")
Back to Journals » Open Access Rheumatology: Research and Reviews » Volume 12
Coexisting Diseases in Patients with Familial Mediterranean Fever
Authors Salehzadeh F , Enteshari Moghaddam A
Received 1 March 2020
Accepted for publication 6 May 2020
Published 28 May 2020 Volume 2020:12 Pages 65—71
DOI https://doi.org/10.2147/OARRR.S252071
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Chuan-Ju Liu
Farhad Salehzadeh,1 Afsaneh Enteshari Moghaddam2
1Pediatric Department, Bouali Children`s Hospital, Ardabil University of Medical Sciences (ARUMS), Ardabil, Iran; 2Internal Medicine Department, Imam Khomeini Hospital, Ardabil University of Medical Sciences (ARUMS), Ardabil, Iran
Correspondence: Afsaneh Enteshari Moghaddam
Internal Medicine Department, Imam Khomeini Hospital, Ardabil University of Medical Sciences (ARUMS), No. 105 Shahrak Azadi, Azerbaijan Streets, Ardabil 56157, Iran
Tel +989141511607
Fax +984533721199
Email [email protected]
Background and Aims: Familial Mediterranean fever (FMF) is a prototype of autoinflammatory disease and mainly associated with MEFV gene mutations. This single-center study as an experience represents FMF-coexisting disease in the FMF registration database.
Methods: Four hundred patients who had FMF based on clinical criteria (Tel-Hashomer) and/or MEFV mutations enrolled the study. Twelve most common MEFV mutations (P369S, F479L, M680I (G/C), M680I (G/A), I692del, M694V, M694I, K695R, V726A, A744S, R761H, E148Q) were analyzed if needed by the reverse hybridization assay. Any co-existed disease had been confirmed by a related subspecialist. All data were analyzed by a simple analytical method.
Results: Fifty-seven (14%) patients had associated disease, 32 patients were male and 24 patients were under 10 years old. They included 92 MEFV variant alleles and only in five patients there were not any mutations. The most common variant alleles were M694V (36%), E148Q (22%), V726A (17%), M680I (1%) and M694I (0.07%) respectively. Rheumatologic disorders were the most common coexisting disease, then followed by gastrointestinal and neurological disorders. Some rare diseases such as TTP, growth hormone deficiency, multiple sclerosis, idiopathic ascites, Leiden factor V deficiency and Felty syndrome have been detected. Homozygote mutations of (M694V-M694V) were associated with idiopathic ascites, orchitis and pericarditis.
Conclusion: Coexisting disease in patients with FMF is presented with positive MEFV gene mutations particularly with these five common variant alleles: M694V, E148Q, V726A, M680I, and M694I. The commonly associated diseases are rheumatologic, gastrointestinal and CNS disorders.
Keywords: familial mediterranean fever, MEFV mutation, FMF-coexisting disease
Introduction
FMF is an autoinflammatory and autosomal recessive disease mainly affecting ethnic groups living around the Mediterranean Sea: Jews, Armenians, Turks, Arabs,1 with a prevalence ranging from 1/200 to 1/1000.2
Early manifestations have usually appeared by the first decade and are characterized by recurrent, self-limiting attacks of polyserositis and fever. Serositis is presented by abdominal and chest pain as peritonitis or pleuritis, and, with less frequency pericarditis and recurrent painful orchitis can be seen.3
Arthritis in periodic and nondestructive form or persistent with chronic destructive pattern usually in large joints may occur. Severe prolonged myalgia or myositis due to vasculitis is seen.4,5 Frequency of attacks is variable and asymptomatic periods that last also a few years have been reported. Laboratory evaluation shows positive results of acute-phase reactant that may be detected moreover in asymptomatic periods, particularly serum amyloid A.6,7
Until 1998, the diagnosis of FMF was based on clinical criteria alone. The Tel Hashomer criteria generally form the basis of the clinical diagnosis. These criteria contain three major (recurrent febrile episodes accompanied by serositis; amyloidosis of AA type; favorable response to colchicine) and three minor criteria (FMF in first-degree relatives; erysiplase like erythema; recurrent febrile episodes); for diagnosis of FMF, it needs two major or one major plus two minor criteria.8 In 1992, the gene responsible for FMF, (MEDITERRANEAN FEVER)MEFV, was found on the short arm of chromosome 16.9 Five years later, the MEFV gene locus was discovered that encode the protein named marenostrin or pyrin.10 This protein probably has an important role in the downregulation of inflammation in innate immune response.
In populations with a high FMF, prevalence clinical criteria have a high specificity of 95–99% for the presence of genetically confirmed FMF, but sensitivity is much lower. In a recent study, FMF was genetically confirmed in 60% of patients who fulfilled clinical criteria in Mediterranean origin,11 while it was much lower in patients from non-Mediterranean areas (10%).12
Although until the last decade, the MEFV gene was considered to be responsible only for FMF; however, it is now known that it can also be associated with other clinical conditions with a main effect on the course and severity of the disease.13
Coexistence of FMF with rheumatoid and autoimmune conditions like seronegative spondyloarthropathy (SpA)14 Bechet’s disease15 rheumatoid arthritis (RA)16 Sjögren’s syndrome.17 Juvenile idiopathic arthritis,18,19 inflammatory bowel disease (IBD)20 and polyarteritis nodosa (PAN)21 have been reported.
In this study, we aimed to evaluate the frequency of comorbid disorders in a large FMF cohort of FMF registration center with relatively long follow-up duration. Additionally, we aimed to assess the association between FMF and other co-existed diseases and conditions with genotype-phenotype correlation survey. (WWW.FMFIRAN.IR)
Methods
Study Population
This is a case series study. The data of 400 FMF patients, who were diagnosed based on Tel- Hashomer criteria at the rheumatologic clinics of Bouali Hospital and from FMF Registration Center database (http://www.fmfiran.ir) were collected.
Demographic information of patients, such as age, race, gender, and their extra FMF disease, which have been confirmed by adult or pediatric subspecialist were collected.
MEFG Gene Analysis Study
Blood samples were screened for the 12 common pathogenic variants (E148Q, P369S, F479L, I692del, M680I (G/C), M680I (G/A), M694V, M694I, K695R, V726A, A 744S and R 761H) according to manufacturer’s instructions (FMF Strip Assay, Vienna lab, Vienna, Austria). The study is complaint with the Helsinki Declaration and was approved by the local Ethics.
Ethical and Legal Aspect
Committee under number IR.ARUMS. REC.1396.95. Written Informed consent was obtained from all the participants and/or their parents.
Comorbidity Diagnosis
Among them, 57 patients had associated disease that had been confirmed by related subspecialist and clinic of the hospital.
Statistical Analysis
Analysis was mainly descriptive, we have done all the statistical analyses with IBM SPSS 20 program (SPSS Inc., Chicago, IL, USA). Categorical variables were reported as numbers and percentages. Fisher’s exact test was used when the sample size was small (expected cell sizes < 5). The statistical significance defined as p value <0.05.
Results
Among the patients, 57 (14%) had associated disease other than FMF manifestations. Thirty-two patients were male and 24 patients were under 10 years old. Tables 1 and 2 show the patient’s profile as inflammatory and non-inflammatory conditions.
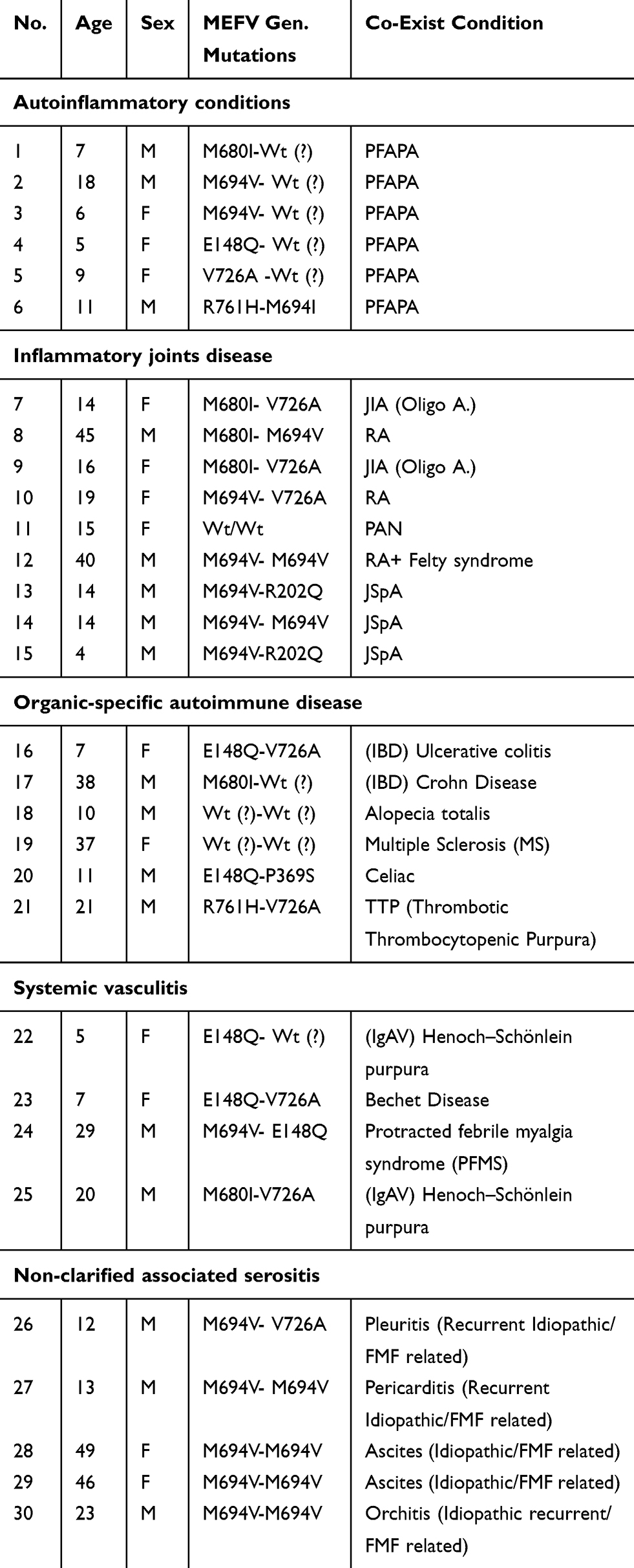 |
Table 1 Autoinflammatory and Autoimmune Disorders Co-Existed |
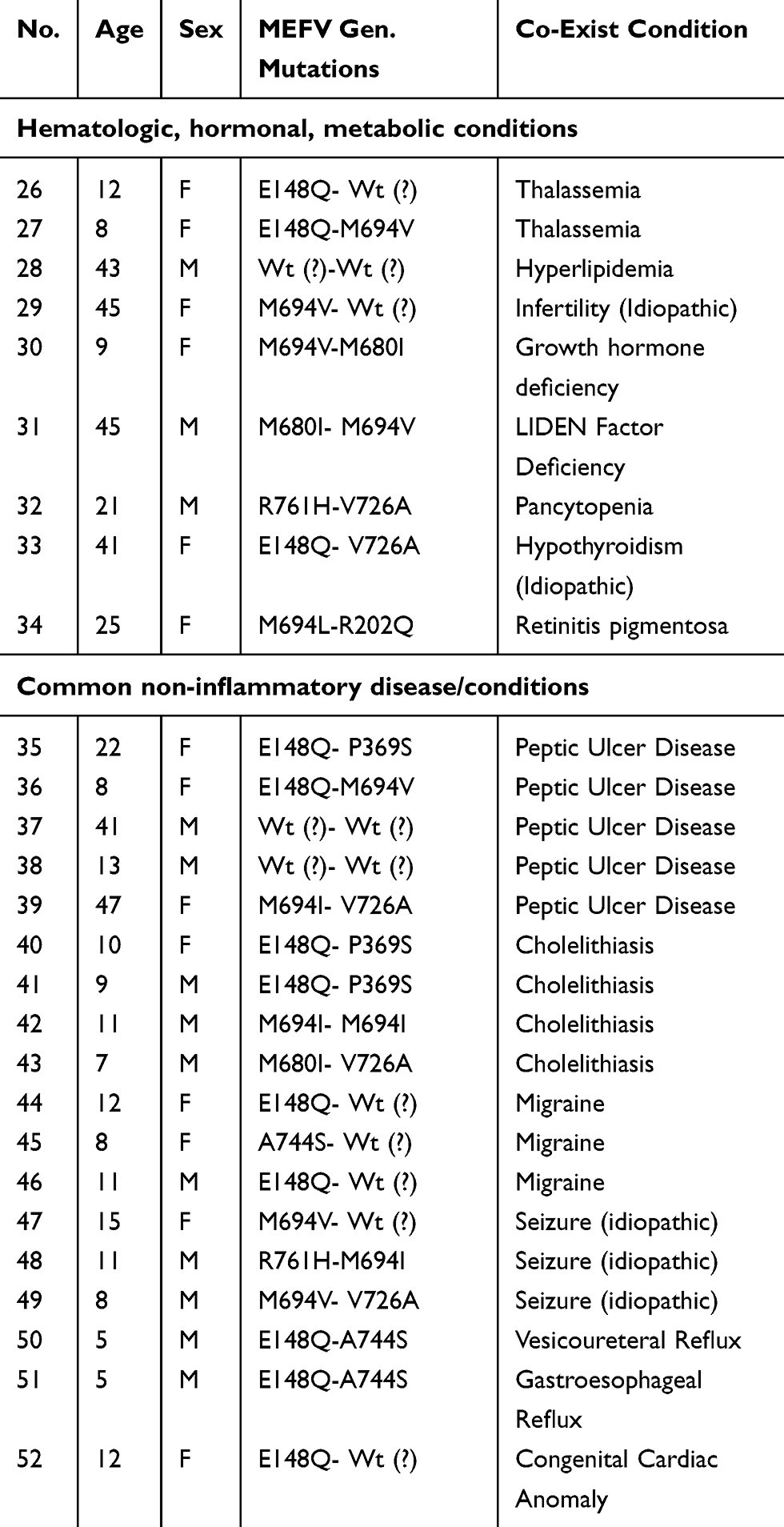 |
Table 2 Non-Inflammatory Coexisting Disorders |
There were 92 MEFV gene mutations. The most common were M694V (36%), E148Q (22%), V726A (17%), M680I (1%) and M694I (0.07%), respectively, and other mutations (R761H, P369S, A744S, M694L, R202Q) were the rest. Rheumatologic disorders were the most common co-exist disease (Arthritis, PFAPA, Vasculitis), followed by gastrointestinal GI (Peptic ulcer, cholelithiasis) and CNS (migraine, seizure) disorder. Some rare diseases such as thrombotic thrombocytopenic purpura TTP, growth hormone deficiency, multiple sclerosis MS, ascites and Leiden factor V deficiency and retinitis pigmentosa have been shown. JIA had M680I-V726A mutations and in RA M694V-M680I or V726A mutations have been shown. These homozygote mutations (M694V-M694V) were associated with idiopathic ascites, orchitis and pericarditis. There were three cases of JSpA and one case of Felty syndrome and one patient with childhood PAN. There was not a meaningful association between MEFV mutations and non-inflammatory disease. (P value 0.05%)
Discussion
Vasculitis
Vasculitis is found at a higher incidence in FMF patients than in the unaffected population.13 In our series, we had just two cases of IgA-V (0.5%) with positive MEFV mutations, while HSP has been reported in 3% even to 11% of FMF patients. Occult FMF cases were identified from Israel, commonly in children with IgA-V.22
PAN also occurs more commonly in patients with FMF usually with a younger age of onset.23 We detected one case of PAN among the patients however with negative MEFV mutations. The prevalence of PAN in FMF patients is about 1% (24) Hypertension and nephritis are more likely to occur in PAN than in FMF-PAN patients25 it seems that PAN is less severe in FMF patients.26 In patients with PAN-FMF, the prevalence of antistreptolysin O antibody elevation is high27.Data are insufficient to determine whether this disorder is more common in FMF patients than in the general population.28
Arthritis
We had two cases of JIA with Oligo-type and same mutations (M680I-V726A) in both and two cases of RA which showed combined heterozygote mutations. This collection contains three cases of Juvenile Spondylo-Arthropathy (JSpA) and one case of RA with neutropenia as a Felty syndrome; in our knowledge FMF association with Felty syndrome has not been reported already. Recurrent monoarthritis can be the sole manifestation of FMF; in such cases, the true diagnosis may not be established for some time. Lidar et al conducted a study to clinically and genetically characterize patients with FMF in whom arthritis constituted the only manifestation of FMF. The authors concluded those FMF groups were febrile with short duration arthritis, positive family history of FMF and MEFV mutations with good response to colchicine.29 We reported a rare form of relapsing arthritis as palindromic rheumatism that had been related to MEFV gene mutations without apparent FMF manifestation.30
Serositis
In our study, there was a patient with recurrent febrile chest pain as the only manifestation of FMF and combined MEFV mutations (M694V-V726A). Pleuritis can rarely present as the sole manifestation of FMF. As a rule in patients with paroxysmal febrile chest pain, especially in the Mediterranean area, FMF should be considered.3
Recurrent pericarditis, though rare, can present as the single manifestation of FMF. Okutur et al described a 25-year-old Turkish woman who presented with recurrent pericarditis of no obvious cause.31 In our series, there was one case with the same problem and M694V-M694V mutation analysis.
Skin Disease
In our series, there was not any especial skin disease except a case of alopecia totalis without MEFV gene mutation, but recurrent urticarial has been reported as a rare manifestation of FMF. Alonso et al described a patient with recurrent urticarial and final diagnosis of FMF.32
Neurological Disease
Neurological involvement has been reported and it varies from headache to aseptic meningitis. Meningitis can occur rarely in FMF as Mollaret meningitis. In each of the reported cases, the patients’ attacks of recurrent aseptic meningitis resolved after treatment with colchicine.33–35 We reported recently neurological manifestation of familial Mediterranean fever as a separate study.36 FMF-associated central nervous system (CNS) involvement includes demyelinating lesions, stroke, and posterior reversible leukoencephalopathy syndrome (PRES). Different studies showed that MS patients with MEFV mutations seem to develop a more progressive disease and it seems that MEFV mutations may increase the risk of MS progression.37–39
Inflammatory Disorder
In this work, there is a Bechet child with E148Q-V726A mutation analysis. An increased frequency of MEFV mutations has been reported in individuals with Bechet disease. FMF carriers with Bechet disease have been found to have an increased risk for venous thrombosis.40 Both FMF and Bechet disease are observed all around the Mediterranean area. From different studies,41–43 BD patients have a higher frequency of MEFV mutations than controls, and this high prevalence provides a further argument to support the role of MEFV mutations in the manifestation of different inflammatory disorders other than FMF.44
This study contains two cases of IBD with E148Q-V726A mutations as Ulcerative Colitis (UC) and M680I-Wt (?) in Crohn disease (CD). Some studies have found an increased frequency of MEFV mutations in patients with UC.20 Other studies have found that CD seems to be more prevalent in FMF patients.45,46 MEFV gene mutations may act as modifiers and affecting the expression of IBD.13
Patients with FMF certainly have an exaggerated response to streptococcal antigens and may be more prone to the late complications of streptococcal infection.20 Although there was not any cases in our patients, Streptococcus-associated diseases with the presence of high levels of antistreptolysin O (ASO) antibodies and streptococcus-associated diseases, such as acute poststreptococcal glomerulonephritis and acute rheumatoid fever, have been reported in patients with FMF.47
Non-Inflammatory Diseases
In this work, there were four cases of peptic ulcer disease (PUD) and four patients with cholelithiasis (CL), which in most of them there were combined heterozygote mutations (Table 2).
Here, we represent a patient with celiac disease coexisted with FMF and E148Q-P369S mutations. FMF and celiac disease (CD) may show different genetic and environmental factors as well as certain clinical features;48 however, the association between CD and FMF remains controversial.
Two sisters had idiopathic ascites and FMF with homozygote mutations as M694V-M694V. A female patient with FMF who developed chronic ascites has been reported by Ureten et al. She was a compound heterozygote for the mutations M694V and M680I, and after dose adjustment of colchicine, the amount of ascites decreased.49 There was one patient with recurrent orchitis and M694V-M694V mutations, although acute scrotum is rarely seen as a complication of FMF.50
Miscellaneous Disorder
Patients with growth hormone deficiency, TTP, Leiden Factor deficiency and retinitis pigmentosa and Felty syndrome were additional and probably unreported association in this study. We have discussed about the MEFV gene and PFAPA in a distinct article.51 However, here we report six FMF patients with co-existed PFAPA. In all of these patients, MEFV mutations are positive and one of them had compound heterozygote mutations (R761H-M694I). In half of these patients, FMF had been developed earlier than PFAPA.
Initially, we thought these patients are colchicine-resistant FMF, but more workup and closed observation with good response to a single dose of prednisolone during attacks revealed their co-existed PFAPA. On the basis of this finding particularly in young patients with colchicine resistant FMF, we recommend the probability of PFAPA as a possible associated condition.
In a recently published study by Yildiz,52 they showed that frequency of certain inflammatory conditions such as juvenile idiopathic arthritis, juvenile spondyloarthropathies, Henoch–Schönlein purpura, uveitis and inflammatory bowel disease was increased in their pediatric FMF patients; in contrary, asthma was less commonly detected in compared to general prevalence. The results of their study are relatively similar to our findings; however, in our experience uveitis is not a feature of FMF patients.53
The most important limitation of our study is the lack of a healthy control group in which we can compare the frequencies of the diseases. The other limitation was including adult and pediatric population in the same study.
Conclusion
Associated diseases in FMF usually are presented in patients with positive MEFV gene mutations particularly with these five mutations M694V, E148Q, V726A, M680I, and M694I. Rheumatologic, gastrointestinal and CNS disorders are common co-existed disease.
Ethical Approval
All procedures performed in studies involving human participants were in accordance with the ethical standards of the institutional and/or national research committees.
Author Contributions
FS and AE work at the rheumatology clinic and planned the study and diagnosis of the FMF patients. AE wrote the final copy. FS wrote the draft copy of the manuscript. All authors made substantial contributions to conception and design, acquisition of data or analysis and interpretation of data; took part in drafting the article or revising it critically for important intellectual content; gave final approval of the version to be published; and agree to be accountable for all aspects of the work.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Lidar M, Yaqubov M, Zaks N, Ben-Horin S, Langevitz P, Livneh A. The prodrome: a prominent yet overlooked pre-attack manifestation of familial Mediterranean fever. J Rheumatol. 2006;33(6):1089–1092.
2. Heller H, Sohar E, Pras M. Ethnic distribution and amyloidosis in Familial Mediterranean fever (FMF). Pathobiology. 1961;24(4):718–723. doi:10.1159/000161188
3. Salehzadeh F. Familial mediterranean fever in Iran: a report from FMF registration center. Int J Rheumatol. 2015;2015:912137. doi:10.1155/2015/912137
4. Soylu A, Kasap B, Türkmen M, Saylam GS, Kavukçu S. Febrile myalgia syndrome in familial Mediterranean fever. J Clin Rheumatol. 2006;12(2):93–96.
5. Özkaya O, Bek K, Alaca N, Ceyhan M, Açıkgöz Y, Taşdemir HA. Cerebral vasculitis in a child with Henoch–Schönlein purpura and familial Mediterranean fever. Clin Rheumatol. 2007;26(10):1729–1732. doi:10.1007/s10067-006-0485-x
6. Topaloglu R, Ozaltin F, Yilmaz E, et al. E148Q is a disease-causing MEFV mutation: a phenotypic evaluation in patients with familial Mediterranean fever. Ann Rheum Dis. 2005;64(5):750–752. doi:10.1136/ard.2004.026963
7. Lachmann HJ, Şengül B, Yavuzşen TU, et al. Clinical and subclinical inflammation in patients with familial Mediterranean fever and in heterozygous carriers of MEFV mutations. Rheumatology. 2006;45(6):746–750. doi:10.1093/rheumatology/kei279
8. Livneh A, Langevitz P, Zemer D, et al. Criteria for the diagnosis of familial Mediterranean fever. Arthritis Rheum. 1997;40(10):1879–1885. doi:10.1002/art.1780401023
9. Pras E, Aksentijevich I, Gruberg L, et al. Mapping of a gene causing familial Mediterranean fever to the short arm of chromosome 16. N Engl J Med. 1992;326(23):1509–1513. doi:10.1056/NEJM199206043262301
10. International FMF Consortium. Ancient missense mutations in a new member of the RoRet gene family are likely to cause familial Mediterranean fever. Cell. 1997;90(4):797–807. doi:10.1016/S0092-8674(00)80539-5
11. Salehzadeh F, Asl MJ, Asl SH, Jahangiri S, Habibzadeh S. MEFV gene profile in northwest of Iran, twelve common MEFV gene mutations analysis in 216 patients with familial mediterranean fever. Iran J Med Sci. 2015;40(1):68.
12. Federici L, Rittore-Domingo C, Koné-Paut I, et al. A decision tree for genetic diagnosis of hereditary periodic fever in unselected patients. Ann Rheum Dis. 2006;65(11):1427–1432. doi:10.1136/ard.2006.054304
13. Shohat M, Halpern GJ. Familial Mediterranean fever—a review. Genet Med. 2011;13(6):487–498. doi:10.1097/GIM.0b013e3182060456
14. Bodur H, Seçkin Ü, Eser F, Ergül G, Seçkin S. Coexistence of familial Mediterranean fever and psoriasis in a patient with seronegative spondyloarthropathy. Rheumatol Int. 2008;29(1):107–110. doi:10.1007/s00296-008-0616-6
15. Matsuda M, Nakamura A, Tsuchiya S, Yoshida T, Horie S, Ikeda SI. Coexistence of familial Mediterranean fever and Behçet’s disease in a Japanese patient. Internal Med. 2006;45(12):799–800. doi:10.2169/internalmedicine.45.1560
16. Gösteren AA. Coexistence of familial Mediterranean fever and rheumatoid arthritis in a case. Turk J Rheumatol. 2010;25:44–46.
17. Tanaka M, Migita K, Miyashita T, et al. Coexistence of familial Mediterranean fever and Sjogren’s syndrome in a Japanese patient. Clin Exp Rheumatol. 2007;25(5):792.
18. Keleş I, Aydın G, Tosun A, İnal E, Keleş H, Orkun S. Familial Mediterranean fever and ankylosing spondylitis in a patient with juvenile idiopathic arthritis: a case report and review of the literature. Rheumatol Int. 2006;26(9):846–851. doi:10.1007/s00296-005-0080-5
19. Rozenbaum M, Rosner I. Severe outcome of juvenile idiopathic arthritis (JIA) associated with familial Mediterranean fever (FMF). Clin Exp Rheumatol. 2004;22:S75–8.
20. Yurtcu E, Gokcan H, Yilmaz U, Sahin FI. Detection of MEFV gene mutations in patients with inflammatory bowel disease. Genet Test Mol Biomarkers. 2009;13(1):87–90. doi:10.1089/gtmb.2008.0094
21. Ben-Chetrit E, Bakkaloglu A, Gur H, et al. Polyarteritis nodosa in patients with Familial Mediterranean Fever (FMF): a concomitant disease or a feature of FMF? Semin Arthritis Rheum. 2001;30(4):281–7. doi:10.1053/sarh.2001.19958
22. Schlesinger M, Rubinow A, Vardy PA. Henoch-Schönlein purpura and familial Mediterranean fever. Isr J Med Sci. 1985;21(1):83.
23. Sachs D, Langevitz P, Morag B, Pras M. Polyarteritis nodosa and familial Mediterranean fever. Rheumatology. 1987;26(2):139–141. doi:10.1093/rheumatology/26.2.139
24. Ozdogan H, Arisoy N, Kasapcapur O, et al. Vasculitis in familial Mediterranean fever. J Rheumatol. 1997;24(2):323–327.
25. Said R, Hamzeh Y, Said S, Tarawneh M, Al-Khateeb M. Spectrum of renal involvement in familial Mediterranean fever. Kidney Int. 1992;41(2):414–419. doi:10.1038/ki.1992.57
26. Hatemi G, Masatlioglu S, Gogus F, Ozdogan H. Necrotizing vasculitis associated with familial Mediterranean fever. Am J Med. 2004;117(7):516–519. doi:10.1016/j.amjmed.2004.02.050
27. Tekin M, Yalcinkaya F, Tumer N, Akar N, Misirlioǧlu M, Çakar N. Clinical, laboratory and molecular characteristics of children with Familial Mediterranean Fever‐associated vasculitis. Acta Paediatr. 2000;89(2):177–182. doi:10.1111/j.1651-2227.2000.tb01212.x
28. Padeh S, Berkun Y. Periodic fever syndromes. In: Shoenfeld Y, Cervera R, Gershwin M.E, editors. Diagnostic Criteria in Autoimmune Diseases. Totowa, NJ: Humana Press; 2008:201–207.
29. Lidar M, Kedem R, Mor A, Levartovsky D, Langevitz P, Livneh A. Arthritis as the sole episodic manifestation of familial Mediterranean fever. J Rheumatol. 2005;32(5):859–862.
30. Salehzadeh F, Barak M, Nematdoust Haghi R. Relapsing periodic arthritis, palindromic rheumatism and MEFV gene-related variants alleles in children. Pediatr Rheumatol Online J. 2019;17(1):28. PubMed PMID: 31171010; PubMed Central PMCID: PMC6555729. doi:10.1186/s12969-019-0329-2
31. Okutur K, Seber S, Oztekin E, Bes C, Borlu F. Recurrent pericarditis as the initial manifestation of Familial Mediterranean fever. Med Sci Monitor. 2008;14(12):CS139–41.
32. Alonso R, Cisteró-Bahima A, Enrique E, San MM. Recurrent urticaria as a rare manifestation of familial Mediterranean fever. J Invest Allergol Clin Immunol. 2002;12(1):60–61.
33. Vilaseca J, Tor J, Guardia J, Bacardi R. Periodic meningitis and familial Mediterranean fever. Arch Intern Med. 1982;142(2):378–379. doi:10.1001/archinte.1982.00340150178032
34. Collard M, Sellal F, Hirsch E, Mutschler V, Marescaux C. Recurrent aseptic meningitis in periodic disease or Mollaret’s meningitis? Rev Neurol (Paris). 1991;147(5):403–405.
35. Karachaliou I, Karachalios G, Charalabopoulos A, Charalabopoulos K. Meningitis associated with familial Mediterranean fever. Int J Clin Pract. 2005;59:60–61. doi:10.1111/j.1368-504X.2005.00290.x
36. Salehzadeh F, Azami A, Motezarre M, Haghi RN, Ahmadabadi F. Neurological manifestations in familial mediterranean fever: a genotype-phenotype correlation study. Open Access Rheumatol. 2020;12:15. doi:10.2147/OARRR.S238649
37. Kalyoncu U, Eker A, Oguz KK, et al. Familial Mediterranean fever and central nervous system involvement: a case series. Medicine. 2010;89(2):75–84. doi:10.1097/MD.0b013e3181d5dca7
38. Unal A, Dursun A, Emre U, Tascilar NF, Ankarali H. Evaluation of common mutations in the Mediterranean fever gene in Multiple Sclerosis patients: is it a susceptibility gene? J Neurol Sci. 2010;294(1–2):38–42. doi:10.1016/j.jns.2010.04.008
39. Topcuoglu MA, Karabudak R. Familial Mediterranean fever and multiple sclerosis. J Neurol. 1997;244(8):510–514. doi:10.1007/s004150050134
40. Rabinovich E, Shinar Y, Leiba M, Ehrenfeld M, Langevitz P, Livneh A. Common FMF alleles may predispose to development of Behcet’s disease with increased risk for venous thrombosis. Scand J Rheumatol. 2007;36(1):48–52. doi:10.1080/03009740600759639
41. Touitou I, Magne X, Molinari N, et al. MEFV mutations in Behçet’s disease. Hum Mutat. 2000;16(3):271–272. doi:10.1002/1098-1004(200009)16:3<271::AID-HUMU16>3.0.CO;2-A
42. Ben-Chetrit E, Cohen R, Chajek-Shaul T. Familial mediterranean fever and Behçet’s disease–are they associated? J Rheumatol. 2002;29(3):530–534.
43. Atagunduz P, Ergun T, Direskeneli H. MEFV mutations are increased in Behçet’s disease (BD) and are associated with vascular involvement. Clin Exp Rheumatol. 2003;21(4; SUPP/30):S35–S37.
44. Livneh A, Aksentijevich I, Langevitz P, et al. A single mutated MEFV allele in Israeli patients suffering from familial Mediterranean fever and Behcet’s disease (FMF-BD). Eur J Human Genet. 2001;9(3):191–196. doi:10.1038/sj.ejhg.5200608
45. Fidder HH, Chowers Y, Lidar M, Sternberg M, Langevitz P, Livneh A. Crohn disease in patients with familial Mediterranean fever. Medicine. 2002;81(6):411–416. doi:10.1097/00005792-200211000-00001
46. Kulog˘ Lu Z, Kansu A, Ustundag˘ G, Burcin Ozcan¸akar Z, Ensari A, Ekim M. An infant with severe refractory Crohn’s disease and homozygous MEFV mutation who dramatically responded to colchicines. Rheumatol Int. 2012;32(3):783–785. doi:10.1007/s00296-009-1326-4
47. Yalçınkaya F, Ince E, Uçar T, et al. Antistreptococcal response is exaggerated in children with familial Mediterranean fever. Clin Rheumatol. 2002;21(5):378–381. doi:10.1007/s100670200101
48. Mor A, Mekori YA, Livneh A. Familial Mediterranean fever and celiac sprue–are they related? Clin Exp Rheumatol. 2004;22(4 Suppl 34):82.
49. Üreten K, Bostancı A, Akbal E, Gönülalan G, Özbek M, Öztürk MA. Familial Mediterranean fever with chronic ascites: a case report and a review of literature. Rheumatol Int. 2009;29(12):1477–1480. doi:10.1007/s00296-009-0842-6
50. Eshel G, Zemer D, Bar-Yochai A. Acute orchitis in familial Mediterranean fever. Ann Intern Med. 1988;109(2):164–165. doi:10.7326/0003-4819-109-2-164
51. Salehzadeh F, Vahedi M, Hosseini-Asl S, Jahangiri S, Habibzadeh S, Hosseini-Khotbesara M. PFAPA and 12 common MEFV gene mutations our clinical experience. Iran J Pediatr. 2014;24(1):64.
52. Yildiz M, Adrovic A, Tasdemir E, et al. Evaluation of co-existing diseases in children with familial Mediterranean fever. Rheumatol Int. 2020;40(1):57–64. doi:10.1007/s00296-019-04391-9 Epub 2019 Jul 27.
53. Salehzadeh F, Yasrebi O, Hosseini Khotbesara M. Hosseini Khotbesara MIdiopathic uveitis and familial Mediterranean fever: is there any relationship? Autoimmune Dis. 2014;2014:238931. doi:10.1155/2014/238931 Epub 2014 Jan 30.
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.