Back to Journals » Neuropsychiatric Disease and Treatment » Volume 13
Cognitive dysfunction in major depression and Alzheimer's disease is associated with hippocampus–prefrontal cortex dysconnectivity
Authors Sampath D ![]() , Sathyanesan M, Newton SS
, Sathyanesan M, Newton SS ![]()
Received 4 March 2017
Accepted for publication 11 May 2017
Published 12 June 2017 Volume 2017:13 Pages 1509—1519
DOI https://doi.org/10.2147/NDT.S136122
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Roger Pinder
Dayalan Sampath,1 Monica Sathyanesan,1,2 Samuel S Newton1,2
1Division of Basic Biomedical Sciences, Sanford School of Medicine, University of South Dakota, Vermillion, 2Sioux Falls VA Healthcare System, Sioux Falls, SD, USA
Abstract: Cognitive dysfunction is prevalent in psychiatric disorders. Deficits are observed in multiple domains, including working memory, executive function, attention, and information processing. Disability caused by cognitive dysfunction is frequently as debilitating as the prominent emotional disturbances. Interactions between the hippocampus and the prefrontal cortex are increasingly appreciated as an important link between cognition and emotion. Recent developments in optogenetics, imaging, and connectomics can enable the investigation of this circuit in a manner that is relevant to disease pathophysiology. The goal of this review is to shed light on the contributions of this circuit to cognitive dysfunction in neuropsychiatric disorders, focusing on Alzheimer’s disease and depression.
Keywords: hippocampus, prefrontal cortex, cognition, depression, Alzheimer’s disease
Introduction
A major goal of preclinical investigations has been to parse the relative importance of specific brain regions and subregions in the context of neuropsychiatric disorders. While this approach is useful in understanding the contribution of specific cellular phenotypes, identifying key signal-transduction pathways and individual molecules, it does not integrate disease-induced pathophysiological changes that influence multiple brain regions and their functional interactions. The ability to define mental health diseases as circuit disorders and identify the circuits that are relevant to particular diseases can lead us to a perspective that is closer to the clinical disease state and enable us to consider interventions that can repair these circuits and restore the brain-activity network. A potential advantage with the circuit approach is that the same circuit is likely to be impaired across disorders when there is overlap in the expression of behavioral deficits.
The hippocampal formation (HPF) and prefrontal cortex (PFC) interact in a bidirectional manner to regulate several cognitive functions and process emotional information. They are critically important structures in the brain’s memory system, facilitating fast encoding of new information, consolidation, retrieval, and organization of the memory network.1 There is strong neural synchrony between the two regions during behavior, and their functional interaction is regulated by oscillations.2,3 The neural ensemble formed by the cells in the hippocampus and PFC mutually influences each other in a circuit-like fashion in modulating emotional and cognitive processes. Connections between the hippocampus and PFC have been demonstrated in rodents,4–7 primates,8 and humans.9 The rodent and human hippocampal prefrontal connections are illustrated schematically in Figures 1 and 2 respectively. The HPF and PFC are connected by both monosynaptic and polysynaptic connections.10 Retrograde-labeling studies revealed that projections from the medial PFC (mPFC) robustly label neurons in the ventral hippocampus and subiculum.10,11 Anterograde labeling revealed connections between the CA1 region of the hippocampus and the subiculum with the mPFC.4 Indirect multisynaptic connections between the HPF to mPFC include projections through the nucleus accumbens, ventral tegmental area, amygdala, entorhinal cortex, and midline thalamus.12–14 These complex multisynaptic pathways are involved in higher cognitive functions and dysregulated in various neuropsychiatric disorders.15 We review the influence of aberrations in the hippocampus–PFC circuit on cognitive dysfunction in depression and Alzheimer’s disease (AD).
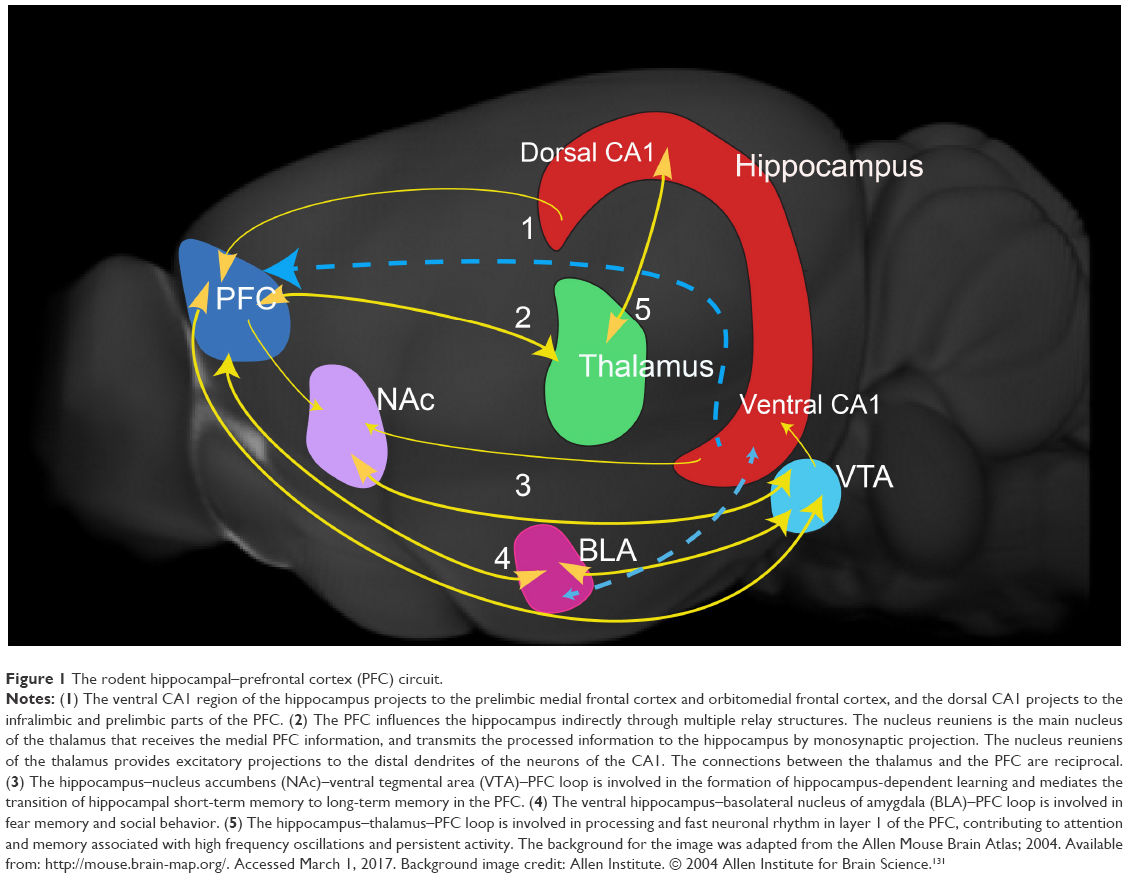 | Figure 1 The rodent hippocampal–prefrontal cortex (PFC) circuit. |
 | Figure 2 Human prefrontal hippocampal connections. |
Hippocampal–PFC circuit in depression
Depression afflicts 298 million people globally.16 Untreated depression is the leading cause of suicide in the 15- to 24-year age-group.17 A better understanding of disease pathology and more efficacious treatments are needed, as currently available prescription antidepressants are effective in only about 60% of affected individuals.18 Although chronic stress and trauma, particularly in early life, has been connected with vulnerability to depression,19,20 the precise neurobiological underpinnings are yet to be understood. A complex interaction between genetic and environmental factors is also involved in disease pathophysiology.
Multiple brain regions have been studied to understand the etiology of depression, including the hippocampus, amygdala, striatum, insula, medial thalamus, and several frontal cortex (FC) regions.21 Significant attention has been focused on the hippocampus, due to its central role in the stress-induced dysregulation of the hypothalamic–pituitary–adrenal axis.22 Additionally, imaging studies in patients with depression have shown a reduction in hippocampal volume.22–24 It is also the most frequently noted brain structure alteration in depression.25 Smaller but consistent reductions have also been reported in the PFC.26 Elevated corticosterone levels and dysregulation of the hypothalamic–pituitary–adrenal axis are ascribed as contributing factors in the hippocampal shrinkage that occurs in major patients with depressive disorder (MDD), but the precise molecular and cellular mechanisms involved are not known. Support for examining the hippocampus also comes from preclinical antidepressant studies demonstrating that hippocampal neurogenesis, which occurs in the subgranular zone of the dentate gyrus (DG), is important for the behavioral effects of antidepressants.27,28 Interestingly, the DG is also vulnerable to the effects of stress, evidenced by a decline in subgranular zone neurogenesis.29
The fact that the hippocampus is also central in memory functions makes it a crucial brain region that mediates both dysregulated mood and cognitive dysfunction in depression. A rapidly accumulating body of evidence indicates that a segmentation of the hippocampus along its axis, dorsoventral in rodents and posterioanterior in primates, is responsible for its dual functionality in regulating both emotion and cognition, the dorsal hippocampus being involved in cognitive function and the ventral in mood and anxiety.30
Hippocampal subfields in depression
There is significant interest in understanding the role of specific hippocampal subfields in depression, as these have the potential to inform the precision of brain stimulation-based therapeutic interventions,31 and deciphering hippocampal neuroplasticity in disease states and response to antidepressant treatment.32,33 Immunohistochemical analysis of postmortem brains from depressed patients revealed lower granule-cell number and smaller volume of the granule-cell layer in the anterior DG of untreated patients.32 No significant changes were noted in the posterior DG. Structural analysis reported the strongest volumetric decreases in the DG of unmedicated MDD patients, which were partially rescued by antidepressant treatment.34 Imaging brain regions as a function of depressive episodes showed that illness progression led to reduction in DG volume and thinning of the left mPFC, with no effect on amygdala volume.35 A consensus picture that emerges from clinical imaging studies is that the DG is the most vulnerable hippocampal subfield in depression. It is however not known whether this is purely disease-related or if there are also predisposing genetic factors that result in the DG being preferentially affected in depression. Animal-stress studies have demonstrated that the DG is particularly sensitive to elevations in glucocorticoid levels and reduced neurogenesis in the subgranular zone. Although a reduction in neurogenesis could contribute to a reduction in DG volume, it is unlikely to be the only factor.36 The findings give rise to several questions that will require clinical and preclinical investigation. As antidepressants (primarily selective serotonin-reuptake inhibitors) are able partially to rescue volumetric deficits, it would be important to investigate the cellular and molecular mechanisms involved in volume recovery. The correlation between volume recovery and mood improvement also needs to be examined.
Cognitive deficits in depression
Cognitive dysfunction in depression has become a topic of recent interest as the field has increasingly recognized that these deficits impede functional recovery and are independent of emotional disturbances.37,38 Patients with MDD frequently exhibit difficulty with attention,39 information processing,40 working memory,41 and executive function.42 It should be stressed that these deficits are not uniformly present in all depressed patients, as some abnormalities have been reported in specific MDD subgroups. However, the persistence of these deficits despite improvement in mood43,44 calls for efforts to understand how cognitive dysfunction is intertwined with depression and to develop treatments that specifically target this aspect of the illness.
Rodent studies have provided support for the hypothesis that functional interactions between the hippocampus and PFC are reflected in synchronized neuronal activity.3,4,45 Interestingly, there is an increase in θ-frequency synchronization between the ventral hippocampus and the mPFC in mice exposed to an anxiogenic environment, implicating this circuit in behavioral inhibition, which is characterized by reduced exploration during anxiety.46 Impaired synaptic plasticity of this circuit was suggested in another neural oscillation study that employed the chronic unpredictable stress paradigm to generate a depression-like phenotype.47 Hippocampus–PFC connectivity-dependent memory consolidation was impaired in depressed patients in a functional magnetic resonance imaging (fMRI) study that showed reduced hippocampal connectivity to the PFC.48 Early-life stress exposure is a known risk factor for developing depression later in life.19 The heightened vulnerability of the brain to emotional and psychological insults during childhood likely results in long-lasting changes in the brain that increase the risk of being afflicted by depression later in life. Understanding the link between childhood-stress exposure and brain function can thus provide critical insight into disease etiology. A significant interaction was found between childhood emotional neglect and reductions in hippocampal and PFC volumes in MDD patients,49 which could signify an association between circuit and structural abnormalities. Can a faulty circuit lead to structural abnormalities? Perhaps they are independent defects that coexist under certain conditions, resulting in higher levels of functional impairment.
Autobiographical memory in depression
A bias toward recall of negative events is frequently seen in depressed patients. In particular, autobiographical memory (AM), the recollection of personal experiences, is impaired in depression. AM is important for effective interaction with the world, as it involves an individual’s perception of self and the ability to solve problems using information from past personal events.50,51 Suicidal patients have been noted to generalize their responses when queried about past events, and unable to recall event specifics.52 This characteristic memory, termed “overgeneral” AM, is a persistent feature of major depression.50 Due to the complex nature of AM, involving episodic memory, self-reflection, emotional valence, and visual recall, the activation of multiple brain regions would be expected.53 High-resolution fMRI using multivoxel pattern analysis firmly implicated the ventromedial PFC and the hippocampus in AM representation.54 A meta-analysis of AM functional neuroimaging studies noted that PFC activation was consistently reported, and over half the studies showed hippocampal activation.53 Hippocampal activation frequently occurred in a core AM network that includes the parahippocampus, perirhinal, and entorhinal cortices. Event-related fMRI shed additional light on the various parameters of AM that influence hippocampal activation, and indicated that recollective qualities were an important component.55 Neuroimaging fMRI analysis of AM retrieval in epilepsy patients with left hippocampal atrophy revealed significant deficits in task-related AM-network activity and strength of connections, suggesting that the hippocampus is a critical node in the AM network.56
Hippocampal–PFC circuit in Alzheimer’s disease
The HPF is strongly implicated in AD pathology, with evidence from imaging, psychological testing, and postmortem analysis. The emergence of initial symptoms in AD corresponds to pathological changes in the hippocampus. A meta-analysis of structural imaging studies utilizing data from 700 patients concluded that at the time of AD diagnosis, hippocampal volume loss was at 23% in comparison to similar-age controls.57 Longitudinal studies have shown that a specific reduction in the volume of the left amygdala–hippocampal complex is evident 5 years before AD diagnosis.58 The AD Neuroimaging Initiative (ADNI), a major multicenter neuroimaging study, reported a reduction in hippocampal volume in patients with AD and mild cognitive impairment (MCI), a precursor to AD development.59 It also documented substantial reduction in volume within a span of 6 months, and more accelerated volume loss in AD carriers of the ApoEε4 allele.59 Furthermore, the rate of volume loss correlated with cognitive decline.25,29 The presence of the ApoEε4 allele is the strongest genetic risk factor for AD, producing higher AD pathology and structural and functional alterations in the brain.60 Structural imaging revealed a specific reduction in left hippocampal volume in ApoEε4 carriers at early-stage AD.61 An MRI marker capable of determining hippocampal texture was developed using scans from the large ADNI database and tested in three separate cohorts.62 From the available prognostic results, it appears that hippocampal texture might be more sensitive than volume reduction in predicting CI and progression from MCI to AD.62
Hippocampal subfields in AD
A recent approach to improve diagnostic predictive power is the imaging of hippocampal subfields. Extensive volumetric and shape analyses employing a variety of methods, such as manual segmentation of the entire hippocampus, voxel-based morphometry, and surface reconstruction, have yielded a consensus that subfield atrophy in AD is primarily in CA1.63 This finding from live imaging investigations correlates well with previous histological examination of postmortem AD brain tissue showing highest neuronal loss in CA1 (68%), followed by the subiculum (47%) and hilus (25%) compared to the age-matched control group.64 CA1 and subiculum atrophy were detectable in cognitively normal individuals 6 years prior to AD diagnosis,65 indicating involvement of specific hippocampal subfields early in the disease process.
There are broad similarities in hippocampal volume loss in AD and depression, which could underlie the overlap in affective and cognitive deficits. Structural imaging of hippocampal subfields and immunohistochemical and histological analyses have shown that there is divergence in the specific subfields and cell types that are affected. Volumetric reduction in the DG is consistently reported in depression, although the precise mechanisms remain to be elucidated. In contrast, hippocampal atrophy in AD is caused by neuronal cell loss in the CA1 region and the entorhinal cortex, while the DG is mostly well preserved.66 Whether these differences also influence how information flows through the hippocampal circuit in these diseases would be worthy of investigation.
Amyloid and neurofibrillary tangles, abnormally folded protein structures in the AD brain, initially accumulate in the entorhinal cortex and then spread to the hippocampus.67 This phenomenon has the potential to disrupt local and global hippocampal-dependent connectivity.68 Connectivity can also be impaired before the emergence of landmark pathology, such as in MCI patients who are at risk for AD, exhibiting significantly reduced connectivity between hippocampi.69 The mPFC and lateral PFC have emerged as an AD-hub region responsible for linking functionally specialized regions.70 As a site of high amyloid-β accumulation, its ability to process information would be increasingly affected with disease progression. Positron-emission tomography imaging of blood flow during a face-memory task indicated that memory deficits early in AD were due to a reduction in integrated network activity, with the PFC and hippocampus as principal components.71 Functional connectivity of the hippocampus with several cortical regions was shown to be impaired in ApoEε4 carriers by a combination of neuropsychological testing and imaging analyses designed to decipher context-dependent alterations in hippocampal connectivity.72 Early-stage AD ApoEε4 carriers also exhibited decreased functional connectivity between the hippocampus and the medial FC and parietal cortex.61
Resting-state fMRI analysis in AD patients found that right hippocampal connectivity with the mPFC and the ventral anterior cingulate cortex was reduced, but connectivity of the left hippocampus with the dorsolateral PFC was enhanced.73 The increased connectivity is likely to be a compensatory adjustment for recruiting additional resources to balance the decline in cognitive function in AD. A functional relationship between the posterior cingulate cortex and the hippocampus was seen in episodic memory fMRI in MCI patients. Interestingly, posterior cingulate cortex activation during episodic memory encoding was connected with right hippocampal activation, while episodic memory recognition was associated with left hippocampal activation, indicating the presence of hemisphere specialization.74 In an AD study that found a strong correlation between hippocampal volume and memory impairment, left hippocampal volume predicted verbal recall while right hippocampal volume predicted spatial recall, providing additional support for hemispheric specialization of memory in AD.75
Neuroimaging studies in AD and depression have progressed from focused analysis of specific anatomical regions involved in disease pathology to incorporating a network approach that can yield insight into global relationships between spatially distinct brain structures, spawning the field of macrolevel circuit analysis – connectomics.76,77 The understanding of brain wiring gained by clinical connectomic investigations can be complemented with preclinical optogenetic technology to examine specific circuits and cell types with high spatial and temporal resolution.78 We expand on the applications of connectomic and optogenetic approaches to interrogate the hippocampal–PFC circuit in cognitive function.
Optogenetics sheds light on the hippocampal–PFC circuit
Optogenetics is rapidly becoming the method of choice to regulate neuronal circuits precisely in preclinical experiments. The method involves the use of light and light-sensitive opsin proteins to exert fine control over in vivo neuronal activity.79,80 Based on the goals of the investigation, specific opsin proteins can be chosen, either to depolarize or excite neurons (channelrhodopsin)81 or hyperpolarize and inhibit (halorhodopsin and archaerhodopsin).82 By utilizing animal models of neuropsychiatric disorders, optogenetics can enable the identification of disrupted brain circuits and complement conventional electrophysiological analysis. An elegant recent study employed optogenetic technology to facilitate the recall of old memories in a mouse model of AD.83 It successfully achieved this by activating the cells involved in the formation of memory engrams, in the hippocampal circuit.83 A similar approach was used by the same group to recall previous positive memories in a mouse model of depression. Optogenetic activation of the positive memory-engram cells in the hippocampal circuit was able to acutely reverse the depressed phenotype.84 This experiment draws attention to the therapeutic potential of positive memory recall in depression, as activating memory of the positive experience was more effective in overcoming the adverse behavioral consequences of stress exposure than the experience itself.85
The existence of an mPFC–thalamic nucleus reuniens–hippocampal CA1 loop for goal-directed spatial navigation was determined by optogenetically manipulating this circuit at multiple nodes to demonstrate the relative importance of subregions within the hippocampus.86 The important role played by direct ventral hippocampus–mPFC afferents in encoding spatial cues during the performance of spatial working memory tasks was shown by employing a projection-specific optogenetic approach.87 The ventral hippocampus–mPFC input was required only for encoding cues, not for retrieval or maintenance. γ-Frequency but not θ-frequency synchrony was essential in this circuit for successful encoding of cues, reinforcing previous work indicating that γ-synchrony could be essential for proper long-range connectivity.88
Optogenetic stimulation of somatostatin and parvalbumin containing interneurons in the PFC helped clarify the distinct contribution of these interneuron subtypes in working memory tasks and reward processing. This provided new insight of prefrontal circuitry in cognitive function.89 The combination of optogenetics and electrophysiology sheds light on the mechanism whereby the PFC regulates the processing of information for attention. This study strongly implicates fast-spiking mPFC parvalbumin neurons in guiding successful attention behavior.90 The therapeutic potential of optogenetics for cognitive deficits was demonstrated by γ-frequency stimulation of PFC interneurons specifically at 40 or 60 Hz.91 Interestingly, a single intervention was sufficient to produce cognitive enhancement that lasted over a week. Precise and selective activation of glutamatergic neurons in the mPFC improved associative recognition memory, an important aspect of cognitive function.92 However, it is important to note that stimulation was effective only if it occurred during the delay phase, and simply elevating glutamate release was ineffective. This observation illustrates how the precision of optogenetic stimulation can inform drug development by providing key details for consideration. A major application of optogenetic research in neuropsychiatry will be to translate the important preclinical findings into useful noninvasive therapies in the clinic. When examining the success of circuit-level optogenetic analyses and behavioral function, it is quite clear that a high degree of spatial and temporal resolution is involved. Currently, therapeutic interventions with comparable precision are not available for clinical use. Several novel approaches have recently been proposed for potential clinical translation.93 These include viral transduction, where virus particles are used to overexpress proteins of interest in a spatially restricted manner. Paramagnetic proteins, primarily ferritin, fused to a channel receptor function as an endogenous iron nanoparticle and enable the use of a magnetic field to modulate channel activity temporally.94,95 Low-intensity focused ultrasound has been recently used to non-invasively alter neuronal activity with high spatial resolution.96 Dysregulation of specific molecular targets has been identified in the entorhinal–hippocampal circuit.93,97,98 It is however likely that the most promising therapies would require some testing in primates before commencing human clinical trials.
Connectomics
In contrast to the highly precise, neuronal subtype-level resolution of optogenetics, connectomics focuses on the major neural highways in the brain. The Human Connectome Project aims to map neural connections in the brain at a scale that has not been previously attempted. The goal is to obtain insight into the anatomical and functional complexity in the human brain at the level of long-range connections. A combination of diffusion MRI to trace white-matter tracts for structural connectivity and resting-state fMRI for functional activity is being employed.77 The hope is that conducting these analyses in over a thousand individuals will provide accurate baselines and enable identification of disease-induced alterations in brain-network connectivity with high confidence.77 Connectomic analyses have begun to provide new insight into the pathophysiology of neuropsychiatric disorders, and it is now possible to define certain illnesses as connectivity disorders. Reduced functional connectivity, determined by functional connectivity MRI, has been reported in advanced AD, with patients demonstrating a striking decrease in connectivity between the hippocampus and FC.99 Altered functional connectivity is also evident early in AD,73 when plaque accumulation would be minimal. Long-distance connectivity was shown to be selectively vulnerable in AD, and deterioration correlated with cognitive decline. Graph theory-based topological analysis revealed that attenuation in long-distance connectivity also reduced the efficiency of the global brain network, leading to more widespread clinical cognitive deficits.100
In patients afflicted with CI and depression, there is substantial overlap of connectivity abnormalities. Network-topology analyses reveal a reduction in network strength, efficiency, and regional connectivity in FC structures, collectively indicating disruption of white-matter integrity.54 In patients with a first episode of untreated depression, white-matter abnormalities can be a potential biomarker of pathophysiology. If compromised white-matter integrity and function are indeed precipitating factors in depression, it would be important to examine the role of antidepressants in rescuing or reversing these deficits. Ketamine has attracted much attention in recent years, due to its rapid-acting antidepressant properties.101 Its psychomimetic effects and abuse potential can however interfere with its use as a mainstream antidepressant in the clinic. Understanding its precise antidepressant mechanism is thus a high priority to develop next-generation compounds that do not carry undesirable side effects. A recent study on the role of ketamine in MDD patients demonstrated its ability to normalize disconnectivity between the PFC and the rest of the brain, suggesting that actions on white matter could be involved in its antidepressant mechanism.102 Although this suggests that impaired connectivity can be rescued, it is critical to understand ketamine’s impact on white matter. Efforts should be aimed at distinguishing between acute and chronic effects, as chronic ketamine usage has been shown to disrupt connectivity between the caudate and PFC.103 Focusing attention on understanding the molecular substrates that influence white-matter integrity would be an important research avenue. It can yield useful information regarding circuit neurobiology, and also potentially identify molecules that can restore connectivity deficits in multiple psychiatric and neurodegenerative disorders.
Studying brain-network connectivity is likely to be challenging when developmental changes are ongoing, as the complex and dynamic maturation windows can influence the consistency of neuroimaging results. However, it is important to obtain structural and functional connectivity information of the brain during its most vulnerable state.104 Depression frequently surfaces during adolescence,105 a critical developmental period where an increase in white matter and reduction in cortical gray matter occurs.106 In unmedicated, first-episode adolescent patients with depression, both structural and functional connectivity in the PFC–hippocampal circuit was abnormal, with decreased functional connectivity in multiple PFC regions.107 Adolescent depression studies have also reported hyperconnectivity of the default network and better functional connectivity than healthy controls between the mPFC and posterior cingulate cortex during the processing of cognitive information.108 Hyperconnectivity was also noted during goal-directed emotional processing.108 The adolescent brain can be influenced by hormones, reward valence, and social interactions, which are also likely to impact network connectivity.104 It will thus be interesting to see the results of follow-up studies designed to examine treatment effects on connectivity.
A common molecular target in AD and depression
Dysregulation of the hippocampus–PFC circuit and similarities in hippocampal volume reduction in AD and depression could point to common deficits in cellular signaling. Identifying disease-related molecules that regulate key nodal points in the intracellular signaling network can yield deep insight into disease pathophysiology and provide opportunities to test the link between molecular dysfunction and behavioral deficits. Transcription factors that function as crucial molecules downstream of cell signaling have an important role in the investigation of molecular mechanisms of neuropsychiatric disorders. They are functionally positioned at critical nodal points where disease and drug-induced signal transduction converge. They are also capable of regulating an entire program of gene expression when activated, and thereby influence several distinct cellular processes and mechanisms. The cAMP response element binding (CREB) protein transcription factor has been actively investigated in memory,109,110 depression,111,112 and antidepressant activity,113,114 providing important understanding into the molecular basis of disease-relevant behavioral responses. CREB is activated by phosphorylation and drives the expression of different target genes based on the brain region where it is activated.115 It is interesting to note that CREB is also emerging as an important molecule in AD research.116,117 Postmortem studies on AD brain tissue have reported downregulation of CREB in the hippocampus.118 Recent postmortem analysis found levels of CREB and activated phospho-CREB to be reduced in the PFC, and rather intriguingly discovered that CREB/phospho-CREB levels were also similarly reduced in peripheral blood mononuclear cells.119 Since reduction in CREB occurred prior to amyloid deposition and can also be detected in blood, it would be worthwhile to investigate CREB levels in clinical populations. CREB could emerge as an important molecular regulator of cognition in both depression and AD, and also serve as a useful biomarker.
Conclusion
The recent shift in focus from imaging gray-matter volumes to integrating brain-network data and analysis has created considerable excitement, and promises to reveal new insight into brain development, neuropsychiatric disorders, and brain function. The macrolevel understanding gained by connectomic studies can be investigated at the level of individual neurons and neural ensembles via optogenetic methodologies. These conceptual and technological advances have enabled neuroscientists to consider psychiatric disorders as circuit malfunctions. The value of investigating a particular circuit such as the hippocampus–PFC is enhanced by the fact that it involves regions that are strongly implicated in depression and AD and also share overlap in cognitive deficits. Determining precise molecular and genetic mechanisms will provide additional resolution and strengthen the rationale for utilizing circuit and connectivity information in disease diagnosis and treatment. Major dividends can ensue in terms of treatments that effectively target primary circuit deficits involved in multiple neuropsychiatric disorders.
Acknowledgments
This work was supported by US Public Health Service grant MH106640 and the use of facilities at the Sioux Falls VA Healthcare system.
Disclosure
The authors report no conflicts of interest in this work.
References
Preston AR, Eichenbaum H. Interplay of hippocampus and prefrontal cortex in memory. Curr Biol. 2013;23(17):R764–R773. | ||
Colgin LL. Oscillations and hippocampal-prefrontal synchrony. Curr Opin Neurobiol. 2011;21(3):467–474. | ||
Gordon JA. Oscillations and hippocampal-prefrontal synchrony. Curr Opin Neurobiol. 2011;21(3):486–491. | ||
Jay TM, Witter MP. Distribution of hippocampal CA1 and subicular efferents in the prefrontal cortex of the rat studied by means of anterograde transport of Phaseolus vulgaris-leucoagglutinin. J Comp Neurol. 1991;313(4):574–586. | ||
Swanson LW, Köhler C. Anatomical evidence for direct projections from the entorhinal area to the entire cortical mantle in the rat. J Neurosci. 1986;6(10):3010–3023. | ||
Thierry AM, Gioanni Y, Dégénétais E, Glowinski J. Hippocampo-prefrontal cortex pathway: anatomical and electrophysiological characteristics. Hippocampus. 2000;10(4):411–419. | ||
Varela C, Kumar S, Yang JY, Wilson MA. Anatomical substrates for direct interactions between hippocampus, medial prefrontal cortex, and the thalamic nucleus reuniens. Brain Struct Funct. 2014;219(3):911–929. | ||
Zhong YM, Yukie M, Rockland KS. Distinctive morphology of hippocampal CA1 terminations in orbital and medial frontal cortex in macaque monkeys. Exp Brain Res. 2006;169(4):549–553. | ||
Croxson PL, Johansen-Berg H, Behrens TE, et al. Quantitative investigation of connections of the prefrontal cortex in the human and macaque using probabilistic diffusion tractography. J Neurosci. 2005;25(39):8854–8866. | ||
Hoover WB, Vertes RP. Anatomical analysis of afferent projections to the medial prefrontal cortex in the rat. Brain Struct Funct. 2007;212(2):149–179. | ||
Jay TM, Glowinski J, Thierry AM. Selectivity of the hippocampal projection to the prelimbic area of the prefrontal cortex in the rat. Brain Res. 1989;505(2):337–340. | ||
Russo SJ, Nestler EJ. The brain reward circuitry in mood disorders. Nat Rev Neurosci. 2013;14(9):609–625. | ||
Maren S. Seeking a spotless mind: extinction, deconsolidation, and erasure of fear memory. Neuron. 2011;70(5):830–845. | ||
Wolff M, Alcaraz F, Marchand AR, Coutureau E. Functional heterogeneity of the limbic thalamus: from hippocampal to cortical functions. Neurosci Biobehav Rev. 2015;54:120–130. | ||
Godsil BP, Kiss JP, Spedding M, Jay TM. The hippocampal-prefrontal pathway: the weak link in psychiatric disorders? Eur Neuropsychopharmacol. 2013;23(10):1165–1181. | ||
Ferrari AJ, Charlson FJ, Norman RE, et al. Burden of depressive disorders by country, sex, age, and year: findings from the global burden of disease study 2010. PLoS Med. 2013;10(11):e1001547. | ||
Galaif ER, Sussman S, Newcomb MD, Locke TF. Suicidality, depression, and alcohol use among adolescents: a review of empirical findings. Int J Adolesc Med Health. 2007;19(1):27–35. | ||
Rush AJ, Trivedi MH, Wisniewski SR, et al. Acute and longer-term outcomes in depressed outpatients requiring one or several treatment steps: a STAR*D report. Am J Psychiatry. 2006;163(11):1905–1917. | ||
Heim C, Newport DJ, Mletzko T, Miller AH, Nemeroff CB. The link between childhood trauma and depression: insights from HPA axis studies in humans. Psychoneuroendocrinology. 2008;33(6):693–710. | ||
Hammen C. Stress and depression. Annu Rev Clin Psychol. 2005;1:293–319. | ||
Pandya M, Altinay M, Malone DA Jr, Anand A. Where in the brain is depression? Curr Psychiatry Rep. 2012;14(6):634–642. | ||
Campbell S, Macqueen G. The role of the hippocampus in the pathophysiology of major depression. J Psychiatry Neurosci. 2004;29(6):417–426. | ||
Sheline YI, Sanghavi M, Mintun MA, Gado MH. Depression duration but not age predicts hippocampal volume loss in medically healthy women with recurrent major depression. J Neurosci. 1999;19(12):5034–5043. | ||
Opel N, Redlich R, Zwanzger P, et al. Hippocampal atrophy in major depression: a function of childhood maltreatment rather than diagnosis? Neuropsychopharmacology. 2014;39(12):2723–2731. | ||
Videbech P, Ravnkilde B. Hippocampal volume and depression: a meta-analysis of MRI studies. Am J Psychiatry. 2004;161(11):1957–1966. | ||
Koolschijn PC, van Haren NE, Lensvelt-Mulders GJ, Hulshoff Pol HE, Kahn RS. Brain volume abnormalities in major depressive disorder: a meta-analysis of magnetic resonance imaging studies. Hum Brain Mapp. 2009;30(11):3719–3735. | ||
Santarelli L, Saxe M, Gross C, et al. Requirement of hippocampal neurogenesis for the behavioral effects of antidepressants. Science. 2003;301(5634):805–809. | ||
Malberg JE, Eisch AJ, Nestler EJ, Duman RS. Chronic antidepressant treatment increases neurogenesis in adult rat hippocampus. J Neurosci. 2000;20(24):9104–9110. | ||
Gould E, McEwen BS, Tanapat P, Galea LA, Fuchs E. Neurogenesis in the dentate gyrus of the adult tree shrew is regulated by psychosocial stress and NMDA receptor activation. J Neurosci. 1997;17(7):2492–2498. | ||
Fanselow MS, Dong HW. Are the dorsal and ventral hippocampus functionally distinct structures? Neuron. 2010;65(1):7–19. | ||
Samuels BA, Leonardo ED, Hen R. Hippocampal subfields and major depressive disorder. Biol Psychiatry. 2015;77(3):210–211. | ||
Boldrini M, Santiago AN, Hen R, et al. Hippocampal granule neuron number and dentate gyrus volume in antidepressant-treated and untreated major depression. Neuropsychopharmacology. 2013;38(6):1068–1077. | ||
Abbott CC, Jones T, Lemke NT, et al. Hippocampal structural and functional changes associated with electroconvulsive therapy response. Transl Psychiatry. 2014;4:e483. | ||
Huang Y, Coupland NJ, Lebel RM, et al. Structural changes in hippocampal subfields in major depressive disorder: a high-field magnetic resonance imaging study. Biol Psychiatry. 2013;74(1):62–68. | ||
Treadway MT, Waskom ML, Dillon DG, et al. Illness progression, recent stress, and morphometry of hippocampal subfields and medial prefrontal cortex in major depression. Biol Psychiatry. 2015;77(3):285–294. | ||
Stockmeier CA, Mahajan GJ, Konick LC, et al. Cellular changes in the postmortem hippocampus in major depression. Biol Psychiatry. 2004;56(9):640–650. | ||
Jaeger J, Berns S, Uzelac S, Davis-Conway S. Neurocognitive deficits and disability in major depressive disorder. Psychiatry Res. 2006;145(1):39–48. | ||
Marazziti D, Consoli G, Picchetti M, Carlini M, Faravelli L. Cognitive impairment in major depression. Eur J Pharmacol. 2010;626(1):83–86. | ||
Keilp JG, Gorlyn M, Oquendo MA, Burke AK, Mann JJ. Attention deficit in depressed suicide attempters. Psychiatry Res. 2008;159(1–2):7–17. | ||
Simons CJ, Jacobs N, Derom C, et al. Cognition as predictor of current and follow-up depressive symptoms in the general population. Acta Psychiatr Scand. 2009;120(1):45–52. | ||
Tavares JV, Clark L, Cannon DM, Erickson K, Drevets WC, Sahakian BJ. Distinct profiles of neurocognitive function in unmedicated unipolar depression and bipolar II depression. Biol Psychiatry. 2007;62(8):917–924. | ||
Castaneda AE, Marttunen M, Suvisaari J, et al. The effect of psychiatric co-morbidity on cognitive functioning in a population-based sample of depressed young adults. Psychol Med. 2010;40(1):29–39. | ||
Paelecke-Habermann Y, Pohl J, Leplow B. Attention and executive functions in remitted major depression patients. J Affect Disord. 2005;89(1–3):125–135. | ||
Fava M, Graves LM, Benazzi F, et al. A cross-sectional study of the prevalence of cognitive and physical symptoms during long-term antidepressant treatment. J Clin Psychiatry. 2006;67(11):1754–1759. | ||
Siapas AG, Lubenov EV, Wilson MA. Prefrontal phase locking to hippocampal theta oscillations. Neuron. 2005;46(1):141–151. | ||
Adhikari A, Topiwala MA, Gordon JA. Synchronized activity between the ventral hippocampus and the medial prefrontal cortex during anxiety. Neuron. 2010;65(2):257–269. | ||
Zheng C, Zhang T. Synaptic plasticity-related neural oscillations on hippocampus-prefrontal cortex pathway in depression. Neuroscience. 2015;292:170–180. | ||
Genzel L, Dresler M, Cornu M, et al. Medial prefrontal-hippocampal connectivity and motor memory consolidation in depression and schizophrenia. Biol Psychiatry. 2015;77(2):177–186. | ||
Frodl T, Reinhold E, Koutsouleris N, Reiser M, Meisenzahl EM. Interaction of childhood stress with hippocampus and prefrontal cortex volume reduction in major depression. J Psychiatr Res. 2010;44(13):799–807. | ||
Williams JM, Barnhofer T, Crane C, et al. Autobiographical memory specificity and emotional disorder. Psychol Bull. 2007;133(1):122–148. | ||
Conway MA, Pleydell-Pearce CW. The construction of autobiographical memories in the self-memory system. Psychol Rev. 2000;107(2):261–288. | ||
Williams JM, Broadbent K. Autobiographical memory in suicide attempters. J Abnorm Psychol. 1986;95(2):144–149. | ||
Svoboda E, McKinnon MC, Levine B. The functional neuroanatomy of autobiographical memory: a meta-analysis. Neuropsychologia. 2006;44(12):2189–2208. | ||
Bai F, Shu N, Yuan Y, et al. Topologically convergent and divergent structural connectivity patterns between patients with remitted geriatric depression and amnestic mild cognitive impairment. J Neurosci. 2012;32(12):4307–4318. | ||
Addis DR, Moscovitch M, Crawley AP, McAndrews MP. Recollective qualities modulate hippocampal activation during autobiographical memory retrieval. Hippocampus. 2004;14(6):752–762. | ||
Addis DR, Moscovitch M, McAndrews MP. Consequences of hippocampal damage across the autobiographical memory network in left temporal lobe epilepsy. Brain. 2007;130(Pt 9):2327–2342. | ||
Shi F, Liu B, Zhou Y, Yu C, Jiang T. Hippocampal volume and asymmetry in mild cognitive impairment and Alzheimer’s disease: meta-analyses of MRI studies. Hippocampus. 2009;19(11):1055–1064. | ||
Bernard C, Helmer C, Dilharreguy B, et al. Time course of brain volume changes in the preclinical phase of Alzheimer’s disease. Alzheimers Dement. 2014;10(2):143–151.e1. | ||
Schuff N, Woerner N, Boreta L, et al. MRI of hippocampal volume loss in early Alzheimer’s disease in relation to ApoE genotype and biomarkers. Brain. 2009;132(Pt 4):1067–1077. | ||
Liu CC, Liu CC, Kanekiyo T, Xu H, Bu G. Apolipoprotein E and Alzheimer disease: risk, mechanisms and therapy. Nat Rev Neurol. 2013;9(2):106–118. | ||
Wang X, Wang J, He Y, et al. Apolipoprotein E ε4 modulates cognitive profiles, hippocampal volume, and resting-state functional connectivity in Alzheimer’s disease. J Alzheimers Dis. 2015;45(3):781–795. | ||
Sørensen L, Igel C, Hansen NL, et al. Early detection of Alzheimer’s disease using MRI hippocampal texture. Hum Brain Mapp. 2016;37(3):1148–1161. | ||
de Flores R, La Joie R, Chetelat G. Structural imaging of hippocampal subfields in healthy aging and Alzheimer’s disease. Neuroscience. 2015;309:29–50. | ||
West MJ, Coleman PD, Flood DG, Troncoso JC. Differences in the pattern of hippocampal neuronal loss in normal ageing and Alzheimer’s disease. Lancet. 1994;344(8925):769–772. | ||
Apostolova LG, Mosconi L, Thompson PM, et al. Subregional hippocampal atrophy predicts Alzheimer’s dementia in the cognitively normal. Neurobiol Aging. 2010;31(7):1077–1088. | ||
Mueller SG, Schuff N, Yaffe K, Madison C, Miller B, Weiner MW. Hippocampal atrophy patterns in mild cognitive impairment and Alzheimer’s disease. Hum Brain Mapp. 2010;31(9):1339–1347. | ||
Braak H, Braak E. Neuropathological stageing [sic] of Alzheimer-related changes. Acta Neuropathol. 1991;82(4):239–259. | ||
Supekar K, Menon V, Rubin D, Musen M, Greicius MD. Network analysis of intrinsic functional brain connectivity in Alzheimer’s disease. PLoS Comput Biol. 2008;4(6):e1000100. | ||
Sorg C, Riedl V, Muhlau M, et al. Selective changes of resting-state networks in individuals at risk for Alzheimer’s disease. Proc Natl Acad Sci U S A. 2007;104(47):18760–18765. | ||
Buckner RL, Sepulcre J, Talukdar T, et al. Cortical hubs revealed by intrinsic functional connectivity: mapping, assessment of stability, and relation to Alzheimer’s disease. J Neurosci. 2009;29(6):1860–1873. | ||
Grady CL, Furey ML, Pietrini P, Horwitz B, Rapoport SI. Altered brain functional connectivity and impaired short-term memory in Alzheimer’s disease. Brain. 2001;124(Pt 4):739–756. | ||
Harrison TM, Burggren AC, Small GW, Bookheimer SY. Altered memory-related functional connectivity of the anterior and posterior hippocampus in older adults at increased genetic risk for Alzheimer’s disease. Hum Brain Mapp. 2016;37(1):366–380. | ||
Wang L, Zang Y, He Y, et al. Changes in hippocampal connectivity in the early stages of Alzheimer’s disease: evidence from resting state fMRI. Neuroimage. 2006;31(2):496–504. | ||
Papma JM, Smits M, de Groot M, et al. The effect of hippocampal function, volume and connectivity on posterior cingulate cortex functioning during episodic memory fMRI in mild cognitive impairment. Eur Radiol. Epub 2017 Mar 13. | ||
de Toledo-Morrell L, Dickerson B, Sullivan MP, Spanovic C, Wilson R, Bennett DA. Hemispheric differences in hippocampal volume predict verbal and spatial memory performance in patients with Alzheimer’s disease. Hippocampus. 2000;10(2):136–142. | ||
Friston KJ, Frith CD, Liddle PF, Frackowiak RS. Functional connectivity: the principal-component analysis of large (PET) data sets. J Cereb Blood Flow Metab. 1993;13(1):5–14. | ||
Fornito A, Zalesky A, Breakspear M. The connectomics of brain disorders. Nat Rev Neurosci. 2015;16(3):159–172. | ||
Lee JH, Durand R, Gradinaru V, et al. Global and local fMRI signals driven by neurons defined optogenetically by type and wiring. Nature. 2010;465(7299):788–792. | ||
Zemelman BV, Lee GA, Ng M, Miesenböck G. Selective photostimulation of genetically chARGed neurons. Neuron. 2002;33(1):15–22. | ||
Boyden ES, Zhang F, Bamberg E, Nagel G, Deisseroth K. Millisecond-timescale, genetically targeted optical control of neural activity. Nature Neurosci. 2005;8(9):1263–1268. | ||
Zhang F, Vierock J, Yizhar O, et al. The microbial opsin family of optogenetic tools. Cell. 2011;147(7):1446–1457. | ||
Han X, Boyden ES. Multiple-color optical activation, silencing, and desynchronization of neural activity, with single-spike temporal resolution. PloS One. 2007;2(3):e299. | ||
Roy DS, Arons A, Mitchell TI, Pignatelli M, Ryan TJ, Tonegawa S. Memory retrieval by activating engram cells in mouse models of early Alzheimer’s disease. Nature. 2016;531(7595):508–512. | ||
Ramirez S, Liu X, MacDonald CJ, et al. Activating positive memory engrams suppresses depression-like behaviour. Nature. 2015;522(7556):335–339. | ||
Dranovsky A, Leonardo ED. Neuroscience: the power of positivity. Nature. 2015;522(7556):294–295. | ||
Ito HT, Zhang SJ, Witter MP, Moser EI, Moser MB. A prefrontal-thalamo-hippocampal circuit for goal-directed spatial navigation. Nature. 2015;522(7554):50–55. | ||
Spellman T, Rigotti M, Ahmari SE, Fusi S, Gogos JA, Gordon JA. Hippocampal-prefrontal input supports spatial encoding in working memory. Nature. 2015;522(7556):309–314. | ||
Yamamoto J, Suh J, Takeuchi D, Tonegawa S. Successful execution of working memory linked to synchronized high-frequency gamma oscillations. Cell. 2014;157(4):845–857. | ||
Kim D, Jeong H, Lee J, et al. Distinct roles of parvalbumin- and somatostatin-expressing interneurons in working memory. Neuron. 2016;92(4):902–915. | ||
Kim H, Ahrlund-Richter S, Wang X, Deisseroth K, Carlén M. Prefrontal parvalbumin neurons in control of attention. Cell. 2016;164(1–2):208–218. | ||
Cho KK, Hoch R, Lee AT, Patel T, Rubenstein JL, Sohal VS. Gamma rhythms link prefrontal interneuron dysfunction with cognitive inflexibility in Dlx5/6+/− mice. Neuron. 2015;85(6):1332–1343. | ||
Benn A, Barker GR, Stuart SA, et al. Optogenetic stimulation of prefrontal glutamatergic neurons enhances recognition memory. J Neurosci. 2016;36(18):4930–4939. | ||
Rajasethupathy P, Ferenczi E, Deisseroth K. Targeting neural circuits. Cell. 2016;165(3):524–534. | ||
Stanley SA, Kelly L, Latcha KN, et al. Bidirectional electromagnetic control of the hypothalamus regulates feeding and metabolism. Nature. 2016;531(7596):647–650. | ||
Stanley SA, Sauer J, Kane RS, Dordick JS, Friedman JM. Remote regulation of glucose homeostasis in mice using genetically encoded nanoparticles. Nat Med. 2015;21(1):92–98. | ||
Yuan Y, Yan J, Ma Z, Li X. Noninvasive focused ultrasound stimulation can modulate phase-amplitude coupling between neuronal oscillations in the rat hippocampus. Front Neurosci. 2016;10:348. | ||
Gazzaley AH, Siegel SJ, Kordower JH, Mufson EJ, Morrison JH. Circuit-specific alterations of N-methyl-D-aspartate receptor subunit 1 in the dentate gyrus of aged monkeys. Proc Natl Acad Sci U S A. 1996;93(7):3121–3125. | ||
Smith TD, Adams MM, Gallagher M, Morrison JH, Rapp PR. Circuit-specific alterations in hippocampal synaptophysin immunoreactivity predict spatial learning impairment in aged rats. J Neurosci. 2000;20(17):6587–6593. | ||
Allen G, Barnard H, McColl R, et al. Reduced hippocampal functional connectivity in Alzheimer disease. Arch Neurol. 2007;64(10):1482–1487. | ||
Liu Y, Yu C, Zhang X, et al. Impaired long distance functional connectivity and weighted network architecture in Alzheimer’s disease. Cereb Cortex. 2014;24(6):1422–1435. | ||
Monteggia LM, Zarate C Jr. Antidepressant actions of ketamine: from molecular mechanisms to clinical practice. Curr Opin Neurobiol. 2015;30:139–143. | ||
Abdallah CG, Averill LA, Collins KA, et al. Ketamine treatment and global brain connectivity in major depression. Neuropsychopharmacology. 2017;42(6):1210–1219. | ||
Roberts RE, Curran HV, Friston KJ, Morgan CJ. Abnormalities in white matter microstructure associated with chronic ketamine use. Neuropsychopharmacology. 2014;39(2):329–338. | ||
Menon V. Developmental pathways to functional brain networks: emerging principles. Trends Cogn Sci. 2013;17(12):627–640. | ||
Kessler RC, Walters EE. Epidemiology of DSM-III-R major depression and minor depression among adolescents and young adults in the National Comorbidity Survey. Depress Anxiety. 1998;7(1):3–14. | ||
Giedd JN, Blumenthal J, Jeffries NO, et al. Brain development during childhood and adolescence: a longitudinal MRI study. Nature Neurosci. 1999;2(10):861–863. | ||
Geng H, Wu F, Kong L, et al. Disrupted structural and functional connectivity in prefrontal-hippocampus circuitry in first-episode medication-naïve adolescent depression. PloS One. 2016;11(2):e0148345. | ||
Ho TC, Connolly CG, Henje Blom E, et al. Emotion-dependent functional connectivity of the default mode network in adolescent depression. Biol Psychiatry. 2015;78(9):635–646. | ||
Kida S, Serita T. Functional roles of CREB as a positive regulator in the formation and enhancement of memory. Brain Res Bull. 2014;105:17–24. | ||
Silva AJ, Kogan JH, Frankland PW, Kida S. CREB and memory. Annu Rev Neurosci. 1998;21:127–148. | ||
Muschamp JW, Van’t Veer A, Parsegian A, et al. Activation of CREB in the nucleus accumbens shell produces anhedonia and resistance to extinction of fear in rats. J Neurosci. 2011;31(8):3095–3103. | ||
Breuillaud L, Rossetti C, Meylan EM, et al. Deletion of CREB-regulated transcription coactivator 1 induces pathological aggression, depression-related behaviors, and neuroplasticity genes dysregulation in mice. Biol Psychiatry. 2012;72(7):528–536. | ||
Newton SS, Thome J, Wallace TL, et al. Inhibition of cAMP response element-binding protein or dynorphin in the nucleus accumbens produces an antidepressant-like effect. J Neurosci. 2002;22(24):10883–10890. | ||
Nibuya M, Nestler EJ, Duman RS. Chronic antidepressant administration increases the expression of cAMP response element binding protein (CREB) in rat hippocampus. J Neurosci. 1996;16(7):2365–2372. | ||
Tanis KQ, Duman RS, Newton SS. CREB binding and activity in brain: regional specificity and induction by electroconvulsive seizure. Biol Psychiatry. 2008;63(7):710–720. | ||
Müller M, Cárdenas C, Mei L, Cheung KH, Foskett JK. Constitutive cAMP response element binding protein (CREB) activation by Alzheimer’s disease presenilin-driven inositol trisphosphate receptor (InsP3R) Ca2+ signaling. Proc Natl Acad Sci U S A. 2011;108(32):13293–13298. | ||
Bartolotti N, Segura L, Lazarov O. Diminished CRE-induced plasticity is linked to memory deficits in familial Alzheimer’s disease mice. J Alzheimers Dis. 2015;50(2):477–489. | ||
Pugazhenthi S, Wang M, Pham S, Sze CI, Eckman CB. Downregulation of CREB expression in Alzheimer’s brain and in Aβ-treated rat hippocampal neurons. Mol Neurodegener. 2011;6:60. | ||
Bartolotti N, Bennett DA, Lazarov O. Reduced pCREB in Alzheimer’s disease prefrontal cortex is reflected in peripheral blood mononuclear cells. Mol Psychiatry. 2016;21(9):1158–1166. | ||
Phan KL, Orlichenko A, Boyd E, et al. Preliminary evidence of white matter abnormality in the uncinate fasciculus in generalized social anxiety disorder. Biol Psychiatry. 2009;66(7):691–694. | ||
Yasmin H, Nakata Y, Aoki S, et al. Diffusion abnormalities of the uncinate fasciculus in Alzheimer’s disease: diffusion tensor tract-specific analysis using a new method to measure the core of the tract. Neuroradiology. 2008;50(4):293–299. | ||
Taylor WD, MacFall JR, Gerig G, Krishnan RR. Structural integrity of the uncinate fasciculus in geriatric depression: relationship with age of onset. Neuropsychiatr Dis Treat. 2007;3(5):669–674. | ||
Ungerleider LG, Gaffan D, Pelak VS. Projections from inferior temporal cortex to prefrontal cortex via the uncinate fascicle in rhesus monkeys. Exp Brain Res. 1989;76(3):473–484. | ||
Makris N, Pandya DN. The extreme capsule in humans and rethinking of the language circuitry. Brain Struct Funct. 2009;213(3):343–358. | ||
Petrides M, Pandya DN. Association fiber pathways to the frontal cortex from the superior temporal region in the rhesus monkey. J Comp Neurol. 1988;273(1):52–66. | ||
Schmahmann JD, Pandya DN. The complex history of the fronto-occipital fasciculus. J Hist Neurosci. 2007;16(4):362–377. | ||
Makris N, Kennedy DN, McInerney S, et al. Segmentation of subcomponents within the superior longitudinal fascicle in humans: a quantitative, in vivo, DT–MRI study. Cereb Cortex. 2005;15(6):854–869. | ||
Poletti CE, Creswell G. Fornix system efferent projections in the squirrel monkey: an experimental degeneration study. J Comp Neurol. 1977;175(1):101–128. | ||
Klein JC, Rushworth MFS, Behrens TE, et al. Topography of connections between human prefrontal cortex and mediodorsal thalamus studied with diffusion tractography. Neuroimage. 2010;51(2):555–564. | ||
Barbas H, Blatt GJ. Topographically specific hippocampal projections target functionally distinct prefrontal areas in the rhesus monkey. Hippocampus. 1995;5(6):511–533. | ||
Allen Institute for Brain Science. Allen Mouse Brain Atlas; 2004. Available from: http://mouse.brain-map.org/. Accessed March 1, 2017. | ||
Allen Institute for Brain Science. Allen Human Brain Atlas; 2010. Available from: http://human.brain-map.org/. Accessed March 1, 2017. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.