Back to Journals » OncoTargets and Therapy » Volume 14
Combination Therapy with iRGD-antiCD3 and PD-1 Blockade Enhances Antitumor Potency of Cord Blood-Derived T Cells
Authors Zhu M, Wang H, Zhou S, Wei J, Ding N, Shao J, Yu L, Feng Z, Liu B
Received 10 November 2020
Accepted for publication 19 January 2021
Published 5 February 2021 Volume 2021:14 Pages 835—844
DOI https://doi.org/10.2147/OTT.S291086
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Prof. Dr. Gaetano Romano
Mei Zhu,1– 3,* Hongmei Wang,1,2,* Shujuan Zhou,4,* Jia Wei,2 Naiqing Ding,2 Jie Shao,2 Lixia Yu,2 Zhenqing Feng,5,6 Baorui Liu1,2
1The Comprehensive Cancer Centre of Nanjing Drum Tower Hospital, Clinical College of Nanjing Medical University, Nanjing, Jiangsu, 210008, People’s Republic of China; 2The Comprehensive Cancer Centre of Drum Tower Hospital, Medical School of Nanjing University and Clinical Cancer Institute of Nanjing University, Nanjing, Jiangsu, 210008, People’s Republic of China; 3Department of Oncology, Xuzhou Cancer Hospital, Xuzhou, Jiangsu, 221005, People’s Republic of China; 4The Department of Radiation Oncology, Fudan University Shanghai Cancer Centre, Shanghai, 200032, People’s Republic of China; 5Department of Pathology, Nanjing Medical University, Nanjing, Jiangsu, 211166, People’s Republic of China; 6Key Laboratory of Antibody Technique of Ministry of Health, Nanjing Medical University, Nanjing, Jiangsu, 211166, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Zhenqing Feng
Department of Pathology, Nanjing Medical University, 101 Longmian Avenue, Jiangning, Nanjing, Jiangsu, 211166, People’s Republic of China
Email [email protected]
Baorui Liu
The Comprehensive Cancer Centre of Nanjing Drum Tower Hospital, Clinical College of Nanjing Medical University, 321 Zhongshan Road, Nanjing, Jiangsu, 210008, People’s Republic of China
Email [email protected]
Background: T cell-redirecting bispecific antibodies (BsAbs) are emerging as a potent cancer therapy that crosslinks tumor cells and T cells by simultaneously binding to tumor-associated antigen and CD3ϵ. However, immune inhibitory molecules can be remarkably upregulated after BsAbs treatment, leading to a suppressive tumor microenvironment and treatment resistance. This can be partially reversed by combination with immune checkpoint inhibitors. In our previous work, we successfully constructed the recombinant protein iRGD-antiCD3 and demonstrated that it promoted antitumor efficacy of transferred T cells by promoting T cell activation and infiltration.
Methods: We detected the levels of both PD-1 and PD-L1 as resistance to iRGD-antiCD3 treatment. Using cord blood-derived T cells, we assessed the activation and effects of iRGD-antiCD3 combined with PD-1 as evidenced by activation markers, Th1/Th2-cytokines, and killing capability against tumor cells in vitro. Moreover, to better mimic the physiological characteristics of in vivo solid tumors, we generated 3D spheroids from target cell lines. Spheroids were stained with a Viability/Cytotoxicity Assay Kit and examined by confocal microscopy to study the in vitro antitumor effect of T cells co-administered with combination iRGD-antiCD3 and PD-1 blockade. The mouse peritoneal metastatic gastric tumor model was employed. The synergistic antitumor effect and safety profiles in vivo were evaluated by tumor and body weight of tumor-bearing mice.
Results: We found that expression of both PD-1 and PD-L1 were increased as resistance to iRGD-antiCD3 treatment. We found that PD-1 blockade partially restored T cell activation as evidenced by elevated activation markers, Th1-cytokines, and killing capability against tumor cells in vitro. The combination of PD-1 blockade consistently and significantly increased cord blood-derived T cell cytotoxicity against 3D tumor spheroids. In vivo, we observed synergistic antitumor activity without obvious side effects.
Conclusion: These results demonstrated that combining iRGD-antiCD3 with PD-1 blockade could further improve antitumor efficacy of T cells, and this strategy holds great potential for the treatment of solid malignancies.
Keywords: cord blood-derived T cell, iRGD-antiCD3, PD-1 blockade, combination therapy
Introduction
Over the last decade, remarkable developments in cancer immunotherapies and particularly immune checkpoint blockers (ICBs) have already revolutionized cancer treatment paradigms. However, most patients with malignant solid tumors, such as gastric cancer, remain unresponsive and do not demonstrate a durable long-term response.1 In contrast to other tumors such as lung cancer and breast cancer, gastrointestinal tumors have mesenchymal traits that hinder the infiltration of immune cells and thereby cripple the immune response to PD-1/PD-L1 blockade.2 Thus, it is vital to improve T cell infiltration to broaden the success of ICBs in gastric cancer.
It has been reported that a tumor-penetrating peptide, iRGD (sequence: CRGDKGPDC), can function to promote extravasation and tumor-specific penetration of small molecules and nanoparticles.3 The mechanism behind this process is thought to depend on the RGD domain and CendR motif. iRGD first binds to αvβ3 or αvβ5 integrins that are highly expressed in the tumor vascular endothelium and on various tumor cells.4 Subsequently, it is proteolytically cleaved by a cell-surface-associated protease to produce CRGDK/R, exposing the CendR motif. The truncated peptide loses its affinity for the primary receptor integrin and binds to neuropilin-1, triggering the tissue penetration pathway.5,6 We previously demonstrated for the first time that iRGD can facilitate the infiltration of lymphocytes in both 3D tumor spheroids and several xenograft mouse models. In addition, combining iRGD modification with PD-1-knockout lymphocytes revealed superior antitumor efficacy.7
BsAbs have emerged as an innovative and promising immunotherapy approach. One of the main strategies in the development of BsAbs is the recruitment and activation of effector T cells by simultaneously targeting both the CD3 domain of the TCR complex and antigens abnormally expressed on the tumor cell surface. The approval of Blinatumomab (anti-CD19/anti-CD3) was a major milestone in the development of BsAbs.8,9 Based on BsAbs and iRGD, we successfully constructed a novel fusion protein, iRGD-antiCD3. We demonstrated that iRGD-antiCD3 promoted both infiltration and activation of tumor-specific T cells. T cells modified with iRGD-antiCD3 exhibited significant tumor growth control in several xenograft mouse models. However, tumor cell expression of immune checkpoint molecules such as PD-L1 can be dramatically upregulated due to the secretion of IFN-γ and TNF-α after exposure to BsAbs, which induces the formation of an immunosuppressive tumor microenvironment with exhausted T cells.10,11 Meanwhile, several studies have shown that combining ICBs with BsAb treatment can significantly augment anti-tumor responses compared to BsAb as a single agent.12,13
Obtaining a sufficient number of effector T cells is a key point for cancer adoptive cell immunotherapy.14 In recent years, cord blood has been increasingly used in cancer immunotherapy, especially in hematologic malignancies. Cord blood is easy to obtain and contains plentiful naïve T cells, many more than in peripheral blood. These naïve T cells have a strong expansion ability, as well as good differentiation ability and low immunogenicity.15,16 Our team has also compared T cells from cord blood with those from peripheral blood. Both the proportions of naïve T cells and CD8+ T cells after activation were higher in cord blood (unpublished). Therefore, cord blood is a good source of effector cells and an effective supplement to peripheral blood for immunotherapy of patients with advanced tumors.
In the current study, we demonstrate that iRGD-antiCD3 therapy elevated the expression of PD-1 and PD-L1. The addition of PD-1 blockade to the treatment regimen partially mitigated these inhibitory signals and released T cell activation. Moreover, we show that combining iRGD-antiCD3 with PD-1 blockade further potentiated the antitumor efficacy of cord blood-derived T cells against target tumor cells, not only when grown in vitro as monolayer cultures or as three-dimensional multicellular tumor spheroids, but also in vivo in BALB/c nude mice.
Materials and Methods
Cell Culture
Human gastric adenocarcinoma cell lines MKN45, HGC27, and MGC803 were purchased from the Cell Bank of Shanghai Institute of Biochemistry and Cell Biology and cultured in RPMI 1640 medium supplemented with 10% fetal calf serum, 100 U mL−1 penicillin, and 100 μg mL−1 streptomycin. All cell lines were confirmed to be Mycoplasma negative.
Animals
Male BALB/c nude mice weighing 18–20 g (4–5 weeks old) were supplied by the Department of Experimental Animals, Nanjing Medical University (Nanjing, People’s Republic of China). The temperature and relative humidity were maintained at 25°C and 45–55%, respectively. All animal procedures were carried out in compliance with guidelines set by the Animal Care Committee at Drum Tower Hospital (Nanjing, the People’s Republic of China). The Ethics Committee of Drum Tower Hospital approved all experiments in this study.
Isolation and Culture of Primary Human Cord Blood T Lymphocytes
Fresh core blood was collected from 3 healthy donors. The core blood collection procedure was carried out in accordance with the guidelines verified and approved by the Ethics Committee of Drum Tower Hospital. All donors signed an informed consent for scientific research statement. The study was conducted in accordance with the Declaration of Helsinki. Human cord blood mononuclear cells (HCBMCs) were isolated from samples of healthy volunteers by centrifugation on a Ficoll density gradient and suspended in AIM-V medium (Gibco, USA) containing 10% fetal bovine serum (Gibco, NY, USA). HCBMC were cultured for 2 hr to permit adherence; non-adherent T lymphocytes were then incubated at 37°C and 5% CO2 and authenticated by checking their microscopic morphology after plating at different concentrations.
Flow Cytometry Analysis
To detect expression changes of PD-1 on T cells and PD-L1 on tumor cells, gastric cancer MKN45 cells were incubated with T cells alone (2.5 × 105 cells/well) at an effector-to-target (E:T) ratio of 5:1 or with T cells and iRGD-antiCD3 (10 μg mL−1) for 24 hr. T cells and tumor cells were harvested and stained for 30 min at 4°C in the dark using these fluorescent-labeled mouse anti-human antibodies: CD3-FITC (UCHT1, BD Bioscience, CA, USA), PD-1-APC (EH12.1, BD Bioscience, CA, USA), and PD-L1-PE (M1H4, BD Bioscience, CA, USA).
For T cell activation assays, gastric cancer MKN45 cells were incubated for 6 hr and 24 hr with T cells alone (2.5 × 105 cells/well) at an E:T ratio of 5:1, T cells with iRGD-antiCD3 (10 μg mL−1), T cells with PD-1 blockade (10 μg mL−1), or T cells with iRGD-antiCD3 and PD-1 blockade. T cells were harvested and stained for 30 m at 4°C in the dark using these fluorescent-labeled mouse anti-human antibodies: CD3-FITC (UCHT1, BD Bioscience, CA, USA), CD25-APC (BD Bioscience, CA, USA), and CD69-PE (BD Bioscience, CA, USA). The cells were then washed twice and resuspended in FACS buffer before analysis. Flow cytometry data were collected on a BD Accuri C6 (BD Bioscience, CA, USA) and analyzed with FlowJo 10.4 software.
For cytokine detection, gastric cancer MKN45 cells were incubated for 24 hr with T cells alone (2 × 106 cells/well) at an E:T ratio of 40:1, T cells with iRGD-antiCD3 (10 μg mL−1), T cells with PD-1 blockade (10 μg mL−1), or T cells with iRGD-antiCD3 and PD-1 blockade. The supernatants were harvested for cytokine quantification using the BD CBA human Th1/Th2 kit (BD Bioscience, NZ, USA) according to the manufacturer’s instructions.
In vitro Cytotoxicity Assays
CFSE/PI. MKN45 target cells were collected and labeled with 4 μM CFSE (carboxyfluorescein succinimidyl ester, Invitrogen) for 10 min at 37°C in PBS. Labeling was stopped by adding a 10-fold volume of PBS, followed by extensive washing with PBS before seeding into 24-well plates. CFSE-labeled cells (5 × 104 cells/well) were then incubated with T cells alone, T cells with iRGD-antiCD3 (10 μg mL−1), T cells with PD-1 blockade (10 μg mL−1), or T cells with iRGD-antiCD3 and PD-1 blockade at different E:T ratios (5:1, 10:1, 20:1, and 40:1) for 24 hr. Propidium iodide (PI) (Sigma) was added to determine cell death ratios, and the samples were analyzed by flow cytometry.
LDH cytotoxicity assays. Lytic activity was also determined in a non-radioactive colorimetric assay using the Cytotoxicity Detection Kit PLUS (Roche Cat. No. 04744934 001), which is based on measurement of lactate dehydrogenase (LDH) activity released from the cytosol of damaged cells. Briefly, target cells (5000) at 50 μL/well were plated and incubated with effector T cells at varying E:T ratios. To calculate percent cytotoxicity, we used the following four controls: background control, low control, high control, and substance control. We added 5 μL of lysis solution to wells of the high control group at least 15 min before harvesting. After 24 hr incubation, supernatants were harvested, 100 μL of reaction mixture (freshly prepared) was added to each well, and the plate was incubated for up to 30 min. The absorbance of the samples was measured at 490 nm after quenching the reaction. The mean percentage lysis was calculated using the following formula: cytotoxicity (%) = [(effector and target cell mix – effector cell control – low control)/(high control – low control)] × 100%.
Multicellular Tumor Spheroids (MCTSs) and Confocal Microscopy
HGC27 cells (1000 in 100 μL of complete media) were gently added to 96-well clear round bottom ultra low attachment microplates (Corning, NY, USA) and allowed to grow for 72 hr at 37°C to attain a diameter of about 300 μm. MCTSs were monitored with a light microscope on days 0, 1, 2, and 3. Uniform compact tumor spheroids were selected for the subsequent studies.
For the cytotoxicity assay, T cells alone (at an E:T ratio of 10:1), T cells with iRGD-antiCD3 (10 μg mL−1), T cells with PD-1 blockade (10 μg mL−1), or T cells with iRGD-antiCD3 and PD-1 blockade were added to spheroids and incubated for another 24 hr. Spheroids were then stained using a viability/cytotoxicity kit (Biotium, CA, USA) in the dark for 45 min. Consecutive images were captured by ZEN 880 confocal microscope using the Z-stack projecting from the top to the middle of spheroids using 5-μm intervals. Maximum intensity projection (MIP) images were obtained with Zeiss processing software. Quantification of live and dead cell areas was performed by measuring total cell area for each dye in Image J.
An additional cytotoxicity experiment for MCTSs was conducted by exposing MCTSs to T cells alone (at an E:T ratio of 10:1), T cells with iRGD-antiCD3 (10 μg mL−1), T cells with PD-1 blockade (10 μg mL−1), or T cells with iRGD-antiCD3 and PD-1 blockade and incubated for another 72 hr. The spheroid diameters were measured, and morphological changes were monitored by brightfield microscopy (Leica, Germany).
In vivo Antitumor Activity and Safety
For peritoneal metastasis tumor model, 4-5-week-old male BALB/c nude mice were injected intraperitoneally with 0.2 mL suspension of MGC803 cells (1 × 107). After 2 weeks, tumor-bearing mice were randomly assigned to 5 groups (each group containing at least five animals): PBS, T cell, T+antiPD-1, T+iRGD-antiCD3, and T+iRGD-antiCD3+antiPD-1. The group-dependent treatment schedules were as follows: intraperitoneal administration of 3 × 107 T cells took place on days 1 and 3; intraperitoneal administration of 100 μg of iRGD-antiCD3 took place daily from day 1 to day 5; and intraperitoneal administration of 250 μg PD-1 took place on days 1, 3, and 5. The activity of the mice were observed during the treatment. Body weights were measured every 2 days. Two weeks after the start of treatment, the mice were euthanized, and their tumors were excised and weighed to evaluate antitumor efficacy.
Statistics Analysis
GraphPad Prism 8.0 software was used for statistical analysis. Data are presented as mean ± SEM. Student’s t-test was used to determine the significances between groups. Values were considered to be statistically significant if P < 0.05, as indicated with asterisks (* P < 0. 05, ** P < 0. 01, *** P <0. 001, or **** P <0.0001).
Results
iRGD-antiCD3 Upregulated PD-1 on Cord Blood-Derived T Cells and PD-L1 on Tumor Cells
The induction of immune co-inhibitory pathways, such as the PD-1/PD-L1 axis, is a major resistance mechanism of BsAbs treatment. Here, after 24 hr incubation with iRGD-antiCD3 and MKN45 cells, PD-1 expression on gated CD3 T cells was significantly increased, and almost 4-fold higher than that of the T+MKN45 group (P < 0.01) (Figure 1A). Likewise, PD-L1 expression on tumor cells was also upregulated in that group (P < 0.05) (Figure 1B).
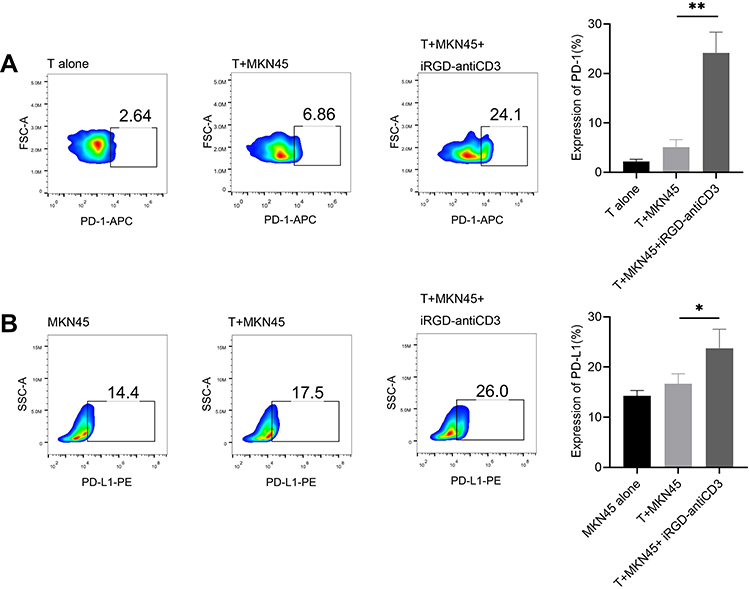 |
Figure 1 iRGD-antiCD3 upregulated PD-1 on CD3+ cord blood-derived T cells and PD-L1 on tumor cells. Flow cytometry results displaying the relative expression level of PD-1 on gated CD3 T cells (A) and PD-L1 on gastric cancer cells (B). Effector T cells and target MKN45 tumor cells were incubated at an E:T ratio of 5:1 for 24 hr in the presence or absence of iRGD-anti-CD3. The experiments were repeated three times with cells from 3 healthy donors, and a representative result is shown. **P < 0.01, *P < 0.05. |
The Addition of PD-1 Blockade Restored Activation of Cord Blood-Derived T Cells
Cytokine release was analyzed with a BD CBA human Th1/Th2 kit to determine the level of T cell activation. As shown in Figure 2A, three Th1-type cytokines (IL-2, TNF-γ, and TNF-α) were significantly upregulated in the iRGD-antiCD3 plus PD-1 blockade group compared with the other groups. We found less secretion of IL-4 in the combination group. No significant differences in IL-6 and IL-10 secretion were found between any groups. In addition, activation markers CD69 after 6 hr of incubation and CD25 after 24 hr on gated CD3 T cells subsets were elevated upon treatment with iRGD-antiCD3 and PD-1 blockade (Figure 2). Compared with the other groups, what needs to be emphasized was that the expression level of CD69 increased about 2-3-fold in the combined group. These data collectively demonstrate that combining iRGD-antiCD3 with PD-1 blockade could reverse T cell activity to some extent and thereby enhance antitumor immune responses.
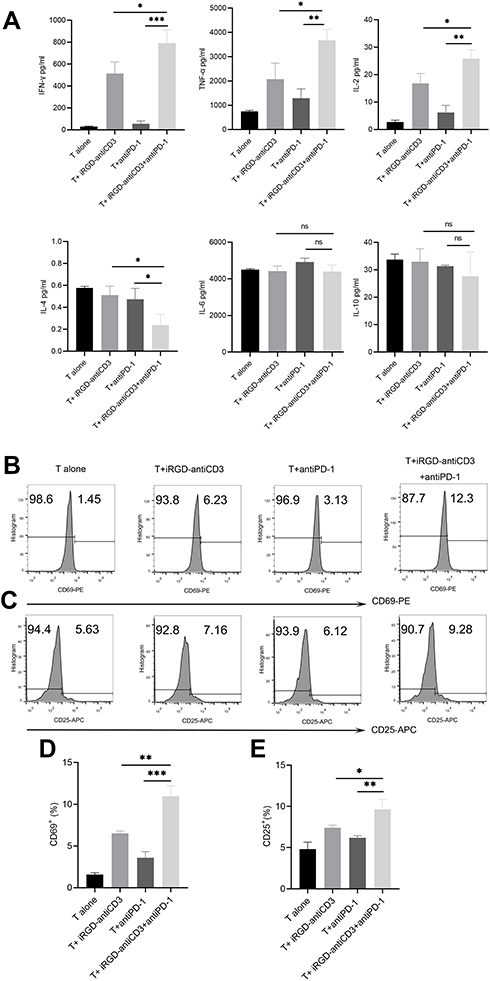 |
Figure 2 The addition of PD-1 blockade restored activation of cord blood-derived T cells. (A) The co-culture supernatants were harvested and used for Th1/Th2 cytokines measurements using the BD CBA human Th1/Th2 flex set. Effector T cells and target MKN45 cells were incubated at an E:T ratio of 40:1 for 24 hr. All values were shown as mean ± the SEM of triplicate measurements and repeated three times with similar results. T cell activation was assessed by measuring the expression level of CD69 after 6 hr of incubation (B) and CD25 after 24 hr of incubation (C) on the gated CD3 T cells by flow cytometry. (D) Bar graphs of CD69 expression. (E) Bar graphs of CD25 expression. Data shown are shown as mean ± SEM of 3 independent experiments, and a representative result is shown. n.s, not significant,***P < 0.001, **P < 0.01, *P < 0.05. |
Combining iRGD-antiCD3 with PD-1 Blockade Improved Tumor Cell Killing of Cord Blood-Derived T Cells
To confirm whether the addition of PD-1 blockade could promote T cell cytotoxicity, MKN45 monolayer cells were first tested as target cells at increasing E:T ratios. We used two assays, CFSE/PI and LDH release, to evaluate tumor cell lysis. As shown in Figure 3, both assays similarly revealed that adding PD-1 blockade enhanced T cell cytotoxicity at all indicated E:T ratios. About 60% lysis of target tumor cells was achieved with combination therapy at an E:T ratio of 40:1, which was almost 1.5-fold higher than that of the iRGD-antiCD3 group. These results indicated that combining iRGD-antiCD3 with PD-1 blockade further improved the antitumor activity of T cells.
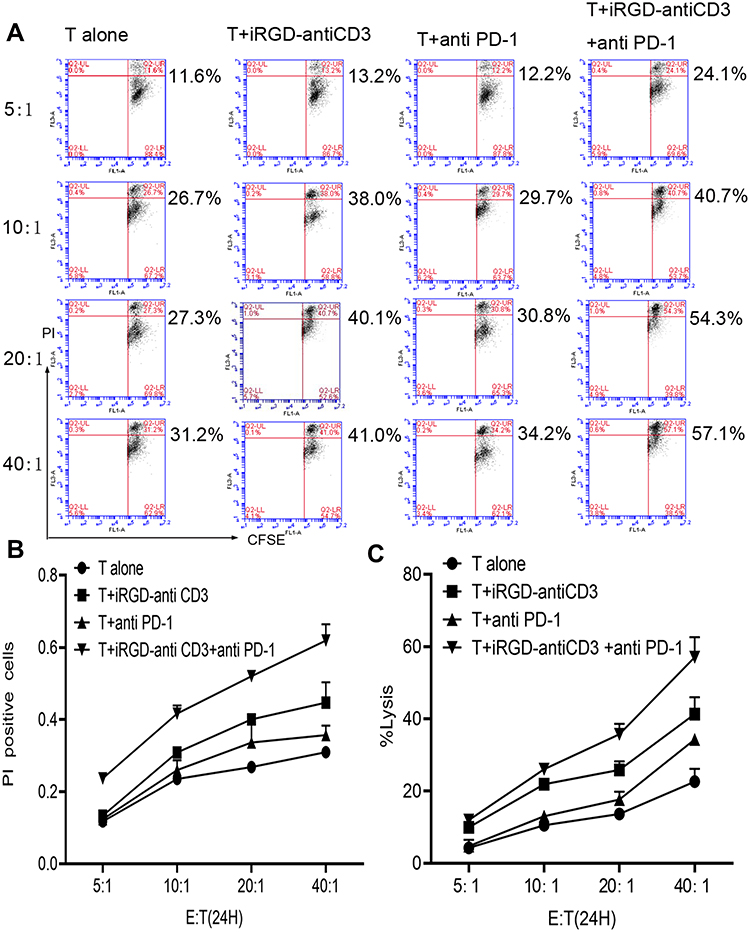 |
Figure 3 Combining iRGD-antiCD3 with PD-1 blockade improved tumor cell killing of cord blood-derived T cells. (A) The cytotoxic activity of effector T cells was measured using the CFSE/PI cytotoxicity assay. Effector T cells were co-cultured with CFSE-labeled tumor cells at E:T ratios of 5:1, 10:1, 20:1, and 40:1 for 24 hr. Then PI was added, and the cells were analyzed by flow cytometry. (B) Line graph of flow cytometry results in A. (C) The cytotoxic activity was also measured using the LDH assay. The experiments were repeated three times with cells from 3 healthy donors, and a representative result is shown. |
Combining iRGD-antiCD3 with PD-1 Blockade Enhanced Cytotoxicity of Cord Blood-Derived T Cells Against MCTSs
MCTSs, which can more precisely simulate in vivo solid tumors, are becoming favored models for evaluating penetration and cytotoxicity of immune cells. Here we generated MCTSs using the gastric cancer HGC27 cell line to study the antitumor efficacy of combining iRGD-antiCD3 with PD-1 blockade, judging by spheroid morphology, volume, and tumor cell death.
In live/dead assays, we observed a limited number of dead cells in MCTSs treated with T cells or PD-1 blockade alone. The addition of iRGD-antiCD3 slightly increased T cell cytotoxicity, whereas the combination of PD-1 blockade and iRGD-antiCD3 yielded robust killing activity and dramatically decreased the viability of MCTSs (Figure 4A and C). Additionally, spheroids incubated with T cells alone maintained their integrity, whereas spheroids in other groups lost their spherical shape and compactness. This was especially true with combined iRGD-antiCD3 and PD-1 blockade, which led to almost complete spheroid destruction and resulted in much smaller average diameters (Figure 4B and D).
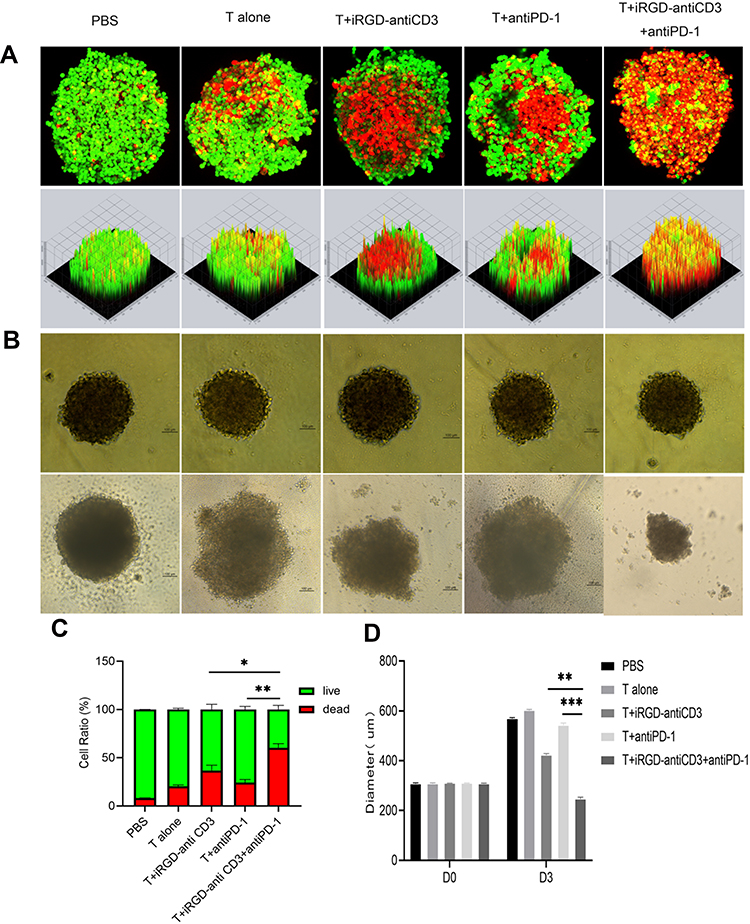 |
Figure 4 Combining iRGD-antiCD3 with PD-1 blockade enhanced cytotoxicity of cord blood-derived T cells against MCTSs. The effect of combining PD-1 blockade and iRGD-antiCD3 on T-cell cytotoxicity against HGC27 tumor spheroids. (A) The representative maximum intensity projection images by confocal microscopy of HGC27 spheroids after treatments for 24 hr. Dead tumor cells in MCTSs fluoresce red (EthD-1), and live tumor cells fluoresce green (calcein-AM). (B) Growth inhibition assays of HGC27 MCTSs. (C) MCTSs lysis was evaluated by comparing the total area of live/dead cells in maximum intensity projection images. (D) Diameters of MCTSs after 72 hr treatment. Magnification, ×200; Scale bars, 100 μm. Data were represented as mean ± SEM; n = 3. ***P < 0.001, **P < 0.01, *P < 0.05. |
Combining iRGD-antiCD3 with PD-1 Blockade Improved the in vivo Antitumor Efficacy of Cord Blood-Derived T Cells
We next studied the antitumor efficacy of combing iRGD-antiCD3 with PD-1 blockade in mice bearing disseminated MGC803 peritoneal tumors. The treatment schedule is shown in Figure 5A. All mice in the first three groups developed massive tumors, indicating that PD-1 blockade alone was not effective enough to control tumor growth. iRGD-antiCD3 slightly reduced tumor burden, while its combination with PD-1 blockade yielded a much greater antitumor effect, as evidenced by a nearly 3-fold tumor reduction compared to the iRGD-antiCD3 group and a nearly 5-fold reduction compared to the PBS control group (Figure 5B). No significant body weight alterations were observed in any of the studied groups (Figure 5C). These results demonstrated that combining iRGD-antiCD3 with PD-1 blockade promoted antitumor efficacy of cord blood-derived T cells in vivo with a favorable safety profile.
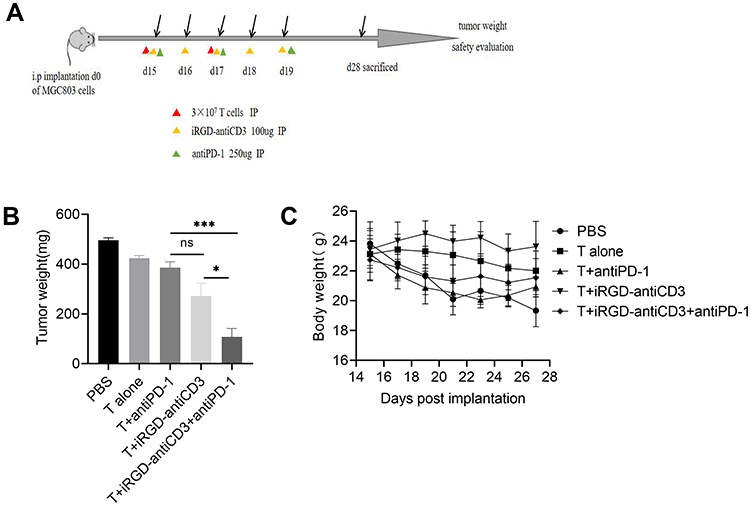 |
Figure 5 Combining iRGD-antiCD3 with PD-1 blockade enhanced antitumor efficacy of cord blood-derived T cells in the peritoneal metastasis tumor model. (A) Schematic illustration of the treatment process in the peritoneal metastasis tumor model. (B) Quantification of tumor weights in all groups. (C) Body weight changes during the treatment process. Data represent mean ± SEM; n = 5. n.s, not significant; ***P < 0.001, *P < 0.05. |
Discussion
BsAbs capable of redirecting and activating T cells to kill malignant tumor cells have shown therapeutic promise in both preclinical and clinical studies.17 However, poor T cell infiltration in solid tumors limits the therapeutic efficacy of BsAbs in solid tumors. Based on this, we successfully constructed a tumor-penetrating BsAb, iRGD-antiCD3, that can simultaneously promote tumor-specific T-cell infiltration and activation. As with other BsAbs, there is an obvious obstacle to the antitumor efficacy of iRGD-antiCD3 with the upregulation of immunosuppressive molecules such as PD-L1 in the tumor microenvironment, due mainly to the secretion of IFN-γ by activated T cells, and leading to tumor immune escape.11,18 As seen in previous studies, both PD-1 on T cells and PD-L1 on tumor cells are upregulated upon treatment with BsAbs.19,20 BsAbs are being tested in clinical trials, and potential synergistic activity with ICBs is also being evaluated. ICBs are not effective for most solid tumors, and a positive outcome may require a high mutational burden and accumulation of effector T cells against mutation-derived neoantigens.21,22 BsAb therapy represents an alternative anticancer immunotherapy that can bypass neoantigen-MHC/TCR complex recognition. Therefore, combing ICBs with BsAbs is a very promising strategy to produce synergistic antitumor effects.
In this study, as iRGD-antiCD3 potently activated T cells, PD-1 expression on T cells and PD-L1 on tumor cells were upregulated. Adding PD-1 blockade significantly enhanced T cell killing against monolayer gastric cancer cells compared to iRGD-antiCD3 alone, indicating that PD-1 blockade could be a potential combinatorial approach with iRGD-antiCD3 treatment. This was further confirmed in the well-recognized model of MCTS. Compared with two-dimensional cell cultures, spheroids can simulate in vivo solid tumors more precisely because of their three-dimensional structure and ability to maintain tumor cell interactions with other spheroids and extracellular matrix secretions.23–25 We have already demonstrated that tumor spheroids provide a novel and excellent platform to evaluate the effects of PD-1 blockade and may be further utilized to develop promising combination therapies.26 Here, confocal images of spheroids with live/dead cell staining revealed superior killing capacity when iRGD-antiCD3 and PD-1 blockade were combined, consistent with cytotoxicity assays using two-dimensional tumor cell cultures. The current study provided a basis for utilizing three-dimensional spheroids as more reliable surrogates for solid tumors in vivo to evaluate T cell functions, facilitate preclinical development of effective combinational immunotherapies, and drive translational research efforts toward clinical application. Our results also demonstrate the potent in vivo antitumor efficacy of the combination therapy. No obvious toxicity and side effects were observed in the mice bearing disseminated peritoneal tumors till the end of treatment.
However, the general value of adding ICBs to BsAb treatment remains uncertain and may depend on the BsAb molecule used as well as on the tumor type. In another study, PD-1/PD-L1 inhibition also enhanced lysis mediated by an anti-CEA/anti-CD3 BsAb but was unable to restore T cell activity completely upon induction of T cell exhaustion, pointing toward additional mechanisms impairing T cell activity.10 Studies have also demonstrated that dual blockade of PD-1 and PD-L1 could maximize T cell killing directed by the anti-CEA/anti-CD3 BsAb, and that combination therapy is more effective if applied earlier.
Taken together, we demonstrate that combination of PD-1 blockade with BsAb treatment could prevent the loss of T cell activity and thereby promote antitumor potency of cord blood-derived T cells in vitro and in vivo. We also support MCTSs as a novel model to drive clinical translational efforts to develop effective combinational immunotherapies. This combinational strategy holds excellent therapeutic potential for multiple solid tumor types. We are currently investigating the coadministration of iRGD-antiCD3 and other anticancer treatments for gastric cancer and other solid tumors. The mechanisms underlying the antitumor efficacy of combination strategies will be further studied to guarantee improved clinical application.
Acknowledgments
This work was supported by grants from the National Natural Science Foundation of China (No. 81930080, No. 81903128), the Natural Science Foundation of Jiangsu Province (No. BE2017607).
Disclosure
The authors have no financial conflicts of interest and report no conflicts of interest in this work.
References
1. Sackstein R, Schatton T, Barthel SR. T-lymphocyte homing: an underappreciated yet critical hurdle for successful cancer immunotherapy. Lab Invest. 2017;97:669–697. doi:10.1038/labinvest.2017.25
2. Quail DF, Joyce JA. Microenvironmental regulation of tumor progression and metastasis. Nat Med. 2013;19(11):1423–1437. doi:10.1038/nm.3394
3. Teesalu T, Sugahara KN, Kotamraju VR, Ruoslahti E. C-end rule peptides mediate neuropilin-1-dependent cell, vascular, and tissue penetration. Proc Natl Acad Sci U S A. 2009;106:16157–16162. doi:10.1073/pnas.0908201106
4. Marelli UK, Rechenmacher F, Sobahi TR, Mas-Moruno C, Kessler H. Tumor Targeting via Integrin Ligands. Front Oncol. 2013;3:222. doi:10.3389/fonc.2013.00222
5. Sugahara KN, Teesalu T, Karmali PP, et al. Tissue-penetrating delivery of compounds and nanoparticles into tumors. Cancer Cell. 2009;16:510–520. doi:10.1016/j.ccr.2009.10.013
6. Sugahara KN, Teesalu T, Karmali PP, et al. Coadministration of a tumor-penetrating peptide enhances the efficacy of cancer drugs. Science. 2010;328:1031–1035. doi:10.1126/science.1183057
7. Ding N, Zou Z, Sha H, et al. iRGD synergizes with PD-1 knockout immunotherapy by enhancing lymphocyte infiltration in gastric cancer. Nat Commun. 2019;10:1336. doi:10.1038/s41467-019-09296-6
8. Brischwein K, Schlereth B, Guller B, et al. MT110: a novel bispecific single-chain antibody construct with high efficacy in eradicating established tumors. Mol Immunol. 2006;43:1129–1143. doi:10.1016/j.molimm.2005.07.034
9. Kontermann RE, Brinkmann U. Bispecific antibodies. Drug Discov Today. 2015;20:838–847. doi:10.1016/j.drudis.2015.02.008
10. Osada T, Patel SP, Hammond SA, Osada K, Morse MA, Lyerly HK. CEA/CD3-bispecific T cell-engaging (BiTE) antibody-mediated T lymphocyte cytotoxicity maximized by inhibition of both PD1 and PD-L1. Cancer Immunol Immunother. 2015;64(6):677–688. doi:10.1007/s00262-015-1671-y
11. Taube JM, Anders RA, Young GD, et al. Colocalization of inflammatory response with B7-h1 expression in human melanocytic lesions supports an adaptive resistance mechanism of immune escape. Sci Transl Med. 2012;4(127):127ra37. doi:10.1126/scitranslmed.3003689
12. Junttila TT, Li J, Johnston J, et al. Antitumor efficacy of a bispecific antibody that targets HER2 and activates T cells. Cancer Res. 2014;74(19):5561–5571. doi:10.1158/0008-5472.CAN-13-3622-T
13. Chang CH, Wang Y, Li R, et al. Combination Therapy with Bispecific Antibodies and PD-1 Blockade Enhances the Antitumor Potency of T Cells. Cancer Res. 2017;77:5384–5394. doi:10.1158/0008-5472.CAN-16-3431
14. Rosenberg SA, Restifo NP. Adoptive cell transfer as personalized immunotherapy for human cancer. Science. 2015;348(6230):62–68. doi:10.1126/science.aaa4967
15. Hiwarkar P, Qasim W, Ricciardelli I, et al. Cord blood T cells mediate enhanced antitumor effects compared with adult peripheral blood T cells. Blood. 2015;126(26):2882–2891. doi:10.1182/blood-2015-06-654780
16. Berglund S, Gertow J, Magalhaes I, Mattsson J, Uhlin M. Cord blood T cells cultured with IL-7 in addition to IL-2 exhibit a higher degree of polyfunctionality and superior proliferation potential. J Immunother. 2013;36(8):432–441. doi:10.1097/CJI.0b013e3182a802f6
17. Wu Z, Cheung NV. T cell engaging bispecific antibody (T-BsAb): from technology to therapeutics. Pharmacol Ther. 2018;182:161–175. doi:10.1016/j.pharmthera.2017.08.005
18. Sun C, Mezzadra R, Schumacher TN. Regulation and Function of the PD-L1 Checkpoint. Immunity. 2018;48(3):434–452. doi:10.1016/j.immuni.2018.03.014
19. Bacac M, Fauti T, Sam J, et al. A novel carcinoembryonic antigen T-cell bispecific antibody (CEA TCB) for the Treatment of Solid Tumors. Clin Cancer Res. 2016;22(13):3286–3297. doi:10.1158/1078-0432.CCR-15-1696
20. Ishiguro T, Sano Y, Komatsu SI, et al. An anti-glypican 3/CD3 bispecific T cell-redirecting antibody for treatment of solid tumors. Sci Transl Med. 2017:9. doi:10.1126/scitranslmed.aal4291.
21. Snyder A, Makarov V, Merghoub T, et al. Genetic basis for clinical response to CTLA-4 blockade in melanoma. N Engl J Med. 2014;371:2189–2199. doi:10.1056/NEJMoa1406498
22. McGranahan N, Furness AJ, Rosenthal R, et al. Clonal neoantigens elicit T cell immunoreactivity and sensitivity to immune checkpoint blockade. Science. 2016;351:1463–1469. doi:10.1126/science.aaf1490
23. Balko JM, Sosman JA. A critical need for better cancer immunotherapy models: are organotypic tumor spheroid cultures the answer. Cancer Discov. 2018;8:143–145. doi:10.1158/2159-8290.CD-17-1356
24. Deng J, Wang ES, Jenkins RW, et al. CDK4/6 inhibition augments antitumor immunity by enhancing T-cell activation. Cancer Discov. 2018;8:216–233. doi:10.1158/2159-8290.CD-17-0915
25. Fadeev R, Chekanov A, Solovieva M, et al. Improved anticancer effect of recombinant protein izTRAIL Combined with Sorafenib and Peptide iRGD. Int J Mol Sci. 2019:20. doi:10.3390/ijms20030525.
26. Zhou S, Zhu M, Meng F, et al. Evaluation of PD-1 blockade using a multicellular tumor spheroid model. Am J Transl Res. 2019;11:7471–7478. doi:10.1126/science.aaf1490
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.