activity of caspase-3, which leads to dendritic spine loss, alteration in synaptic plasticity, and impaired working memory.182 Previous studies have reported that donepezil reduced caspase-3 activity following cholinergic depletion.183 Donepezil induces functional upregulation and increases the expression of nicotinic acetylcholine receptors (nAChRs), increases the level and binding of ACh with α7-nAChRs, which activate the anti-apoptotic PI3K/Akt/Bcl-2 pathway.184,185 Elbaz et al186 demonstrated that donepezil decreased the expression of caspase-3 and the Bax/Bcl-2 ratio and inhibited lipid peroxidation and inflammation.
Caspase-3 is used as a marker for apoptosis. In the present study, the induced neuronal degeneration and apoptosis in rats by AlCl3 was approved by the significantly higher cortical and hippocampal caspase-3 activity in the AD-like untreated rats than in the normal group. Treatment with DPL–NLCs showed significantly lower caspase-3 activity than the untreated group in the cortex and hippocampus. Treatment with DPL/AST–NLCs significantly decreased the cortical and hippocampal caspase-3 activity compared to DPL–NLCs, and completely normalized the cortical caspase-3 activity. The significantly higher anti-apoptotic effect of DPL/AST–NLCs compared to DPL–NLCs may be due to the caspase-3 inhibitory effect of both astaxanthin and donepezil, their neuroprotective effects, and the increased levels of ACh, which stimulate the anti-apoptotic α7-nAChR/PI3K/Akt/Bcl-2 pathway.
Several pathways are involved in neurodegeneration in AD, including the formation of Aβ and hyperphosphorylated tau, glutamate excitotoxicity, inflammation, oxidative stress, and cholinergic depletion.3 Astaxanthin has potent neuroprotective properties due to its ability to reduce the production of Aβ1-42, mitochondrial ROS, and pro-inflammatory cytokines, restore cholinergic neurotransmission, increase the BDNF levels and nerve cell regeneration, inhibit NF-κB, AChE, GSK-3β, and caspase-3 activity, activate the neuroprotective PI3K/Akt/Nrf2/ARE pathway, maintain the BBB integrity, and decrease dendritic spine loss, the expression of N-methyl-D-aspartate (NMDA) receptors, glutamate excitotoxicity, and peripheral inflammation, resulting in marked improvement in behavioral and cognitive deficits in AD.22,187,188
Previous studies have shown that donepezil might have disease modification or neuroprotective potential against Aβ- and glutamate-induced neurotoxicity, neuroinflammation, and tau pathology, especially in early stages of AD.19,189 In AD, Aβ1-42 peptides decrease the levels of ACh and strongly bind to α4- and α7-nAChRs causing neurotoxicity.190 Aβ1-42 increases the release of glutamate and decreases γ-aminobutyric acid (GABA) release, which causes excitation-inhibition imbalance and induces glutamate excitotoxicity.191,192 Donepezil activates and upregulates α4- and α7-nAChRs that provide neuroprotection against Aβ- and glutamate-induced neurotoxicity.193 α4- and α7-nAChRs activate the neuroprotective PI3K/Akt pathway, which plays a central role in neuronal survival, via activation of Janus kinase 2.194,195 Donepezil decreases glutamatergic excitotoxicity, the activation of NMDA receptors and apoptosis.196,197 Noh et al108 reported that donepezil inhibited GSK-3β, stimulated PP2A, activated nAChRs and the PI3K/Akt pathway. Yoshiyama et al110 reported that donepezil treatment ameliorated tau pathology, decreased tau insolubility and phosphorylation, reduced neuroinflammation, synaptic loss, and neuronal loss. Sigma-1 (σ-1) receptors are found in many areas of the brain, and they have anti-apoptotic and neuroprotective roles.198 Solntseva et al199 and Meunier et al200 reported that donepezil had high binding capacity to σ-1 receptors and provided neuroprotection against Aβ-toxicity. Donepezil upregulated the BDNF levels in the brain, which increases synaptic plasticity, and it has neuroprotective functions.201,202 Nakano et al203 reported that donepezil improved cerebral blood flow and enhanced the hippocampal function, which has been associated with behavioral and cognitive improvement. Donepezil decreased the release of lactate dehydrogenase, which is a marker of neuronal damage.204 Donepezil has been shown to slow the progression of hippocampal atrophy, which is a reliable marker of disease progression.205 Zimmermann et al206 reported that donepezil increased the ratio of the neuroprotective AChE-R (AChE read-through isoform) to the neurodegenerative AChE-S (AChE-synaptic isoform), contributing to the neuroprotective effects of donepezil. Ongnok et al18 reported that donepezil decreased the levels of MDA, ROS, Aβ, and pro-inflammatory mediators in the brain, ameliorated brain apoptosis, and reduced dendritic spine loss.
Histopathological Examination
As illustrated in Figures 13 and 14, histopathological assessment of rats using hematoxylin and eosin (H and E) staining showed that the control rats had a normal histoarchitecture of the cortical and hippocampal tissues. Distorted cellular morphology and various histopathological alterations (misaligned degenerated, shrunken darkly stained, pyknotic and necrotic neurons with neuronophagia and satellitosis, more glial cells and pericellular spaces) were observed in the cortex and hippocampus of the AD-like untreated group, which was consistent with the studies in the literature and was consistent with AlCl3-induced neurotoxicity.47,207 Treatment of rats with DPL/AST–NLCs showed a marked improvement in the cortical and hippocampal histopathological parameters compared to the AD-like untreated rats and groups treated with DPL–NLCs or DPL/AST–Solution and almost normalized the histopathological changes in the cortex and hippocampus. This may be due to the higher anti-amyloidogenic, anti-inflammatory, antioxidant, anti-acetylcholinesterase, anti-apoptotic, and neuroprotective effects of both astaxanthin and donepezil, especially in NLCs, as observed in the biochemical and behavioral testing. Wu et al83 reported that astaxanthin significantly improved the histopathological changes in the brain of aging rats. Pattanashetti et al74 reported that pretreatment of rats with a combination of donepezil and quercetin improved the brain histopathology and had higher neuroprotective effects than the individual therapy. Keshk et al75 demonstrated that pretreatment with a combination of donepezil and losartan improved the histopathological changes in rats and provided a better anti-amnestic effect than either treatment alone. The combination of donepezil with other neuroprotective agents has beneficial therapeutic effects. Therefore, donepezil was combined with astaxanthin in the present study.
Correlation Study
As illustrated in Table 5, Pearson correlation analysis showed that the markers of the amyloidogenic pathway (BACE-1 expression and Aβ1-42 content) were negatively correlated with GSH and ACh and positively correlated with caspase-3, MDA, AChE, and NF-κB in the cortex and hippocampus. ACh levels were negatively correlated with NF-κB, caspase-3, MDA, and AChE, and positively correlated with GSH. GSH showed a similar correlation pattern to ACh, whereas NF-κB, caspase-3, MDA, and AChE showed opposite correlation patterns. The correlation data confirmed a possible link between the cholinergic and inflammatory pathways, as there was a positive correlation between NF-kB and AChE and a negative correlation between NF-kB and ACh. BACE-1 and Aβ1-42 were negatively correlated with ACh and positively correlated with AChE, confirming the link between the amyloidogenic pathway progression and disturbance in the cholinergic pathway. There was a positive correlation between the amyloidogenic pathway progression and apoptotic cell death, as BACE-1 and Aβ1-42 were positively correlated with caspase-3. NF-κB was positively correlated with MDA and negatively correlated with GSH, confirming the link between the induction of oxidative stress and neuroinflammation.
 |
Table 5 Pearson Correlation Study Between the Different Biochemical Parameters in the AD-Like Untreated, DPL–Sol, DPL/AST–Sol, Free NLCs, DPL–NLCs, and DPL/AST–NLCs Groups (n = 36) in the Cortex and Hippocampus |
Alzheimer’s disease is a devastating progressive neurodegenerative disorder.3 Donepezil is a potent selective AChEI that improves cognitive deficits in AD and might have a disease-modifying effect.18,19 Astaxanthin is a natural potent antioxidant carotenoid with anti-amyloidogenic, anti-inflammatory, anti-acetylcholinesterase, anti-apoptotic, and neuroprotective effects that have been proved in several studies including our previous study, and it has the ability to cross the BBB.20,30 Oral administration of donepezil produces gastrointestinal adverse events, and in addition it has poor ability to penetrate the BBB as it is a hydrophilic drug, and it suffers from hepatic first-pass metabolism.8,16 Astaxanthin shows low oral bioavailability due to its high lipophilicity.27 The intranasal (IN) route was used in the present study as an alternative non-invasive approach to restrict the limitations associated with oral administration of donepezil and astaxanthin for better brain targeting of both drugs. The IN route allows direct delivery of drugs to the brain, which has been proven in several previous studies, avoiding the BBB and hepatic first-pass metabolism, minimizing systemic exposure, restricting the side effects associated with oral administration of donepezil, avoiding the problem of poor oral bioavailability of astaxanthin, and enhancing drug distribution, absorption, and bioavailability in the brain.12,208 However, the IN drug delivery is affected by several factors, such as enzymatic drug degradation, mucociliary clearance, short residence time, and small-administered volume.209
In the present study, nanostructured lipid carriers (NLCs) were used to overcome the IN limitations and improve the nose to brain drug delivery, as they decrease the rapid removal of drugs from the nose due to mucociliary clearance, increase the nasal residence time, protect the encapsulated drugs from enzymatic metabolism in the nasal cavity, and improve the drug delivery to the brain and therapeutic outcomes in AD.210 NLCs are safe, biocompatible and biodegradable lipid-based nanosystems, and they are rapidly absorbed by the brain due to their natural lipid composition and flexible nano-sized particles. NLCs can provide sustained release of drugs and site-specific targeting ability. The lipid matrix in NLCs contains solid and liquid lipids, the incorporation of liquid lipid results in an imperfect crystal structure and therefore provides more room for drug accommodation, allowing higher drug loading efficiency of NLCs, decreased drug expulsion during storage, and increased the NLC stability.211,212 Furthermore, NLCs can accommodate both hydrophilic and lipophilic drugs, and dual drug-loaded NLCs have been successfully prepared in previous studies.33,34,213 In previous studies, NLCs have been shown to successfully deliver hydrophilic donepezil16 or lipophilic astaxanthin29 individually to the brain through the IN route, and they showed sustained drug release and high encapsulation efficiency and stability. In our previous study, astaxanthin was loaded alone in NLCs for IN delivery to the brain, and we examined the different neuroprotective effects of astaxanthin in AD-like model.30 In the present study, we formulated NLCs co-loaded with donepezil and astaxanthin (DPL/AST–NLCs), for better management of AD, and we examined their in vivo efficacy in AD-like rats 30 days after daily IN administration compared to donepezil alone in NLCs (DPL–NLCs). Dual-drug targeting was significantly more beneficial than single-drug targeting. The results of the behavioral MWM test and biochemical parameters showed that DPL/AST–NLCs had significantly better in vivo effects than DPL–NLCs and the free drug solution (DPL/AST–Solution). NLCs improve the nasal permeability of drugs more than the free drug solution, reduce mucociliary clearance, and protect drugs from enzymatic degradation within the nasal cavity, they are rapidly absorbed by the brain, and, therefore, they increase the bioavailability and therapeutic efficacy of drugs in the brain more than the free drug solution.212,214
In the present study, AD was induced in rats by oral administration of AlCl3 that induced disturbance in the cholinergic pathway, neuro-inflammation, activation of the amyloidogenic pathway, oxidative stress, and apoptosis, which led to cortical and hippocampal histopathological alterations, memory and cognitive deficits as observed in the MWM test.47 As observed in the biochemical testing, the AD-like untreated rats showed significantly higher levels of Aβ1-42, BACE-1, caspase-3, AChE, MDA, and NF-κB, and significantly lower levels of GSH and ACh in the cortex and hippocampus than the control rats. Treatment of the AD-like rats with DPL–NLCs or DPL/AST–NLCs significantly improved these changes, and combined treatment with donepezil and astaxanthin in NLCs (DPL/AST–NLCs) showed significantly better in vivo effects than the individual therapy with DPL–NLCs. The results of the pharmacodynamics study revealed that treatment of the AD-like rats with DPL/AST–NLCs showed significantly higher anti-amyloidogenic, antioxidant, anti-anti-inflammatory, anti-apoptotic, and AChE inhibitory effects than DPL–NLCs. Treatment with DPL–NLCs showed significantly lower cortical and hippocampal levels of AChE, Aβ1-42, MDA, NF-κB, and caspase-3, significantly lower hippocampal BACE-1 expression, significantly higher cortical and hippocampal ACh levels, and significantly higher hippocampal GSH levels than the AD-like untreated rats. Treatment with DPL/AST–NLCs showed significantly higher levels of ACh and GSH, and significantly lower levels of Aβ1-42, BACE-1, AChE, MDA, NF-κB, and caspase-3 in the cortex and hippocampus than DPL–NLCs and the AD-like untreated group, and also completely normalized the cortical and hippocampal levels of ACh, AChE, Aβ1-42, GSH and MDA, the cortical BACE-1 expression, NF-κB, and caspase-3 activity. The superiority of co-administration of donepezil and astaxanthin in NLCs for attenuation of AlCl3-induced AD could be explained by higher neuroprotective actions of both astaxanthin and donepezil in NLCs that may be mediated through the activation of the cholinergic pathway, suppression of inflammation, oxidative stress, amyloidogenesis, and apoptosis. The results of the biochemical testing were related to the behavioral and histopathological results. Treatment of rats with DPL/AST–NLCs showed significantly shorter escape latency time and significantly higher percentage of retention time than DPL–NLCs, and also completely normalized the escape latency time in the MWM test and the percentage of retention time in the probe trial test. Treatment with DPL/AST–NLCs resulted in a marked improvement in the histopathology of the rat cortex and hippocampus compared to DPL–NLCs. This study demonstrated a marked neuroprotective effect of the combination of donepezil and astaxanthin in NLCs after their daily IN administration for 30 days in AD-like rats, and suggests that the nose-to-brain delivery of the developed DPL/AST–NLCs is a promising strategy for the management of AD.
Conclusion
The use of the intranasal route together with nanostructured lipid carriers is advantageous for targeting drugs to the brain, and it is a promising approach to manage Alzheimer’s disease. These lipid-based nanosystems encompass a great potential to improve the bioavailability of drugs used to treat AD using the intranasal administration that allows drugs to be transported directly from the nasal cavity to the brain, and they have demonstrated higher effectiveness upon nose-to-brain delivery than the intranasal administration of solutions of free drugs. In this study, NLCs co-loaded with donepezil and astaxanthin were successfully prepared, and they showed a small uniform particle size, optimum entrapment efficiency, and in vitro sustained release pattern of both donepezil and astaxanthin for up to 24 h, and they were stable for six months at 4–8 ± 2°C. The intranasal treatment of the AD-like rats with the developed DPL/AST–NLCs for 30 days significantly decreased the production of Aβ1-42, inflammation, oxidative stress, and apoptosis, and significantly increased the cholinergic neurotransmission, resulting in marked improvement in the histopathological parameters in the rat cortex and hippocampus, and significantly attenuated cognitive impairment induced in rats by AlCl3, as compared to the AD-like untreated rats and that treated with either donepezil-NLCs or solution of free drugs. In this study, we observed the beneficial role of the developed dual drug-loaded NLCs in several targets of AD pathology. The future directions for the potential clinical use of the developed DPL/AST–NLCs include more extensive in vivo studies of animal models of AD, and translation to the clinic in order to play an essential role in the future management of AD and improve patients’ quality of life.
Acknowledgments
This study did not receive any specific grant from funding agencies in the public, commercial, or not-for-profit sectors.
Disclosure
The authors report no conflicts of interest in this study.
References
1. Oliyaei N, Moosavi-Nasab M, Tanideh N, Iraji A. Multiple roles of fucoxanthin and astaxanthin against Alzheimer’s disease: their pharmacological potential and therapeutic insights. Brain Res Bull. 2023;193:11–21. doi:10.1016/j.brainresbull.2022.11.018
2. Atri A. Current and Future Treatments in Alzheimer’s Disease. Semin Neurol. 2019;39(2):227–240. doi:10.1055/s-0039-1678581
3. Sharma P, Srivastava P, Seth A, Tripathi PN, Banerjee AG, Shrivastava SK. Comprehensive review of mechanisms of pathogenesis involved in Alzheimer’s disease and potential therapeutic strategies. Prog Neurobiol. 2019;174:53–89. doi:10.1016/j.pneurobio.2018.12.006
4. Bekdash RA. The Cholinergic System, the Adrenergic System and the Neuropathology of Alzheimer’s Disease. Int J Mol Sci. 2021;22(3). doi:10.3390/ijms22031273
5. Sharma P, Tripathi MK, Shrivastava SK. Cholinesterase as a Target for Drug Development in Alzheimer’s Disease. Methods Mol Biol. 2020;2089:257–286. doi:10.1007/978-1-0716-0163-1_18
6. Walczak-Nowicka LJ, Herbet M. Acetylcholinesterase Inhibitors in the Treatment of Neurodegenerative Diseases and the Role of Acetylcholinesterase in their Pathogenesis. Int J Mol Sci. 2021;22(17). doi:10.3390/ijms22179290
7. Li DD, Zhang YH, Zhang W, Zhao P. Meta-Analysis of Randomized Controlled Trials on the Efficacy and Safety of Donepezil, Galantamine, Rivastigmine, and Memantine for the Treatment of Alzheimer’s Disease. Front Neurosci. 2019;13:472. doi:10.3389/fnins.2019.00472
8. Sutthapitaksakul L, Dass CR, Sriamornsak P. Donepezil—an updated review of challenges in dosage form design. J Drug Deliv Sci Technol. 2021;63:102549. doi:10.1016/j.jddst.2021.102549
9. Birks JS, Harvey RJ. Donepezil for dementia due to Alzheimer’s disease. Cochrane Database Syst Rev. 2018;6(6):Cd001190. doi:10.1002/14651858.CD001190.pub3
10. Li Q, He S, Chen Y, Feng F, Qu W, Sun H. Donepezil-based multi-functional cholinesterase inhibitors for treatment of Alzheimer’s disease. Eur J Med Chem. 2018;158:463–477. doi:10.1016/j.ejmech.2018.09.031
11. Deardorff WJ, Grossberg GT. A fixed-dose combination of memantine extended-release and donepezil in the treatment of moderate-to-severe Alzheimer’s disease. Drug Des Devel Ther. 2016;10:3267–3279. doi:10.2147/dddt.s86463
12. Alexander A, Agrawal M, Chougule M, Saraf S. Nose-to-brain drug delivery: an alternative approach for effective brain drug targeting. In: Shegokar R, editor. Nanopharmaceuticals. London: Elsevier; 2020:175–2000.
13. Kaur A, Nigam K, Bhatnagar I, et al. Treatment of Alzheimer’s diseases using donepezil nanoemulsion: an intranasal approach. Drug Deliv Transl Res. 2020;10(6):1862–1875. doi:10.1007/s13346-020-00754-z
14. Yasir M, Sara UVS, Chauhan I, et al. Solid lipid nanoparticles for nose to brain delivery of donepezil: formulation, optimization by Box–Behnken design, in vitro and in vivo evaluation. Artific Cells Nanomed Biotechnol. 2018;46(8):1838–1851. doi:10.1080/21691401.2017.1394872
15. Al Asmari AK, Ullah Z, Tariq M, Fatani A. Preparation, characterization, and in vivo evaluation of intranasally administered liposomal formulation of donepezil. Drug Des Devel Ther. 2016;10:205–215. doi:10.2147/dddt.s93937
16. Butani S. Fabrication of an ion-sensitive in situ gel loaded with nanostructured lipid carrier for nose to brain delivery of donepezil. Asian J Pharmaceut. 2018;12(4):293–302. doi:10.22377/ajp.v12i04.2838
17. Espinoza LC, Silva-Abreu M, Clares B, et al. Formulation Strategies to Improve Nose-to-Brain Delivery of Donepezil. Pharmaceutics. 2019;11(2). doi:10.3390/pharmaceutics11020064
18. Ongnok B, Khuanjing T, Chunchai T, et al. Donepezil provides neuroprotective effects against brain injury and Alzheimer’s pathology under conditions of cardiac ischemia/reperfusion injury. Biochim Biophys Acta Mol Basis Dis. 2021;1867(1):165975. doi:10.1016/j.bbadis.2020.165975
19. Kim SH, Kandiah N, Hsu JL, Suthisisang C, Udommongkol C, Dash A. Beyond symptomatic effects: potential of donepezil as a neuroprotective agent and disease modifier in Alzheimer’s disease. Br J Pharmacol. 2017;174(23):4224–4232. doi:10.1111/bph.14030
20. Bahbah EI, Ghozy S, Attia MS, et al. Molecular Mechanisms of Astaxanthin as a Potential Neurotherapeutic Agent. Mar Drugs. 2021;19(4):201. doi:10.3390/md19040201
21. Hongo N, Takamura Y, Nishimaru H, et al. Astaxanthin Ameliorated Parvalbumin-Positive Neuron Deficits and Alzheimer’s Disease-Related Pathological Progression in the Hippocampus of App(NL-G-F/NL-G-F) Mice. Front Pharmacol. 2020;11:307. doi:10.3389/fphar.2020.00307
22. Fakhri S, Aneva IY, Farzaei MH, Sobarzo-Sánchez E. The Neuroprotective Effects of Astaxanthin: therapeutic Targets and Clinical Perspective. Molecules. 2019;24(14). doi:10.3390/molecules24142640
23. Régnier P, Bastias J, Rodriguez-Ruiz V, et al. Astaxanthin from Haematococcus pluvialis Prevents Oxidative Stress on Human Endothelial Cells without Toxicity. Mar Drugs. 2015;13(5):2857–2874. doi:10.3390/md13052857
24. Liu X, Luo Q, Rakariyatham K, et al. Antioxidation and anti-ageing activities of different stereoisomeric astaxanthin in vitro and in vivo. J Funct Food. 2016;25:50–61. doi:10.1016/j.jff.2016.05.009
25. Turck D, Castenmiller J, de Henauw S, et al. Safety of astaxanthin for its use as a novel food in food supplements. EFSA j. 2020;18(2):e05993. doi:10.2903/j.efsa.2020.5993
26. Fakhri S, Abbaszadeh F, Dargahi L, Jorjani M. Astaxanthin: a mechanistic review on its biological activities and health benefits. Pharmacol Res. 2018;136:1–20. doi:10.1016/j.phrs.2018.08.012
27. Mercke Odeberg J, Lignell A, Pettersson A, Höglund P. Oral bioavailability of the antioxidant astaxanthin in humans is enhanced by incorporation of lipid based formulations. Eur J Pharm Sci. 2003;19(4):299–304. doi:10.1016/s0928-0987(03)00135-0
28. Bhatt PC, Srivastava P, Pandey P, Khan W, Panda BP. Nose to brain delivery of astaxanthin-loaded solid lipid nanoparticles: fabrication, radio labeling, optimization and biological studies. RSC Adv. 2016;6(12):10001–10010. doi:10.1039/C5RA19113K
29. Gautam D, Singh S, Maurya P, Singh M, Kushwaha S, Saraf SA. Appraisal of Nano-Lipidic Astaxanthin cum Thermoreversible Gel and its Efficacy in Haloperidol Induced Parkinsonism. Curr Drug Deliv. 2021;18(10):1550–1562. doi:10.2174/1567201818666210510173524
30. Shehata MK, Ismail AA, Kamel MA. Nose to Brain Delivery of Astaxanthin-Loaded Nanostructured Lipid Carriers in Rat Model of Alzheimer’s Disease: preparation, in vitro and in vivo Evaluation. Int J Nanomedicine. 2023;18:1631–1658. doi:10.2147/ijn.s402447
31. Cunha S, Costa CP, Loureiro JA, et al. Double Optimization of Rivastigmine-Loaded Nanostructured Lipid Carriers (NLC) for Nose-to-Brain Delivery Using the Quality by Design (QbD) Approach: formulation Variables and Instrumental Parameters. Pharmaceutics. 2020;12(7):599. doi:10.3390/pharmaceutics12070599
32. Costa CP, Moreira JN, Sousa Lobo JM, Silva AC. Intranasal delivery of nanostructured lipid carriers, solid lipid nanoparticles and nanoemulsions: a current overview of in vivo studies. Acta Pharm Sin B. 2021;11(4):925–940. doi:10.1016/j.apsb.2021.02.012
33. Sood S, Jain K, Gowthamarajan K. Curcumin-donepezil–loaded nanostructured lipid carriers for intranasal delivery in an Alzheimer’s disease model. Alzheimers Dement. 2013;9(4):299. doi:10.1016/j.jalz.2013.05.609
34. Lin YK, Huang ZR, Zhuo RZ, Fang JY. Combination of calcipotriol and methotrexate in nanostructured lipid carriers for topical delivery. Int J Nanomedicine. 2010;5:117–128. doi:10.2147/ijn.s9155
35. Li X, Jia X, Niu H. Nanostructured lipid carriers co-delivering lapachone and doxorubicin for overcoming multidrug resistance in breast cancer therapy. Int J Nanomedicine. 2018;13:4107–4119. doi:10.2147/ijn.s163929
36. Zhao X, Chen Q, Liu W, et al. Codelivery of doxorubicin and curcumin with lipid nanoparticles results in improved efficacy of chemotherapy in liver cancer. Int J Nanomedicine. 2015;10:257–270. doi:10.2147/ijn.s73322
37. Youssef AAA, Dudhipala N, Majumdar S. Dual Drug Loaded Lipid Nanocarrier Formulations for Topical Ocular Applications. Int J Nanomedicine. 2022;17:2283–2299. doi:10.2147/ijn.s360740
38. Uner M. Preparation, characterization and physico-chemical properties of solid lipid nanoparticles (SLN) and nanostructured lipid carriers (NLC): their benefits as colloidal drug carrier systems. Pharmazie. 2006;61(5):375–386.
39. Jores K, Mehnert W, Drechsler M, Bunjes H, Johann C, Mäder K. Investigations on the structure of solid lipid nanoparticles (SLN) and oil-loaded solid lipid nanoparticles by photon correlation spectroscopy, field-flow fractionation and transmission electron microscopy. J Control Release. 2004;95(2):217–227. doi:10.1016/j.jconrel.2003.11.012
40. Yasir M, Sara U. Development of UV spectrophotometric method for the analysis of acetylcholinesterase inhibitor. Int J Pharm Pharm Sci. 2014;6(9):128–131.
41. Sun W, Xing L, Leng K, et al. Determination of astaxanthin in Antarctic krill and its products by high performance liquid chromatography. J Food Safety Quality. 2017;8(4):1248–1253.
42. Tamjidi F, Shahedi M, Varshosaz J, Nasirpour A. Design and characterization of astaxanthin-loaded nanostructured lipid carriers. Innov Food Sci Emerg Technol. 2014;26:366–374. doi:10.1016/j.ifset.2014.06.012
43. Bunjes H, Unruh T. Characterization of lipid nanoparticles by differential scanning calorimetry, X-ray and neutron scattering. Adv Drug Deliv Rev. 2007;59(6):379–402. doi:10.1016/j.addr.2007.04.013
44. Yasir M, Sara UV. Solid lipid nanoparticles for nose to brain delivery of haloperidol: in vitro drug release and pharmacokinetics evaluation. Acta Pharm Sin B. 2014;4(6):454–463. doi:10.1016/j.apsb.2014.10.005
45. Korsmeyer RW, Gurny R, Doelker E, Buri P, Peppas NA. Mechanisms of solute release from porous hydrophilic polymers. Int J Pharm. 1983;15(1):25–35. doi:10.1016/0378-5173(83)90064-9
46. Makoni PA, Wa Kasongo K, Walker RB. Short Term Stability Testing of Efavirenz-Loaded Solid Lipid Nanoparticle (SLN) and Nanostructured Lipid Carrier (NLC) Dispersions. Pharmaceutics. 2019;11(8):397. doi:10.3390/pharmaceutics11080397
47. Ali AA, Ahmed HI, Abu-Elfotuh K. Modeling stages mimic Alzheimer’s disease induced by different doses of aluminum in rats: focus on progression of the disease in response to time. J Alzheimer’s Parkinsonism Dementia. 2016;1(1):2.
48. Vorhees CV, Williams MT. Morris water maze: procedures for assessing spatial and related forms of learning and memory. Nat Protoc. 2006;1(2):848–858. doi:10.1038/nprot.2006.116
49. Draper HH, Hadley M. Malondialdehyde determination as index of lipid peroxidation. Methods Enzymol. 1990;186:421–431. doi:10.1016/0076-6879(90)86135-i
50. Griffith OW. Determination of glutathione and glutathione disulfide using glutathione reductase and 2-vinylpyridine. Anal Biochem. 1980;106(1):207–212. doi:10.1016/0003-2697(80)90139-6
51. Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001;25(4):402–408. doi:10.1006/meth.2001.1262
52. Lilli R, Bancroft J, Gamble M. The Hematoxylin and Eosin, Histopathologic Technique and Practical Biochemistry.
53. Madane RG, Mahajan HS. Curcumin-loaded nanostructured lipid carriers (NLCs) for nasal administration: design, characterization, and in vivo study. Drug Deliv. 2016;23(4):1326–1334. doi:10.3109/10717544.2014.975382
54. Das S, Ng WK, Tan RB. Are nanostructured lipid carriers (NLCs) better than solid lipid nanoparticles (SLNs): development, characterizations and comparative evaluations of clotrimazole-loaded SLNs and NLCs? Eur J Pharm Sci. 2012;47(1):139–151. doi:10.1016/j.ejps.2012.05.010
55. Kovacevic A, Savic S, Vuleta G, Müller RH, Keck CM. Polyhydroxy surfactants for the formulation of lipid nanoparticles (SLN and NLC): effects on size, physical stability and particle matrix structure. Int J Pharm. 2011;406(1–2):163–172. doi:10.1016/j.ijpharm.2010.12.036
56. Gänger S, Schindowski K. Tailoring Formulations for Intranasal Nose-to-Brain Delivery: a Review on Architecture, Physico-Chemical Characteristics and Mucociliary Clearance of the Nasal Olfactory Mucosa. Pharmaceutics. 2018;10(3):116. doi:10.3390/pharmaceutics10030116
57. Zambito Y, Pedreschi E, Di Colo G. Is dialysis a reliable method for studying drug release from nanoparticulate systems?-A case study. Int J Pharm. 2012;434(1–2):28–34. doi:10.1016/j.ijpharm.2012.05.020
58. Ritger PL, Peppas NA. A simple equation for description of solute release II. Fickian and anomalous release from swellable devices. J Control Release. 1987;5(1):37–42. doi:10.1016/0168-3659(87)90035-6
59. Agrawal R, Tyagi E, Saxena G, Nath C. Cholinergic influence on memory stages: a study on scopolamine amnesic mice. Indian J Pharmacol. 2009;41(4):192–196. doi:10.4103/0253-7613.56072
60. Marighetto A, Valerio S, Desmedt A, Philippin JN, Trocmé-Thibierge C, Morain P. Comparative effects of the alpha7 nicotinic partial agonist, S 24795, and the cholinesterase inhibitor, donepezil, against aging-related deficits in declarative and working memory in mice. Psychopharmacology. 2008;197(3):499–508. doi:10.1007/s00213-007-1063-x
61. Shin CY, Kim HS, Cha KH, et al. The Effects of Donepezil, an Acetylcholinesterase Inhibitor, on Impaired Learning and Memory in Rodents. Biomol Ther (Seoul). 2018;26(3):274–281. doi:10.4062/biomolther.2017.189
62. Cutuli D, Foti F, Mandolesi L, et al. Cognitive performances of cholinergically depleted rats following chronic donepezil administration. J Alzheimers Dis. 2009;17(1):161–176. doi:10.3233/jad-2009-1040
63. Yamada K, Takayanagi M, Kamei H, et al. Effects of memantine and donepezil on amyloid beta-induced memory impairment in a delayed-matching to position task in rats. Behav Brain Res. 2005;162(2):191–199. doi:10.1016/j.bbr.2005.02.036
64. Riedel G, Kang SH, Choi DY, Platt B. Scopolamine-induced deficits in social memory in mice: reversal by donepezil. Behav Brain Res. 2009;204(1):217–225. doi:10.1016/j.bbr.2009.06.012
65. Su D, Zhao Y, Wang B, et al. Isoflurane-induced spatial memory impairment in mice is prevented by the acetylcholinesterase inhibitor donepezil. PLoS One. 2011;6(11):e27632. doi:10.1371/journal.pone.0027632
66. Manczak M, Kandimalla R, Yin X, Reddy PH. Hippocampal mutant APP and amyloid beta-induced cognitive decline, dendritic spine loss, defective autophagy, mitophagy and mitochondrial abnormalities in a mouse model of Alzheimer’s disease. Hum Mol Genet. 2018;27(8):1332–1342. doi:10.1093/hmg/ddy042
67. Grimmig B, Kim SH, Nash K, Bickford PC, Douglas Shytle R. Neuroprotective mechanisms of astaxanthin: a potential therapeutic role in preserving cognitive function in age and neurodegeneration. Geroscience. 2017;39(1):19–32. doi:10.1007/s11357-017-9958-x
68. Che H, Li Q, Zhang T, et al. Effects of Astaxanthin and Docosahexaenoic-Acid-Acylated Astaxanthin on Alzheimer’s Disease in APP/PS1 Double-Transgenic Mice. J Agric Food Chem. 2018;66(19):4948–4957. doi:10.1021/acs.jafc.8b00988
69. Taksima T, Chonpathompikunlert P, Sroyraya M, Hutamekalin P, Limpawattana M, Klaypradit W. Effects of Astaxanthin from Shrimp Shell on Oxidative Stress and Behavior in Animal Model of Alzheimer’s Disease. Mar Drugs. 2019;17(11):628. doi:10.3390/md17110628
70. Rahman SO, Panda BP, Parvez S, et al. Neuroprotective role of astaxanthin in hippocampal insulin resistance induced by Aβ peptides in animal model of Alzheimer’s disease. Biomed Pharmacother. 2019;110:47–58. doi:10.1016/j.biopha.2018.11.043
71. Al-Amin MM, Mahmud W, Pervin MS, Ridwanul Islam SM, Ashikur Rahman M, Zinchenko A. Astaxanthin ameliorates scopolamine-induced spatial memory deficit via reduced cortical-striato-hippocampal oxidative stress. Brain Res. 2019;1710:74–81. doi:10.1016/j.brainres.2018.12.014
72. Manabe Y, Komatsu T, Seki S, Sugawara T. Dietary astaxanthin can accumulate in the brain of rats. Biosci Biotechnol Biochem. 2018;82(8):1433–1436. doi:10.1080/09168451.2018.1459467
73. Akinyemi AJ, Oboh G, Oyeleye SI, Ogunsuyi O. Anti-amnestic Effect of Curcumin in Combination with Donepezil, an Anticholinesterase Drug: involvement of Cholinergic System. Neurotox Res. 2017;31(4):560–569. doi:10.1007/s12640-017-9701-5
74. Pattanashetti LA, Taranalli AD, Parvatrao V, Malabade RH, Kumar D. Evaluation of neuroprotective effect of quercetin with donepezil in scopolamine-induced amnesia in rats. Indian J Pharmacol. 2017;49(1):60–64. doi:10.4103/0253-7613.201016
75. Keshk A, ElFiky S, AlSharaky D, ElBatsh M. Antiamnestic effect of co-administration of losartan and donepezil in l-methionine-induced vascular dementia in rats. Menoufia Med J. 2022;35(1):48–54. doi:10.4103/mmj.mmj_49_21
76. Cachard-Chastel M, Devers S, Sicsic S, et al. Prucalopride and donepezil act synergistically to reverse scopolamine-induced memory deficit in C57Bl/6j mice. Behav Brain Res. 2008;187(2):455–461. doi:10.1016/j.bbr.2007.10.008
77. Wang X, Zhang T, Chen X, et al. Simultaneous Inhibitory Effects of All-Trans Astaxanthin on Acetylcholinesterase and Oxidative Stress. Mar Drugs. 2022;20(4):247. doi:10.3390/md20040247
78. Chen MH, Wang TJ, Chen LJ, et al. The effects of astaxanthin treatment on a rat model of Alzheimer’s disease. Brain Res Bull. 2021;172:151–163. doi:10.1016/j.brainresbull.2021.04.020
79. Nai Y, Liu H, Bi X, Gao H, Ren C. Protective effect of astaxanthin on acute cerebral infarction in rats. Hum Exp Toxicol. 2018;37(9):929–936. doi:10.1177/0960327117745693
80. Tiwari V, Mishra A, Singh S, et al. Protriptyline improves spatial memory and reduces oxidative damage by regulating NFκB-BDNF/CREB signaling axis in streptozotocin-induced rat model of Alzheimer’s disease. Brain Res. 2021;1754:147261. doi:10.1016/j.brainres.2020.147261
81. Tong L, Prieto GA, Kramár EA, et al. Brain-derived neurotrophic factor-dependent synaptic plasticity is suppressed by interleukin-1β via p38 mitogen-activated protein kinase. J Neurosci. 2012;32(49):17714–17724. doi:10.1523/jneurosci.1253-12.2012
82. Damodara Gowda KM, Suchetha Kumari N, Ullal H. Role of astaxanthin in the modulation of brain-derived neurotrophic factor and spatial learning behavior in perinatally undernourished Wistar rats. Nutr Neurosci. 2020;23(6):422–431. doi:10.1080/1028415x.2018.1515301
83. Wu W, Wang X, Xiang Q, et al. Astaxanthin alleviates brain aging in rats by attenuating oxidative stress and increasing BDNF levels. Food Funct. 2014;5(1):158–166. doi:10.1039/c3fo60400d
84. Castellani RJ, Plascencia-Villa G, Perry G. The amyloid cascade and Alzheimer’s disease therapeutics: theory versus observation. Lab Invest. 2019;99(7):958–970. doi:10.1038/s41374-019-0231-z
85. Jabir NR, Rehman MT, Alsolami K, et al. Concatenation of molecular docking and molecular simulation of BACE-1, γ-secretase targeted ligands: in pursuit of Alzheimer’s treatment. Ann Med. 2021;53(1):2332–2344. doi:10.1080/07853890.2021.2009124
86. Kent SA, Spires-Jones TL, Durrant CS. The physiological roles of tau and Aβ: implications for Alzheimer’s disease pathology and therapeutics. Acta Neuropathol. 2020;140(4):417–447. doi:10.1007/s00401-020-02196-w
87. Gouras GK, Olsson TT, Hansson O. β-Amyloid peptides and amyloid plaques in Alzheimer’s disease. Neurotherapeutics. 2015;12(1):3–11. doi:10.1007/s13311-014-0313-y
88. Fanaee-Danesh E, Gali CC, Tadic J, et al. Astaxanthin exerts protective effects similar to bexarotene in Alzheimer’s disease by modulating amyloid-beta and cholesterol homeostasis in blood-brain barrier endothelial cells. Biochim Biophys Acta Mol Basis Dis. 2019;1865(9):2224–2245. doi:10.1016/j.bbadis.2019.04.019
89. Han JH, Lee YS, Im JH, et al. Astaxanthin Ameliorates Lipopolysaccharide-Induced Neuroinflammation, Oxidative Stress and Memory Dysfunction through Inactivation of the Signal Transducer and Activator of Transcription 3 Pathway. Mar Drugs. 2019;17(2):123. doi:10.3390/md17020123
90. Lobos P, Bruna B, Cordova A, et al. Astaxanthin Protects Primary Hippocampal Neurons against Noxious Effects of Aβ-Oligomers. Neural Plast. 2016;2016:3456783. doi:10.1155/2016/3456783
91. Kellar D, Craft S. Brain insulin resistance in Alzheimer’s disease and related disorders: mechanisms and therapeutic approaches. Lancet Neurol. 2020;19(9):758–766. doi:10.1016/s1474-4422(20)30231-3
92. Babalola JA, Lang M, George M, et al. Astaxanthin enhances LRP1-modulated insulin sensitivity and amyloid beta clearance in an in vitro blood-brain barrier model. Alzheimers Dement. 2021;17(S3):e053736. doi:10.1002/alz.053736
93. Huang C, Wen C, Yang M, et al. Astaxanthin Improved the Cognitive Deficits in APP/PS1 Transgenic Mice Via Selective Activation of mTOR. J Neuroimmune Pharmacol. 2021;16(3):609–619. doi:10.1007/s11481-020-09953-4
94. Alghazwi M, Smid S, Musgrave I, Zhang W. In vitro studies of the neuroprotective activities of astaxanthin and fucoxanthin against amyloid beta (Aβ(1-42)) toxicity and aggregation. Neurochem Int. 2019;124:215–224. doi:10.1016/j.neuint.2019.01.010
95. Inestrosa NC, Urra S, Colombres M. Acetylcholinesterase (AChE)--amyloid-beta-peptide complexes in Alzheimer’s disease. the Wnt signaling pathway. Curr Alzheimer Res. 2004;1(4):249–254. doi:10.2174/1567205043332063
96. Ye CY, Lei Y, Tang XC, Zhang HY. Donepezil attenuates Aβ-associated mitochondrial dysfunction and reduces mitochondrial Aβ accumulation in vivo and in vitro. Neuropharmacology. 2015;95:29–36. doi:10.1016/j.neuropharm.2015.02.020
97. Dong H, Yuede CM, Coughlan CA, Murphy KM, Csernansky JG. Effects of donepezil on amyloid-beta and synapse density in the Tg2576 mouse model of Alzheimer’s disease. Brain Res. 2009;1303:169–178. doi:10.1016/j.brainres.2009.09.097
98. Takada-Takatori Y, Nakagawa S, Kimata R, et al. Donepezil modulates amyloid precursor protein endocytosis and reduction by up-regulation of SNX33 expression in primary cortical neurons. Sci Rep. 2019;9(1):11922. doi:10.1038/s41598-019-47462-4
99. Kimura M, Akasofu S, Ogura H, Sawada K. Protective effect of donepezil against Abeta(1-40) neurotoxicity in rat septal neurons. Brain Res. 2005;1047(1):72–84. doi:10.1016/j.brainres.2005.04.014
100. Zhang N, Gordon ML. Clinical efficacy and safety of donepezil in the treatment of Alzheimer’s disease in Chinese patients. Clin Interv Aging. 2018;13:1963–1970. doi:10.2147/CIA.S159920
101. Cai Z, Zhao Y, Zhao B. Roles of Glycogen Synthase Kinase 3 in Alzheimer’s Disease. Curr Alzheimer Res. 2012;9(7):864–879. doi:10.2174/156720512802455386
102. Ly PT, Wu Y, Zou H, et al. Inhibition of GSK3β-mediated BACE1 expression reduces Alzheimer-associated phenotypes. J Clin Invest. 2013;123(1):224–235. doi:10.1172/jci64516
103. Terwel D, Muyllaert D, Dewachter I, et al. Amyloid activates GSK-3beta to aggravate neuronal tauopathy in bigenic mice. Am J Pathol. 2008;172(3):786–798. doi:10.2353/ajpath.2008.070904
104. Toral-Rios D, Pichardo-Rojas PS, Alonso-Vanegas M, Campos-Peña V. GSK3β and Tau Protein in Alzheimer’s Disease and Epilepsy. Front Cell Neurosci. 2020;14:19. doi:10.3389/fncel.2020.00019
105. Zhou XW, Gustafsson JA, Tanila H, et al. Tau hyperphosphorylation correlates with reduced methylation of protein phosphatase 2A. Neurobiol Dis. 2008;31(3):386–394. doi:10.1016/j.nbd.2008.05.013
106. Koh SH, Noh MY, Kim SH. Amyloid-beta-induced neurotoxicity is reduced by inhibition of glycogen synthase kinase-3. Brain Res. 2008;1188:254–262. doi:10.1016/j.brainres.2007.10.064
107. Wen X, Huang A, Hu J, et al. Neuroprotective effect of astaxanthin against glutamate-induced cytotoxicity in HT22 cells: involvement of the Akt/GSK-3β pathway. Neuroscience. 2015;303:558–568. doi:10.1016/j.neuroscience.2015.07.034
108. Noh MY, Koh SH, Kim SM, Maurice T, Ku SK, Kim SH. Neuroprotective effects of donepezil against Aβ42-induced neuronal toxicity are mediated through not only enhancing PP2A activity but also regulating GSK-3β and nAChRs activity. J Neurochem. 2013;127(4):562–574. doi:10.1111/jnc.12319
109. Noh MY, Koh SH, Kim Y, Kim HY, Cho GW, Kim SH. Neuroprotective effects of donepezil through inhibition of GSK-3 activity in amyloid-beta-induced neuronal cell death. J Neurochem. 2009;108(5):1116–1125. doi:10.1111/j.1471-4159.2008.05837.x
110. Yoshiyama Y, Kojima A, Ishikawa C, Arai K. Anti-inflammatory action of donepezil ameliorates tau pathology, synaptic loss, and neurodegeneration in a tauopathy mouse model. J Alzheimers Dis. 2010;22(1):295–306. doi:10.3233/jad-2010-100681
111. Rekatsina M, Paladini A, Piroli A, Zis P, Pergolizzi JV, Varrassi G. Pathophysiology and Therapeutic Perspectives of Oxidative Stress and Neurodegenerative Diseases: a Narrative Review. Adv Ther. 2020;37(1):113–139. doi:10.1007/s12325-019-01148-5
112. Tönnies E, Trushina E. Oxidative Stress, Synaptic Dysfunction, and Alzheimer’s Disease. J Alzheimers Dis. 2017;57(4):1105–1121. doi:10.3233/jad-161088
113. Islam BU, Jabir NR, Tabrez S. The role of mitochondrial defects and oxidative stress in Alzheimer’s disease. J Drug Target. 2019;27(9):932–942. doi:10.1080/1061186x.2019.1584808
114. Bhatt S, Puli L, Patil CR. Role of reactive oxygen species in the progression of Alzheimer’s disease. Drug Discov Today. 2021;26(3):794–803. doi:10.1016/j.drudis.2020.12.004
115. Ayala A, Muñoz MF, Argüelles S. Lipid peroxidation: production, metabolism, and signaling mechanisms of malondialdehyde and 4-hydroxy-2-nonenal. Oxid Med Cell Longev. 2014;2014:360438. doi:10.1155/2014/360438
116. Barros MP, Rodrigo MJ, Zacarias L. Dietary Carotenoid Roles in Redox Homeostasis and Human Health. J Agric Food Chem. 2018;66(23):5733–5740. doi:10.1021/acs.jafc.8b00866
117. Peña-Bautista C, Durand T, Oger C, Baquero M, Vento M, Cháfer-Pericás C. Assessment of lipid peroxidation and artificial neural network models in early Alzheimer Disease diagnosis. Clin Biochem. 2019;72:64–70. doi:10.1016/j.clinbiochem.2019.07.008
118. Focsan AL, Polyakov NE, Kispert LD. Carotenoids: importance in Daily Life-Insight Gained from EPR and ENDOR. Appl Magn Reson. 2021;52(8):1093–1112. doi:10.1007/s00723-021-01311-8
119. Nishida Y, Nawaz A, Hecht K, Tobe K. Astaxanthin as a Novel Mitochondrial Regulator: a New Aspect of Carotenoids, beyond Antioxidants. Nutrients. 2021;14(1):107. doi:10.3390/nu14010107
120. Balendra V, Singh SK. Therapeutic potential of astaxanthin and superoxide dismutase in Alzheimer’s disease. Open Biol. 2021;11(6):210013. doi:10.1098/rsob.210013
121. Goto S, Kogure K, Abe K, et al. Efficient radical trapping at the surface and inside the phospholipid membrane is responsible for highly potent antiperoxidative activity of the carotenoid astaxanthin. Biochim Biophys Acta. 2001;1512(2):251–258. doi:10.1016/s0005-2736(01)00326-1
122. Kohandel Z, Farkhondeh T, Aschner M, Samarghandian S. Nrf2 a molecular therapeutic target for Astaxanthin. Biomed Pharmacother. 2021;137:111374. doi:10.1016/j.biopha.2021.111374
123. Lu MC, Ji JA, Jiang ZY, You QD. The Keap1-Nrf2-ARE Pathway As a Potential Preventive and Therapeutic Target: an Update. Med Res Rev. 2016;36(5):924–963. doi:10.1002/med.21396
124. Davinelli S, Saso L, D’Angeli F, Calabrese V, Intrieri M, Scapagnini G. Astaxanthin as a Modulator of Nrf2, NF-κB, and Their Crosstalk: molecular Mechanisms and Possible Clinical Applications. Molecules. 2022;27(2):502. doi:10.3390/molecules27020502
125. Zarneshan SN, Fakhri S, Farzaei MH, Khan H, Saso L. Astaxanthin targets PI3K/Akt signaling pathway toward potential therapeutic applications. Food Chem Toxicol. 2020;145:111714. doi:10.1016/j.fct.2020.111714
126. Cuadrado A. Structural and functional characterization of Nrf2 degradation by glycogen synthase kinase 3/β-TrCP. Free Radic Biol Med. 2015;88(Pt B):147–157. doi:10.1016/j.freeradbiomed.2015.04.029
127. Rada P, Rojo AI, Chowdhry S, McMahon M, Hayes JD, Cuadrado A. SCF/{beta}-TrCP promotes glycogen synthase kinase 3-dependent degradation of the Nrf2 transcription factor in a Keap1-independent manner. Mol Cell Biol. 2011;31(6):1121–1133. doi:10.1128/mcb.01204-10
128. Sztretye M, Dienes B, Gönczi M, et al. Astaxanthin: a Potential Mitochondrial-Targeted Antioxidant Treatment in Diseases and with Aging. Oxid Med Cell Longev. 2019;2019:3849692. doi:10.1155/2019/3849692
129. Kim SH, Kim H. Inhibitory Effect of Astaxanthin on Oxidative Stress-Induced Mitochondrial Dysfunction-A Mini-Review. Nutrients. 2018;10(9):1137. doi:10.3390/nu10091137
130. Yang Y, Seo JM, Nguyen A, et al. Astaxanthin-rich extract from the green alga Haematococcus pluvialis lowers plasma lipid concentrations and enhances antioxidant defense in apolipoprotein E knockout mice. J Nutr. 2011;141(9):1611–1617. doi:10.3945/jn.111.142109
131. Umukoro S, Adewole FA, Eduviere AT, Aderibigbe AO, Onwuchekwa C. Free radical scavenging effect of donepezil as the possible contribution to its memory enhancing activity in mice. Drug Res. 2014;64(5):236–239. doi:10.1055/s-0033-1357126
132. Atukeren P, Cengiz M, Yavuzer H, et al. The efficacy of donepezil administration on acetylcholinesterase activity and altered redox homeostasis in Alzheimer’s disease. Biomed Pharmacother. 2017;90:786–795. doi:10.1016/j.biopha.2017.03.101
133. Munishamappa V, Seethalakshmi VA, Rajathilagam T. Evaluation of the antioxidant activity of donepezil-in vitro study. Nat J Physiol Pharm Pharmacol. 2019;9(2):108–110. doi:10.5455/njppp.2019.9.1134624112018
134. Jiang L, Wang Y, Su L, et al. Donepezil Attenuates Obesity-Associated Oxidative Stress and Central Inflammation and Improves Memory Deficit in Mice Fed a High-Fat Diet. Dement Geriatr Cogn Disord. 2019;48(3–4):154–163. doi:10.1159/000504800
135. Obafemi TO, Olasehinde OR, Olaoye OA, et al. Metformin/Donepezil combination modulates brain antioxidant status and hippocampal endoplasmic reticulum stress in type 2 diabetic rats. J Diabetes Metab Disord. 2020;19(1):499–510. doi:10.1007/s40200-020-00541-0
136. Rao YL, Ganaraja B, Marathe A, et al. Comparison of malondialdehyde levels and superoxide dismutase activity in resveratrol and resveratrol/donepezil combination treatment groups in Alzheimer’s disease induced rat model. Biotech. 2021;11(7):329. doi:10.1007/s13205-021-02879-5
137. Akkoyun HT, Bengu AS, Ulucan A, et al. Effect of astaxanthin on rat brains against oxidative stress induced by cadmium: biochemical, histopathological evaluation. J Ins Sci Technol. 2018;8(4):33–39. doi:10.21597/jist.412070
138. Ahmad MA, Kareem O, Khushtar M, et al. Neuroinflammation: a Potential Risk for Dementia. Int J Mol Sci. 2022;23(2):616. doi:10.3390/ijms23020616
139. Leng F, Edison P. Neuroinflammation and microglial activation in Alzheimer disease: where do we go from here? Nat Rev Neurol. 2021;17(3):157–172. doi:10.1038/s41582-020-00435-y
140. Zhao J, Bi W, Xiao S, et al. Neuroinflammation induced by lipopolysaccharide causes cognitive impairment in mice. Sci Rep. 2019;9(1):5790. doi:10.1038/s41598-019-42286-8
141. Anaeigoudari A, Soukhtanloo M, Shafei MN, et al. Neuronal nitric oxide synthase has a role in the detrimental effects of lipopolysaccharide on spatial memory and synaptic plasticity in rats. Pharmacol Rep. 2016;68(2):243–249. doi:10.1016/j.pharep.2015.09.004
142. Leonoudakis D, Zhao P, Beattie EC. Rapid tumor necrosis factor alpha-induced exocytosis of glutamate receptor 2-lacking AMPA receptors to extrasynaptic plasma membrane potentiates excitotoxicity. J Neurosci. 2008;28(9):2119–2130. doi:10.1523/jneurosci.5159-07.2008
143. Agrawal I, Jha S. Mitochondrial Dysfunction and Alzheimer’s Disease: role of Microglia. Front Aging Neurosci. 2020;12:252. doi:10.3389/fnagi.2020.00252
144. Simpson DSA, Oliver PL. ROS Generation in Microglia: understanding Oxidative Stress and Inflammation in Neurodegenerative Disease. Antioxidants. 2020;9(8):743. doi:10.3390/antiox9080743
145. Webers A, Heneka MT, Gleeson PA. The role of innate immune responses and neuroinflammation in amyloid accumulation and progression of Alzheimer’s disease. Immunol Cell Biol. 2020;98(1):28–41. doi:10.1111/imcb.12301
146. Reed-Geaghan EG, Savage JC, Hise AG, Landreth GE. CD14 and toll-like receptors 2 and 4 are required for fibrillar A{beta}-stimulated microglial activation. J Neurosci. 2009;29(38):11982–11992. doi:10.1523/jneurosci.3158-09.2009
147. Kaur D, Sharma V, Deshmukh R. Activation of microglia and astrocytes: a roadway to neuroinflammation and Alzheimer’s disease. Inflammopharmacology. 2019;27(4):663–677. doi:10.1007/s10787-019-00580-x
148. Liu T, Zhang L, Joo D, Sun SC. NF-κB signaling in inflammation. Signal Transduct Target Ther. 2017;2:17023. doi:10.1038/sigtrans.2017.23
149. Kohandel Z, Farkhondeh T, Aschner M, Pourbagher-Shahri AM, Samarghandian S. Anti-inflammatory action of astaxanthin and its use in the treatment of various diseases. Biomed Pharmacother. 2022;145:112179. doi:10.1016/j.biopha.2021.112179
150. Yang C, Hassan YI, Liu R, et al. Anti-Inflammatory Effects of Different Astaxanthin Isomers and the Roles of Lipid Transporters in the Cellular Transport of Astaxanthin Isomers in Caco-2 Cell Monolayers. J Agric Food Chem. 2019;67(22):6222–6231. doi:10.1021/acs.jafc.9b02102
151. Chen Y, Zhao S, Jiao D, et al. Astaxanthin Alleviates Ochratoxin A-Induced Cecum Injury and Inflammation in Mice by Regulating the Diversity of Cecal Microbiota and TLR4/MyD88/NF-κB Signaling Pathway. Oxid Med Cell Longev. 2021;2021:8894491. doi:10.1155/2021/8894491
152. Kim YH, Koh HK, Kim DS. Down-regulation of IL-6 production by astaxanthin via ERK-, MSK-, and NF-κB-mediated signals in activated microglia. Int Immunopharmacol. 2010;10(12):1560–1572. doi:10.1016/j.intimp.2010.09.007
153. Choi SK, Park YS, Choi DK, Chang HI. Effects of astaxanthin on the production of NO and the expression of COX-2 and iNOS in LPS-stimulated BV2 microglial cells. J Microbiol Biotechnol. 2008;18(12):1990–1996.
154. Wen X, Xiao L, Zhong Z, et al. Astaxanthin acts via LRP-1 to inhibit inflammation and reverse lipopolysaccharide-induced M1/M2 polarization of microglial cells. Oncotarget. 2017;8(41):69370–69385. doi:10.18632/oncotarget.20628
155. Ahmed SM, Luo L, Namani A, Wang XJ, Tang X. Nrf2 signaling pathway: pivotal roles in inflammation. Biochim Biophys Acta Mol Basis Dis. 2017;1863(2):585–597. doi:10.1016/j.bbadis.2016.11.005
156. Farruggia C, Kim MB, Bae M, et al. Astaxanthin exerts anti-inflammatory and antioxidant effects in macrophages in NRF2-dependent and independent manners. J Nutr Biochem. 2018;62:202–209. doi:10.1016/j.jnutbio.2018.09.005
157. Wu HY, Hudry E, Hashimoto T, et al. Amyloid beta induces the morphological neurodegenerative triad of spine loss, dendritic simplification, and neuritic dystrophies through calcineurin activation. J Neurosci. 2010;30(7):2636–2649. doi:10.1523/jneurosci.4456-09.2010
158. Cavallucci V, Berretta N, Nobili A, Nisticò R, Mercuri NB, D’Amelio M. Calcineurin inhibition rescues early synaptic plasticity deficits in a mouse model of Alzheimer’s disease. Neuromolecular Med. 2013;15(3):541–548. doi:10.1007/s12017-013-8241-2
159. Tewari D, Sah AN, Bawari S, et al. Role of Nitric Oxide in Neurodegeneration: function, Regulation, and Inhibition. Curr Neuropharmacol. 2021;19(2):114–126. doi:10.2174/1570159x18666200429001549
160. Trollor JN, Smith E, Agars E, et al. The association between systemic inflammation and cognitive performance in the elderly: the Sydney Memory and Ageing Study. Age. 2012;34(5):1295–1308. doi:10.1007/s11357-011-9301-x
161. Zhou X, Zhang J, Li Y, Cui L, Wu K, Luo H. Astaxanthin inhibits microglia M1 activation against inflammatory injury triggered by lipopolysaccharide through down-regulating miR-31-5p. Life Sci. 2021;267:118943. doi:10.1016/j.lfs.2020.118943
162. Kim HG, Moon M, Choi JG, et al. Donepezil inhibits the amyloid-beta oligomer-induced microglial activation in vitro and in vivo. Neurotoxicology. 2014;40:23–32. doi:10.1016/j.neuro.2013.10.004
163. Guo HB, Cheng YF, Wu JG, et al. Donepezil improves learning and memory deficits in APP/PS1 mice by inhibition of microglial activation. Neuroscience. 2015;290:530–542. doi:10.1016/j.neuroscience.2015.01.058
164. Hori K, Hosoi M, Konishi K. Cholinesterase inhibitors as a disease-modifying therapy for Alzheimer’s disease: the anticholinergic hypothesis. Austin J Clin Neurol. 2016;3:1091–1094.
165. Kim J, Lee HJ, Park SK, et al. Donepezil Regulates LPS and Aβ-Stimulated Neuroinflammation through MAPK/NLRP3 Inflammasome/STAT3 Signaling. Int J Mol Sci. 2021;22(19). doi:10.3390/ijms221910637
166. Haraguchi Y, Mizoguchi Y, Ohgidani M, et al. Donepezil suppresses intracellular Ca(2+) mobilization through the PI3K pathway in rodent microglia. J Neuroinflammation. 2017;14(1):258. doi:10.1186/s12974-017-1033-0
167. Kummer MP, Hermes M, Delekarte A, et al. Nitration of tyrosine 10 critically enhances amyloid β aggregation and plaque formation. Neuron. 2011;71(5):833–844. doi:10.1016/j.neuron.2011.07.001
168. Maroli A, Di Lascio S, Drufuca L, et al. Effect of donepezil on the expression and responsiveness to LPS of CHRNA7 and CHRFAM7A in macrophages: a possible link to the cholinergic anti-inflammatory pathway. J Neuroimmunol. 2019;332:155–166. doi:10.1016/j.jneuroim.2019.04.012
169. Martelli D, McKinley MJ, McAllen RM. The cholinergic anti-inflammatory pathway: a critical review. Auton Neurosci. 2014;182:65–69. doi:10.1016/j.autneu.2013.12.007
170. Cox MA, Bassi C, Saunders ME, et al. Beyond neurotransmission: acetylcholine in immunity and inflammation. J Intern Med. 2020;287(2):120–133. doi:10.1111/joim.13006
171. Liu EYL, Xia Y, Kong X, et al. Interacting with α 7 nAChR is a new mechanism for AChE to enhance the inflammatory response in macrophages. Acta Pharm Sin B. 2020;10(10):1926–1942. doi:10.1016/j.apsb.2020.05.005
172. de Jonge WJ, van der Zanden EP, The FO, et al. Stimulation of the vagus nerve attenuates macrophage activation by activating the Jak2-STAT3 signaling pathway. Nat Immunol. 2005;6(8):844–851. doi:10.1038/ni1229
173. De Simone R, Ajmone-Cat MA, Carnevale D, Minghetti L. Activation of alpha7 nicotinic acetylcholine receptor by nicotine selectively up-regulates cyclooxygenase-2 and prostaglandin E2 in rat microglial cultures. J Neuroinflammation. 2005;2(1):4. doi:10.1186/1742-2094-2-4
174. Wei P, Yang F, Zheng Q, Tang W, Li J. The Potential Role of the NLRP3 Inflammasome Activation as a Link Between Mitochondria ROS Generation and Neuroinflammation in Postoperative Cognitive Dysfunction. Front Cell Neurosci. 2019;13:73. doi:10.3389/fncel.2019.00073
175. Ke P, Shao BZ, Xu ZQ, Chen XW, Wei W, Liu C. Activating α7 nicotinic acetylcholine receptor inhibits NLRP3 inflammasome through regulation of β-arrestin-1. CNS Neurosci Ther. 2017;23(11):875–884. doi:10.1111/cns.12758
176. Arikawa M, Kakinuma Y, Noguchi T, Todaka H, Sato T. Donepezil, an acetylcholinesterase inhibitor, attenuates LPS-induced inflammatory response in murine macrophage cell line RAW 264.7 through inhibition of nuclear factor kappa B translocation. Eur J Pharmacol. 2016;789:17–26. doi:10.1016/j.ejphar.2016.06.053
177. Combs CK, Karlo JC, Kao SC, Landreth GE. beta-Amyloid stimulation of microglia and monocytes results in TNFalpha-dependent expression of inducible nitric oxide synthase and neuronal apoptosis. J Neurosci. 2001;21(4):1179–1188. doi:10.1523/jneurosci.21-04-01179.2001
178. Czabotar PE, Lessene G, Strasser A, Adams JM. Control of apoptosis by the BCL-2 protein family: implications for physiology and therapy. Nat Rev Mol Cell Biol. 2014;15(1):49–63. doi:10.1038/nrm3722
179. D’Amelio M, Sheng M, Cecconi F. Caspase-3 in the central nervous system: beyond apoptosis. Trends Neurosci. 2012;35(11):700–709. doi:10.1016/j.tins.2012.06.004
180. Ikeda Y, Tsuji S, Satoh A, Ishikura M, Shirasawa T, Shimizu T. Protective effects of astaxanthin on 6-hydroxydopamine-induced apoptosis in human neuroblastoma SH-SY5Y cells. J Neurochem. 2008;107(6):1730–1740. doi:10.1111/j.1471-4159.2008.05743.x
181. El-Agamy SE, Abdel-Aziz AK, Wahdan S, Esmat A, Azab SS. Astaxanthin Ameliorates Doxorubicin-Induced Cognitive Impairment (Chemobrain) in Experimental Rat Model: impact on Oxidative, Inflammatory, and Apoptotic Machineries. Mol Neurobiol. 2018;55(7):5727–5740. doi:10.1007/s12035-017-0797-7
182. D’Amelio M, Cavallucci V, Middei S, et al. Caspase-3 triggers early synaptic dysfunction in a mouse model of Alzheimer’s disease. Nat Neurosci. 2011;14(1):69–76. doi:10.1038/nn.2709
183. Cutuli D, De Bartolo P, Caporali P, et al. Neuroprotective effects of donepezil against cholinergic depletion. Alzheimers Res Ther. 2013;5(5):50. doi:10.1186/alzrt215
184. Takada-Takatori Y, Kume T, Izumi Y, et al. Roles of nicotinic receptors in acetylcholinesterase inhibitor-induced neuroprotection and nicotinic receptor up-regulation. Biol Pharm Bull. 2009;32(3):318–324. doi:10.1248/bpb.32.318
185. Kihara T, Shimohama S, Sawada H, et al. alpha 7 nicotinic receptor transduces signals to phosphatidylinositol 3-kinase to block A beta-amyloid-induced neurotoxicity. J Biol Chem. 2001;276(17):13541–13546. doi:10.1074/jbc.M008035200
186. Elbaz EM, Essam RM, Ahmed KA, Safwat MH. Donepezil halts acetic acid-induced experimental colitis in rats and its associated cognitive impairment through regulating inflammatory/oxidative/apoptotic cascades: an add-on to its anti-dementia activity. Int Immunopharmacol. 2023;116:109841. doi:10.1016/j.intimp.2023.109841
187. Ye Q, Zhang X, Huang B, Zhu Y, Chen X. Astaxanthin suppresses MPP(+)-induced oxidative damage in PC12 cells through a Sp1/NR1 signaling pathway. Mar Drugs. 2013;11(4):1019–1034. doi:10.3390/md11041019
188. Zhang J, Ding C, Zhang S, Xu Y. Neuroprotective effects of astaxanthin against oxygen and glucose deprivation damage via the PI3K/Akt/GSK3β/Nrf2 signalling pathway in vitro. J Cell Mol Med. 2020;24(16):8977–8985. doi:10.1111/jcmm.15531
189. Cui X, Guo YE, Fang JH, et al. Donepezil, a drug for Alzheimer’s disease, promotes oligodendrocyte generation and remyelination. Acta Pharmacol Sin. 2019;40(11):1386–1393. doi:10.1038/s41401-018-0206-4
190. Wang HY, Lee DH, D’Andrea MR, Peterson PA, Shank RP, Reitz AB. beta-Amyloid(1-42) binds to alpha7 nicotinic acetylcholine receptor with high affinity. Implications for Alzheimer’s disease pathology. J Biol Chem. 2000;275(8):5626–5632. doi:10.1074/jbc.275.8.5626
191. Snyder EM, Nong Y, Almeida CG, et al. Regulation of NMDA receptor trafficking by amyloid-beta. Nat Neurosci. 2005;8(8):1051–1058. doi:10.1038/nn1503
192. Xi D, Keeler B, Zhang W, Houle JD, Gao WJ. NMDA receptor subunit expression in GABAergic interneurons in the prefrontal cortex: application of laser microdissection technique. J Neurosci Methods. 2009;176(2):172–181. doi:10.1016/j.jneumeth.2008.09.013
193. Akaike A, Takada-Takatori Y, Kume T, Izumi Y. Mechanisms of neuroprotective effects of nicotine and acetylcholinesterase inhibitors: role of alpha4 and alpha7 receptors in neuroprotection. J Mol Neurosci. 2010;40(1–2):211–216. doi:10.1007/s12031-009-9236-1
194. Crowder RJ, Freeman RS. Phosphatidylinositol 3-kinase and Akt protein kinase are necessary and sufficient for the survival of nerve growth factor-dependent sympathetic neurons. J Neurosci. 1998;18(8):2933–2943. doi:10.1523/jneurosci.18-08-02933.1998
195. Shaw S, Bencherif M, Marrero MB. Janus kinase 2, an early target of alpha 7 nicotinic acetylcholine receptor-mediated neuroprotection against Abeta-(1-42) amyloid. J Biol Chem. 2002;277(47):44920–44924. doi:10.1074/jbc.M204610200
196. Shen H, Kihara T, Hongo H, et al. Neuroprotection by donepezil against glutamate excitotoxicity involves stimulation of alpha7 nicotinic receptors and internalization of NMDA receptors. Br J Pharmacol. 2010;161(1):127–139. doi:10.1111/j.1476-5381.2010.00894.x
197. Lin H, Vicini S, Hsu FC, et al. Axonal α7 nicotinic ACh receptors modulate presynaptic NMDA receptor expression and structural plasticity of glutamatergic presynaptic boutons. Proc Natl Acad Sci U S A. 2010;107(38):16661–16666. doi:10.1073/pnas.1007397107
198. Jin JL, Fang M, Zhao YX, Liu XY. Roles of sigma-1 receptors in Alzheimer’s disease. Int J Clin Exp Med. 2015;8(4):4808–4820.
199. Solntseva EI, Kapai NA, Popova OV, Rogozin PD, Skrebitsky VG. The involvement of sigma1 receptors in donepezil-induced rescue of hippocampal LTP impaired by beta-amyloid peptide. Brain Res Bull. 2014;106:56–61. doi:10.1016/j.brainresbull.2014.06.002
200. Meunier J, Ieni J, Maurice T. The anti-amnesic and neuroprotective effects of donepezil against amyloid beta25-35 peptide-induced toxicity in mice involve an interaction with the sigma1 receptor. Br J Pharmacol. 2006;149(8):998–1012. doi:10.1038/sj.bjp.0706927
201. Jian WX, Zhang Z, Zhan JH, et al. Donepezil attenuates vascular dementia in rats through increasing BDNF induced by reducing HDAC6 nuclear translocation. Acta Pharmacol Sin. 2020;41(5):588–598. doi:10.1038/s41401-019-0334-5
202. Leyhe T, Stransky E, Eschweiler GW, Buchkremer G, Laske C. Increase of BDNF serum concentration during donepezil treatment of patients with early Alzheimer’s disease. Eur Arch Psychiatry Clin Neurosci. 2008;258(2):124–128. doi:10.1007/s00406-007-0764-9
203. Nakano S, Asada T, Matsuda H, Uno M, Takasaki M. Donepezil hydrochloride preserves regional cerebral blood flow in patients with Alzheimer’s disease. J Nucl Med. 2001;42(10):1441–1445.
204. Akasofu S, Kosasa T, Kimura M, Kubota A. Protective effect of donepezil in a primary culture of rat cortical neurons exposed to oxygen-glucose deprivation. Eur J Pharmacol. 2003;472(1–2):57–63. doi:10.1016/s0014-2999(03)01865-x
205. Dubois B, Chupin M, Hampel H, et al. Donepezil decreases annual rate of hippocampal atrophy in suspected prodromal Alzheimer’s disease. Alzheimers Dement. 2015;11(9):1041–1049. doi:10.1016/j.jalz.2014.10.003
206. Zimmermann M. Neuronal AChE splice variants and their non-hydrolytic functions: redefining a target of AChE inhibitors? Br J Pharmacol. 2013;170(5):953–967. doi:10.1111/bph.12359
207. Buraimoh A, Ojo S. Effects of aluminium chloride exposure on the histology of the stomach of Wistar rats. Int J Pharm Bio Sci. 2012;3(1):266–276. doi:10.5296/jbls.v3i1.1421
208. Lee D, Minko T. Nanotherapeutics for Nose-to-Brain Drug Delivery: an Approach to Bypass the Blood Brain Barrier. Pharmaceutics. 2021;13(12):2049. doi:10.3390/pharmaceutics13122049
209. Erdő F, Bors LA, Farkas D, Bajza Á, Gizurarson S. Evaluation of intranasal delivery route of drug administration for brain targeting. Brain Res Bull. 2018;143:155–170. doi:10.1016/j.brainresbull.2018.10.009
210. Jojo GM, Kuppusamy G, De A, Karri V. Formulation and optimization of intranasal nanolipid carriers of pioglitazone for the repurposing in Alzheimer’s disease using Box-Behnken design. Drug Dev Ind Pharm. 2019;45(7):1061–1072. doi:10.1080/03639045.2019.1593439
211. Haider M, Abdin SM, Kamal L, Orive G. Nanostructured Lipid Carriers for Delivery of Chemotherapeutics: a Review. Pharmaceutics. 2020;12(3):288. doi:10.3390/pharmaceutics12030288
212. Akel H, Ismail R, Csóka I. Progress and perspectives of brain-targeting lipid-based nanosystems via the nasal route in Alzheimer’s disease. Eur J Pharm Biopharm. 2020;148:38–53. doi:10.1016/j.ejpb.2019.12.014
213. Zhang K, Lv S, Li X, et al. Preparation, characterization, and in vivo pharmacokinetics of nanostructured lipid carriers loaded with oleanolic acid and gentiopicrin. Int J Nanomedicine. 2013;8:3227–3239. doi:10.2147/ijn.s45031
214. Pottoo FH, Sharma S, Javed MN, et al. Lipid-based nanoformulations in the treatment of neurological disorders. Drug Metab Rev. 2020;52(1):185–204. doi:10.1080/03602532.2020.1726942
 © 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.