Back to Journals » Journal of Inflammation Research » Volume 19
Colchicine Alleviates Non-Atherosclerotic Vascular Aging by Targeting Endothelial Dysfunction and Inflammatory Status
Authors Mohammadi Jouabadi S ![]() , Jüttner AA, Golshiri K, Ataei Ataabadi E, de Boer M
, Jüttner AA, Golshiri K, Ataei Ataabadi E, de Boer M ![]() , De Vries R, Van Veghel R, Goos Y
, De Vries R, Van Veghel R, Goos Y ![]() , Boon Y, Musterd-Bhaggoe U, Duncker DJ
, Boon Y, Musterd-Bhaggoe U, Duncker DJ ![]() , MaassenVanDenBrink A, Danser AHJ, Bax WA, Cornel JH, Roks AJM
, MaassenVanDenBrink A, Danser AHJ, Bax WA, Cornel JH, Roks AJM
Received 3 June 2025
Accepted for publication 14 January 2026
Published 7 February 2026 Volume 2026:19 538430
DOI https://doi.org/10.2147/JIR.S538430
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Durga Misra
Soroush Mohammadi Jouabadi,1,2 Annika A Jüttner,1 Keivan Golshiri,1 Ehsan Ataei Ataabadi,1 Martine de Boer,3 Rene De Vries,1 Richard Van Veghel,1 Yoëlle Goos,1 Youri Boon,1 Usha Musterd-Bhaggoe,1 Dirk J Duncker,3 Antoinette MaassenVanDenBrink,1 AH Jan Danser,1 Willem A Bax,4 Jan H Cornel,5,6 Anton JM Roks1
1Division of Pharmacology and Vascular Medicine, Department of Internal Medicine, Erasmus MC, University Medical Center Rotterdam, Rotterdam, the Netherlands; 2Department of Epidemiology, Erasmus MC, University Medical Center Rotterdam, Rotterdam, the Netherlands; 3Division of Experimental Cardiology, Department of Cardiology, Erasmus MC, University Medical Center Rotterdam, Rotterdam, the Netherlands; 4Department of Internal Medicine, Northwest Clinics, Alkmaar, the Netherlands; 5Department of Cardiology, Radboud University Nijmegen Medical Centre, the Netherlands; 6Department of Cardiology, Northwest Clinics, Alkmaar, the Netherlands
Correspondence: Anton JM Roks, Division of Pharmacology and Vascular Medicine, Department of Internal Medicine, Erasmus MC, University Medical Center Rotterdam, Wytemaweg 80, 3015 CN, Rotterdam, the Netherlands, Email [email protected]
Background: Non-atherosclerotic vascular aging (NAVA) contributes to cardiovascular risk through progressive arterial stiffening and endothelial dysfunction. Colchicine, best known for anti-inflammatory activity, has been proposed to protect vascular structure and function. We evaluated whether chronic colchicine mitigates NAVA in a smooth-muscle–specific ERCC1 knockout (SMC-KO) mouse model of DNA-damage–driven vascular aging.
Methods: We performed experiments in the SMC-KO treated with colchicine (0.1mg/kg/day) or vehicle from the age of 10 to 22 weeks. Endothelial function was assessed by acetylcholine-induced vasorelaxation, and vascular structure by pulse-wave velocity (PWV), carotid intima–media thickness (cIMT), and elastin integrity.
Results: SMC-KO mice developed increased arterial stiffness and impaired acetylcholine-mediated relaxation. Chronic colchicine significantly (p< 0.01) lowered PWV, preserved elastin architecture, and improved endothelium-dependent relaxation, while sodium-nitroprusside responses and systemic cytokine levels remained unchanged.
Conclusion: Chronic colchicine treatment preserves endothelial function and vascular elastin structure and reduces arterial stiffness in a DNA-damage–driven model of non-atherosclerotic vascular aging. These findings highlight colchicine’s pleiotropic vascular benefits beyond anti-inflammation, supporting its potential repurposing for primary prevention in vascular aging and cardiovascular health or the development of colchicine-mimicking drugs with greater selectivity and improved safety profiles.
Keywords: vascular aging, colchicine, stiffness, endothelial function, beta-adrenoreceptors
Introduction
Vascular aging is a significant contributor to global morbidity and mortality, impacting cardiovascular health and organ function.1,2 Traditionally, efforts to address vascular diseases have focused on atherosclerosis, the primary driver of conditions like myocardial ischemia and infarction. However, with advances in atherosclerosis prevention and treatment, there is a growing focus on age-related vascular dysfunction characterized by progressive stiffness and loss of vascular elasticity, which we refer to as non-atherosclerotic vascular aging (NAVA).3 Unlike atherosclerotic vascular disease, NAVA primarily involves changes in vascular structure and function, often driven by intrinsic cellular aging processes within the vascular wall. A critical feature of NAVA is vascular stiffness, which increases the load on the heart and compromises blood flow to vital organs, including the brain. Mechanistically, vascular aging is associated with DNA damage accumulation-either due to internal or external factors.4 Furthermore, it features with endothelial cell (EC) and smooth muscle cell (SMC) dysfunction, leading to structural changes in the extracellular matrix (ECM), diminished nitric oxide (NO) responsiveness, and endothelial dysfunction.3,5–9 The management of NAVA poses a challenge due to limited availability of pharmacological interventions. Hence, addressing one of the pathological changes (wherein targeting one hallmark may impact others) represents a promising therapeutic target to mitigate the effects of vascular aging and improve systemic vascular health.10 Colchicine, a microtubular de-polymerizer, has shown potential beyond inflammation control, particularly in vascular health. By targeting cellular pathways implicated in aging, colchicine may influence vascular stiffness and hemodynamics.11–15 Furthermore, recent studies suggest that colchicine’s interaction with β-adrenergic receptors (β-AR) may also play a role in its vascular effects, potentially contributing to its cardioprotective properties.16 Yet, to date, no study has tested colchicine in a plaque-free model of DNA-damage–driven vascular aging, leaving its potential in NAVA unknown. In this study, we first investigated ex vivo the direct vasorelaxation response to colchicine in mice, accompanied by further investigation whether chronic colchicine treatment could attenuate features of NAVA induced in mice by using a previously phenotyped model of smooth muscle cell (SMC)-specific knockout of the ERCC1 (Excision Repair Cross Complementation Group 1) gene, shown to induce accelerated vascular aging and inflammation7 through DNA damage (as a primary hallmark of vascular aging).7–9,17 The SMC-specific model was chosen over the endothelial-cell–specific model because vascular SMCs are the principal drivers of extracellular-matrix remodeling and medial stiffening, key determinants of arterial compliance, the primary outcome of this study.
This model exhibits notable features of human NAVA,7 including vascular stiffness and NO hypo-responsiveness, without the confounding effects of atherosclerosis. By focusing on non-atherosclerotic vascular dysfunction, our study provides the first mechanistic and translational evidence that colchicine mitigates DNA-damage–associated vascular aging, laying the groundwork for future development of colchicine-mimetic therapies.
Materials And Methods
Animals
Generation and characterization of SM22αCre+ Ercc1fl/- (SMC-KO), along with their corresponding littermates SM22αCre+ Ercc1fl/+ (SMC-LM), were previously detailed.7 All methods were applied to both sex, housed in individually ventilated filter top cages. We provided the mice with a 12-hour light/dark cycle at 20–22 °C. Daily welfare assessments, including weight measurements and visual inspections, were conducted to identify and score any signs of discomfort in the animals. Animal breeding and care, including ad libitum access to water and food (standard chow), were carried out at the Erasmus MC animal facility. Mice were anesthetized by using isoflurane inhalation (2%–3%). In all experiments, mice were euthanized by cervical dislocation under isoflurane inhalation. These procedures adhered to the guidelines outlined (in Directive (2010/63/EU) of the European Parliament) regarding the protection of vertebrate animals used for academic and scientific purposes. The study received approval from both the National Animal Care Committee (License #2216204) and the local administration within Erasmus MC, University Medical Centre, Rotterdam.
Study Design
A total of 27 SMC-KO mice (13 female and 14 male) and 25 age-matched SMC-LM mice (14 female and 11 male) were divided into four experimental groups at the age of 10 weeks. Within each genotype, one received normal drinking water, while another group received drinking water containing colchicine (0.1 mg/kg/day, Sigma-Aldrich) over a 12-week period. This 12-week colchicine regimen was selected to span the period of early and progressive vascular changes in SMC-KO mice, which takes place from the age of 10 weeks until full development at 22 weeks, as was reported in our previous studies.7 At 21 weeks of age, ultrasound imaging of abdominal aorta was performed. Tissues were collected following sacrifice at 22 weeks of age, and immediately snap-frozen, and post-hoc measurements were executed on freshly obtained arterial tissue.
Ultrasound Imaging
At 21 weeks of age, mice were sedated in an anesthesia box with isoflurane (4%), subsequently mice were intubated, and respiration was monitored with a pressure-controlled ventilator.18 Anesthesia was sustained using isoflurane 2.5% with the body temperature at 37°C by placing the mouse on a heating pad. Imaging conducted using the Vevo 3100 (High-Resolution Imaging System from FUJIFILM VisualSonics, Inc)., equipped with the MX550 transducer. We conducted non-invasive assessments of the local geometry and stiffness of the abdominal aorta using ultrasound.19 Local pulse wave velocity (PWV) was assessed from ECG-triggered kilohertz visualization (EKV) sequences using the VevoVasc software. Longitudinal images of the abdominal aorta were acquired, and the pulse transmission time (ΔT) between two predefined arterial segments, separated by a known distance (ΔD), was measured. PWV was then computed as the ratio of distance to time (ΔD/ΔT).
Pressure-Volume Loop Measurements
At the age of 22 weeks, just before sacrifice, calibrated close-chest Pressure-Volume Loop (PV-Loop) measurement was performed as described previously.20 The anaesthetized mouse was positioned in supine orientation, and a 15 mm midline incision was made to expose the trachea. Intubation was performed through the trachea, connecting the mouse to a positive-pressure volume-controlled ventilator (SAR-830/P; CWE, USA). A 1.2F micro conductance pressure catheter (Transonic Science 1.2F PV, Canada) was advanced into the left ventricle through the right side of the carotid artery. The catheter was calibrated using parallel-conductance correction (hypertonic saline bolus) and α-calibration according to manufacturer instructions. Because the goal was to characterize steady-state cardiac performance, no preload reduction or vena-cava occlusion was performed. Real-time PV data were acquired and analyzed with LabChart/ADV500 software (Transonic, USA) to derive standard systolic and diastolic indices, including heart rate, stroke volume, cardiac output, ejection fraction, end-systolic elastance (Ees), arterial elastance (Ea), and maximal and minimal dP/dt as well as the relaxation constant (τ). This experiment was terminal and animals were immediately sacrificed.
Ex Vivo Vascular Function Assessment (Wire Myography)
To examine both acute and chronic effects of colchicine on vascular relaxation, we performed two complementary experiments:
Acute Exposure to Colchicine
To assess the direct vasorelaxation effect of the colchicine, thoracic aortic rings (1.5–2.0 mm in length, cleaned of perivascular fat) from non-treated mice (both SMC-LM and SMC-KO) were mounted in a small-vessel wire myograph (Danish Myograph Technology, Aarhus, Denmark) containing 6mL of oxygenated Krebs–Henseleit buffer (118 mmol/L NaCl, 4.7 mmol/L KCl, 25 mmol/L NaHCO3, 1.2 mmol/L MgSO4, 1.2 mmol/L KH2PO4, 2.5 mmol/L CaCl2, 8.3 mmol/L glucose; pH 7.4) maintained at 37 °C and gassed with 95% O2/5% CO2. Tension normalization involved step-wise stretching until 90% of the estimated diameter, corresponding to a biologically effective transmural pressure of 100 mmHg.21 Viability and maximum contractile response to 100 mmol/L KCl were assessed. After triple washing with 10-minute intervals, segments were first pre-constricted with U46619 compound (30 nmol/L)-a thromboxane A2 analogue to 80–100% of the maximal KCl contraction. A concentration–response curve (CRC) for colchicine (10−13–10−5 mol/L) was generated after pre-contraction with U46619 (30 nmol/L). For each ring, relaxation was calculated as the percentage decrease in tension relative to the U46619 pre-constriction level (set as 100%). Involvement of the β-AR was evaluated using a non-selective β-blocker (propranolol, Sigma-Aldrich; 1 µmol/L), and selective β2-adrenoceptor (butoxamine Sigma-Aldrich; 1µmol/L) and β3-adrenoceptor (SR-59230A Sigma-Aldrich; 1µmol/L) antagonists. Furthermore, to rule out the pleiotropic effects of colchicine, we investigated whether its relaxation effect is associated with its microtubule disruption activity by incubating the rings with a microtubule stabilizer (Paclitaxel, Sigma-Aldrich; 10 µmol/L). Finally, to determine whether adrenergic stimulation is dependent on endothelial Nitric Oxide Synthase (eNOS), we repeated the colchicine CRC, this time in the presence of L-NAME (100 µmol/L).
Chronic in Vivo Colchicine Treatment
Immediately following animal sacrifice, the thoracic segment of the aorta was isolated and immersed in a cold and oxygenated (95% O2 and 5% CO2) Krebs-Henseleit buffer solution. Vessel segments cleaned from surrounding fat, measuring 1.5 to 2 mm in length, were installed on small wire myograph organ baths with a 6-mL volume (Danish Myograph Technology, Aarhus, Denmark). Similar to the previous section, tension normalization involved step-wise stretching until 90% of the estimated diameter, corresponding to a biologically effective transmural pressure of 100 mmHg.21 Viability and maximum contractile response to 100 mmol/L KCl were assessed. After triple washing with 10-minute intervals, segments were first pre-constricted with U46619 compound (30 nmol/L). Endothelium-dependent vasorelaxation response to acetylcholine (ACh, 10−9-10−5 mol/L) and endothelial-independent vasorelaxation response to sodium nitroprusside (SNP, 10−11-10−4 mol/L) were measured in a concentration-response manner after reaching a contraction plateau. To explore the contribution of the nitric oxide-cyclic guanosine monophosphate (NO-cGMP) and endothelium-dependent hyperpolarization (EDH) pathways, pathway inhibitors (L-NAME 100 µmol/L and TRAM-34,100 nmol/L plus apamin 100 nmol/L, respectively) were added 15 minutes before the pre-contraction with U46619. For the endothelium-independent response to SNP, L-NAME (100 µmol/L) was added to prevent potential bias from intrinsic NO release. Exact doses, exposure times, and concentrations for colchicine and all pharmacologic agents (U46619, L-NAME, propranolol, butoxamine, SR-59230A, and paclitaxel) are provided in Supplementary Table S1 for quick reference.
Mechanical Properties of the Vascular Wall (Pressure Myography)
The carotid artery of the mice was used to measure the passive mechanical properties of the vasculature. Fat-free carotids were mounted on a pressure myograph device (Danish Myograph Technology, Aarhus, Denmark) within a calcium-free buffer (data are in mmol/L: NaCl (120), KCl (120), EGTA (2), MgCl2 (3.6), NaH2PO4 (1.2), glucose (11.4), NaHCO3 (26.3); pH 7.4). The intraluminal pressure of vessels underwent incremental elevation, commencing at 10 mmHg, with subsequent increases occurring at 3-minute intervals until reaching 120 mmHg. Measurements of lumen diameter and wall thickness were conducted at each step after the vessel achieving equilibrium.
Histology
Right carotid artery was embedded in paraffin and subsequently sectioned into 4 µm slices. Carotid sections underwent picro-sirius red staining for collagen content assessment and the obtained images were analysed utilizing a quantitative image analysis system (BioPix iQ software, BioPix AB). Furthermore, Van Gieson staining was employed to visualize elastin fragmentation and measure the carotid intima-media thickness (cIMT). The cIMT was quantified using the NDP.view2 software. For each measurement, the surface of the inner boundary of the vessel, corresponding to the lumen-intima interface, and the outer boundary, corresponding to the media-adventitia interface, were manually traced. The difference between these two surface boundaries was calculated to determine the cIMT. All histology analyses, including cIMT measurements, were performed blinded to both genotype and treatment group.
Immunofluorescence
For the immunofluorescence staining in carotid arteries, slides were incubated in heated citrate buffer (pH 6.0) to retrieve antigens. After blocking with 1% [w/v] BSA and 0.5% [v/v] normal goat serum which was in Tris-buffered saline with 1% [v/v] Tween 20 (1x TBS-T), samples were washed with 1x TBS-T and incubated with the Actin (1:100, Merck, A3853), iNOS (1:250, Santa Cruz, sc-650), COX-2 (1:250, Cell signaling, 12282S), NFkB-p65 (1:100, Cell signaling, 8242S), AKT-Phospho (ser473) (1:100, Cell signalling, 9060S), Phospho-p38 MAPK (1:100, Cell signalling, 9251S) antibodies at 4 °C overnight. On the next day, samples were washed with 1x TBS-T and incubated with the secondary antibody (1:100, donkey anti-rabbit antibody, Alexa FluorTM 488, AB_2535792 was used for all primary antibodies except for Actin and AKT-Phospho where 1:100 donkey anti-mouse antibody, Alexa FlurTM 555, A31570 was used). To minimize non-specific signal, vessel autofluorescence was reduced using the Vector® TrueVIEW® Quenching Kit (SP-8400-15, Vector Laboratories), followed by nuclear counterstaining with DAPI. Fluorescence intensity was quantified with ImageJ following a standardized analysis pipeline: images were imported as 16-bit TIFFs, background was subtracted using the rolling-ball method (radius 50 px), regions of interest (ROI) encompassing the vessel wall were manually traced, and mean fluorescence intensity was calculated and normalized to ROI area minus mean background. All analyses were performed blinded to genotype and treatment.
Western Blotting
Proteins from lung tissue (as a vascular enriched tissue) were isolated in homogenization buffer (0.3 mol/L sucrose, 50 mmol/L Tris-HCL pH 7.5, 1 mmol/L EDTA, 1 mmol/L EGTA, 1 mmol/L sodium-orthovanadate, 50 mmol/L sodium fluride, 1 mmol/L DTT, 1 mmol/L PMSF, 1% (v/v) Triton x-100, phosphatase inhibitor cocktail 3 (Sigma-Aldrich) and cOmpleteTM protease inhibitor cocktail (Roche)), loaded (25 µg) on gels (4–20% Criterion TGX Gel, Bio-Rad), and transferred to membranes (Trans-Blot Turbo 0.2 µm PVDF Transfer Pack, Bio-Rad) with the Tans-Blot Turbo Transfer system (Bio-Rad). Membranes were blocked with either 5% (w/v) BSA or non-fat milk according to the primary antibody, then subjected to overnight incubation with primary antibody (Supplementary Table S2). Subsequently, after secondary antibody incubation, membrane visualization was carried out utilizing Clarity Western ECL substrate (Bio-Rad) in conjunction with an Amersham Al600. Quantitative analysis was conducted using Image Studio software, employing background stain subtraction through the built-in module.
Plasma Cytokines Levels
Protein levels of Interleukin 6 (Il-6), Monocyte Chemoattractant Protein-1 (MCP1), Tumour Necrosis Factor alpha (TNF-α), and Growth Differentiation Factor 15 (GDF-15) in plasma were measured using a Luminex multiplex bead immunoassay system (R&D Systems Europe, Ltd; UK). Measurement of these markers was performed primarily to rule out systemic inflammation as a confounding factor and to monitor safety of chronic colchicine exposure. The assays were performed according to the instructions provided from manufacturer. Bio-Plex MAGPIX machine was used for the measurements, and Bio-Plex Manager MP software for the analysis.
Statistical Analysis
The reported data are in mean and standard error of the mean (mean ± SEM), unless stated otherwise. Depending on the number of variables included, group differences were assessed using either one-way ANOVA or two-way ANOVA. The relaxation responses to both ACh and SNP were relative to the maximum contraction induced by 30 nmol/L U46619. Differences between CRCs in repeated measures were examined using a general linear model. Repeated-measures data (eg, CRCs) were first tested for the assumption of sphericity using Mauchly’s test. When sphericity was violated (p < 0.05), we reported the Greenhouse–Geisser–corrected degrees of freedom and p values. A P-value threshold of <0.05 was set for statistical significance. Statistical analyses were conducted using IBM SPSS Statistics (IBM Corporation, version 25) and GraphPad Prism (GraphPad Software Inc., version 8.0.1; San Diego, CA), and R Studio V4.3.2 was used for the Pearson correlation analysis.
Results
Colchicine Acutely (Ex Vivo) Relaxes Mouse Aorta Through NO-cGMP Axis
To investigate the acute ex vivo effect of colchicine on vasorelaxation in mice, thoracic aortas from both untreated SMC-KO mice and their littermate controls (SMC-LM) were mounted on a wire myograph, and a CRC for colchicine was performed. Colchicine directly induced relaxation in the thoracic aortas of both vehicle-treated SMC-KO and SMC-LM mice (Figure 1A). This effect was more strongly mediated by the NO-cGMP pathway compared to the EDH pathway, as indicated by the effect of NOS vs Ca2+- activated K channel blockade (Figure 1B for SMC-LM and Figure 1C for SMC-KO). The colchicine-induced relaxation was reduced in SMC-KO mice compared to SMC-LM mice, likely due to the impaired NO-cGMP pathway in SMC-KO mice, as demonstrated in our previous study.7 Additional mechanistic pharmacology experiments, performed in SMC-LM control rings, confirmed that colchicine’s vasorelaxant effect (at both clinical serum range (7.5 nmol/L) and upper pharmacological levels (75 µmol/L) is potentially mediated through microtubule disruption and β-adrenergic signaling. Detailed results and a representative trace of the colchicine relaxation kinetics are provided in the Supplementary Figures S1–S3A.
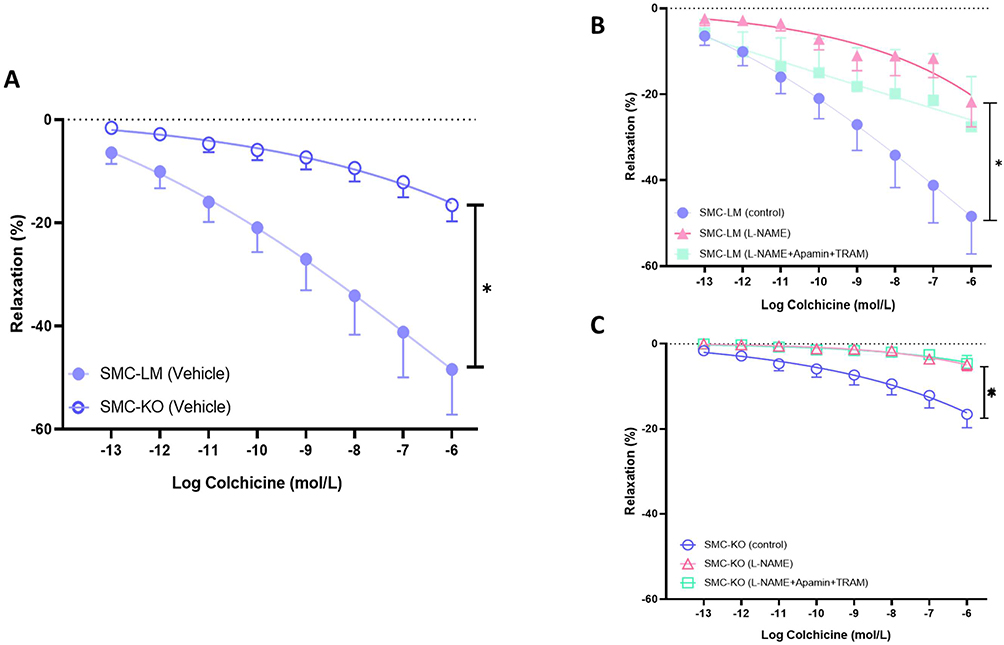 |
Figure 1 Colchicine induces direct vasorelaxation in mouse aorta via NO–cGMP and EDH pathways. (A), Colchicine concentration–response curves (CRCs) in thoracic aortic rings from SMC-KO and SMC-LM mice. (B), Effect of nitric-oxide synthase inhibition (L-NAME, 100 µmol/L) on colchicine-induced relaxation in SMC-KO rings. (C), Effect of combined EDH blockade (apamin + TRAM-34, each 100 nmol/L) on colchicine-induced relaxation in SMC-LM rings. All experiments were performed in wire-mounted thoracic aortic rings (1.5–2 mm) pre-contracted with U46619 to 80–100% of maximal KCl response. Data are expressed as % relaxation of the pre-constriction. Concentration–response curves were analyzed by repeated-measures ANOVA with Greenhouse–Geisser correction for sphericity and Bonferroni post-hoc tests. *P < 0.05 vs vehicle; n = 6 rings per condition. |
Chronic Colchicine Treatment Mitigates Vascular Stiffening
Given the beneficial direct vasorelaxation effects of colchicine observed ex vivo, we decided to further investigate its chronic effects on the vasculature. To investigate the potential impact of colchicine on vascular stiffness, we initially conducted in vivo ultrasound imaging to quantify local stiffness of the abdominal aorta in mice at 21 weeks of age. PWV was higher in SMC-KO mice compared to the SMC-LM mice (P<0.01), indicating increased vessel stiffness. Chronic administration of colchicine effectively mitigated PWV, normalizing it to SMC-LM levels (P<0.001; Figure 2A and B). Vascular stiffness is known to be dependent on blood pressure. However, in our previous studies using tail cuff measurements, we did not observe any changes in blood pressure between the SMC-KO mice and their littermates.7 To further confirm these findings, we also measured end-systolic pressure directly via catheter insertion in the current study, which further validated our previous observations regarding the absence of blood pressure phenotype (Supplementary Figure S4A). Other cardiac indices measured via catheter insertion and ultrasound imaging are reported in Supplementary Table S3. Furthermore, Ultrasound measurements of abdominal aortic diameters showed no significant differences between vehicle- and colchicine-treated SMC-KO mice (Supplementary Figure S4B), indicating that the reduction in PWV reflects structural preservation rather than baseline vasodilation.
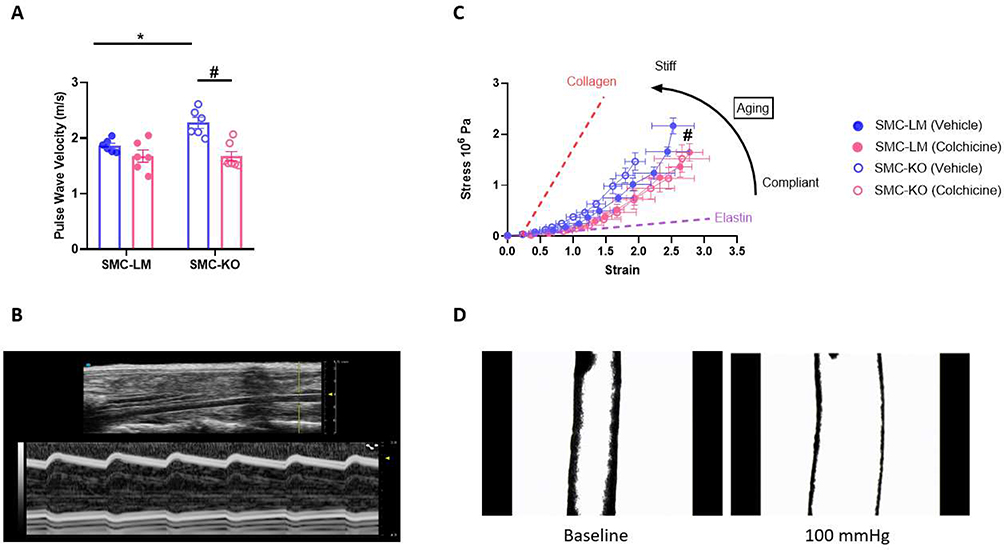 |
Figure 2 Colchicine treatment protects SMC-KO mice against vascular stiffness. (A), Colchicine decreased vascular stiffness in SMC-KO mice (N=6 per group). Data are mean ± SEM. Statistical analysis was by two-way ANOVA followed by Bonferroni post-hoc test, *P<0.05 for SMC-LM vehicle Vs SMC-KO vehicle,and #P<0.05 for SMC-KO vehicle Vs SMC-KO colchicine. (B), M-Mode (Lower wavy image) and B-Mode (top) images of Pulse Wave Velocity (PWV) measured by ultrasound imaging of mouse abdominal aorta. (C), Strain differences plotted against stress differences in mouse carotid artery suggesting potential shift to more elastin content after colchicine treatment (N=6 per group, except for SMC-LM (vehicle); N=5). Statistical analysis was by a general linear model for repeated measurement. #P<0.05 comparing SMC-KO vehicle Vs SMC-KO colchicine. (D), Corresponding image of mouse carotid artery in pressure myograph at the baseline pressure and physiological pressure of 100 mmHg. |
Subsequently, we assessed in vitro vascular stiffness and mechanical characteristics of the mouse carotid artery using pressure myography. SMC-KO mice exhibited a significant elevation in media stress at comparable levels of media strain compared to SMC-LM mice confirming the increased stiffness that was also observed in vivo, and suggesting a flow-, pressure- and Ca2+-independent contributing component. Chronic in vivo colchicine treatment resulted in improved vessel compliance in the isolated carotid artery (Figure 2C and D). Vascular stiffening is a consequence of intricate interactions involving structural, vasomotor, and molecular constituents within the vascular bed.22 Hence, we delved deeper into these elements to discern whether one predominates or if they act synergistically in models used in this study.
Colchicine Ameliorates the Structural Changes in the Vessel Wall of SMC-KO Mice
To investigate the role of structural components in mediating the effects of colchicine on vascular stiffness reduction, we assessed hypertrophy and ECM changes. In SMC-KO mice, we observed an increased cIMT to lumen ratio, which was normalized by colchicine treatment (P=0.02, Figure 3A). Although collagen deposition was elevated in the intima-media layer of SMC-KO mice, colchicine treatment did not affect this parameter (Figure 3B). Additionally, colchicine reduced elastin fragmentation in SMC-KO mice (Figure 3C).
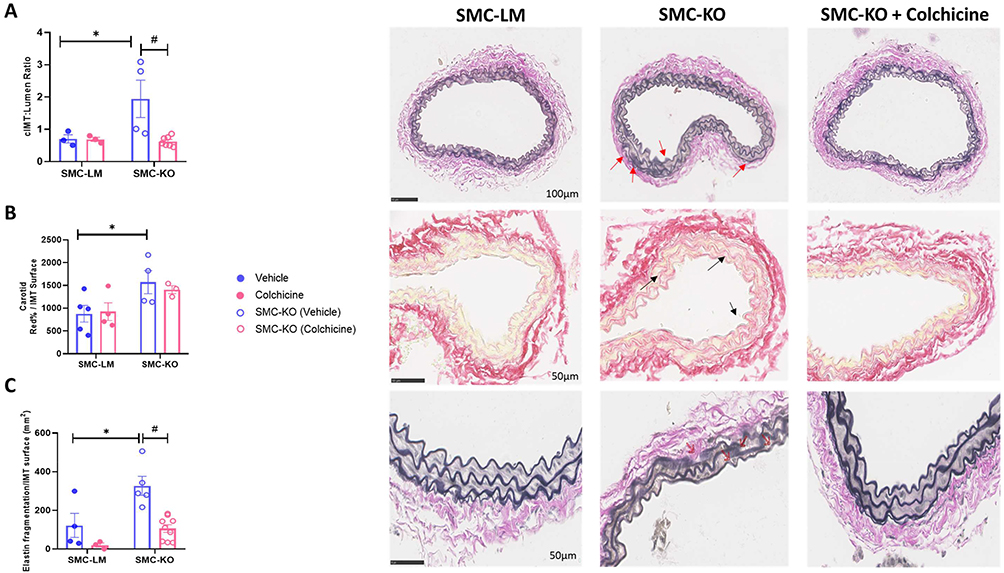 |
Figure 3 Colchicine treatment protects SMC-KO mice against vascular remodeling. (A), Colchicine treatment restored the increased carotid intima-media thickness (cIMT) in SMC-KO mice (N=3-7 per group as indicated by dots in the graph, scale bar = 100μm, Red arrows shows the area where the intima-media is thickened). (B), Collagen content, measured by Sirius Red staining, was elevated in SMC-KO mice compared to their littermate controls (N=3-5 per group as indicated by dots in the graph, scale bar = 50μm, Black arrows show increased collagen deposition in the intima-media layer). (C), Colchicine treatment reduced elastin fragmentation in SMC-KO mice (N=3-8 per group as indicated by dots in the graph, scale bar = 50μm, Red arrows shows location of breaks). All the measurements were performed manually by an independent observer blinded to genotype and treatment. Data are presented as mean ± SEM. Statistical analysis was performed using two-way ANOVA followed by Bonferroni post-hoc test; *P<0.05 indicates a significant genotype effect, and #P<0.05 indicates a significant colchicine treatment effect. |
Colchicine Protects Endothelium-Dependent Relaxation via the NO Pathway
Both endothelium-independent (SNP-mediated) and -dependent (ACh-mediated) vasodilation responses were decreased in SMC-KO mice. Chronic colchicine treatment significantly improved the vasodilatory response to ACh (Figure 4A). In contrast, the response to SNP was not affected by colchicine treatment (Figure 4B). The lack of effect on SMC (via SNP) is consistent with the absence of an effect of colchicine on KCl-induced contractility in SMC-KO mice or SMC-LM, supporting that colchicine’s effects are primarily on endothelium-dependent effects rather than through a generalized change in smooth muscle motor function (Supplementary Figure S3B). To further investigate the underlying vasorelaxation pathway, we conducted experiments using various pharmacological inhibitors. The results indicate that inhibition of NOS by L-NAME led to a reduction in the enhanced ACh response after colchicine treatment, while TRAM34/Apamin had no additional blocking effect (Figure 4C and D). Finally, the improvement in ACh response remained significant also after adjusting for the maximum SNP response (10−4 mol/L), indicating that colchicine improves endothelial NO signaling without evidence of a synergistic effect on smooth-muscle responsiveness (Figure 4E). The corresponding effect sizes (mean differences with 95% confidence intervals) for all these comparisons are provided in Supplementary Table 4.
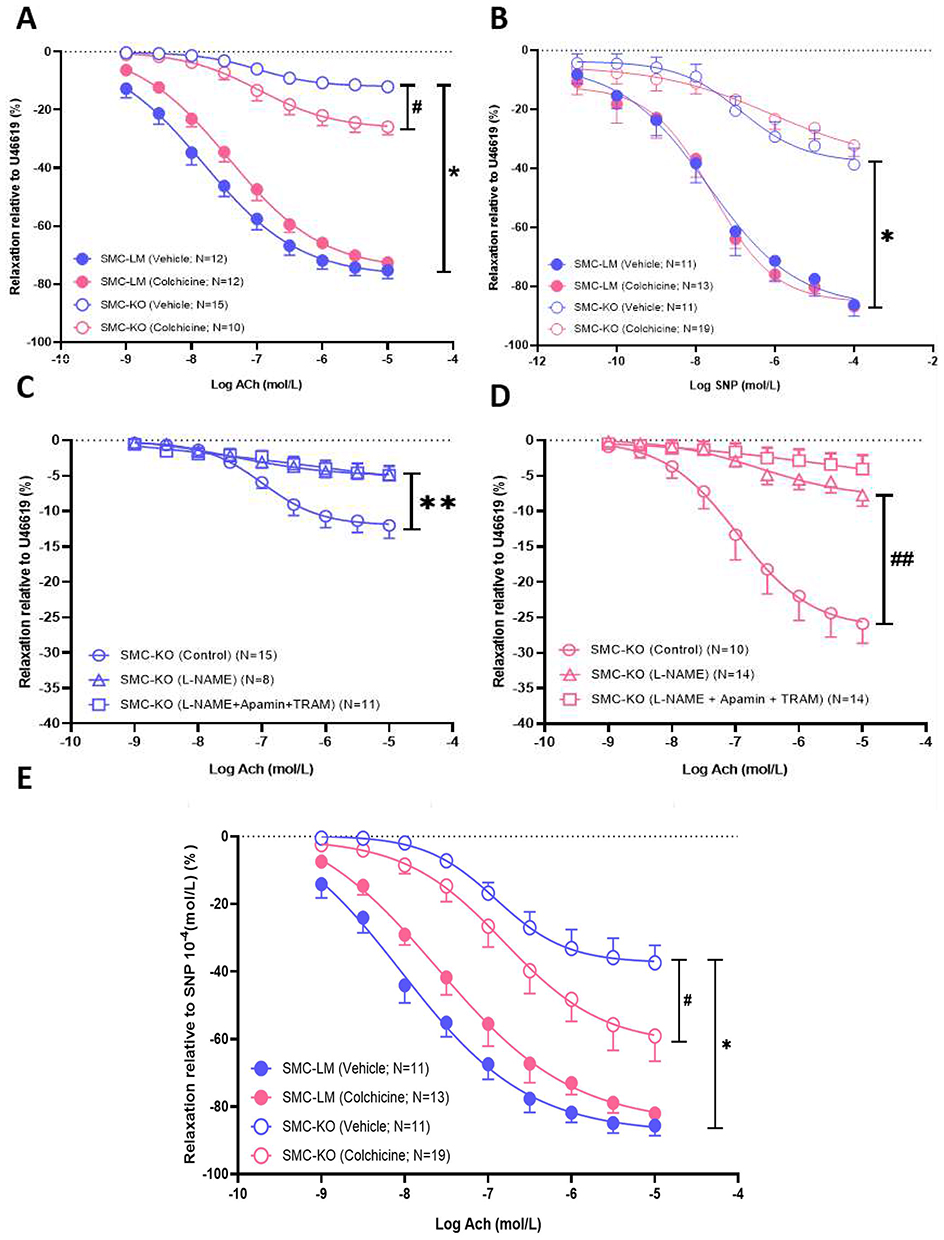 |
Figure 4 Colchicine improves aging-related endothelial dysfunction by upregulating NO. (A and B), Response to sodium nitroprusside (SNP) and acetylcholine (ACh) in aortic rings of SMC-KO and SMC-LM mice treated with vehicle or colchicine (N=10-19 per group as indicated in each graph). (C and D), contribution of the NO-cGMP (blocked by L-NAME) and Endothelium-Dependent Hyperpolarization EDH (blocked by Apamin + TRAM34) pathways to the vasorelaxant effects of ACh in aortas of vehicle- and colchicine-treated SMC-KO mice (N=10-19 per group as indicated in each graph). (E), ACh-mediated relaxation expressed relative to maximal SNP-induced relaxation (10−4 mol/L) to assess potential synergy between endothelial NO signaling and smooth-muscle sensitivity (N = 11–19). Relaxations (mean ± SEM) have been expressed as a percentage of the preconstriction induced by U46619. *:P<0.05 for genotype, #:P<0.05 for effect of colchicine, **:P<0.05 for pathway contribution for genotype, ##:P<0.05 for pathway contribution for colchicine as analyzed by repeated-measures ANOVA with Greenhouse–Geisser correction for sphericity and Bonferroni post-hoc tests. |
Colchicine Reduces Local Inflammation Through Inhibition of COX-2 and iNOS
Local NO and prostaglandin have been recognized as important pro-inflammatory pathways connecting to Mitogen-activated protein kinases (MAPK-p38), with iNOS and cyclooxygenase-2 (COX-2) as important mediating enzymes of which activity is increased with the protein expression level. As further evidence for the impact of colchicine on NO signalling, iNOS proteins levels, which were elevated in the carotid arteries of SMC-KO mice, were partially restored to normal levels by colchicine treatment (Figure 5A). Western blotting of lung tissue, a vascular-rich organ, confirmed elevated iNOS expression in SMC-KO and a lower iNOS/p-eNOS ratio in colchicine-treated SMC-KO mice (Supplementary Figure S5), supporting the immunohistochemical evidence of attenuated local inflammatory signaling. Additionally, we found that COX-2 was increased in the carotid artery of SMC-KO and reduced by colchicine (Figure 5B). Subsequently, we inspected the main pro-inflammatory signalling pathways MAPK-p38 and AKT-NFκB (Protein kinase B - nuclear factor-κB) in vascular tissue. Colchicine selectively normalized the hyperactivated MAPK-p38 pathway, while no significant effect was observed on AKT-NFκB signalling (Figure 5C–E).
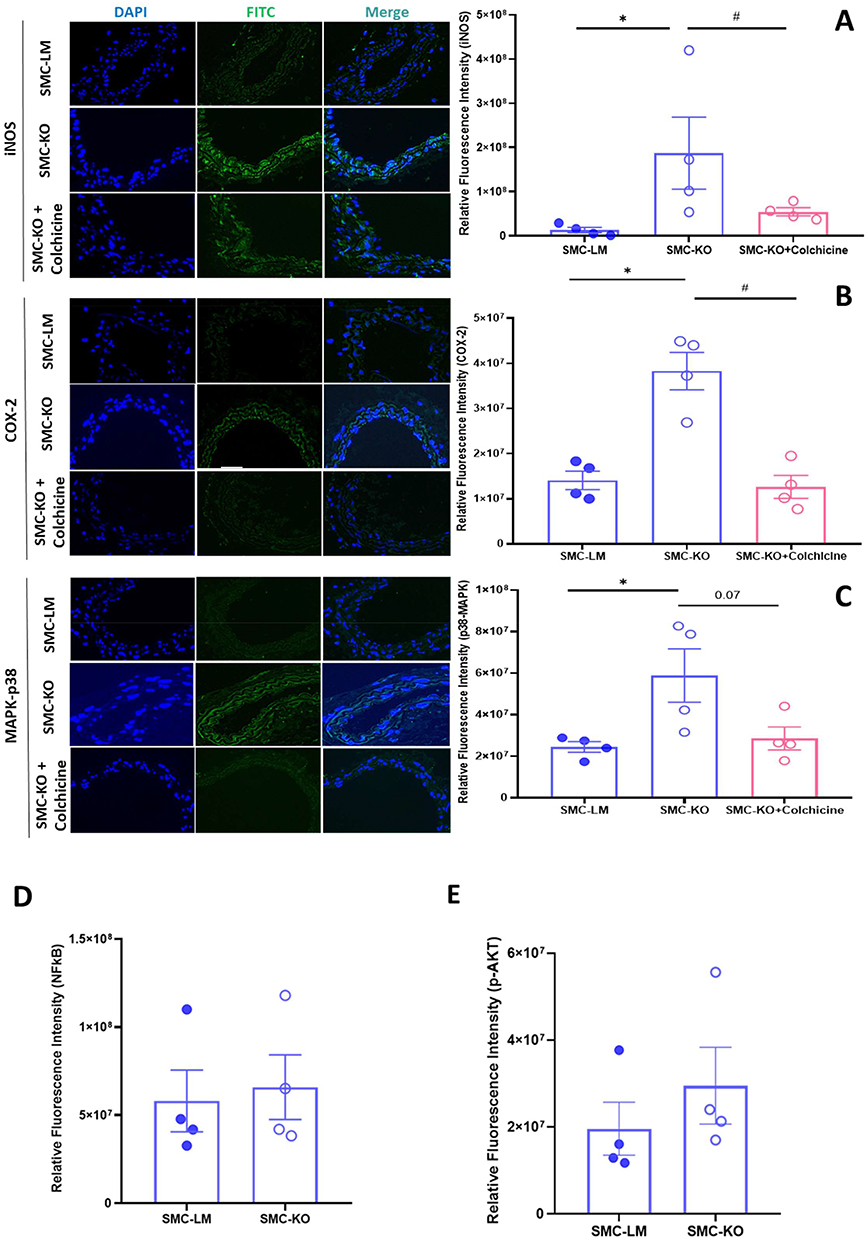 |
Figure 5 Colchicine reduces inducible NOS and COX-2 in the carotid arterial bed of SMC-KO mice. (A and B), Immunofluorescent staining of the carotid arteries in mice shows elevated iNOS and COX-2 expression, indicated by increased fluorescence intensity. Colchicine treatment effectively restored these markers to baseline levels (N=4). (C), MAPK-p38 was hyperactivated in SMC-KO animals, which was partially reversed by colchicine treatment (N=4), Zoom = 20μm. (D), SMC-KO mice show no changes in AKT-p and (E), NFκB expression. Statistical analysis was performed using one-way ANOVA for panels A-C and unpaired t-test for panels D and E. *:P<0.05 for genotype, #:P<0.05 for effect of colchicine. iNOS; inducible Nitric Oxide Synthase, COX-2; Cyclooxygenase-2, MAPK; Mitogen-activated protein kinases, NFκB, nuclear factor κB; AKT, protein kinase B. |
To determine whether colchicine could have effects through modulation of circulating inflammatory factors IL-6, MCP-1, and TNF-α were measured. No significant differences were observed between SMC-KO and SMC-LM mice, nor between control and treated animals in these plasma markers. Surprisingly, the inflammatory hepatokine, GDF-15, increased following colchicine treatment in SMC-KO mice (Supplementary Figure S6).
Discussion
Our study demonstrates that colchicine exerts acute vasorelaxation effects in mice which are comparable to those previously reported in humans16 ie, involving NO signaling, β-adrenergic receptor activation, and microtubule polymerization. These findings highlight the translational relevance of mouse models for investigating colchicine’s vascular effects. Additionally, in our smooth muscle-specific mouse model of accelerated aging, chronic colchicine treatment effectively mitigates vascular stiffening and preserves endothelium-dependent vasorelaxation. This protective effect is partly mediated by reductions in local vascular inflammation, preservation of elastin structure, and sustained NO signaling. Collectively, our results reveal a novel therapeutic potential for colchicine in addressing vascular pathologies, supporting its possible repurposing as a treatment option in vascular disease.
Given the potential pleiotropy of colchicine’s effects, the acute ex vivo experiment helped to better understand consequences thereof for concentration-dependent responses, i.e. there are no indications for a clear-cut, single binding site-effectuated response. The acute vasorelaxation in response to colchicine in mouse aorta mirrors effects observed in humans. Unlike classical receptor-mediated agonists, colchicine produced a shallow, non-sigmoidal concentration–response curve and a delayed onset of relaxation (5 min to initial effect). This lack of a classical dose-response curve suggests that the colchicine effect may be time-dependent, rather than concentration-dependent. Colchicine might target cytoskeletal structures over time at various binding sites with various affinity. Thus, its effects may evolve gradually, with delayed responses reflecting the kinetics of the interaction with various binding sites, rather than showing abrupt, stepwise concentration-dependent effects at a specific binding site. Regarding the concentrations used in this setup, we chose a range from 10−13 to 10−4 M to assess both lower and higher doses around the human plasma concentration (7.5 nM).
Evidence suggests that colchicine induces β-AR activation in human arteries in vivo, plausibly mediated via activation of voltage-gated Kv7 channels downstream of cAMP16, the second messenger for β-AR activation, rather than through direct allosteric binding to β-AR. This mechanism, involving microtubules, has been previously demonstrated in rat mesenteric and renal arteries.23,24 In our study, we replicated these responses in mouse aorta, employing β-AR subtype-specific antagonists and the microtubule stabilizer paclitaxel, further supporting cross-species validity in humans, rats, and mice. Additionally, our findings highlight the involvement of NO signaling, culminating in downstream cGMP production.25 The potential cross-talk between cAMP and cGMP signaling pathways, possibly mediated by nanodomain phosphodiesterase (PDE) interactions,26,27 presents an intriguing avenue for further research. The detailed elucidation of these interactions remains a critical, albeit long-term, objective.
Our study is the first to directly demonstrate that colchicine treatment mitigates vascular stiffness, an effect that previous epidemiological and clinical investigations, primarily focused on specific populations (e.g., familial Mediterranean fever and HIV patients), have reported with mixed results.28,29 By measuring PWV, we provide a more precise, localized assessment of vascular stiffness, revealing that colchicine reduces stiffness in both the aorta and carotid arteries. A recent randomized controlled trial demonstrated that low-dose colchicine attenuates progression of arterial stiffness in patients with type 2 diabetes, while explicitly acknowledging that the underlying mechanisms remain to be clarified.30 In the present study, we provide potential mechanistic evidence that may account for these vascular effects. This reduction appears linked to colchicine’s prevention of elastin fragmentation, while collagen levels remain unaffected. Stiffness changes can be attributed to elastin fragmentation and hypertrophy, common contributors to modulation of passive stiffness.31 Apparently, in the SMC-KO mouse model, increased collagen does not translate to heightened stiffness when elastin is preserved, as confirmed by our ex vivo pressure myograph analyses showing a compliance shift toward the elastin axis: compliance is effectively determined by the elastin fibers.31,32 Consistently, echocardiography showed no change in abdominal aortic diameter with chronic colchicine treatment, indicating that the reduction in PWV reflects structural preservation rather than sustained vasodilation, consistent with plasma colchicine levels during oral dosing, which are within the lower range of the concentrations used in the acute ex-vivo assays and subject to normal physiological regulation of vascular tone.
Interestingly though, NO plays a critical role in passive stiffness through its effects on vascular remodeling. NO achieves this by inhibiting the migration and proliferation of VSMCs and modulating extracellular matrix dynamics.33 Therefore, the preservation of NO signaling observed with colchicine might importantly link to reduction of stiffness through modulation of passive components. Since chronic colchicine treatment had no effect on VSMC contractility, colchicine’s impact on stiffness seems to be primarily through endothelial mechanisms and structural remodeling rather than through modulation of smooth muscle contractile function. Arterial stiffness can also be modulated by active components, particularly vasomotor function, with NO and β-AR signaling playing key roles.31,34,35 This active component was excluded in our ex vivo carotid artery experiments by the absence of Ca2+. However, we cannot rule out its potential role in the PWV measurements. Our findings suggest that chronic colchicine therapy protects against the loss of VSMC response to endothelial NO. This effect does not appear to be due to modulation of cGMP production by the ubiquitously present soluble guanylyl cyclase (sGC), the primary NO-responsive enzyme that produces cGMP, since this would have led to restored responses to SNP. Several mechanisms could explain the colchicine effect, including a reduction in local reactive oxygen species pools, alterations in nanodomain interactions between sGC and PDE, and effects on NO signaling via cellular junctions between endothelial and smooth muscle cells. Determining the exact mechanism(s) by which colchicine exerts this effect will be an intriguing yet not straightforward goal for future studies.
Inflammation and increased oxidative stress are widely recognized as key mechanisms in vascular pathogenesis associated with aging,5,36 with iNOS playing an important role in peroxynitrite formation.37 In the lung, the iNOS to p-eNOS ratio was markedly elevated in SMC-KO mice and normalized by colchicine, indicating a shift from an NO-producing, vasoprotective environment toward one favoring peroxynitrite generation and oxidative injury; colchicine’s ability to restore this balance likely contributes to preserved NO bioavailability and improved endothelial function. Furthermore, the observed increase in iNOS and MAPK-p38 levels in SMC-KO mice, along with their reduction following colchicine treatment, aligns with pro- and anti-inflammatory profiles, respectively. Although it remains speculative in this study, peroxynitrite can decrease NO bioavailability, potentially explaining the loss of vasodilation in SMC-KO mice and its preservation with colchicine treatment.38,39 Despite these local changes Plasma IL-6, MCP-1, and TNF-α levels remained unchanged across groups, indicating that the vascular effects of colchicine arose from local vessel-wall mechanisms rather than systemic inflammation, and confirming that chronic treatment did not induce systemic immunosuppression or toxicity. Furthermore, we incidentally observed a meaningful increase in the anti-inflammatory hepatokine GDF-15 following the colchicine treatment. Pharmacokinetically, colchicine accumulates in hepatocytes, leading to an increase in GDF-15 secretion.40 Evidence suggests that elevated circulating GDF-15 is integral to colchicine’s anti-inflammatory effects in the cardiovascular system.40 Further investigation of GDF-15 in models of vascular aging is warranted.
Some limitations of the current work should be acknowledged. Acute ex vivo pharmacology with β-blockers and paclitaxel was performed primarily in control SMC-LM rings because colchicine-induced relaxation in SMC-KO vessels was too small to permit reliable multi-inhibitor testing; consequently, direct confirmation of these pathways in the disease model remains to be addressed. We assessed global NO bioavailability using ACh/SNP ratios and L-NAME inhibition and directly evaluated endothelial signaling surrogates in lung tissue (e.g., eNOS phosphorylation/NOS isoform expression); however, these assays were not performed in the aortic/carotid beds studied functionally and structurally, which limits tissue-specific mechanistic attribution. Furthermore, although we documented local vascular inflammatory signaling (iNOS, COX-2, MAPK-p38) and excluded systemic cytokine changes, other inflammatory mediators and endothelial-senescence pathways were not examined. The 12-week colchicine regimen was chosen to span the critical window of vascular remodeling in SMC-KO mice, but alternative dosing schedules could reveal additional effects. The manual cIMT tracing—although performed blinded—remains semi-quantitative. Finally, while we addressed sphericity violations in our repeated-measures ANOVA using Greenhouse–Geisser corrections, future analyses employing linear mixed-effects models may offer an even more flexible framework for handling within-animal clustering and enhancing precision. Despite these caveats, our findings provide robust evidence that colchicine attenuates non-atherosclerotic vascular aging through preserved NO signaling, reduced local inflammation, and protection of elastin architecture, supporting future mechanistic and translational investigations.
Conclusion
In conclusion, our study identifies smooth muscle cell aging as a key driver of vascular stiffness in an accelerated vascular aging mouse model. Colchicine’s ability to modulate vascular stiffness holds promising implications for central hemodynamics, potentially improving organ function beyond the cardiac system to benefit for instance also cerebral microvasculature and possibly even cognitive function. Additionally, colchicine’s direct effects on β-adrenergic receptors in this model offer further insights into its cardio protective properties. This study lays a foundational basis for the positive outcomes observed in the LoDoCo2 and other clinical colchicine trials and supports repurposing colchicine for primary prevention in vascular diseases. It also highlights the potential for developing colchicine-mimicking drugs with higher selectivity and improved safety profiles.
Abbreviations
Ach, Acetylcholine; AKT, Protein kinase B; β-AR, Beta-adrenoreceptors; CRC, Concentration response curve; COX-2, Cyclooxygenase-2; EKV, ECG-triggered kilohertz visualization; ECM, Extracellular matrix; ECs, Endothelial cells; EDH, Endothelial dependent hyperpolarization; Ercc1, Excision Repair Cross Complement-1; GDF-15, Growth Differentiation Factor 15; iNOS, Inducible Nitric oxide synthases; IMT, Intima media thickness; IL-6, Interleukin 6; NAVA, Non atherosclerotic vascular aging; MAPK, Mitogen-activated protein kinases; MCP-1Monocyte Chemoattractant Protein 1; NO, Nitric oxide; NO-cGMP, Nitric oxide- cyclic guanosine monophosphate; NFkB, Nuclear factor kappa B; SGc, Soluble guanylate cyclase; PDE, Phosphodiesterases; PKA, Phosphokinase A; PeNOS, Phosphorylated endothelial Nitric oxide synthases; PWV, Pulse wave velocity; TNF-α, Tumour Necrosis Factor alpha; ROS, Reactive oxygen species; SMC-KO, Smooth muscle cell-selective knockout; SMC-LM, Smooth muscle cell-selective littermates; SNP, Sodium nitroprusside; VSMCs, Vascular smooth muscle cells.
Acknowledgments
SMJ is supported by the Topconsortium Kennis en Innovatie (TKI) – Topsector Life Science and Health (LSH) grant EMCLSH 23035. DJD was supported by grants from the Dutch Cardiovascular Alliance, an initiative with support of the Dutch Heart Foundation (2020B008 RECONNEXT), and the ERA4Health CARDINNOV programme with support of the Dutch Research Council (NWO-era4healthcvd-100)
Funding
This work was funded by the Alkmaar Vascular Research Foundation.
Disclosure
Authors have no conflicts of interest to disclose.
References
1. Roth GA, Johnson C, Abajobir A, et al. Global, regional, and national burden of cardiovascular diseases for 10 causes, 1990 to 2015. J Am Coll Cardiol. 2017;70(1):1–15. doi:10.1016/j.jacc.2017.04.052
2. Mozaffarian D, Benjamin EJ, Go AS, et al. Heart disease and stroke statistics—2015 update: a report from the American Heart Association. circulation. 2015;131(4):e29–e322. doi:10.1161/CIR.0000000000000152
3. Jouabadi SM, Ataabadi EA, Golshiri K, et al. Clinical impact and mechanisms of non-atherosclerotic vascular aging: the new kid to be blocked. Cana J Cardiol. 2023;39:1839–1858. doi:10.1016/j.cjca.2023.07.022
4. Incalza MA, D’Oria R, Natalicchio A, Perrini S, Laviola L, Giorgino F. Oxidative stress and reactive oxygen species in endothelial dysfunction associated with cardiovascular and metabolic diseases. Vasc. Pharmacol. 2018;100:1–19.
5. López-Otín C, Blasco MA, Partridge L, Serrano M, Kroemer G. Hallmarks of aging: an expanding universe. Cell. 2023;186:243–278. doi:10.1016/j.cell.2022.11.001
6. Ataei Ataabadi E, Golshiri K, Jüttner AA, et al. Soluble guanylate cyclase activator BAY 54–6544 improves vasomotor function and survival in an accelerated ageing mouse model. Aging Cell. 2022;21(9):e13683. doi:10.1111/acel.13683
7. Ataei Ataabadi E, Golshiri K, van der Linden J, et al. Vascular ageing features caused by selective dna damage in smooth muscle cell. Oxid Med Cell Longev. 2021;2021:2308317. doi:10.1155/2021/2308317
8. Durik M, Kavousi M, van der Pluijm I, et al. Nucleotide excision DNA repair is associated with age-related vascular dysfunction. Circulation. 2012;126(4):468–478. doi:10.1161/CIRCULATIONAHA.112.104380
9. Bautista-Niño PK, Portilla-Fernandez E, Rubio-Beltrán E, et al. Local endothelial DNA repair deficiency causes aging-resembling endothelial-specific dysfunction. Clin Sci. 2020;134(7):727–746. doi:10.1042/CS20190124
10. Kirkland JL, Tchkonia T. Senolytic drugs: from discovery to translation. J Internal Med. 2020;288(5):518–536. doi:10.1111/joim.13141
11. Youm Y-H, Grant RW, McCabe LR, et al. Canonical Nlrp3 inflammasome links systemic low-grade inflammation to functional decline in aging. Cell Metab. 2013;18(4):519–532. doi:10.1016/j.cmet.2013.09.010
12. Nidorf SM. Seeing colchicine in a new light: repurposing low-dose colchicine for secondary prevention of cardiovascular disease. Clin. Ther. 2023;45:1029–1033. doi:10.1016/j.clinthera.2023.07.007
13. Nidorf SM, Ben-Chetrit E, Ridker PM. Low-dose colchicine for atherosclerosis: long-term safety. Eur Heart J. 2024;45:1596–1601. doi:10.1093/eurheartj/ehae208
14. Nidorf SM, Fiolet ATL, Mosterd A, et al. Colchicine in patients with chronic coronary disease. N Engl J Med. 2020;383(19):1838–1847. doi:10.1056/NEJMoa2021372
15. Tardif J-C, Kouz S, Waters DD, et al. Efficacy and safety of low-dose colchicine after myocardial infarction. N Engl J Med. 2019;381(26):2497–2505. doi:10.1056/NEJMoa1912388
16. Ehlers TS, van der Horst J, Møller S, et al. Colchicine enhances β adrenoceptor‐mediated vasodilation in men with essential hypertension. Br. J. Clin. Pharmacol. 2023;89:2179–2189. doi:10.1111/bcp.15688
17. Bloom SI, Tucker JR, Machin DR, et al. Reduction of double-strand DNA break repair exacerbates vascular aging. Aging. 2023;15(19):9913–9947. doi:10.18632/aging.205066
18. de Boer M, Te Lintel Hekkert M, de Boer M, et al. DNA repair in cardiomyocytes is critical for maintaining cardiac function in mice. Aging Cell. 2023;22(3):e13768. doi:10.1111/acel.13768
19. Sharma N, Sun Z, Hill MA, Hans CP. Measurement of pulse propagation velocity, distensibility and strain in an abdominal aortic aneurysm mouse model. J Vis Exp.
20. Clark JE, Kottam A, Motterlini R, Marber MS. Measuring left ventricular function in the normal, infarcted and CORM-3-preconditioned mouse heart using complex admittance-derived pressure volume loops. J Pharmacol Toxicol Methods. 2009;59(2):94–99. doi:10.1016/j.vascn.2008.10.007
21. Bridges LE, Williams CL, Pointer MA, Awumey EM. Mesenteric artery contraction and relaxation studies using automated wire myography. JoVE. 2011;(55):e3119.
22. Zieman SJ, Melenovsky V, Kass DA. Mechanisms, pathophysiology, and therapy of arterial stiffness. Arterioscler Thromb Vasc Biol. 2005;25(5):932–943. doi:10.1161/01.ATV.0000160548.78317.29
23. van der Horst J, Rognant S, Hellsten Y, Aalkjær C, Jepps TA. Dynein coordinates β2-adrenoceptor-mediated relaxation in normotensive and hypertensive rat mesenteric arteries. Hypertension. 2022;79(10):2214–2227. doi:10.1161/HYPERTENSIONAHA.122.19351
24. Lindman J, Khammy MM, Lundegaard PR, Aalkjær C, Jepps TA. Microtubule regulation of kv7 channels orchestrates camp-mediated vasorelaxations in rat arterial smooth muscle. Hypertension. 2018;71(2):336–345. doi:10.1161/HYPERTENSIONAHA.117.10152
25. Ataei Ataabadi E, Golshiri K, Jüttner A, Krenning G, Danser AHJ, Roks AJM. Nitric oxide-cGMP signaling in hypertension: current and future options for pharmacotherapy. Hypertension. 2020;76(4):1055–1068. doi:10.1161/HYPERTENSIONAHA.120.15856
26. Golshiri K, Ataei Ataabadi E, Portilla Fernandez EC, Jan Danser AH, Roks AJM. The importance of the nitric oxide‐cGMP pathway in age‐related cardiovascular disease: focus on phosphodiesterase‐1 and soluble guanylate cyclase. Basic Clin. Physiol. Pharmacol. 2020;127(2):67–80. doi:10.1111/bcpt.13319
27. Kelly MP, Nikolaev VO, Gobejishvili L, et al. Cyclic nucleotide phosphodiesterases as drug targets. Pharmacol Rev. 2025:100042. doi:10.1016/j.pharmr.2025.100042.
28. Sgouropoulou V, Stabouli S, Trachana M. Arterial stiffness in Familial Mediterranean Fever: correlations with disease-related parameters and colchicine treatment. Clin Rheumatol. 2019;38(9):2577–2584. doi:10.1007/s10067-019-04601-6
29. Hays AG, Schär M, Barditch-Crovo P, et al. A randomized, placebo-controlled, double-blinded clinical trial of colchicine to improve vascular health in people living with HIV. Aids. 2021;35(7):1041–1050. doi:10.1097/qad.0000000000002845
30. Baier JM, Funck KL, Vernstrøm L, Gullaksen S, Laugesen E, Poulsen PL. Colchicine mitigates arterial stiffness in high-risk patients with type 2 diabetes: a randomized placebo-controlled trial. Eur. J. Prev. Cardiol. 2025. doi:10.1093/eurjpc/zwaf143
31. Wesley CD, Neutel CHG, De Meyer GRY, Martinet W, Guns P-J. Unravelling the impact of active and passive contributors to arterial stiffness in male mice and their role in vascular aging. Sci Rep. 2024;14(1):18337. doi:10.1038/s41598-024-68725-9
32. Glasser SP, Arnett DK, McVeigh GE, et al. Vascular compliance and cardiovascular disease: a risk factor or a marker? Am J Hypertens. 1997;10(10):1175–1189. doi:10.1016/s0895-7061(97)00311-7
33. Fernández-Varo G, Ros J, Morales-Ruiz M, et al. Nitric oxide synthase 3-dependent vascular remodeling and circulatory dysfunction in cirrhosis. Am J Pathol. 2003;162(6):1985–1993. doi:10.1016/S0002-9440(10)64331-3
34. Butlin M, Tan I, Spronck B, Avolio AP. Measuring arterial stiffness in animal experimental studies. Arteriosclerosis Thrombosis Vasc Biol. 2020;40(5):1068–1077. doi:10.1161/ATVBAHA.119.313861
35. van Bussel FCG, van Bussel BCT, Hoeks APG, et al. A control systems approach to quantify wall shear stress normalization by flow-mediated dilation in the brachial artery. PLoS One. 2015;10(2):e0115977. doi:10.1371/journal.pone.0115977
36. Mohammadi Jouabadi S, Claringbould A, Danser AHJ, et al. High-sensitivity C-reactive protein mediates age-related vascular dysfunction: the Rotterdam study. Eur. J. Prev. Cardiol. 2025. doi:10.1093/eurjpc/zwaf370
37. Pacher P, Beckman JS, Liaudet L. Nitric oxide and peroxynitrite in health and disease. Physiol Rev. 2007;87(1):315–424.
38. Roy R, Wilcox J, Webb AJ, O’Gallagher K. Dysfunctional and dysregulated nitric oxide synthases in cardiovascular disease: mechanisms and therapeutic potential. Int J Mol Sci. 2023;24(20):15200. doi:10.3390/ijms242015200
39. Nakano M, Denda N, Matsumoto M, et al. Interaction between cyclooxygenase (COX)-1-and COX-2-products modulates COX-2 expression in the late phase of acute inflammation. Eur. J. Pharmacol. 2007;559(2–3):210–218. doi:10.1016/j.ejphar.2006.11.080
40. Weng JH, Koch PD, Luan HH, et al. Colchicine acts selectively in the liver to induce hepatokines that inhibit myeloid cell activation. Nat Metab. 2021;3(4):513–522. doi:10.1038/s42255-021-00366-y
 © 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.