")
Back to Journals » Journal of Inflammation Research » Volume 13
Coagulation Disorders in COVID-19: Role of Toll-like Receptors
Received 11 July 2020
Accepted for publication 27 August 2020
Published 29 October 2020 Volume 2020:13 Pages 823—828
DOI https://doi.org/10.2147/JIR.S271768
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Ning Quan
Indranil Biswas,1 Gausal A Khan2
1Cardiovascular Biology Research Program, Oklahoma Medical Research Foundation, Oklahoma City, OK 73104, USA; 2Department of Physiology & Physiotherapy, College of Medicine, Nursing and Health Sciences, Fiji National University, Suva, Fiji Islands
Correspondence: Gausal A Khan
Department of Physiology & Physiotherapy, College of Medicine, Nursing and Health Sciences, Fiji National University, Suva, Fiji Islands
Email [email protected]
Abstract: Coronavirus disease 2019 (COVID-19) has spread rapidly throughout the world. The range of the disease is broad but among hospitalized patients with COVID-19 are coagulation disorders, pneumonia, respiratory failure, and acute respiratory distress syndrome (ARDS). The excess production of early response proinflammatory cytokines results in what has been described as a cytokine storm, leading to an increased risk of thrombosis, inflammations, vascular hyperpermeability, multi-organ failure, and eventually death over time. As the pandemic is spreading and the whole picture is not yet clear, we highlight the importance of coagulation disorders in COVID-19 infected subjects and summarize it. COVID-19 infection could induce coagulation disorders leading to clot formation as well as pulmonary embolism with detrimental effects in patient recovery and survival. Coagulation and inflammation are closely related. In this review, we try to establish an association between virus infections associated with innate immune activation, inflammation and coagulation activation.
Keywords: COVID-19, coagulation disorders, TLR3, tissue factor
Introduction
Patients with coronavirus disease 2019 (COVID-19) show a diverse level of severity that ranges from asymptomatic to acute phase of multi-organ dysfunctions.1 Although most of the COVID-19 patients have respiratory infection,2 about 60–70% of the hospitalized patients develop coagulation abnormalities such as thrombocytopenia, hypercoagulation, disseminated intravascular coagulation (DIC) and venous thrombosis (VT).3 Coagulation activation as well as widespread vascular inflammation related diseases have been shown to be present in COVID-19 patients with severe outcome, and autopsy revealed almost 58% of patients died due to venous thrombosis and pulmonary embolism.4,5 Emerging reports show that a subset of COVID-19 infected patients, who develop severe disease, are commonly shown with prolonged activated partial thromboplastin time (APTT), delayed prothrombin time (PT), higher D-dimer and fibrin degradation products (FDP) levels, increased thrombin-antithrombin (TAT) complex and decreased antithrombin (AT) in plasma samples.6 These changes clearly indicated that coagulation activation, increased thrombin generation and consumptive coagulopathy are evident in sudden decrease in plasma fibrinogen level in nonsurvivors.6 Another study, involving Caucasian patients showed diffuse pulmonary inflammation along with pulmonary-specific vasculopathy. A study also reported elevated levels of vWF (von Willebrand factor) in circulation and increased activity of vWF and factor VIII.7
The pathophysiology of severe acute respiratory syndrome (SARS)-CoV-2-mediated vascular complications are similar to that of severe pneumonia caused by other viruses or bacteria.8,9 But the detailed molecular mechanism of SARS-CoV-2-mediated hypercoagulation is far from clear. Catanzaro et al hypothesized that SARS-CoV-2 may downregulate ACE-2 expression, thus regulating over production of angiotensin II and concomitant enhancement of IL-6. IL-6 in positive inflammatory feedback loop inactivates ACE-2, enhancing angiotensin II retention lead to endothelial activation and inflammation.10 Several factors including endothelial activation,11 monocyte infiltration,12 complement activation,13 increased plasma cytokine14 may contribute to viral infection mediated systemic coagulation activation.
COVID-19 and Extrinsic Coagulation Pathway
Immune response towards microorganism activates extrinsic coagulation pathway, which is vital for host defense but also responsible for multi-organ failure.15 In normal physiological conditions, tissue factor (TF) resides within the endothelial cells.16 However, during trauma, tissue injury or inflammation, TF gets exposed to the external surface and comes into contact with coagulation factor VII, thereby activating the extrinsic coagulation pathway.17 Leukocyte infiltration and TF expression in inflammatory monocytes further worsen the thrombotic complications.18 A recent report demonstrates the presence of viral elements within endothelial cells of renal vessels of COVID-19 patients.19 This study also showed the endothelial activation, accumulation of inflammatory cells in lung vasculatures in COVID-19 patients with the evidence of endothelial and inflammatory cell death.19 The potential involvement of complement activation mediated coagulation activation in COVID-19 patients has been documented in another case study, where the authors demonstrated coagulation abnormalities, endothelial injury, microvascular thrombotic complications mediated by intense complement activation and terminal complement complex (C5b-9) deposition.20 Complement and coagulation pathways are closely related and complement activation may lead to coagulation activation.
Inflammation and Coagulation Activation in COVID-19
Coagulation and inflammation are interrelated. It is well known that overwhelmed innate immune response activates coagulation, whereas coagulation activation and thrombin generation further lead to augmented inflammation.21 Other than promoting clot formation by activating platelets and by cleaving fibrinogen to fibrin,22 thrombin also exerts multiple cellular effects via protease activated receptors (PARs).23 Thrombin generation and its activity is tightly regulated by anticoagulant molecules like AT,24 tissue factor pathway inhibitor (TFPI) and activated protein C (APC).25 During inflammation, most of these anticoagulant protective mechanisms are impaired, as noted in COVID-19 patients.15 As a result of impaired regulation of procoagulant–anticoagulant molecules, the natural homeostasis further shifts towards more procoagulant and pro-inflammatory phenotype that predisposes to the development of intravascular thrombosis, DIC and multi-organ failure. Overproduction of early response pro-inflammatory cytokines like tumor necrosis factor α (TNFα), IL-6, and IL-1β as found in COVID-19 patients results in cytokine storm, leading to hypercoagulation, platelet activation, leukocyte infiltration and vascular hyperpermeability.26 Acute increase of pro-inflammatory molecules in lung, lead to simultaneous formation of pulmonary edema and pulmonary embolism as observed in COVID-19 patients.4,27 Treatment with natural anticoagulant molecule, APC and AT which is known to have anticoagulant, anti-inflammatory cytoprotective effect may rescue the COVID-19 patients with hyperinflammation and coagulopathy.28,30
Neutrophil Extracellular Trap (NET) and Platelet Activation in COVID-19
Neutrophil activation and NET formation lead to release of myeloperoxidase (MPO), neutrophil elastase and chromatin content (DNA and histones), which are responsible for coagulation activation, DIC and thrombosis.31 In a recent finding, Zuo et al found increased citrullinated histone H3 (CitH3), MPO-DNA complex in COVID-19 patients. Increase in NET parameters also correlated with inflammation (C-reactive protein) and coagulation (D-dimer) markers.32 Interestingly, sera from COVID-19 patients also triggered NET formation, suggesting a critical link between COVID-19 pathophysiology and NET formation. NETosis sequester platelets in microcirculation that are responsible for increased fibrin-platelet interaction and formation of microthrombi.33 Thrombocytopenia has been reported in COVID-19 patients.34 Megakaryocytes and platelets associated with fibrin microthrombi were found in cardiac microvasculature, fibrin associated and fibrin independent platelet aggregates were seen in lung parenchyma, renal circulation (glomerular platelet aggregate) and in hepatic sinusoids in autopsy sample of COVID-19.35
COVID-19 and Fibrinolysis Pathway
Fibrinolysis, cleavage of fibrin polymers is one of the rescue mechanisms to combat hyper-coagulation and excessive fibrin deposition. Plasmin, a central regulator of fibrinolysis also plays an important role in COVID-19 pathophysiology through increase in virulence and infectivity of SARS-CoV-2. Increased D-dimer level in COVID-19 patientsnot only shows coagulation activation, but also manifests the altered fibrinolysis.36 This altered fibrinolysis is typically attributed by tissue plasminogen activator (TPA) that released from injured endothelial cells.37 Interesting recent findings showed that furin-like cleavage site (682RRAR/S686) present in the S1/S2 protease cleavage site of the SARS-CoV-2 virus, can also be proteolytically cleaved by higher concentration of plasmin, cathepsin and elastase. Unlike furin, which is predominantly present in the cytoplasm of alveolar type II epithelial cells, plasminogen and TPA that are released from the endothelial cells augment the virus infectivity.38,39
Toll-like Receptors and COVID-19
Generation of proinflammatory cytokines and vascular inflammation is mediated through a number of pattern recognition receptors, known as toll-like receptors (TLRs) and nod-like receptors (NLRs).40,41 TLRs are expressed in different types of innate immune cells including monocyte, macrophages, and dendritic cells present in alveolar microenvironment. Alveolar macrophages along with pro-inflammatory macrophages that infiltrate from circulation provide the immune surveillance in lung. Upon entering the lung, SARS-CoV-2 infects alveolar epithelial cells and lung microvascular endothelial cells.42 Viral infection in these cells directly or indirectly activates TLRs. Viral RNA can directly activate TLRs in these cells or debris from infected dying cells can activate other immune cells, like alveolar macrophages. Macrophages participate in COVID-19 inflammatory response by phagocytic uptake of viral particle or cellular debris containing viral RNA.43 Single stranded viral RNA can be detected by TLR7-8 and positive strand or double stranded secondary structure activates TLR3.44 Downstream of TLR engagement, Bruton's tyrosine kinase (BTK) dependent NFκB activation results in the production of cytokines and chemokines.44 The role of TLR2 and TLR4 is interesting in this context. Traditionally TLR2 and TLR4 recognize peptidoglycan (PGN) and lipopolysaccharides (LPS) from gram-positive and gram-negative bacteria. However, recent studies showed that damage associated molecular pattern (DAMP) like high mobility group box protein (HMGB1), histones, and oxidized phospholipids also can activate these TLRs. Activation of TLRs through oxidized phospholipids or DAMP can activate the production of TNFα, IL6, and other cytokines that are responsible for cytokine storm.42 Interestingly, one in silico interaction study hypothesized that TLR4 may be involved in recognizing molecular pattern from spike protein of corona virus. However, further studies will be required to understand the roles of TLRs in COVID-19.45
COVID-19 TLR3 and Coagulation
TLR3 is classically known to recognize the single stranded and positive stranded viral RNA.46 Previous studies showed that TLR3 exerts protective role via interferon release, thereby inhibiting the coronavirus infection in macrophages.47 TLR3 signaling also contributes in protective innate immune response to SARS corona virus48 and other respiratory viruses.49 These studies show that TLR3 plays an important role in antiviral immunity. However, the role of TLR3 in regulation of infection and inflammation is very complex in nature. Along with the immune cells, endothelial cells also express TLR3 and vividly respond to the viral infection. Recent study showed COVID-19 has been found in endothelial cells, and viral infection may lead to TLR3 activation by viral RNA.19 In the context of antiviral immune response and inflammation, it was reported that activation of TLR3 through viral RNA analogue polyinosinic-polycytidylic acid (poly I: C), can activate TF in endothelial cells.50 Severe hypoxemia due to impaired pulmonary function has been reported in COVID-19 cases.51 Interestingly, hypoxic condition is also known to activate TLR3 activation that leads to monocyte infiltration, TF expression and hypercoagulation.52,53 An earlier study also showed that virus-induced hemorrhagic fever also develops a procoagulant state due to TF activation through TLR3.50 This suggests that viral genetic material could activate TF and coagulation. Several other studies also showed that the RNA released from either damaged tissue or endocytosed cells can serve as ligands for TLR3.52,54 This study further showed that pretreatment of hydroxychloroquine (an TLR3 inhibitor) inhibits hypoxia induced TF activation and intravascular coagulation where extracellular RNA is a ligand for the same. It is interesting that extracellular RNA released from damaged tissues could also activate intrinsic pathways of coagulation.55 Therefore, the viral particles could induce coagulation disorders though TLR3 activation.
Conclusion
The coagulation disorders lead to formation of an intravascular clot in pulmonary or systemic circulation, which is prominent findings in COVID-19 infection associated severe respiratory disease, and has been demonstrated both in humans and in animal models. COVID-19 infection could induce coagulation disorder leading to overt clot formation, pulmonary embolism with detrimental effects in patient recovery and survival. Although the mechanism of COVID-19-mediated coagulation activation is not clearly known. It seems that viral particles may lead to TLR activation directly or indirectly. In this review, we hypothesize that COVID-19-mediated TLRs activation may be responsible for activation of extrinsic coagulation pathway and leukocyte infiltration, inflammatory signaling in endothelial cells, neutrophil granule release, NET formation and platelet aggregation, fibrin deposition that ultimately lead to thrombosis (Figure-1). Most of these phenomena are mediated through TLRs. TLRs can be considered as suitable drug target.
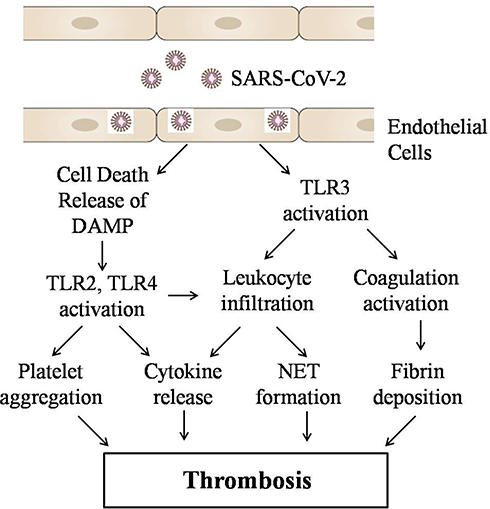 |
Figure 1 Possible mechanism of SARS-CoV-2 medicated thrombosis. |
Acknowledgment
I thank Madiha Khan for helping in manuscript editing. This study was supported by grants from the Grand Challenge Canada to GAK (R-ST-POC-1807- 13914).
Disclosure
The authors report no conflicts of interest in this work.
References
1. Huang C, Wang Y, Li X, et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet. 2020;395(10223):497–506. doi:10.1016/S0140-6736(20)30183-5
2. Chen N, Zhou M, Dong X, et al. Epidemiological and clinical characteristics of 99 cases of 2019 novel coronavirus pneumonia in Wuhan, China: a descriptive study. Lancet. 2020;395(10223):507–513. doi:10.1016/S0140-6736(20)30211-7
3. Connors JM, Levy JH. COVID-19 and its implications for thrombosis and anticoagulation. Blood J Am Society Hematol. 2020;135(23):2033–2040.
4. Wichmann D, Sperhake JP, Lütgehetmann M, et al. Autopsy findings and venous thromboembolism in patients with COVID-19: a prospective cohort study. Ann Intern Med. 2020;173:268–277. doi:10.7326/M20-2003
5. Klok FA, Kruip MJ, Van der Meer NJ, et al. Incidence of thrombotic complications in critically ill ICU patients with COVID-19. Thromb Res. 2020.
6. Tang N, Li D, Wang X, Sun Z. Abnormal coagulation parameters are associated with poor prognosis in patients with novel coronavirus pneumonia. J Thrombosis Haemostasis. 2020;18(4):844–847. doi:10.1111/jth.14768
7. Leonard-Lorant I, Delabranche X, Severac F, et al. Acute pulmonary embolism in COVID-19 patients on CT angiography and relationship to D-dimer levels. Radiology. 2020;201561.
8. Hwang DM, Chamberlain DW, Poutanen SM, Low DE, Asa SL, Butany J. Pulmonary pathology of severe acute respiratory syndrome in Toronto. Modern Pathol. 2005;18(1):1. doi:10.1038/modpathol.3800247
9. Ruuskanen O, Lahti E, Jennings LC, Murdoch DR. Viral pneumonia. Lancet. 2011;377(9773):1264–1275. doi:10.1016/S0140-6736(10)61459-6
10. Catanzaro M, Fagiani F, Racchi M, Corsini E, Govoni S, Lanni C. Immune response in COVID-19: addressing a pharmacological challenge by targeting pathways triggered by SARS-CoV-2. Signal Transduction Targeted Therapy. 2020;5(1):1. doi:10.1038/s41392-020-0191-1
11. Juffrie M, van Der Meer GM, Hack CE, Haasnoot K, Veerman AJ, Thijs LG. Inflammatory mediators in dengue virus infection in children: interleukin-8 and its relationship to neutrophil degranulation. Infect Immun. 2000;68(2):702–707. doi:10.1128/IAI.68.2.702-707.2000
12. Jessie K, Fong MY, Devi S, Lam SK, Wong KT. Localization of dengue virus in naturally infected human tissues, by immunohistochemistry and in situ hybridization. J Infect Dis. 2004;189(8):1411–1418. doi:10.1086/383043
13. O’Brien KB, Morrison TE, Dundore DY, Heise MT, Schultz-Cherry S. A protective role for complement C3 protein during pandemic 2009 H1N1 and H5N1 influenza A virus infection. PLoS One. 2011;6(3):e17377. doi:10.1371/journal.pone.0017377
14. Chaturvedi UC, Shrivastava R, Tripathi RK, Nagar R. Denguevirus-specific suppressor T cells: current perspectives. FEMS Immunol Med Microbiol. 2007;50(3):285–299. doi:10.1111/j.1574-695X.2007.00273.x
15. Tang N, Bai H, Chen X, Gong J, Li D, Sun Z. Anticoagulant treatment is associated with decreased mortality in severe coronavirus disease 2019 patients with coagulopathy. J Thrombosis Haemostasis. 2020;18(5):1094–1099. doi:10.1111/jth.14817
16. Bach RR. Initiation of coagulation by tissue facto. Critical Rev Biochem. 1988;23(4):339–368. doi:10.3109/10409238809082548
17. Owens AP, Mackman N. Tissue factor and thrombosis: the clot starts here. Thromb Haemost. 2010;104(3):432. doi:10.1160/TH09-11-0771
18. Lawson CA, Yan SD, Yan SF, et al. Monocytes and tissue factor promote thrombosis in a murine model of oxygen deprivation. J Clin Invest. 1997;99(7):1729–1738. doi:10.1172/JCI119337
19. Varga Z, Flammer AJ, Steiger P, et al. Endothelial cell infection and endotheliitis in COVID-19. Lancet. 2020;395(10234):1417–1418. doi:10.1016/S0140-6736(20)30937-5
20. Diao B, Feng Z, Wang C, et al. Human kidney is a target for novel severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) infection. MedRxiv. 2020.
21. Strukova S. Blood coagulation-dependent inflammation. Coagulation-dependent inflammation and inflammation-dependent thrombosis. Front Biosci. 2006;11(1):59–80. doi:10.2741/1780
22. Celikel R, McClintock RA, Roberts JR, et al. Modulation of α-thrombin function by distinct interactions with platelet glycoprotein Ibα. Science. 2003;301(5630):218–221. doi:10.1126/science.1084183
23. Coughlin SR. Thrombin signaling and protease-activated receptors. Nature. 2000;407(6801):258–264. doi:10.1038/35025229
24. Uchiba M, Okajima K, Kaun C, Wojta J, Binder BR. Inhibition of the endothelial cell activation by antithrombin in vitro. Thromb Haemost. 2004;92(12):1420–1427. doi:10.1160/TH04-03-0139
25. Riewald M, Petrovan RJ, Donner A, Ruf W. Activated protein C signals through the thrombin receptor PAR1 in endothelial cells. J Endotoxin Res. 2003;9(5):317–321. doi:10.1177/09680519030090050801
26. Liu B, Li M, Zhou Z, Guan X, Xiang Y. Can we use interleukin-6 (IL-6) blockade for coronavirus disease 2019 (COVID-19)-induced cytokine release syndrome (CRS). J Autoimmun. 2020;111:102452. doi:10.1016/j.jaut.2020.102452
27. Han H, Yang L, Liu R, et al. Prominent changes in blood coagulation of patients with SARS-CoV-2 infection. Clin Chem Laboratory Med. 2020;1(ahead–of–print).
28. Griffin JH, Lyden P. COVID-19 hypothesis: activated protein C for therapy of virus‐induced pathologic thromboinflammation. Res Practice Thrombosis Haemostasis. 2020;4(4):506–509. doi:10.1002/rth2.12362
29. Cai X, Biswas I, Panicker SR, Giri H, Rezaie AR. Activated protein C inhibits lipopolysaccharide-mediated acetylation and secretion of high mobility group box 1 in endothelial cells. J Thrombosis Haemostasis. 2019;17(5):803–817. doi:10.1111/jth.14425
30. Dinarvand P, Yang L, Biswas I, Giri H, Rezaie AR. Plasmodium falciparum histidine rich protein HRPII inhibits the anti-inflammatory function of antithrombin. J Thrombosis Haemostasis. 2019;18(6):1473–1483. doi:10.1111/jth.14713
31. Fuchs TA, Brill A, Duerschmied D, Schatzberg D. Extracellular DNA traps promote thrombosis. Proc Natl Acad Sci USA. 2010;107(15):815880. doi:10.1073/pnas.1005743107
32. Zuo Y, Yalavarthi S, Shi H, et al. Neutrophil extracellular traps (NETs) as markers of disease severity in COVID-19. medRxiv. 2020.
33. Thierry A, Roch B. NETs by-products and extracellular DNA may play a key role in COVID-19 pathogenesis: incidence on patient monitoring and therapy. Med Pharmacol. 2020.
34. Lippi G, Plebani M, Henry BM. Thrombocytopenia is associated with severe coronavirus disease 2019 (COVID-19) infections: a meta-analysis. Clinica Chimica Acta. 2020.
35. Rapkiewicz AV, Mai X, Carsons SE, et al. Megakaryocytes and platelet-fibrin thrombi characterize multi-organ thrombosis at autopsy in COVID-19: A case series. E Clin Med. 2020;24:100434. doi:10.1016/j.eclinm.2020.100434
36. Wright FL, Vogler TO, Moore EE, et al. Fibrinolysis shutdown correlates to thromboembolic events in severe COVID-19 Infection. J Am Coll Surg. 2020;231:193–203.e1. doi:10.1016/j.jamcollsurg.2020.05.007
37. Whyte CS, Morrow GB, Mitchell JL, Chowdary P, Mutch NJ. Fibrinolytic abnormalities in acute respiratory distress syndrome (ARDS) and versatility of thrombolytic drugs to treat COVID-19. J Thrombosis Haemostasis. 2020;18:1548–1555. doi:10.1111/jth.14872
38. Ji HL, Zhao R, Matalon S, Matthay MA. Elevated plasmin (ogen) as a common risk factor for COVID-19 Susceptibility. Physiological Reviews. 2020;100(3):1065–1075.
39. Thierry AR. Anti-protease treatments targeting plasmin (ogen) and neutrophil elastase may be beneficial in fighting COVID-19. Physiol Rev. 2020;100(4):1597–1598. doi:10.1152/physrev.00019.2020
40. O’Neill LA, Bowie AG. The family of five: TIR-domain-containing adaptors in Toll-like receptor signaling. Nat Rev Immunol. 2007;7(5):353–364. doi:10.1038/nri2079
41. Schroder K, Tschopp J. The inflammasomes. Cell. 2010;140(6):821–832. doi:10.1016/j.cell.2010.01.040
42. Merad M, Martin JC. Pathological inflammation in patients with COVID-19: a key role for monocytes and macrophages. Nat Rev Immunol. 2020;1–8.
43. Dalpke AH, Helm M. RNA mediated Toll-like receptor stimulation in health and disease. RNA Biol. 2012;9(6):828–842. doi:10.4161/rna.20206
44. Roschewski M, Lionakis MS, Sharman JP, et al. Inhibition of Bruton tyrosine kinase in patients with severe COVID-19. Science Immunol. 2020;5:48. doi:10.1126/sciimmunol.abd0110
45. Choudhury A, Mukherjee S. In silico studies on the comparative characterization of the interactions of SARS-CoV-2 spike glycoprotein with ACE-2 receptor homologs and human TLRs. J Med Virol. 2020;92:2105–2113. doi:10.1002/jmv.25987
46. Cavassani KA, Ishii M, Wen H, et al. TLR3 is an endogenous sensor of tissue necrosis during acute inflammatory events. J Exp Med. 2008;205(11):2609–2621. doi:10.1084/jem.20081370
47. Mazaleuskaya L, Veltrop R, Ikpeze N, Martin-Garcia J, Navas-Martin S. Protective role of Toll-like receptor 3-induced type I interferon in murine coronavirus infection of macrophages. Viruses. 2012;4(5):901–923. doi:10.3390/v4050901
48. Totura AL, Whitmore A, Agnihothram S, et al. Toll-like receptor 3 signaling via TRIF contributes to a protective innate immune response to severe acute respiratory syndrome coronavirus infection. MBio. 2015;6:3. doi:10.1128/mBio.00638-15
49. Wang JP, Kurt P, Jones EA, Finberg RW. Innate immunity to respiratory viruses. Cell Microbiol. 2007;9(7):1641–1646. doi:10.1111/j.1462-5822.2007.00961.x
50. Shibamiya A, Hersemeyer K, Schmidt Wöll T, et al. A key role for Toll-like receptor-3 in disrupting the hemostasis balance on endothelial cells. Blood J Am Society Hematol. 2009;113(3):714–722.
51. Zhu N, Zhang D, Wang W, et al. A novel coronavirus from patients with pneumonia in China, 2019. New England J Med. 2020;382:727–733. doi:10.1056/NEJMoa2001017
52. Biswas I, Singh B, Sharma M, Agrawala PK, Khan GA. Extracellular RNA facilitates hypoxia-induced leukocyte adhesion and infiltration in the lung through TLR3-IFN-γ-STAT1 signaling pathway. Eur J Immunol. 2015;45(11):3158–3173. doi:10.1002/eji.201545597
53. Biswas I, Garg I, Singh B, Khan GA. A key role of toll-like receptor 3 in tissue factor activation through extracellular signal regulated kinase ½ pathway in a murine hypoxia model. Blood Cells Mol Dis. 2012;49(2):92–101. doi:10.1016/j.bcmd.2012.05.001
54. Bhagat S, Biswas I, Ahmed R, Khan GA. Hypoxia induced up-regulation of tissue factor is mediated through extracellular RNA activated Toll-like receptor 3-activated protein 1 signaling. Blood Cells Mol Dis. 2020;102459.
55. Kannemeier C, Shibamiya A, Nakazawa F, et al. Extracellular RNA constitutes a natural procoagulant cofactor in blood coagulation. Proc Nat Acad Sci. 2007;104(15):6388–6393. doi:10.1073/pnas.0608647104
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.