Back to Journals » International Journal of Nanomedicine » Volume 20
Combined ϵ-Toxin Nanovaccine with Enhanced Immunity for Effective Protection in a Murine Model
Authors Li D, Tang L, Fang X, Liu T, Luo X, Li J, Wang J, Li Y, Yuan B, Wang J, Gao S, Kang L, Zhao B, Xin W ![]()
Received 24 March 2025
Accepted for publication 30 October 2025
Published 18 November 2025 Volume 2025:20 Pages 13817—13835
DOI https://doi.org/10.2147/IJN.S530074
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Farooq A. Shiekh
Dongxue Li,1– 3,* Li Tang,1,* Xinyu Fang,1,* Tingting Liu,1,* Xi Luo,1 Jiaxin Li,1 Jing Wang,1 Yanwei Li,1 Bing Yuan,1 Jinglin Wang,1 Shan Gao,1 Lin Kang,1 Baohua Zhao,2 Wenwen Xin1
1State Key Laboratory of Pathogen and Biosecurity, Academy of Military Medical Sciences, Beijing, 100071, People’s Republic of China; 2Hebei Key Laboratory of Animal Physiology, Biochemistry and Molecular Biology, College of Life Sciences, Hebei Normal University, Shijiazhuang, 050010, People’s Republic of China; 3National Key Laboratory of Intelligent Tracking and Forecasting for Infectious Diseases, National Institute for Communicable Disease Control and Prevention, Chinese Center for Disease Control and Prevention, Beijing, 102206, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Baohua Zhao; Email [email protected] Wenwen Xin, Email [email protected]
Introduction: Clostridium perfringens ϵ toxin (ETX), a category B biological weapon, causes fatal enterotoxemia in livestock. Vaccination is an effective way to prevent ETX intoxication.
Methods: A cell membrane–encapsulated nanoparticle vaccine was prepared that adsorbed ASP–ETX (Ov-ASP-1 and epsilon toxin fusion protein) with a targeting effect on B cells and was loaded with the ETXY196E mutant protein (MNP–ASP–ETX). The antigen presentation efficiency of the nanoparticle vaccine was monitored through in vitro cell experiments and in vivo animal experiments, and the biological safety and overall efficacy of the nanoparticle vaccine were evaluated.
Results: The ETX nanoparticle vaccine showed superior biosafety both in vivo and in vitro, favorable encapsulation efficiency, and a particle size that was easily taken up by antigen-presenting cells. The ETX nanoparticle vaccine released antigens more steadily and exhibited greater lymph node drainage capacity than the traditional aluminum adjuvanted group (ETXY196E + Al). MNP–ASP–ETX elicited antibody titers comparable to the ETXY196E + Al group and induced Th1 and Th2 immune responses. However, the protective effect of the ETX nanoparticle vaccine was slightly weaker than that of ETXY196E + Al. We therefore analyzed immune cell production in the spleen. Although MNP–ASP–ETX increased B-cell levels, indicating that ASP–ETX facilitates the ETX nanoparticle vaccine to target B cells, the effect was not statistically significant, likely owing to the less adsorption of ASP–ETX. The antibody titer and protective effect significantly increased after 2 months, with the magnitude of change surpassing that of ETXY196E + Al and indicating the strong potential of ETX nanoparticle vaccine for providing long-term immunity.
Discussion: These findings demonstrate that MNP–ASP–ETX can be considered a novel vaccine for the prevention of ETX intoxication and provide new strategies for designing and developing toxin vaccines.
Keywords: ETX, membrane-encapsulated nanoparticles, targeting B-cell, toxin vaccine
Introduction
Clostridium perfringens is conventionally categorized into seven toxinotypes (types A–G) according to its six primary lethal toxins (α, β, ε, ι, CPE, and Net B).1,2 ε-toxin (ETX) is a major virulence factor produced by C. perfringens types B and D. ETX is the third most potent bacterial toxin following botulinum and tetanus neurotoxins in terms of lethality. Recognized for its significant biothreat potential, ETX has been classified as a category B priority pathogen by the Centers for Disease Control and Prevention (CDC) in the United States.3 ETX is secreted as a prototoxin and is activated after the removal of its N-terminal 11–13 and C-terminal 22–29 amino acids (AAs) by proteases such as trypsin, chymotrypsin, and γ protease.4,5 ETX causes rapidly fatal enterotoxaemia in ruminant livestock, causing death within hours or even minutes and leading to large losses in animal husbandry.6–9 Madin–Darby canine kidney (MDCK) cells demonstrate the highest sensitivity to ETX and consequently serve as the primary in vitro cell model for investigating ETX pathogenesis. Upon binding to its putative receptor, ETX undergoes oligomerization to form a heptameric transmembrane pore complex within the plasma membrane of MDCK cells.10 The absorbed toxin increases vascular permeability, causes severe vascular damage, and induces central nervous system damage.11–14 However, very few ETX-mediated diseases have been reported in humans.15,16 ETX can form pores on human mature red blood cells and exert hemolytic effects in a short period.17,18 Currently, no effective treatment drugs are available for ETX intoxication in clinical practice.
Vaccines are among the most important means of preventing and controlling toxins.8,19,20 Conventional toxoid vaccines, prepared through formaldehyde inactivation of whole-cell cultures or bacterial culture filtrates, have demonstrated efficacy and have been widely implemented in clinical practice over recent decades.21 However, concerns have emerged regarding their potential to induce inflammatory responses post vaccination and their inconsistent immunogenic profiles. Recombinant vaccines have used genetically detoxified toxins, such as toxin mutants, which are not biologically active but retain immunogenicity.22,23 Nevertheless, a significant limitation of mutant-based vaccines is related to the potential for the reversion of safe molecules to toxic forms, raising safety concerns regarding their clinical application.24
In recent years, cell membrane–encapsulated nanoparticles (NPs) have been widely used in biomedical research, integrating the diverse functionalities of cellular membranes with the design flexibility of synthetic nanomaterials.25–27 Cell membrane–encapsulated NPs exhibit immune escape, prolonged circulation, and targeted release capabilities. Poly(lactic-co-glycolic acid) (PLGA) NPs can load antigens in various ways (eg, conjugation, encapsulation, and adsorption) to protect them from environmental influences.28 Cell membrane–encapsulated NPs exhibit a favorable antigen depot effect at the injection site and prolonged antigen retention time in lymph nodes.29–31 Moreover, these NPs demonstrate enhanced in vivo residence time, facilitate uptake by antigen-presenting cells (APCs), and improve T-cell responses.32,33
The strategy of vaccine to activate APCs for robust immune system stimulation is well developed. Dendritic cells (DCs) and macrophages are the primary targets. However, when immunized with virus-like particles or inactivated influenza viruses, B cells are the dominant APCs that activate naive CD4+ T cells.34,35 B cells exhibit specific recognition and binding of antigens and have the advantage of presenting low-dose of antigens, which are useful for designing new type vaccines. Onchocerca volvulus activation-associated secreted protein-1 (Ov-ASP-1) is a nontoxic protein, which demonstrates good adjuvant effects and binds to B cells in vitro, inducing a strong humoral immune response.36,37
In this study, based on the B-cell targeting capability of Ov-ASP-1 and the pore-forming activity of ETX, an ASP–ETX fusion protein was genetically engineered; this fusion protein had pore-forming activity and B-cell targeting capability. The ETXY196E mutant was generated by mutating tyrosine at position 196 to glutamate and ligating with the C terminus of the toxin. ETXY196E achieved soluble expression in the Escherichia coli expression system and maintained good conformation, showing good immunogenicity and protection.38 NPs loaded ETXY196E attenuator protein and MDCK cell membranes-encapsulated NPs vaccine was prepared (MNP). Meanwhile, MNP was used to adsorb recombinant ASP–ETX toxin to prepare a novel ETX NP vaccine that adsorbed structurally intact toxin and loaded attenuated antigen (MNP–ASP–ETX) with the ability to target B cells and exhibit good immunogenicity.
Materials and Methods
Materials
MDCK cells used in this study were preserved by State Key Laboratory of Pathogen and Biosecurity, Academy of Military Medical Sciences, and cultured in Dulbecco’s Modified Eagle Medium supplemented with 10% fetal bovine serum (FBS) (Gibco). In this study, 6–8-week-old female BABL/c mice of Specific Pathogen Free grade were purchased from Sipeifu (Beijing, China). PLGA were purchased from LACTEL Absorbable Polymers (Birmingham, USA). Polyvinyl alcohol (PVA) were purchased from Sigma (St. Louis, MO, USA). Moreover, 200-nm polycarbonate membranes were purchased from Avanti Polar Lipids (Alabama, USA). Cy5.5-antibody conjugation kits were purchased from Bioss (Beijing, China).
Expression and Purification of Recombinant Toxin Proteins
ETXY196E (Figure S1), HIS-ETX (His-tagged recombinant epsilon toxin) (Figure S2), and GST-ETX (GST-tagged recombinant epsilon toxin) (Figures S3 and S4) recombinant strains were stored in our laboratory, and the target proteins were purified via affinity chromatography following established laboratory protocols. The ASP–ETX DNA fragment was subcloned into a pET-28a (+) expression vector (Figure S5). The constructed plasmid was transformed into BL21 (DE3) using heat-shock method and expressed and purified as previously described (Figure S6).39
Preparation of the Nanoparticle Vaccines
According to previous laboratory methods, NPs were prepared using double emulsion-solvent evaporation method.17,40 In brief, 1.2 g polyvinyl alcohol (PVA) was dissolved in 320 mL of ultrapure water and 0.24 g PLGA and 10 mg purified ETXY196E protein were dissolved in 24 mL of dichloromethane (DCM). The dissolved PLGA-ETXY196E solution was thoroughly mixed with the PVA solution, and homogenized into a stable emulsion via sonication. The mixture was then stirred overnight in a fume hood, and centrifugation was performed at 7000 × g for 10 min to remove precipitates. The supernatant was centrifuged at 20,000 × g for 40 min to obtain PLGA NPs. The NPs were washed three times with ultrapure water to remove residual PVA by centrifugation at 20,000×g for 30 min. Finally, the NPs were resuspended in 10 mL of ultrapure water and lyophilized overnight in a vacuum freeze-drying process to obtain dried PLGA NPs.
MDCK cells were collected using a trypsin treatment and scraper. Cells were centrifuged at 1000 × g at 4°C for 10 min, washed three times using 1 × phosphate-buffered saline (PBS), then resuspended in 2 mL of ultrapure water. MDCK cells were broken under osmotic pressure, and the broken MDCK cells membranes were washed three times with 1 × PBS to remove residual cellular inclusions. MDCK cell membranes were resuspended in 1 mL of 1 × PBS and mixed with 10 mg NP. The mixture was then extruded through 200 nm polycarbonate membranes to prepare MDCK cell membrane–encapsulated NPs (MNPs).
Encapsulation Efficiency of Nanoparticles
The encapsulation efficiency of NP was calculated using the following equations:
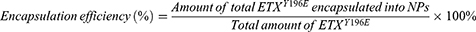
The NP encapsulation efficiency of antigen was determined via extractive hydrolysis.41 In brief, 10 mg of NP were added to 1 mL of acetonitrile, mixed until completely dissolved, and centrifuged at 16,000 × g for 10 min. Then, the supernatant was discarded. The precipitate was dried overnight in a 37°C vacuum drying oven then resuspended in 1 mL of PBS and centrifuged at 1000 × g at 4°C for 10 min to collected supernatant. Then, 1 mL of 0.1 M sodium hydroxide (NaOH) was added to precipitate. The supernatant was collected via centrifugation at 16,000 × g for 10 min, combined with the collected PBS supernatant, and supplemented with 100 μL of 0.1 M hydrochloric acid (HCl). The encapsulation of NP was determined using BCA and polyacrylamidegel electrophoresis (PAGE).
The Animal Studies Using Mice Model
Mice were housed at the Academy of Military Medical Sciences and fed sterile commercial mouse food (Beijing, China). In Table 1, based on whether the MDCK cell membrane–encapsulated NP vaccine and the type of adsorbed toxin, the immunization group can be classified into six groups: PBS negative control group and MDCK cell membrane–encapsulated NP adsorbed ASP–ETX toxin group (MNP–ASP–ETX), MDCK cell membrane–encapsulated NP adsorbed HIS-ETX toxin group (MNP–HIS-ETX), MDCK cell membrane–encapsulated NP group (MNP), NP group (NP), and ETXY196E attenuated mutant protein mixed with aluminum adjuvant (ETXY196E + Al) as the immune control group. Immunization was performed via subcutaneous (s.c.) immunization, and the immunization dose was 10 μg at a volume of 200 μL, injected at two points. The amount of antigen of MNP–ASP–ETX and MNP–HIS–ETX group was calculated as the amount of surface-adsorbed ASP–ETX or HIS-ETX + loaded ETXY196E = 10 μg.
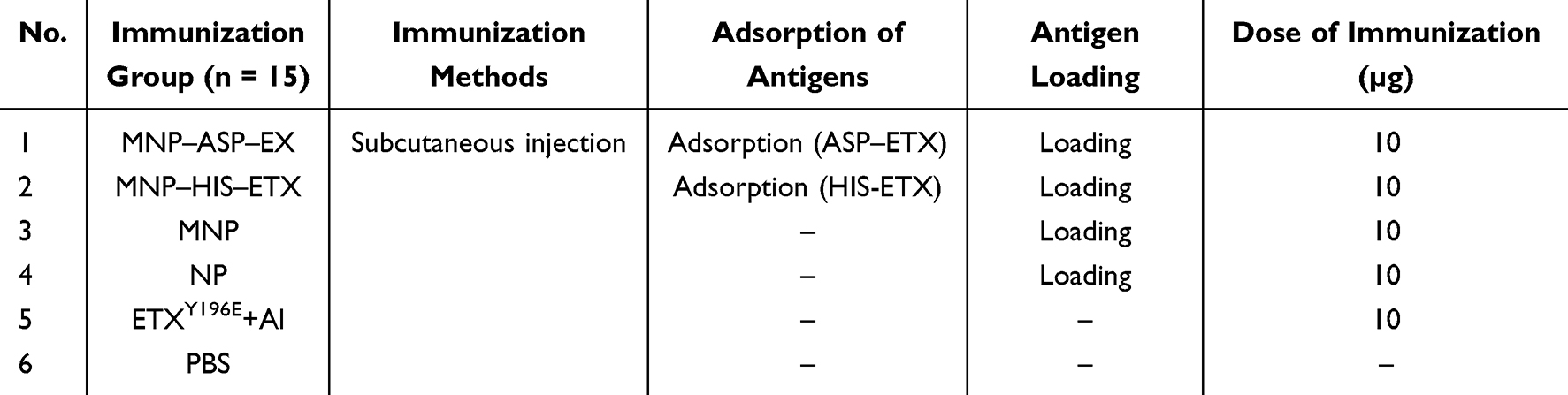 |
Table 1 Immunization Experiment Groups |
Cytotoxicity Assay
The MTS method was used to evaluate cytotoxic activity. MDCK cells were seeded into 96-well plates at a density of 1×105 cells/mL and incubated at 37°C for 24 h. After three washes with PBS, cells were exposed to a series of samples (Figures S7 and S8) with different concentrations and incubated for 1 h at 37°C. Next, the culture medium was removed, and plates were washed three times with PBS. MTS was added to the plate wells, and the toxicity was estimated by measuring absorbance at A492 nm. Each experiment was performed in triplicate.
Enzyme-Linked Immunosorbent Assay (ELISA)
ETX-specific IgG (Solarbio, China), IgG1 (Abcam, UK), and IgG2a (Abcam, UK) levels in the serum were determined via ELISA. In brief, 96-well ELISA plates were coated with 5 μg/mL HIS-ETX in ELISA coating buffer at 4°C. The following day, the plates were washes for three times with PBS-Tween 20 (PBST) and blocked with 5% bovine serum albumin for 1 h at 37°C. Then, the plate was incubated with 100 μL 10-fold serial diluted serum for 1 h at 37°C. After three washes with PBST, the plates were incubated with horseradish peroxidase-coupled goat anti-mouse IgG (1:5000), IgG 1 (1:5000), and IgG 2a (1:5000) at 100 μL/well for 1 h at 37°C. The plates were washed three times with PBST. TMB colorimetric substrate (Solarbio, China) was added at 100 µL/well and incubated for 10 min. Finally, an ELISA stop solution (Solarbio, China) was used to stop the reaction. Absorbance was determined at A450 nm using a microplate reader.
Determination of Neutralizing Antibody Titer (TNA)
MTS was used to evaluate the CT50 of GST-ETX (66.25 ng/mL) (Figure S9) in MDCK cells. Sera collected after the third immunization were serially diluted twofold at a beginning 1:2 dilution for TNA detection. Each sample diluted with 10 × CT50 GST-ETX were mixed in equal volumes and incubated for 1 h at 37°C. MDCK cells were spread on 96-well plates at a density of 1×105 cells/mL and incubated at 37°C for 24 h. After three washes with PBS, toxin-serum mixtures were added to MDCK cells and incubated for 1 h. MTS was added to the plate wells, and toxicity was estimated by measuring absorbance at A492 nm. Each experiment was performed in triplicate. TNA (ED50) were expressed as the reciprocal of the highest serum dilution, showing ≥50% cells.
Challenges of Immunized Mice
One week after the third immunization, immunized mice were challenged with 10 × CT50 and 100 × CT50 GST-ETX via intraperitoneal injection. Body weight and survival were monitored daily.
In vitro Neutralization Assay
In vitro neutralizing animal toxicity assay. Sera were diluted 20-fold and mixed with 2 × LD50 GST-ETX (LD50 = 11.86 μg/kg). This serum–toxin mixture was incubated at 37°C for 1 h and injected intraperitoneally into blank mice at a volume of 500 µL, and survival was monitored daily for 3 days.
In vitro neutralizing cytotoxicity assay. Sera were diluted 20-fold and mixed with 2 × CT50, 4 × CT50, 8 × CT50, 16 × CT50, 32 × CT50 and 64 × CT50 GST-ETX. This serum–toxin mixture was incubated at 37°C for 1 h, added to MDCK cells, and incubated for 1 h at 37°C in a 5% CO2 incubator. After three washes with PBS, MTS was added to the plate and the mixture was incubated for 3 h. Toxicity was estimated by measuring absorbance at A492 nm. Each experiment was performed in triplicate.
Recombinant Toxin Protein Fluorescent Labeling
A Cy5.5-antibody conjugation kit (Beijing, China) was used to label the ETXY196E protein. Cy5.5-ETXY196E was encapsulated into NPs to prepare Cy5.5 fluorescently labeled NPs. ETXY196E was then diluted to a final concentration of 1 mg/mL in 1× PBS. We centrifuged the lyophilized Cy5.5 in a powder tube (provided in the kit) at 8000 × g for 30s to pellet the powder. Next, we added 50 μL of dimethyl sulfoxide (DMSO) to the tube and mixed thoroughly until the Cy5.5 dye was completely dissolved. ETXY196E solution (100 μL) was labeled using 4 μL of Cy5.5 fluorescent dye, and the resulting mixture was incubated in the dark at 37°C and 150 rpm for 1 h. A purification column was then centrifuged at 3000 × g for 2 min before being transferred to a new tube. At this point, we loaded 100 μL of the Cy5.5-ETXY196E mixture onto the column. To remove free dye, the solution was centrifuged twice at 3000 × g for 2 min at 4°C. The purified Cy5.5-ETXY196E solution was then collected and stored at 4°C until further use. MDCK cell membrane was coated with fluorescently labeled NPs to generate fluorescently labeled NP vaccines.
In vivo Imaging
Mice were injected s.c. at a dose of 10 μg of Cy5.5 labeled ETXY196E + Al, NP, and MNP (Cy5.5-labeled ETXY196E encapsulated in both NP and MNP was 10 μg) in a volume of 200 μL and injected at two points. The fluorescence signal intensity at different time points was documented and calculated using IVIS Spectrum (PerkinElmer, Waltham, MA, USA) with the excitation light set at 640 nm and the emission light set at 700 nm.
Mice were injected s.c. with 2 μg of Cy5.5 labeled ETXY196E + Al, NP, and MNP (Cy5.5-labeled ETXY196E encapsulated in both NP and MNP was 10 μg) in an injection volume of 200 μL at 2 points. The spleen and lymph nodes were removed at different time points, and the fluorescence signal intensity was observed.
Histological Examination of the Brain and Kidney
After challenging with 10 × LD50 GST-ETX, the brains and kidneys were separated, fixed in a 4% formaldehyde solution for 24 h, embedded, stained with hematoxylin and eosin (H&E), and observed via slicing.
Immune Cell Levels in the Spleen
The antibodies used in this study were as follows: CD45 (Alexa Fluor 700, 56–0451-82, Thermo Fisher), CD3 (PerCP-eFluor 710, 46–0032-82, Thermo Fisher), CD4 (BV785, 100552, Biolegend), CD8 (BV510,100752, Biolegend), CD44 (BV480, 566116, BD), CD62L (APC-Fire 750, 104450, Biolegend), CD19 (FITC, 115505, Biolegend), CD80 (BV421, 104726, Biolegend), CD273 (PE-Cy7, 107214, Biolegend), CD45R (PerCP-Cy5.5, 103236, Biolegend), CD138 (BV605, 142516, Biolegend), CD95 (PE, 152608, Biolegend), GL7 (AF647, 144606, Biolegend), CD45 (BV510, 103138, Biolegend), CD3 (APC-Fire 810, 100268, Biolegend), CD3 (BV605, 564009, BD), CD4 (R718, 566939, BD), CD8 (PerCP-Cy5.5, 100734, Biolegend), CD80 (PE-Fire640, 104759, BD), CD45R (BV785, 103246, Biolegend), CD138 (BV421, 142508, Biolegend), anti-mouse CD16/32 (BD, 553141, 2.4G2), True-Stain Monocyte Blocker (Biolegend, 426102), Brilliant Stain Buffer (BD, 563794), and anti-mouse CD16/32 (BD, 553141, 2.4G2). All fluorescent antibodies listed above were centrifuged at 10,000× g for 5 min at the relevant quantity before use. They were then prepared with True-Stain Monocyte Blocker (Biolegend, 426102). Brilliant Stain Buffer (BD, 563794) was used to create a staining antibody mixture. An analysis of immune cells in the spleen was performed in mice that survived for 7 days following a 10 × LD50 challenge. After cervical dislocation, the spleen was removed, minced in a biosafety cabinet, gently ground, and suspended in 1640 medium containing 10% FBS, 0.1 mM 2-mercaptoethanol, 100 U/mL penicillin, and 100 U/mL streptomycin to make a single cell suspension. Approximately 2×106 cells were placed in a flow tube, 2 µg Fc was added, and incubated for 10 min at room temperature away from light. The premixed staining antibody mixture was added and incubated for 25 min at room temperature away from light. Then, 2 mL wash solution was added, and the mixture was centrifuged at 400 × g for 5 min to discard the supernatant. Approximately 1 mL of Dead Dye Working Solution (Zombie NIR™ Fixable Viability Kit, Biolegend, 423105) was added, mixed well, and incubated for 10 min at room temperature. Then, 2 mL of wash solution was added and centrifuged at 400 × g for 5 min. Subsequently, 0.2 mL of wash solution was added, and the samples were obtained using an Aurora spectral flow cytometer (Cytek). Data were analyzed using SpectroFlo (Cytek, V3.2.1) software.
Statistical Analyses
All data were analyzed using one-way ANOVA on GraphPad Prism software. All data are expressed as mean ± SD. Statistical significance was indicated by p-values <0.05 (*), <0.01 (**), <0.001 (***), and <0.0001 (****); nonsignificant differences were indicated by p-values ≥0.05 (ns).
Results
Synthesis and Characterization of Nanoparticle
Following the previous method, the PLGA copolymer was used to generate loaded ETXY196E protein (NP) using the water/oil/water (w/o/w) double emulsion method. The extracted MDCK membranes were coated on NP surface via sonication and extrusion through a 200-nm porous polycarbonate membrane (MNP). Transmission electron microscopy (TEM) images revealed that the NP exhibited well-structured spherical structures, and the MNP clearly exhibited a double-layer core-shell structure (Figure 1A and B). The average particle size was determined using a Flow NanoAnalyzer. Results showed that the average particle size of NP was approximately 160 nm, and after MDCK cell membrane coating, the average particle size increased to approximately 190 nm, which was approximately 30 nm larger than that of bare NPs (Figure 1C). Dynamic light scattering (DLS) analysis revealed that the zeta potentials of NP and MNP were −13 mv and −5 mv, respectively (Figure 1D). The polydispersity index (PDI) of NP and MNP was less than 0.2, indicating that they had good dispersion in PBS (Figure 1E). These results confirmed that the MDCK membrane was coated onto NP, and MDCK cell membrane–encapsulated nanoparticles were successfully prepared. To determine the encapsulation efficiency of NP in ETXY196E mutant proteins, extractive hydrolysis was used to determine the concentration of ETXY196E encapsulated in NP. Results showed that 1 mg PLGA nanoparticles could encapsulate 50 μg ETXY196E, and the encapsulation efficiency of NP were ~37.5% (Figure 1F). To test the adsorption of ASP–ETX toxins by MNP, HIS-ETX was used as experimental control group. Approximately 2 mg of MNP was treated with a series of different concentration of ASP–ETX and HIS-ETX. Then, the mixture of MNP–toxin mixture was coincubated with MDCK cells for 1 h, and the PBS–toxin mixtures were used as negative controls. Next, we determined the toxin concentration required to achieve 50% cell viability (CT50) for both the PBS-HIS-ETX control group and MNP–ASP–ETX experimental group. The adsorbed toxin quantity was then derived by calculating the concentration differential between these two groups as per the following equation: Adsorbed toxin (μg) = CT50 (PBS-ASP–ETX) − CT50 (MNP–ASP–ETX). The adsorption capacity of MNP toward HIS-ETX toxin was calculated as per the same protocol described for ASP–ETX (see above). Results showed that 2 mg of MNP adsorbed 187 ng of ASP–ETX and 1.45 ng of HIS-ETX (Figure 1G and H). Adsorption of ASP–ETX by MNP was higher than that by HIS-ETX, which was facilitated to subsequent immunization experiments. The amount of the prepared nanoparticle vaccine was sufficient for a complete immunization procedure and guaranteed the stability and uniformity of vaccine batches.
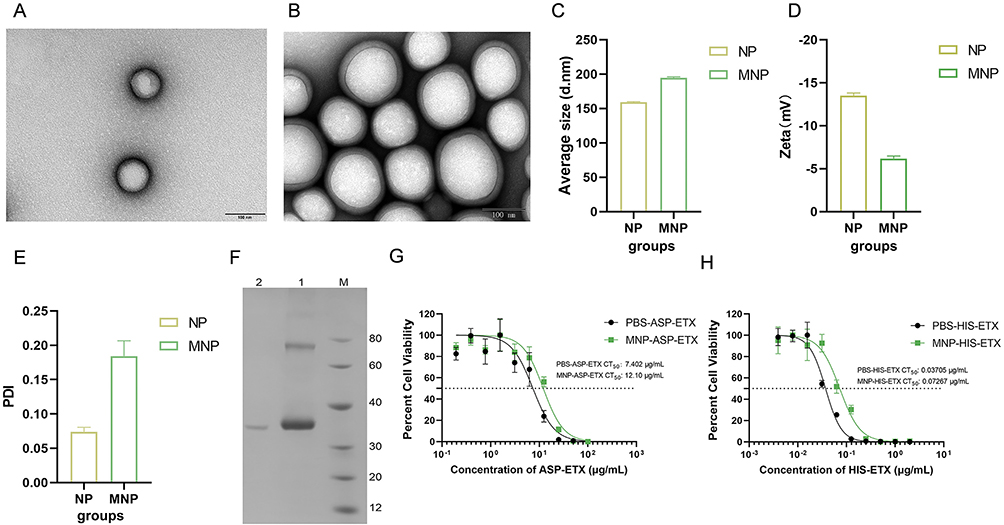 |
Figure 1 Characterization of nanoparticle vaccine. (A and B) Representative TEM images of NPs and MNPs. (C) Nanoparticle diameter measurement using a Flow NanoAnalyzer. (D) DLS measurements of the zeta potential (Zeta, mV) of the nanoparticle vaccine. (E) The PDIs of NP and MNP were measured using DLS. (F) SDS-PAGE analysis of the encapsulation efficiency of ETXY196E. (M) molecular weight marker. Lanes 1, encapsulated ETXY196E in NP, lanes 2, free ETXY196E in supernatant. (G and H) Adsorption rates of MNP to ASP–ETX and HIS–ETX. Different ASP–ETX and HIS–ETX concentrations were treated with 2 mg of MNP or PBS for 1 h at 37°C. Then mixture was coincubated with MDCK cells, and the survival of MDCK cells was measured. |
Antigen Persistence at Injection Sites and Antigen Transport into Draining Lymph Nodes
The antigen depot effect can extend the exposure time of antigen, enhancing its capture by the immunity system.42 It has been shown that PLGA nanoparticles also have a favorable adjuvant effect.30 To investigate whether NP and MNP have a depot effect, the persistence and lymph node drainage ability of antigens in vivo over time were observed using an imaging system (Figure 2A). First, the release of antigens at the injection site in mice was examined. 3 hours after injection, the injection sites of all groups exhibited strong fluorescent signals. At 12 h, the fluorescence signal of ETXY196E + Al and MNP gradually decreased, and the rate of decrease of ETXY196E + Al was more rapid than that of MNP. At 12–48 h, the fluorescence intensity signals of NP and ETXY196E + Al showed a slow decrease, whereas the fluorescence signal intensity of MNP was not changed. Until 72 h, the fluorescence signals of mice showed a slow decline. Nearly half of the antigen in the ETXY196E + Al group was released at 12 h. In contrast, NP and MNP maintained the antigen at the injection site until 72 h, the fluorescence intensity slowly decreased to half the level of initial injection. The trend of fluorescence signal of MNP was more stable than that of the other two groups, indicating MNP has the potential for long-term antigen release. However, ETXY196E + Al could persist at the injection sites for more than 320 h (14 days), whereas NP and MNP remained for 246 h (10 days) (Figure 2B and C).
 |
Figure 2 Antigen release and lymph nodes drainage. (A) Schematic illustration of the experiments. (B) BALB/c mice were injected s.c. with 10 μg MNP, NP, or ETXY196E + Al. The fluorescence intensity at the injection site was detected at different time points. (C) Comparison of release time at the injection site. Antigen persistence at the injection sites was evaluated and documented using an in vivo imaging system at the indicated time points. Trend changes in fluorescence intensity at different times (average fluorescence intensity /first-time average fluorescence intensity × 100%) (n = 3). (D) Comparison of the fluorescence intensity of the lymph nodes and spleen. (E) Changes in the total fluorescence intensity of lymph nodes at different time points. (F) Changes in the total fluorescence intensity of spleen samples at different time points (n = 3). |
NP and MNP exhibit different antigen slow-release effects compared to ETXY196E+Al. Whether these differences affected antigen transport to draining lymph nodes was determined. Mice were injected s.c. with 2 μg ETXY196E + Al, NP, or MNP. Lymph nodes were collected from mice at different time points (Figure 2D) The changes in fluorescence signals showed that the ETXY196E + Al group antigen aggregated rapidly in the spleen at 24 h after injection, and the fluorescence signals in the lymph nodes reached their maximum value at 6 h. The lymph nodes fluorescence signals of the NP and MNP groups reached the maximum values at 48 h, with the MNP exhibited a higher intensity compared to NP and ETXY196E + Al, suggesting improved lymph node drainage potential (Figure 2E and F). Results suggested that MNP antigens can be more efficiently drained to lymph nodes, indicating a longer interaction time with APCs.
Safety Evaluation of the Nanoparticle Vaccine
Vaccine safety is one of the primary prerequisites for vaccine development. In previous studies, ETXY196E triggered a strong immune response in mice, but it had side effects, such as rapid weight loss, loss of appetite, and even death at high doses.38 The safety of ETX nanoparticle vaccine was evaluate in vitro and in vivo. First, the in vitro safety of ETX nanoparticle vaccine was evaluated using a cytotoxicity assay (Figure 3A). MDCK cells are highly sensitive cells to ETX and are commonly used in ETX cytotoxicity assays.43 A series of ETX nanoparticle vaccine were incubated with MDCK cells. Results show that concentrations as high as 125 μg/mL were not toxic to MDCK cells (Figure 3C).
 |
Figure 3 Safety evaluation of ETX nanoparticle vaccine in vitro and in vivo. (A) Schematic illustration of the in vitro cytotoxicity assay. (B) Schematic illustration of in vivo safety experiments. (C) The ETX nanoparticle vaccine and ETXY196E were diluted to 125 μg/mL, sequentially diluted twofold to multiple gradients, then coincubated with MDCK cells for 1 h at 37°C. Survival of MDCK cells was measured. (D and E) Survival and average weight changes of immunized mice. Mice were injected s.c. with 10 μg of ETX nanoparticle vaccine or ETXY196E + Al. Their body weight and survival were recorded at 2- to 3-day intervals. Each point represents the current total weight relative to the original total weight (n=10). (F and G) Chemical composition analysis of serum. Serum was collected from mice injected with 10 μg antigen after 35 days; this was then subjected to analysis chemical composition (n = 3). (H) Histopathological sections of the brain and kidney tissues of immunized mice. Scale bar, 200/50 μm (n = 3). Statistical significance of differences in group means as calculated by one-way ANOVA: p ≥ 0.05 (not significant, ns). |
Based on the MDCK cell membrane-encapsulates nanoparticle and the toxin type adsorbed on MNP, the groups were categorized into MNP–ASP–ETX, MNP–HIS–ETX, MNP, NP, ETXY196E + Al control, and PBS negative control groups. Different groups of antigens were injected into the mice, their body weights were examined for 35 successive days, and their kidneys and brains were examined for pathologically analysis (Figure 3B). Body weights and survival were monitored throughout the immunization period. All mice survived well (Figure 3D) and exhibited a steady increase in body weight (Figure 3E). Notably, the body weight of ETXY196E + Al mice showed a decrease after the first immunization. Furthermore, serum samples were collected from mice after the third immunization for blood biochemical assays. Indicators of urea, Ca, and K in blood are thought to reflect kidney damage (Figure 3F), whereas alanine aminotransferase (ALT), alkaline phosphatase (ALP), aspartate transaminase (AST), and total bilirubin in the blood were thought to reflect liver damage (Figure 3G). All indicators were normal in each group and were not significantly different from those of PBS group. Whether the ETX nanoparticle vaccine caused damage to the brain and kidney of mice was examined using H&E staining in the immunized group. Histopathological results showed no pathological damage (Figure 3H) (Figure S10), indicating that the ETX nanoparticle vaccine and the ETXY196E + Al groups had good and long-term safety in vivo. Observing the condition of mice in each immunization group, there was obvious s.c. nodule generation in ETXY196E + Al, whereas this condition was not found in nanoparticle vaccine group, thereby proving the safety of mice in the nanoparticle vaccine group (Figure S11).
Nanoparticle Vaccine Induced Functional Antibody Responses in Mice
To investigate the immunization effects of ETX nanoparticle vaccine, immunization experiments were performed to detect antibody titers in immunized mice. The immunization groups are shown in Table 1. Immunization was performed on days 0, 14, and 28, the immunization dose was 10 μg (Figure 4A). The titers of ETX-specific IgG, IgG 1, and IgG 2a antibodies were measured using ELISA. The results showed that after the second immunization, serum IgG titers elicited by NP were significantly higher than other groups. After the third immunization, ETX-specific IgG antibody titers of the all groups reached 104, indicating that ETX nanoparticle vaccine have almost as good immunogenicity as ETXY196E + Al control groups and elicit a stronger immune response (Figure 4B). To explore the differentiation direction of helper T cells, ETX-specific IgG 1 and IgG 2a antibody titers were measured after the third immunization. The IgG 2a/IgG 1 ratio represents the polarization effect of the immune response in both Th1 and Th2. All groups exhibited comparable IgG 1 antibody titers (Figure 4C), and the IgG 2a antibody of ETX nanoparticle vaccine was higher than that of ETXY196E + Al groups (Figure 4D). The IgG 2a/IgG 1 ratio of ETXY196E + Al suggested that they elicited Th2-biased immune responses, whereas MNP–ASP–ETX, MNP–HIS–ETX, MNP, and NP exhibited a higher IgG 2a/IgG 1 ratio than ETXY196E + Al, suggesting that they can elicit Th2-biased immune but with a slight Th1 bias (Figure 4E).
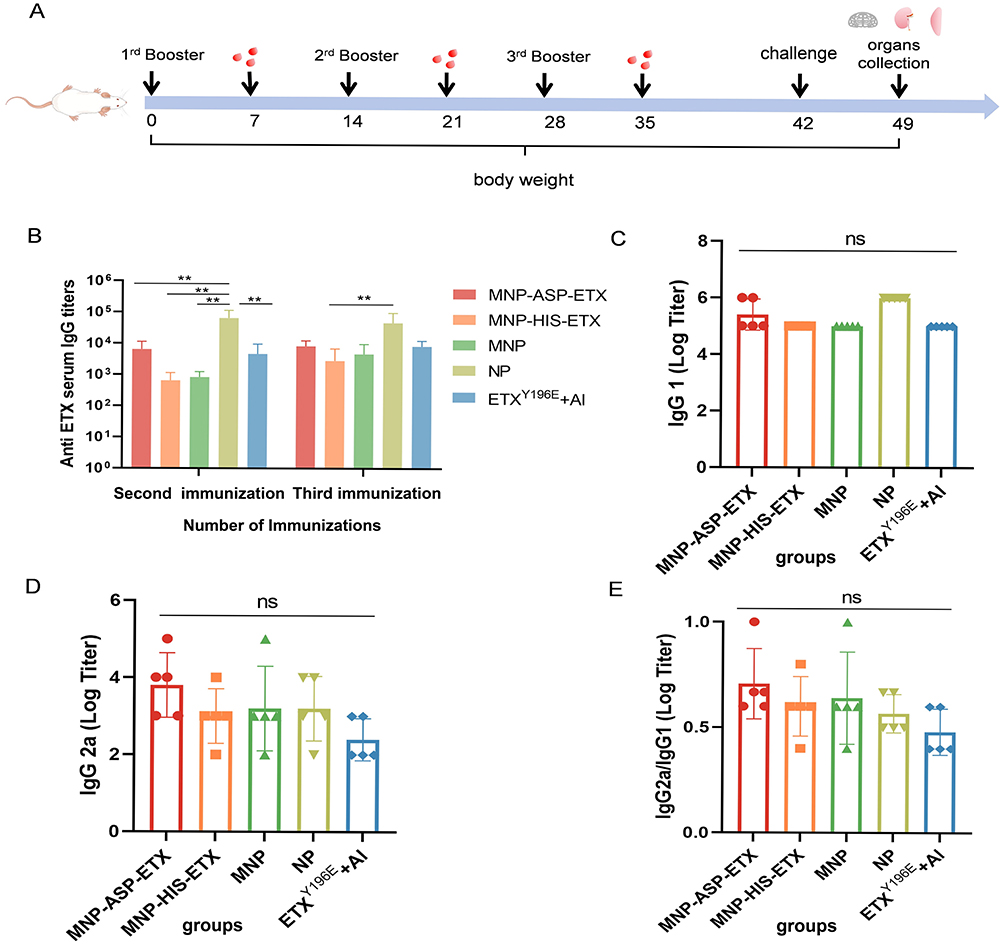 |
Figure 4 Immune response induced by ETX nanoparticle vaccine in mice. (A) Schematic illustration of immune procedure at different time points. Mice was immunized s.c. with the same dose of 10 μg for three immunizations on days 0, 14, and 28, and serum samples from different groups were collected at appropriate time points. (B–D) Detection of ETX-specific antibodies IgG (B), IgG 1 (C) and IgG 2a (D) using ELISA. (E) Ratio of IgG 2a/IgG 1 (n = 5). Statistical significance of differences in group means as calculated using one-way ANOVA: p < 0.001 (**), p ≥ 0.05 (not significant, ns). |
Protective Evaluation of the Nanoparticle Vaccine
Protective efficacy is an important aspect of evaluating vaccine quality. Thus, after the third immunization, immunized mice were challenged with 10 × LD50 and 100 × LD50 GST-ETX and their body weights were monitored for 7 days. The body weights of the surviving mice showed slight or no changes (Figure 5A). Mice in the PBS group died 6 h after challenged; in contrast, MNP–ASP–ETX, MNP–HIS–ETX, NP and ETXY196E + Al immunized mice resisted 100 × LD50 GST-ETX challenge. However, two mice in the MNP group died, with a survival rate of 60% (Figure 5B and C).
 |
Figure 5 Protective evaluation of ETX nanoparticle vaccine. (A) Body weight was monitored for 7 days after challenge. (B and C) Survival of mice challenged with GST-ETX. After the third immunization, mice were randomly divided into two groups, and each group was injected intraperitoneally with 500 μL 10 × LD50 (B) and 100 × LD50 (C) GST-ETX. (D) TNA. (E) In vitro neutralization cytotoxicity assay. Serum collected after the third immunization was diluted 20-fold then incubated with 2 × CT50, 4 × CT50, 8 × CT50, 16 × CT50, 32 × CT50 and 64 × CT50 GST-ETX. The toxin-serum mixture was then coincubated with MDCK cells at 37°C for 1 h, after which the survival rate of MDCK cells was calculated (n = 3). (F) In vitro neutralization of animal toxicity. The serum was diluted 20-fold, then incubated with 2 × LD50 GST-ETX at 37 °C for 1 h. The resulting mixture was then injected intraperitoneally into blank mice, and their survival rate was observed (n = 5). (G) Histopathological sections of brain and kidney tissues from mice challenged with 10 × LD50 GST-ETX. Scale bar, 200/50 μm (n = 3). Statistical significance of differences in group means as calculated using one-way ANOVA: p < 0.0001 (****). Red arrows: Cellular swelling, cytoplasmic laxity; Black arrows (brain): Vascular bruises; Black arrows (kidney): Hydropic degeneration in renal tubular epithelial cells; Green arrows: Interstitial bruising and cellular swelling. |
Neutralizing antibody titer (TNA) levels in sera were determined using a cytotoxicity assay. TNA (ED50) were expressed as the reciprocal of the highest serum dilution, showing ≥50% cells. The TNA of MNP–ASP–ETX, MNP–HIS–ETX, MNP, and NP reached 64, whereas that of ETXY196E + Al reached 512, which was significantly higher than that of the ETX NP vaccine (Figure 5D). Results suggested that the protective effect of the ETX NP vaccine was lower than that of ETXY196E + Al.
Therefore, an in vitro neutralization assay was performed to evaluate the protective effect of the ETX nanoparticle vaccine. In vitro neutralization cytotoxicity assay, the serum was diluted 20-fold and mixed with different concentrations of GST-ETX, and the toxin-serum mixture was coincubate with MDCK cells. MNP–ASP–ETX, MNP–HIS–ETX, and NP completely neutralized 16 × CT50 GST-ETX and protect more than 50% cells from 32 × CT50 GST-ETX. MNP completely neutralized 8 × CT50 GST-ETX and protect 90% of cells from 16 × CT50 GST-ETX. ETXY196E + Al could neutralize 32 × CT50 GST-ETX and protect 90% of cells from 64 × CT50 GST-ETX (Figure 5E).
In vitro neutralization of animal toxicity assay shows that serum in MNP–ASP–ETX, MNP–HIS–ETX, MNP, NP and ETXY196E + Al can protect naive mice from 2 × LD50 GST-ETX after a 20-fold dilution (Figure 5F). The results of the in vitro neutralization assay were consistent with TNA, indicating that the ETX nanoparticle vaccine exert protective effects on cells and animals; however, its effectiveness was lower than that of the ETXY196E + Al immune control group. The brain and kidneys are the main target organs attacked by ETX.15 For histopathological analysis, H&E staining was used to observe the brain and kidneys of mice. Immunized mice subjected to 10 × LD50 GST-ETX challenge were used as the experimental group, and PBS-injected mice were used as negative controls. Blank mice injected intraperitoneally with 10 × LD50 GST-ETX were positive control (Figure 5G) (Figure S12). Mice in the positive control group injected with 10 × LD50 GST-ETX showed pathological lesions in the brain and kidneys. The brain of control group exhibited more vascular bruises (black arrows) across all regions of the brain tissue, and a small number of neurons showed sign of edematous (red arrows), characterized by cellular swelling and cytoplasmic laxity. The kidney of control group contained renal tubular epithelial cells. Hydropic degeneration (black arrows), cellular swelling, and more bruising are seen in the interstitium (green arrows). In contrast, the ETX nanoparticle vaccine and the ETXY196E + Al group showed no lesions in the brain and kidneys, indicating that the ETX NP vaccine group had good in vivo protective properties.
Immune Cell Levels in the Spleens of Vaccinated Mice
The ETX nanoparticle vaccine induced high IgG antibody levels comparable to those elicited by ETXY196E + Al, but its TNA and protective efficacy were lower than those of ETXY196E + Al. To investigate whether ASP–ETX plays a role in targeting B cells, the spleens of surviving mice challenged with 10 × LD50 GST-ETX were isolated and the levels of central memory CD4+ T cells (CM CD4+), central memory CD8+ T cells (CM CD8+), effective memory CD4+ T cells (EM CD4+), effective memory CD8+ T cells (EM CD8+), B lymphocytes, plasma cells, memory B cells (MBCs), and germinal center (GC) B cells were assessed via flow cytometry (Figure 6A–H). The ETX NP vaccine and ETXY196E + Al elicited high levels of CM CD4+ T cells (Figure 6A), and the proportion of EM CD4+ T cells (Figure 6B) and EM CD8+ T cells (Figure 6D) in the MNP and NP immune groups were significantly higher than those in the other groups, suggesting that the trend is to induce a T-cell-based cellular immune response. Following the adsorption of ASP–ETX and HIS-ETX toxin, MNP–ASP–ETX and MNP–HIS–ETX induced the production of high levels of CM CD8+ B cells (Figure 6C) and increased numbers of GC B cells (Figure 6F), plasma cells (Figure 6G), and MBCs (Figure 6H). This trend was consistent with that of ETXY196E + Al, indicating that the adsorption of ASP–ETX activated B cells; however, the effect was not significant and was weaker than that of ETXY196E + Al.
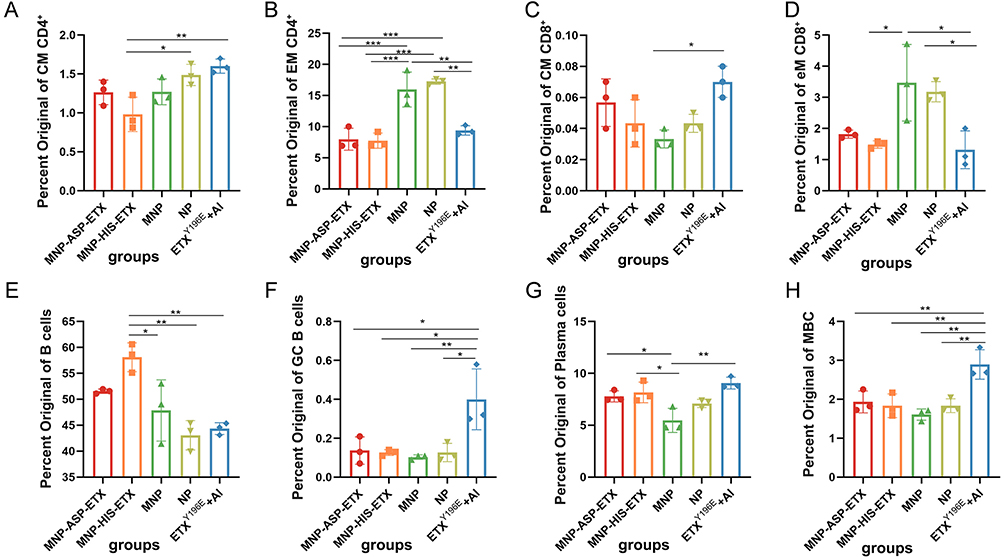 |
Figure 6 Levels of different immune cells in mouse spleens after challenge. (A) Central memory CD4+ T cells. (B) Effective memory CD4+ T cells. (C) Central memory CD8+ T cells. (D) Effective memory CD8+ T cells. (E) B cells. (F) Germinal center B cells. (G) Plasma cells. (H) MBCs (n = 3). Statistical significance of differences in group means as calculated by one-way ANOVA: p <0.05 (*), p<0.01 (**), p<0.001 (***). |
ETXY196E + Al induced a large number of GC B cells (Figure 6F) and MBCs (Figure 6H). The GC is a lymphocyte organ formed during immunization and is the site of clonal proliferation and affinity maturation of B cells. The process of affinity maturation leads to the production of high-affinity antibodies. Therefore, the high number of GC B cells in the ETXY196E + Al group implied that they may have higher levels and affinity for neutralizing antibodies. However, the ETX nanoparticle vaccine had a lower number of GC B cells than ETXY196E + Al, which provides a theoretical basis for the observed TNA and reduced protection of ETX nanoparticles vaccine compared with ETXY196E + Al.
Long-Term Immunization Effects of Nanoparticle Vaccines
In vivo imaging experiments showed that MNP released antigen more slowly and stably. Therefore, mice that survived for 2 months after third immunization were randomly selected, and blood samples from the tail vein were collected to determine antibody titers and protective effects (Figure 7A). First, the IgG antibody titers of ETX NP vaccine increased to 105, whereas that of ETXY196E + Al decreased to 102 (Figure 7B). The IgG 1 antibody titer in all groups reached 105, whereas the IgG 2a antibody titer reached 104 (Figure 7C and D). Results of the IgG 2a/IgG 1 ratio revealed a Th2 bias in each immunization group (Figure 7E). The protection effects were further evaluated. TNA in the ETX nanoparticle vaccine group increased significantly from 64 to 256–512. ETXY196E+Al increased from 512 to 512–1024, but the difference between the ETX nanoparticle vaccine and the ETXY196E + Al immunized control groups was significantly reduced from nearly eightfold to nearly fourfold compared with the third immunization (Figure 7F).
 |
Figure 7 Evaluation of antibody titer and protection in mice surviving 2 months after the third immunization. (A) Schematic illustration of the experiment. Mice were randomly selected, and blood was collected from the tail vein to detect antibody titers and protective effects. (B–D) Detection of ETX-specific antibodies IgG (B), IgG 1 (C), and IgG2a (D) in serum using ELISA. (E) Ratio of IgG 2a/IgG 1 (n = 5). (F) TNA (G) In vitro neutralization cytotoxicity assay results (n=3). (H) In vitro neutralization of animal toxicity (n = 5). Serum was diluted 20-fold then mixed with 10 × LD50 GST-ETX injected intraperitoneally into blank mice. Statistical significance of differences in group means as calculated using one-way ANOVA: p<0.05 (*), p<0.01 (**), p≥ 0.05 (not significant, ns). |
In vitro neutralization cytotoxicity assay showed that the protective effects of the NP and MNP–ASP–ETX groups protected cells from to 32 × CT50 GST-ETX, NP protected approximately 70% of MDCK cells, and the MNP–ASP–ETX group protected approximately 90% MDCK cells from 64 × CT50 GST-ETX challenged. The MNP and MNP–HIS–ETX groups showed a large increase in the protective effect to 64 × CT50 GST-ETX. The protective effect was significantly increased after 2 months (Figure 7G).
In vitro neutralizing animal toxicity assay results showed that the MNP–ASP–ETX and ETXY196E + Al groups completely protected naive mice from 10 × LD50 GST-ETX. The NP group protected 60% of mice, and MNP and MNP–HIS–ETX protected 20% of the mice from 10 × LD50 GST-ETX (Figure 7H). In conclusion, compared with the third immunization, the antibody level and protection of ETX nanoparticle vaccine increased significantly and narrowed the difference compared with ETXY196E + Al, indicating that the ETX nanoparticle vaccine can produce long-lasting immunity.
Discussion
ETX is associated with fatal enterotoxemia in livestock species, mainly sheep and goats, resulting in considerable economic losses.44,45 ETX is one of the most potent known toxins after botulinum toxin and tetanus toxin; it is therefore classified as a category B agent by the CDC.43 In recent studies, toxoid vaccines have been reported to elicit variable immune responses as well as inflammatory responses following vaccination.22 Another study has suggested that site-directed mutants of ETX may significantly reduce the toxicity of the toxin and may therefore be vaccine candidates.46 However, complete elimination of its toxicity is not feasible, and there always remains a possibility of reversion to full toxicity.21
In this study, a novel ETX nanoparticle vaccine was prepared. The size of the different particles determines the type of APC and lymph nodes drainage ability. The size of ETX nanoparticle vaccine is 190 nm, which is susceptible to uptake by DCs.47 The encapsulation efficiency of NP was approximately 37.5%. The high encapsulation efficiency guarantees the consistency of vaccine batches. An in vitro cytotoxicity assay showed that 2 mg of MNP adsorbed 187 ng of ASP–ETX, which was significantly higher than the adsorption of HIS-ETX, laying a good foundation for the calculating antigen levels in immunization experiments and determining the function of targeting B cells.
PLGA can prolong the duration of antigen release and lymph node drainage.48 The antigen release effect was examined via in vivo imaging experiments. Results showed that MNP and NP can significantly prolong the antigen release time and steadily promote release at the injection site, indicated a longer interaction time with APCs in the lymph nodes. The rates of antigen release of MNP and NP were steadier than those of ETXY196E + Al, which was favorable for avoiding acute toxic reactions resulting from the large amount of antigen released in a short period. However, the total release times of MNP and NP were slightly shorter than those of ETXY196E + Al. Based on the observation of mouse skin, a group of ETXY196E + Al mice showed significant nodules at the injection sites on the back, which might be due to the side effect of the aluminum adjuvant.49,50 It was speculated that the antigen was anchored at the injection site by the formation of nodules, leading to the prolonged release of the antigen. In contrast, no nodule formations were observed in the ETX nanoparticle vaccine group. In this study, in vivo and in vitro safety investigations confirmed that ETX nanoparticle vaccine has superior biosafety.
ETX nanoparticle vaccine has favorable immunogenicity and produce high IgG antibody levels. Unlike ETXY196E + Al, which induces only predominantly humoral immunity, ETX NP vaccine produces a predominantly humoral immune response with high levels of cellular immunity in mice. ETX NP vaccine and ETXY196E + Al protected mice against 100 × LD50 GST-ETX challenge, MDCK cells from 16 × CT50 GST-ETX and blank mice from 2 × LD50 toxin challenge in vitro. ETX nanoparticle vaccine confirmed that the 10 × LD50 GST-ETX challenged mice did not show pathological abnormalities in their brain and kidneys. Results of long-term immunization showed that ETX nanoparticle vaccine retained a high level of ETX-specific IgG antibody and showed a significant increase in TNA and protection compared with the third immunization.
However, the in vitro protection ability of the ETX NP vaccine was significantly lower than that of the ETXY196E + Al group. As indicated by the level of immune cells in the spleens of mice, ETX NP vaccine produces high levels of memory T cells, which induce cellular immunity. After the adsorption of ASP–ETX and HIS-ETX, the numbers of B cells, plasma cells, and MBCs increased, indicating that ASP–ETX acted as a target for B cells and induced the body to produce a humoral immune response, consistent with the changes observed in the ETXY196E + Al control group. However, because of the small amount of ASP–ETX adsorbed on the membrane, the humoral immune response is not significant, and cellular immunity is still the primary focus.
As an exotoxin protein, the treatment and prevention of ETX mainly depends on the structural specificity of the antibody–antigen. Therefore, vaccine development against ETX should also aim to generate humoral immunity with specific antibodies. Cell membrane–encapsulated PLGA NPs induce a cellular-based immune response, generating many cytotoxic T cells (CTLs).51–53 PLGA-mediated slow antigen release also promotes the initiation of memory CD8+ T cells, eliciting a prolonged memory T-cell immune response, which is more beneficial for treating diseases such as bacterial or viral infection and tumor or for the preparation of vaccines. In response to structure-specific ETX, the cellular immune response is less effective compared with the humoral immunity, which is based on the production of specific antibodies. Although the ASP–ETX fusion toxin, which targets B cells, is adsorbed to cell membrane, and the type of immunity produced is changed from cellular immunity to humoral immunity, the limited adsorption capacity makes it difficult to fully activate B cells and induce strong humoral immune responses. The high loading capacity of NPs for toxins can cause a stronger humoral immune response.54 Different toxins interact with cell membranes through different mechanisms of action; thus, the loading capacity nanoparticles to toxins is also correlated with the type of toxin. These results also demonstrated that the cell membrane–encapsulated nanoparticles-adsorbing toxins can induce a strong immune response and have a great potential to inducing a high-quality immune response.
In future, the immunization interval can be appropriately extended according to the characterization of the slow-release effect, which is more conducive to the function of cell membrane–encapsulated NP vaccine platforms. In addition, the development of nanoparticle vaccine should focus on modifying the cell membranes to increase the adsorption of the toxin, so that it can induce a high level of cellular immunity and simultaneously induce a high level of neutralizing antibodies. ETX nanoparticle vaccine has flexible functions and advantages that are not available in conventional vaccines, and has a great potential to prevent ETX, which can be further developed as a vaccine candidate for ETX.
Conclusion
In this study, a loaded ETXY196E attenuator protein that adsorbed structurally intact and targeted ASP–ETX was prepared to prevent ETX intoxication based on cell membrane–encapsulated NP platform. The ETX nanoparticle vaccine demonstrated optimal physicochemical properties for APC uptake along with excellent safety and immunogenicity profiles. It induced potent humoral immunity (high ETX-specific IgG titers) and generated an immune response dominated by cellular immunity that is more conducive for treating diseases such as bacterial or viral infections and tumors or for preparing the corresponding vaccine. Although the proportion of B cells in MNP–ASP–ETX increased significantly and the type of immunity changed, the levels of humoral immunity induced were still lower than those of the immunization control group because of the less adsorbed amount, it demonstrates flexibility and superior safety not typically found in traditional vaccines. The ETX NP vaccine has great potential for further research and development as a candidate vaccine for ETX.
Data Sharing Statement
Data from this study are available within the article and its Additional Information files, or from the corresponding author (Wenwen Xin: [email protected]) upon reasonable request.
Ethics Approval and Consent to Participate
All animal work was conducted in strict accordance with recommendations in the Guide for the Care and Use of Laboratory Animals and the American Veterinary Medical Association Guidelines for the Euthanasia of Animals. The protocol was approved by the Institutional Animal Care and Use Committee (IACUC) of the Academy of Military Medical Sciences, review number IACUC-DWZX-2023-036 (approved on: 7 July 2023). All efforts were made to minimize mouse suffering. Consent for publication not applicable.
Consent for Publication
We confirm that all authors have read and agreed to the manuscript before submission.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
This work is sponsored by Beijing Nova Program (Z201100006820028).
Disclosure
The authors declare no conflict of interest.
References
1. Navarro MA, McClane BA, Uzal FA. Mechanisms of action and cell death associated with clostridium perfringens Toxins. Toxins. 2018;10(5):212. doi:10.3390/toxins10050212
2. Rood JI, Adams V, Lacey J, et al. Expansion of the Clostridium perfringens toxin-based typing scheme. Anaerobe. 2018;53:5–10. doi:10.1016/j.anaerobe.2018.04.011
3. Bokori-Brown M, Savva C, Fernandes da Costa SP, et al. Molecular basis of toxicity of clostridium perfringens epsilon toxin. 1742-4658 (Electronic).
4. Bhown AS, Habeerb AF. Structural studies on epsilon-prototoxin of Clostridium perfringens type D. Localization of the site of tryptic scission necessary for activation to epsilon-toxin. Biochem Biophys Res Commun. 1977;78(3):889–896. doi:10.1016/0006-291x(77)90506-x
5. Minami J, Katayama S, Matsushita O, Matsushita C, Okabe A. Lambda-toxin of Clostridium perfringens activates the precursor of epsilon-toxin by releasing its N- and C-terminal peptides. Microbiol Immunol. 1997;41(7):527–535. doi:10.1111/j.1348-0421.1997.tb01888.x
6. Uzal FA, Kelly WR, Morris WE, Assis RA. Effects of intravenous injection of Clostridium perfringens type D epsilon toxin in calves. J Comp Pathol. 2002;126(1):71–75. doi:10.1053/jcpa.2001.0514
7. Uzal FA, Songer JG. Diagnosis of Clostridium perfringens intestinal infections in sheep and goats. J Vet Diagn Invest. 2008;20(3):253–265. doi:10.1177/104063870802000301
8. Alves GG, Machado de Ávila RA, Chávez-Olórtegui CD, Lobato FC. Clostridium perfringens epsilon toxin: the third most potent bacterial toxin known. Anaerobe. 2014;30:102–107. doi:10.1016/j.anaerobe.2014.08.016
9. Awad MM, Bryant AE, Stevens DL, Rood JI. Virulence studies on chromosomal alpha-toxin and theta-toxin mutants constructed by allelic exchange provide genetic evidence for the essential role of alpha-toxin in Clostridium perfringens-mediated gas gangrene. Mol Microbiol. 1995;15(2):191–202. doi:10.1111/j.1365-2958.1995.tb02234.x
10. Linden JR, Ma Y, Zhao B, et al. Clostridium perfringens epsilon toxin causes selective death of mature oligodendrocytes and central nervous system demyelination. mBio. 2015;6(3):e02513. doi:10.1128/mBio.02513-14
11. Chassin C, Bens M, de Barry J, et al. Pore-forming epsilon toxin causes membrane permeabilization and rapid ATP depletion-mediated cell death in renal collecting duct cells. Am J Physiol Renal Physiol. 2007;293(3):F927–37. doi:10.1152/ajprenal.00199.2007
12. Soler-Jover A, Dorca J, Popoff MR, et al. Distribution of Clostridium perfringens epsilon toxin in the brains of acutely intoxicated mice and its effect upon glial cells. Toxicon. 2007;50(4):530–540. doi:10.1016/j.toxicon.2007.04.025
13. Finnie JW. Pathogenesis of brain damage produced in sheep by Clostridium perfringens type D epsilon toxin: a review. Aust Vet J. 2003;81(4):219–221. doi:10.1111/j.1751-0813.2003.tb11474.x
14. Uzal FA, Kelly WR, Morris WE, Bermudez J, Baisón M. The pathology of peracute experimental Clostridium perfringens type D enterotoxemia in sheep. J Vet Diagn Invest. 2004;16(5):403–411. doi:10.1177/104063870401600506
15. Mantis NJ. Vaccines against the category B toxins: staphylococcal enterotoxin B, epsilon toxin and ricin. Adv Drug Deliv Rev. 2005;57(9):1424–1439. doi:10.1016/j.addr.2005.01.017
16. Kohn J, Warrack GH. Recovery of Clostridium welchii type D from man. Lancet. 1955;268(6860):385. doi:10.1016/s0140-6736(55)91276-9
17. Xu J, Li D, Kang L, et al. Systematic evaluation of membrane-camouflaged nanoparticles in neutralizing Clostridium perfringens ε-toxin. J Nanobiotechnology. 2023;21(1):95. doi:10.1186/s12951-023-01852-z
18. Gao J, Xin W, Huang J, et al. Research articleHemolysis in human erythrocytes by Clostridium perfringens epsilon toxin requires activation of P2 receptors. Virulence. 2018;9(1):1601–1614. doi:10.1080/21505594.2018.1528842
19. Bokori-Brown M, Savva CG, Fernandes da Costa SP, Naylor CE, Basak AK, Titball RW. Molecular basis of toxicity of Clostridium perfringens epsilon toxin. FEBS J. 2011;278(23):4589–4601. doi:10.1111/j.1742-4658.2011.08140.x
20. Oyston PCF, Payne DW, Havard HL, Williamson ED, Titball RW. Production of a non-toxic site-directed mutant of Clostridium perfringens epsilon-toxin which induces protective immunity in mice. Microbiology. 1998;144(Pt 2):333–341. doi:10.1099/00221287-144-2-333
21. Titball RW. Clostridium perfringens vaccines. Vaccine. 2009;27(Suppl 4):D44–7. doi:10.1016/j.vaccine.2009.07.047
22. Uzal FA, Bodero DA, Kelly WR, Nielsen K. Variability of serum antibody responses of goat kids to a commercial Clostridium perfringens epsilon toxoid vaccine. Vet Rec. 1998;143(17):472–474. doi:10.1136/vr.143.17.472
23. Uzal FA, Wong JP, Kelly WR, Priest J. Antibody response in goats vaccinated with liposome-adjuvanted Clostridium perfringens type D epsilon toxoid. Vet Res Commun. 1999;23(3):143–150. doi:10.1023/a:1006206216220
24. Li Q, Xin W, Gao S, Kang L, Wang J. A low-toxic site-directed mutant of Clostridium perfringens ε-toxin as a potential candidate vaccine against enterotoxemia. Hum Vaccin Immunother. 2013;9(11):2386–2392. doi:10.4161/hv.25649
25. Swartz MA, Hirosue S, Hubbell JA. Engineering approaches to immunotherapy. Sci Transl Med. 2012;4(148):148rv9. doi:10.1126/scitranslmed.3003763
26. Irvine DJ, Swartz MA, Szeto GL. Engineering synthetic vaccines using cues from natural immunity. Nat Mater. 2013;12(11):978–990. doi:10.1038/nmat3775
27. Angsantikul P, Thamphiwatana S, Gao W, Zhang L. Cell membrane-coated nanoparticles as an emerging antibacterial vaccine platform. Vaccines. 2015;3(4):814–828. doi:10.3390/vaccines3040814
28. Zhao L, Seth A, Wibowo N, et al. Nanoparticle vaccines. Vaccine. 2014;32(3):327–337. doi:10.1016/j.vaccine.2013.11.069
29. Zhang W, Wang L, Liu Y, et al. Immune responses to vaccines involving a combined antigen-nanoparticle mixture and nanoparticle-encapsulated antigen formulation. Biomaterials. 2014;35(23):6086–6097. doi:10.1016/j.biomaterials.2014.04.022
30. Guo Y, Wang D, Song Q, et al. Erythrocyte membrane-enveloped polymeric nanoparticles as nanovaccine for induction of antitumor immunity against melanoma. ACS Nano. 2015;9(7):6918–6933. doi:10.1021/acsnano.5b01042
31. Bachmann MF, Jennings GT. Vaccine delivery: a matter of size, geometry, kinetics and molecular patterns. Nat Rev Immunol. 2010;10(11):787–796. doi:10.1038/nri2868
32. Demento SL, Siefert AL, Bandyopadhyay A, Sharp FA, Fahmy TM. Pathogen-associated molecular patterns on biomaterials: a paradigm for engineering new vaccines. Trends Biotechnol. 2011;29(6):294–306. doi:10.1016/j.tibtech.2011.02.004
33. Nochi T, Yuki Y, Takahashi H, et al. Nanogel antigenic protein-delivery system for adjuvant-free intranasal vaccines. Nat Mater. 2010;9(7):572–578. doi:10.1038/nmat2784
34. Hong S, Zhang Z, Liu H, et al. B cells are the dominant antigen-presenting cells that activate naive CD4+ T cells upon immunization with a virus-derived nanoparticle antigen. Immunity. 2018;49(4):695–708.e4. doi:10.1016/j.immuni.2018.08.012
35. Guo C, Peng Y, Lin L, et al. A pathogen-like antigen-based vaccine confers immune protection against SARS-CoV-2 in non-human primates. Cell Rep Med. 2021;2(11):100448. doi:10.1016/j.xcrm.2021.100448
36. He Y, Barker SJ, MacDonald AJ, et al. Recombinant Ov-ASP-1, a Th1-biased protein adjuvant derived from the helminth Onchocerca volvulus, can directly bind and activate antigen-presenting cells. J Immunol. 2009;182(7):4005–4016. doi:10.4049/jimmunol.0800531
37. Guo J, Yang Y, Xiao W, et al. A truncated fragment of Ov-ASP-1 consisting of the core pathogenesis-related-1 (PR-1) domain maintains adjuvanticity as the full-length protein. Vaccine. 2015;33(16):1974–1980. doi:10.1016/j.vaccine.2015.02.053
38. Yao W, Kang J, Kang L, et al. Immunization with a novel Clostridium perfringens epsilon toxin mutant rETX(Y196E)-C confers strong protection in mice. Sci Rep. 2016;6:24162. doi:10.1038/srep24162
39. Zhao Y, Kang L, Gao S, et al. Expression and purification of functional Clostridium perfringens alpha and epsilon toxins in Escherichia coli. Protein Expr Purif. 2011;77(2):207–213. doi:10.1016/j.pep.2011.02.001
40. Zhang D, Liu L, Wang J, et al. Drug-loaded PEG-PLGA nanoparticles for cancer treatment. Front Pharmacol. 2022;13:990505. doi:10.3389/fphar.2022.990505
41. Diwan M, Park TG. Pegylation enhances protein stability during encapsulation in PLGA microspheres. J Control Release. 2001;73(2–3):233–244. doi:10.1016/s0168-3659(01)00292-9
42. Christensen D, Henriksen-Lacey M, Kamath AT, et al. A cationic vaccine adjuvant based on a saturated quaternary ammonium lipid have different in vivo distribution kinetics and display a distinct CD4 T cell-inducing capacity compared to its unsaturated analog. J Control Release. 2012;160(3):468–476. doi:10.1016/j.jconrel.2012.03.016
43. Popoff MR. Epsilon toxin: a fascinating pore-forming toxin. FEBS J. 2011;278(23):4602–4615. doi:10.1111/j.1742-4658.2011.08145.x
44. Songer JG. Clostridial enteric diseases of domestic animals. Clin Microbiol Rev. 1996;9(2):216–234. doi:10.1128/cmr.9.2.216
45. Nagahama M, Sakurai J. Distribution of labeled Clostridium perfringens epsilon toxin in mice. Toxicon. 1991;29(2):211–217. doi:10.1016/0041-0101(91)90105-z
46. Jiang Z, Chang J, Wang F, et al. Etx-Y71A as a non-toxic mutant of Clostridium perfringens epsilon toxin induces protective immunity in mice and sheep. Vaccine. 2020;38(42):6553–6561. doi:10.1016/j.vaccine.2020.08.002
47. Manolova V, Flace A, Bauer M, Schwarz K, Saudan P, Bachmann MF. Nanoparticles target distinct dendritic cell populations according to their size. Eur J Immunol. 2008;38(5):1404–1413. doi:10.1002/eji.200737984
48. Demento SL, Cui W, Criscione JM, et al. Role of sustained antigen release from nanoparticle vaccines in shaping the T cell memory phenotype. Biomaterials. 2012;33(19):4957–4964. doi:10.1016/j.biomaterials.2012.03.041
49. Han HR, Park HM. Effects of adjuvants on the immune response of staphylococcal alpha toxin and capsular polysaccharide (CPS) in rabbit. J Vet Med Sci. 2000;62(3):237–241. doi:10.1292/jvms.62.237
50. Hoffmann SS, Elberling J, Skamstrup hansen K, et al. Adverse reactions after oral provocation with aluminium in children with vaccination granulomas and aluminium contact allergy. J Eur Acad Dermatol Venereol. 2023;37(5):1028–1035. doi:10.1111/jdv.18811
51. Moon JJ, Suh H, Bershteyn A, et al. Interbilayer-crosslinked multilamellar vesicles as synthetic vaccines for potent humoral and cellular immune responses. Nat Mater. 2011;10(3):243–251. doi:10.1038/nmat2960
52. Blair DA, Turner DL, Bose TO, et al. Duration of antigen availability influences the expansion and memory differentiation of T cells. J Immunol. 2011;187(5):2310–2321. doi:10.4049/jimmunol.1100363
53. Li AV, Moon JJ, Abraham W, et al. Generation of effector memory T cell-based mucosal and systemic immunity with pulmonary nanoparticle vaccination. Sci Transl Med. 2013;5(204):204ra130. doi:10.1126/scitranslmed.3006516
54. Hu CM, Fang RH, Luk BT, Zhang L. Nanoparticle-detained toxins for safe and effective vaccination. Nat Nanotechnol. 2013;8(12):933–938. doi:10.1038/nnano.2013.254
 © 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.