")
Back to Journals » International Journal of Nanomedicine » Volume 19
Co-Delivery of Aceclofenac and Methotrexate Nanoparticles Presents an Effective Treatment for Rheumatoid Arthritis
Authors Negi S, Tandel N, Garg NK, Sharma P, Kumar R, Sharma P, Kumar R , Saini S, Sharma A, Tyagi RK
Received 3 October 2023
Accepted for publication 1 February 2024
Published 5 March 2024 Volume 2024:19 Pages 2149—2177
DOI https://doi.org/10.2147/IJN.S439359
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Yan Shen
Sushmita Negi,1,2,* Nikunj Tandel,3,* Neeraj K Garg,4 Prakriti Sharma,1 Rajinder Kumar,1 Praveen Sharma,1 Reetesh Kumar,5 Sheetal Saini,1 Aman Sharma,6 Rajeev K Tyagi1,2
1Biomedical Parasitology and Translational-Immunology Lab, Division of Cell Biology and Immunology, CSIR-Institute of Microbial Technology (IMTECH), Chandigarh, 160036, India; 2Academy of Scientific and Innovation Research (Acsir), Ghaziabad, 201002, India; 3Institute of Science, Nirma University, Ahmedabad, Gujarat, India; 4University Institute of Pharmaceutical Sciences, Panjab University, Chandigarh, 160014, India; 5Faculty of Agricultural Sciences, Institute of Applied Sciences and Humanities, GLA University, Mathura, Uttar Pradesh, India; 6Clinical Immunology and Rheumatology Wing, Department of Internal Medicine, Postgraduate Institute of Medical Education and Research, Chandigarh, 160012, India
*These authors contributed equally to this work
Correspondence: Rajeev K Tyagi, Biomedical Parasitology and Translational-immunology Lab, Division of Cell Biology and Immunology, CSIR-Institute of Microbial Technology (IMTECH), Sec-39a, Chandigarh, 160036, India, Tel +91-172-6665278; +91-172-6665279, Email [email protected]; [email protected]
Background: Rheumatoid arthritis (RA) is a common acute inflammatory autoimmune connective tissue arthropathy. The genetic studies, tissue analyses, experimental animal models, and clinical investigations have confirmed that stromal tissue damage and pathology driven by RA mounts the chronic inflammation and dysregulated immune events.
Methods: We developed methotrexate (MTX)-loaded lipid-polymer hybrid nanoparticles (MTX-LPHNPs) and aceclofenac (ACE)-loaded nanostructured lipid carriers (ACE-NLCs) for the efficient co-delivery of MTX and ACE via intravenous and transdermal routes, respectively. Bio-assays were performed using ex-vivo skin permeation and transport, macrophage model of inflammation (MMI) (LPS-stimulated THP-1 macrophages), Wistar rats with experimental RA (induction of arthritis with Complete Freund’s adjuvant; CFA and BCG), and programmed death of RA affected cells. In addition, gene transcription profiling and serum estimation of inflammatory, signaling, and cell death markers were performed on the blood samples collected from patients with RA.
Results: Higher permeation of ACE-NLCs/CE across skin layers confirming the greater “therapeutic index” of ACE. The systemic delivery of MTX-loaded LPHNPs via the parenteral (intravenous) route is shown to modulate the RA-induced inflammation and other immune events. The regulated immunological and signaling pathway(s) influence the immunological axis to program the death of inflamed cells in the MMI and the animals with the experimental RA. Our data suggested the CD40-mediated and Akt1 controlled cell death along with the inhibited autophagy in vitro. Moreover, the ex vivo gene transcription profiling in drug-treated PBMCs and serum analysis of immune/signalling markers confirmed the therapeutic role co-delivery of drug nanoparticles to treat RA. The animals with experimental RA receiving drug treatment were shown to regain the structure of paw bones and joints similar to the control and were comparable with the market formulations.
Conclusion: Our findings confirmed the use of co-delivery of drug nanoformulations as the “combination drug regimen” to treat RA.
Keywords: methotrexate, aceclofenac, lipid polymer hybrid nanoparticles, nanostructured lipid nanocarriers, rheumatoid arthritis, MMP-1
Introduction
Rheumatoid arthritis (RA), a heterogeneous systemic inflammatory autoimmune disease, is characterized by the synovitis, progressive bone damage, joint destruction, and early death.1,2 The Genetic analyses and investigations using in vivo models and patient studies have confirmed the RA induced inflammation mediated health complications. The disease-induced inflammation is characterized by the production of inflammatory cytokines.3,4 The aceclofenac (ACE), the non-steroidal anti-inflammatory drug, alleviates pain and suffering due to inflamed joints by blocking the enzymatic activity of cyclo-oxygenase-2 (COX-2) that inhibits the synthesis of prostaglandins.5 The RA-driven immune dysbiosis may be repaired by the combination of ACE and MTX. The low-dose methotrexate (MTX) is known to suppress the inflammation,6 with its cytotoxic and/or anti-proliferative effects. Whereas a higher dose of MTX leads to the irreversible damage,7 as well as lymphoblastic leukaemia.8 The suboptimal therapeutic efficacy of monotherapy with conventional synthetic disease-modifying anti-rheumatic drugs (csDMARDs)9,10 suggests the need for the combination drug therapy to manage and treat RA.11 Briefly, topical application of ACE12 reduces the inflammation, and intravenously injected methotrexate controls the adenosine-driven immune suppression to avoid the joint destruction13 during RA pathology.
Also, the long-term oral intake of ACE and MTX leaves adverse effects, including gastrointestinal ulcers and bleeding, mucosal ulceration, stomatitis, bone marrow suppression, drug-induced hepatic fibrosis and cirrhosis.14–16 Therefore, our group prepared ACE-NLCs,17 MTX-LPHNPs,18,19 formulations for the effective delivery of ACE and MTX, respectively, with the minimal exposure to the healthy tissues. Our nano-formulations exhibited higher loading and entrapment efficiency, the lowest-level drug leakage due to lipid polymorphism, improved biocompatibility, in vivo stability and better drug penetration and permeation across the skin-layers.20,21 Moreover, our previous findings with ACE and MTX when used as “monotherapy” for RA and breast cancer prompted us use ACE and MTX as a combination therapy co-delivered via the nano-formulations in Wistar rats with experimental RA.17,19,22,23 Encouraged by our previous findings, we delineated the distinct therapeutic potential of the co-delivery of ACE and MTX mediated by the nanostructured lipid nanocarriers (NLCs) to treat/manage inflammatory diseases.21 The matrix-metalloproteinases (MMP-1, MMP-8, and MMP-13) and collagenases reportedly shown to cause bone damage,24 and hence inhibitors of MMP-1 were used to avoid connective tissue damage during RA pathology.25,26
The LPS-stimulated human THP-1 macrophages (MMI) showed a reduction in inflammation upon receiving treatment with ACE and MTX individually and in combination. The monotherapy and combination drug therapy in Wistar rats with experimental RA modulated inflammation and repaired immune dysbiosis. Further, the stimulated macrophages treated with a combination of drugs exhibited cell death controlled by the PI3K-Akt signaling. The death of the stimulated cells was characterized by gene transcription profiling and protein expression of inflammatory and signaling mediators. Besides, the molecular docking of the proteins with the drugs/ligands (ACE and MTX) was shown based on their binding affinity.
Ex-vivo assays performed on the samples (PBMCs and serum isolated from blood) collected from the patients with RA. The quantitative PCR was carried out to determine the mRNA expression of the immune markers from blood and serum profiling of inflammatory and signaling markers when treated with the combination of ACE and MTX. Interestingly, the fold regulation (FR) of relative mRNA expression of inflammation and bone degradation (IL-1β, IL-8, IL-6, IL-10, TNF-α, iNOS-1, iNOS-2, COX-2, NF-kβ, and MMP-1), as well as signalling markers (CD40, Akt, NF-kB, and Bim) in the mono- and combination drug-treated and LPS stimulated macrophages was shown to establish the immune homeostasis by balancing the cell death and autophagy axis. The immune axis was tipped towards the death of inflamed cells programmed by the combination of ACE and MTX by repairing the immune dysbiosis. Further, the reactive oxygen species (ROS) and nitric oxide (NO) production correlates with the death of stimulated macrophages. Hence, we validated the in vitro and ex-vivo findings in the animal model of Wistar rats with experimental RA induced by the combination of complete Freund’s adjuvant (CFA) and BCG. The drug ACE and MTX nano-formulations administered via transdermal and intravenous routes, respectively, were shown to reduce RA-induced inflammation. In the end, the drug nanoparticles were able to manage and treat RA comparable to that with the existing market formulations. Overall, the combination of the ACE and MTX nanoparticles presents a better therapeutic option for treating/managing the RA. In the end, our drug nano-formulations could be a better “therapeutic” strategy to treat the RA.
Materials and Methods
Aceclofenac (ACE) and methotrexate (MTX) were received as gift samples from IPCA Laboratories, Mumbai. Glyceryl monostearate (GMS), Cetyl Palmitate (CP), stearic acid (SA), and cetyl alcohol (CA) were purchased from LobaChemie Pvt. Ltd. (Mumbai, India). Market gel (1.5%) for ACE was procured from a local pharmacy. The liquid lipids, Transcutol, Labrafac, Labrazol, and Gelucire 50/13, were supplied by Gateosse (Saint Priest, Cedex, France). Phospolipon-S100 was a gift from Lipoid GmbH (Germany). Capmul® MCM (C10) was obtained as a gift sample from ABITEC Janesville, USA. Poloxamer (Pluronic F-68) was a gift from BASF (Mumbai, India). Tween-80 was purchased from Fischer Scientific Pvt., Ltd. (India). The solvents used for HPLC were HPLC grade. All other chemicals and reagents were of analytical grade unless otherwise specified. MTX (Rheumatrex) and ACE gel (Hifenac) were purchased from a local drug house and injected through subcutaneous (s.c.) and transdermal (t.d.) routes, respectively. Phorbol 12-myristate 13-acetate (P8139), lipopolysaccharide from E. coli O111:B4 (L2630), Histopaque-1077 (10,771), and Griess reagent (G4410) were procured from Sigma-Aldrich, USA. The Annexin V-FITC Apoptosis Detection Kit (cat. No. 88–8005-74) was procured from Invitrogen (USA). An RNeasy Mini Kit (Cat. No 74104), and iScript cDNA kit, were procured from Bio-Rad, USA (Cat no: 1708891). Primary monoclonal antibodies included β-actin (D6A8), Akt1(C73H10), pAkt1 (S473), CD40 (D8W3N), FOXO1 (C29H4), Bim (C34C5), CAS3 (9662S), Beclin-1 (D40C5), iNOS (D6B6S), MMP-1 (E9S9N), NF-kB P65 (D14E12), P-NF-kB P65 (S536), IDO1 (D5J4E), COX-2(D5H5), TLR-4(14358S), LC3AB (4108S), p-mTOR (AP0115), ATG5 (A0203) rabbit mAb, and secondary antibodies, such as mouse anti-rabbit IgG-HRP (sc-2357), were obtained from Cell Signalling Technologies (USA) and Santa Cruz Biotechnology (USA). Hoechst nuclear-staining dye was purchased from Thermo Fisher Scientific (USA). The ECL reagent (WBLUF0100) was purchased from Millipore (USA), and femtoLUCENT PLUS-HRP was procured from G-Biosciences Sigma (USA). The dialysis bags were purchased from HiMedia (India). The ELISA kits for the detection of IFN-γ (Cat. BMS228), IL-17A (Cat. BMS2017), IL-10 (Cat no. 88–7106), TGF-β (Cat no. BMS2065), TNF-α (Cat. BMS223-4), IL-6 (Cat no. 88–7066), and MMP-1 (Cat no. EHMMP1) were purchased from Invitrogen (Carlsbad, CA, USA).
Preparation of ACE and MTX Nano-Formulations
ACE-loaded NLCs were prepared using the hot microemulsion method using a probe sonicator (Sonicator 3000, Misonix) and a high-shear homogenizer (Heidolph, Germany), as previously described.17
ACE and solid lipids (CA: Gelucire; 1:2) were mixed with an ethanolic solution of phospolipon and heated to 55–60°C, after which hot lipid oil-containing drugs were added. Subsequently, Tween 80 was added to the water and sonicated for 30 s at 3W to form a hot micro-emulsion. The emulsion was then transferred to 70 mL aqueous surfactant solution (0.5% poloxamer) using a micro-syringe with continuous homogenization at 8000 rpm for 10 min, followed by magnetic stirring for 2–3 h at 500 rpm. NLC dispersions were dialyzed using a cellulose dialysis bag (MWCO, 10 kDa) against a dialyzing medium with a double distilled water and acetone mixture (2:1) to remove the unentrapped drug. The suspensions were lyophilized and stored for long-term use.
MTX-LPHNPs were prepared using a modified single-step nanoprecipitation method following our published protocol.27 Briefly, MTX and polycaprolactone (PCL) were dissolved in 3 mL DMF and heated at 60–70°C. Phospholipid mixture (20% PCL), phospholipid 90G, cholesterol, and DSPE-PEG-2000 (10% molar ratio of PL) 60:15:12 mass ratio was dissolved in 2 mL DCM: DMF (1:1) and heated at 60–70°C. Both solutions were mixed and stirred continuously for 10 min, and the prepared solution was added drop by drop to a cold surfactant solution of Lutrol® F-87 (0.5%w/v) at a constant flow rate of 1 mL/min and stirred using a magnetic stirrer (Remi, Mumbai, India) at 800 rpm for 2–3 h. The resulting LPHN suspension was collected by centrifugation at 20,000 rpm for 10 min and washed twice with distilled water to remove organic solvent. The optimized NPs were lyophilized (Vir Tis, Wizard 2.0, New York, USA) using a stepwise freeze-drying cycle,28 followed by the application of a condenser temperature (−60°C) at 200Torr at each step of the cycle. Mannitol (5% w/v) was used as the cryo-protectant for storage.
Characterization of MTX Loaded LPHNPs and NLCs Encapsulated ACE
Size, Polydispersity Index (PDI) and Zeta Potential
The size and PDI of LPHNP–NLCs and the zeta potential of LPHNPs and NLCs were determined in folded capillary cells by Laser Doppler Anemometry (LDA) using a Malvern Zetasizer (PCS, Nano ZS90 Zetasizer, Malvern Instruments Corp., UK).
Transmission Electron Microscopy (TEM)
The surface morphologies of LPHNPs and NLCs were assessed using TEM. One drop of diluted sample was placed onto a membrane-coated grid surface and stained with 1% phosphotungstic acid, which was immediately added to the grid surface and excess fluid removed. The grid was then air-dried at room temperature, and the prepared sample was examined using HR-TEM (TECNAI 200 kV TEM, Fei, Electron Optics) at 10×15,000 magnification.
Percentage Drug Encapsulation and Loading Efficiency
Drug encapsulation efficiency was calculated using the direct lysis method, and the measured quantities of NLC and LPHNP suspensions were lysed in chloroform/DMF by brief sonication. The lysed sample was diluted with methanol and filtered through a 0.22 µM filter, followed by HPLC analysis to quantitate the loaded drug. Drug entrapment efficiency was calculated as follows:
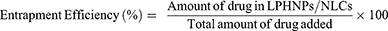
Drug loading was calculated by taking the fixed amount of lyophilized drug-loaded samples and lysing them in chloroform/DMF, and the quantity of drug was calculated by HPLC as follows:
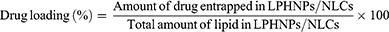
The drug content was determined using an HPLC system (Shimadzu, Japan) equipped with a double reciprocating pump (ThermoScientificTMHypersil BDS C18 Columns) (particle size 5 µm; length 250 nm X 4.6 mm) (250 mm×4.6 mm, 5µ), as reported by our group.29
X-Ray Diffractometry (X-RD) Analysis
The crystalline state of the drug before and after nanoparticle formulation was determined by X-RD. The X-ray diffraction (XRD) patterns of the nanoparticles were recorded on an X’Pert PRO-PANalytical instrument (the Netherlands). Free drugs, empty LPHNPs and NLCs, lipid mixtures of respective LPHNPs and NLCs, ACE-NLCs, and MTX-LPHNPs were analyzed. A known amount of each sample (10–15 mg) was loaded into a 25 mm poly-methyl methacrylate (PMMA) holder, and the diffracto-grams were analyzed using diffraction software (XPERT High Score software).
In vitro Drug Release
The dialysis bag method (MWCO; 12 kDa) was used to study the in vitro drug release profile of formulations with a slight modification of our published protocol.27,30 This method was used to study the in vitro drug release profile of formulations.27,30 In vitro drug release studies were carried out in phosphate buffer saline (pH= 7.4). Briefly, 2 mL of suspension in distilled water was placed in a dialysis bag, tied at both ends, and immersed in 20 mL drug release medium (physiological PBS), followed by continuous stirring at 100 rpm. The entire system was maintained at 37±1°C throughout the experiment. A 0.5 mL sample was withdrawn, and the same volume was replaced with PBS at predetermined time intervals. The collected samples were quantified using HPLC to calculate the quantity of drug present in the samples.
Preparation and Characterization of Aceclofenac-Loaded Hydrogel, Rheology and Texture Behavior
Hydrogels were prepared to obtain adequate viscosity for transdermal applications. ACE-NLC dispersions and chemical enhancer (PEG-200 in 5% ethanol) at a ratio of 70:30 were individually incorporated into previously hydrated Carbopol® 940 gel, neutralized with tri-ethanolamine.31
An NLC-incorporated gel was prepared to achieve 1.5% of total formulation concentration and assessed with respect to its color, grittiness, aesthetic appeal, rheological behavior, and texture analysis. Rheology and texture profile analysis (TPA) of formulations were carried out using Dynamic Rheometer (Anton Paar, MCR-102; Austria) and TA-XTPlus texture analyzer (Stable Microsystems, UK), as previously reported.31,32 Rheology and texture profile analysis (TPA) of the prepared formulations were carried out using a Dynamic Rheometer (Anton Paar, MCR-102; Austria) and a TA-XTPlus texture analyzer (Stable Microsystems, UK), according to the published protocols.31,33 The linear visco-elastic region (LVR) was determined through amplitude sweep tests by measuring G′ (storage modulus) and G″ (loss modulus) as a function of strain (%) ranging from 0.01% to 100% at a constant angular frequency of 1, 10 rad/s LVR, which provides information about the minimum strain needed for oscillation frequency sweep test. The oscillation frequency sweep test was carried out by measuring G′ and G″ as a function of angular frequency (rad/s) ranging from 0.1 to 100 rad/s at constant strain amplitude of 0.05% and 5% in the linear visco-elastic region. This test was performed to monitor the behavior and stability of the sample at a constant strain with varying frequencies.
The prepared NLC gel was examined for TPA prior to the testing, the cone was calibrated (upper: male vs lower: female) and set experiment,17 and the recommended volume of NLC gel was placed over the lower stage of the equipment to note down the readings of the samples for analysis. The texture profile was assessed using a male cone to penetrate and detach from the test formulation present in the female cone.
Cell Uptake of Prepared Gel Formulations by Human Hyper-Proliferative Keratinocyte Cells (HaCaT)
The potency of skin permeation of NLCs and gel-(ACE-NLCs), with and without chemical enhancer (CE) conjugation, was determined by the cell uptake efficiency of HaCaT cells (procured from NCCS, Pune, India). Cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum (FBS), L-glutamine, non-essential amino acids, sodium bicarbonate, sodium pyruvate, 100 U/mL penicillin, and 100 μg/mL streptomycin (PAA Laboratories GmbH, Austria) at 37°C under 5% CO2, and fluorescent NLCs were prepared by co-encapsulation of coumarin-6 (C-6) with ACE (equivalent to 1 μg/mL free C-6). C-6 dye was added in the organic phase and NLCs were prepared (Section 2.2.1); after 24 h, 50,000 cells were seeded in 6-well culture plates (Costars, Corning Inc., NY, USA) overnight for the adherence of cells. Cells were incubated with coumarin-6-ACE-NPs for 6hr, washed with HBSS (5X) to remove extracellular particles, fixed with 3% paraformaldehyde (Merck, India), and permeabilized with 0.2% Triton X-100 (Sigma-Aldrich, USA). The fixed and permeabilized cells were observed under a confocal laser scanning microscope (CLSM) (Olympus FV1000, Japan), and photomicrographs were obtained.
Cell Viability (MTT) Assay with HaCaT Cells
10,000 HaCaT cells reconstituted in 200 μL of medium were seeded in 96-well tissue culture plates and incubated overnight for cell attachment. The growth medium was replaced with an equal volume of complete medium containing ACE and ACE formulations (100 μg/mL; equivalent to free ACE) and placebo NLCs for 72 h. 150 μL MTT solution (0.5 mg/mL reconstituted in PBS) was added to the washed cells in each well, which were then incubated for 3–4 h to facilitate the formation of formazan crystals. MTT formazan crystals were dissolved in 200 μL DMSO, and the plates were read at 550 nm using an ELISA plate reader (BioTek, USA).
Ex-Vivo Permeation and Dermatokinetic Modeling with the Skin of Pig’s Ear Pinnae
Ex vivo permeation studies of the vestigial part of the pig ear pinna (procured from the government’s slaughterhouse, industrial phase, Mohali, Punjab, India) were conducted without heat treatment. The prepared skin samples were examined microscopically for surface irregularities. The skin was clamped on a vertical Franz diffusion cell with a surface area of 3.14 cm2 so that the stratum corneum side faced upward in the donor compartment and the dermal side faced downwards in the receptor compartment. Market gel-(ACE-MKT), gel-(ACE-NLCs), and gel-(ACE-NLCs+CE) formulations (equivalent to 5 mg/cell, i.e.~1.6 mg/cm2) were gently applied to the donor compartment. Aceclofenac-loaded NLC formulations co-encapsulated with coumarin-6 (C-6) dye were applied to the skin at the donor compartment of the Franz diffusion assembly following our published protocol.34 All samples were filtered through a 0.22 µm cellulose membrane filter, and the drug permeated through the skin was quantified. Raw data obtained from in vitro diffusion drug release studies were analyzed by applying correction factors for volume and drug losses using their placement method, and various dermato-kinetic modeling study parameters were studied.35,36 The skin was removed from the Franz cells at different sampling times, and the skin tissue was washed three times with deionized water and allowed to dry. The epidermal and dermal layers were manually separated using tweezers, and the skin layers were chopped into pieces and macerated with a homogenizer in 5 mL methanol for 8–10 h to obtain a complete drug extract in ethanol.
Obtained data were fitted into a one-compartment open model following the equation
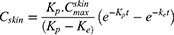
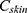 is the concentration of the drug in the skin at time t, Kp is the dermal permeation constant,
is the concentration of the drug in the skin at time t, Kp is the dermal permeation constant, 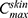 is the maximum concentration achieved in the skin, and Ke is the skin elimination constant. Win-Nonlin software (version 5.0) was used to compute various dermatokinetic parameters: Kp,
is the maximum concentration achieved in the skin, and Ke is the skin elimination constant. Win-Nonlin software (version 5.0) was used to compute various dermatokinetic parameters: Kp, 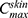 , Ke
, Ke 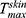 (time required to achieve
(time required to achieve 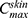 ), and area under the curve (AUC0−24hrs) using the Wagner–Nelson method.
), and area under the curve (AUC0−24hrs) using the Wagner–Nelson method.
Assessment of Cell Toxicity with LPS-Stimulated Human U937 Cells
The percentage viability of ACE-NLC- and MTX-LPHNP-based formulations was determined for human monocytic cells (U-937). U-937 cells (purchased from the National Center for Cell Science, Pune, India) were maintained in RPMI-1640 medium supplemented with 10% heat-inactivated fetal-bovine serum and 50 μg/mL gentamycin (Invitrogen, USA). U937 cells were differentiated into human macrophages by PMA (100nM; 100ng/mL) treatment in 12-well tissue culture plates (Nunclon) for 24 h and stimulated with 1 µg/mL and 5 µg/mL LPS (Sigma) for 24 h. After incubation, the media was aspirated and 1 mL 0.5M EDTA was added to each well; the cells were incubated for 15 min, and centrifuged at 1700 rpm for 10 min. Ten thousand cells were seeded in 96-well plates at a final volume of 100 µL/well. ACE- and MTX-based formulations in experimental control wells were used to achieve a total concentration of 100 μg/mL (equivalent to free ACE and MTX). PBS-washed cells were treated with 150 μL of MTT (0.5 mg/mL) reconstituted in PBS and incubated for 3–4 h to facilitate the formation of formazan crystals. MTT formazan crystals were dissolved in 200 μL DMSO and the plates were read at 550 nm using an ELISA plate reader.
Interaction of ACE and MTX with the Immune Receptors
The pharmacological compounds aceclofenac (C16H13Cl2NO4; PubChem ID:71771) and methotrexate (C20H22N8O5; PubChem ID:126941) were considered drug molecules against specific proteins for the treatment of RA. The 3D proteins used as macromolecules in the molecular docking analysis retrieved from Protein Data Bank/AlphaFold were AKT 1 (PDB:3O96_A), TNF-α (PDB:1TNF), Cox-2 (PDB:5F19_A), pAKT1 (PDB:3O96_A), Cas-3 (PDB:1GFW), MMP-1 (PDB:3SHI), Cas-8 (PDB:3KJQ), TGF-β (PDB:5VQP_A), IL-17 (PDB:4HR9_A), IFN-γ (PDB:1FG9_A), IL-1β (PDB:1I1B), CD40 (PDB:3QD6_R), IL-6 (PDB:1ALU), Bim (PDB:AF_O43521), IL-10 (PDB:1LK3_A), and NF-kB (PDB:1SVC_P). The pAKT1 protein was phosphorylated PDB:3O96_A, which was further energy minimized by the Chiron web server (www.dokhlab.med.psu.edu/Chiron).37 The molecular docking study was performed using AutoDockVina 1.1.38 For AutoDockVina 1.1, a grid box with a spacing of 1 Å and a size of 120 × 120 × 120 was built around the center of the binding site as predicted by the CB-dock server.39 Other parameters of docking were set to default, while exhaustiveness value was adjusted to 8 and energy range to 4. The 3D structure was further analyzed using Pymol software,40 whereas the 2D structure depicting the corresponding protein–ligand interaction was analyzed using Discovery Studio.41
In vitro Cell Culture
Human THP-1 cells (procured from ATCC, USA) were maintained in RPMI-1640 supplemented with heat inactivated fetal bovine serum (FBS 10% v/v), 1% penicillin–streptomycin, 0.05 mM, 2-mercaptoethanol. Cells were maintained in a CO2 incubator at 37°C in a humid atmosphere.
Cell Differentiation, Stimulation and Drug Treatment
THP-1 cells were differentiated into the macrophage phenotype by PMA (100 ng/mL) treatment in 12-well tissue culture plates for 24 h. Differentiated THP-1 macrophages were stimulated with 1µg/mL LPS, followed by treatment with ACE and MTX (100 µg/mL) individually as well as combination of ACE (50 µg/mL) and MTX (50 µg/mL) for 24 h.
Relative mRNA Expression by Quantitative RT-PCR (qRT-PCR)
THP-1 macrophages were stimulated with LPS (1 µg/mL) followed by the treatment with ACE and MTX (100 µg/mL) individually and in combination (ACE, 50 µg/mL; MTX, 50 µg/mL) for 24 h. After incubation, the media was aspirated and 1mL 0.5M EDTA was used to detach the cells, followed by 10–15 min incubation, and centrifugation at 1200 rpm for 10 min. The supernatant was discarded, the cells were washed twice with PBS, total RNA was isolated using an RNeasy Mini Kit (Qiagen, Cat. No. 74104) from LPS stimulated and unstimulated THP-1 macrophages, and quantified RNA was converted into cDNA for qRT-PCR using the iScript cDNA kit (Cat no: 1708891; Bio-Rad, USA). Briefly, 5 µL of 2X SYBER green (Bio-Rad), 0.5 µL forward and reverse primers, 2 µL of diluted cDNA, and nuclease-free water were reconstituted to make 10 µL reaction volume thermal conditions as follows: 95°C for activating the Taq Polymerase, and the remaining 35 cycles were performed at 95°C for 15 s; annealing was carried out at 58°C for 30 s, and extension at 72°C for 30 s. Relative mRNA expression was quantified using the changes in threshold method (ΔΔCT) and normalized to that of the housekeeping control. mRNA expression was quantified using the changes in threshold method (ΔΔCT) and normalized to the expression of the house-keeping ACTB mRNA (encoding β-actin and GAPDH).
Confocal Microscopy-Based Imaging to Detect the Expression of Bim
PMA differentiated and drug-treated/-untreated THP-1 macrophages were fixed with 4% paraformaldehyde (PFA) for 5 min followed by permeabilization with 0.1% Triton X-100 for 5 min. Cells were incubated with BSA (3%) for 1 h at room temperature to block non-specific antibody binding, followed by addition of primary antibody (anti-Bim antibody; 1:1000) or isotype control antibody (1:500 dilution) for 1 h at room temperature. The PBS-washed cells were incubated with the secondary antibody Alexa Fluor 568-conjugated goat anti-rabbit IgG (1:1000 dilution) for 1 h at room temperature. Cells were stained with the nuclear staining dye DAPI, mounted on cover slips with slow fade (Thermo Fisher Scientific, USA), and photographed using a NIKON A1R laser scanning confocal microscope.
Determination of ROS Production
THP-1 cells were treated and harvested, as described above. Cells were washed with 1X PBS buffer and stained with 1 µM CM-H2DCFDA dye (C6827, ThermoFisher Scientific) at 37°C for 15 min, and fluorescence was monitored using a flow cytometer. Ten thousand events were captured, and the data were analyzed using FlowJo software (FlowJo_v10.8.1).42
Measurement of Nitric Oxide (NO)
NO production was evaluated by measuring the nitrite, a stable metabolite of NO, content of the tissue homogenates, or culture media with the Griess reaction.43 Culture media were collected from LPS-treated THP-1 derived macrophage cultures. Duplicates of 50 μL of culture medium were added to 96-well microtiter plates and mixed with 50 μL of modified Griess reagent. The plate was then read on a microtiter plate reader using a 550 nm filter. A standard curve with increasing concentrations of sodium nitrite was generated in parallel and was used for quantitation.
Detection of Cell Death by Annexin V-FITC/Propidium Iodide
THP-1 cells were seeded in 6-well plates at a density of 1.0×106 cells/well. THP-1 cells were treated with ACE and MTX (100 µg/mL) individually or in combination with ACE (50 µg/mL) and MTX (50 µg/mL) for 24 h. Cells were harvested upon completion of incubation, and the percentage of apoptotic cells was analyzed using an Annexin V-FITC Apoptosis Detection kit according to the manufacturer’s recommendations. The cells were acquired using a C-6 Accuri flow cytometer (FlowJo software, BD Biosciences, Franklin Lakes, USA).
Immunoblotting
THP-1 cells were treated with drugs (ACE and MTX) as described above for 24 h, harvested, and washed with 1X-PBS. Protein lysates were prepared using RIPA lysis buffer and quantified using Bradford reagent. About 20 µg of protein sample was subjected to SDS-PAGE at 10% and 15%, which was then transferred to NCM (nitrocellulose membrane) and blocked with 5% BSA (TBST). The membrane was probed overnight with primary antibodies against NF-κB, AKT1, CD40, BIM, LC3-II, β-actin, Cas-3, Beclin, and ATG5 (1:1000 dilution). After overnight incubation, the membranes were washed thrice with TBST buffer and incubated with an HRP-conjugated secondary antibody (1:10000) for 1 h. The blots were washed thrice for 5 min each, followed by incubation with the secondary antibody. Blots were developed using ECL, and images were captured using SyngeneChemiDoc. β-Actin was used as the housekeeping gene.
Animal Studies
Female Wistar rats, aged 4–6 weeks old, were provided water ad libitum and maintained under pathogen-free conditions with convenient access to food and water. Animal protocols were approved by the Institutional Animal Ethical Committee (PU/IAEC/5/14/95) of Panjab University and the Institute of Microbial Technology (IAEC/43/2022/02), Chandigarh, India. We followed the guidelines laid down by the Animal Welfare Board of India (AWBI) and Committee for the Purpose of Control and Supervision of Experiments on Animals (CPCSEA) (Reg No. 55/GO/Re/Rc/Bi/Bt/S/99/CPCSEA) under the Ministry of Environment Forest and Climate Change (MoFE & CC). The experimental animals were euthanized by asphyxiation followed by the cervical dislocation.
Experimental Induction of RA in Wistar Rats
Rheumatoid arthritis was induced using CFA supplemented with Mycobacterium tuberculosis (Mtb) following the published protocols.44–46 1 mg Mtb (H37Ra, ATCC 25177) mixed in 1 mL CFA was heat killed, dried, and reconstituted in 0.85 mL paraffin oil and 0.15 mL mannide mono-oleate. Lyophilized Mtb (10, 20, or 40 mg) was dissolved in 1 mL CFA. Anesthetized female rats were intradermally injected with a 0.1 mL suspension in the right ankle joint. On days 7 and 14 post-sensitization, the rats were intradermally administered 0.1 mL CFA for boosting to stimulate the induction of RA, and the induced inflammation and nociception were studied before and after the induction of arthritis.
Quantification of Methotrexate in Plasma and Synovial Fluid of RA Animals
CFA-induced arthritis (CIA) rats were divided into four groups. The two groups were dedicated primarily to quantifying the drug in the plasma, and the remaining two in the synovial fluid. Experimental RA animals from groups I and III were subcutaneously injected with 0.6 mg/kg MTX, and groups II and IV animals received MTX-loaded LPHNP. Furthermore, an equivalent dose of MTX was administered intravenously, and blood sampling was performed for 7 days to perform pharmacokinetic quantification of MTX in the plasma. Blood samples from groups I and II were drawn at 24-h intervals in heparinized tubes, and an equivalent volume of normal saline was injected into the animal at each blood draw to make up the volume. MTX was quantified in plasma for 7 days (168 h). MTX was quantified in the synovial fluid of euthanized animals from groups III and IV at 12, 24, 48, 72, and 96 h. Synovial joints were flushed with 1 mL saline and the collected fluid was analyzed to quantify free and MTX-loaded LPHNPs.
Anti-Arthritis Activity of ACE and MTX Delivered Through the NLCs and LPHNPs
Anti-arthritis treatment was started on day 2 post-booster dose, and animals were categorized into eight groups: group 1, single injection of gel-(ACE-NLCs + CE) (t.d.); group 2, MTX-LPHNPs (i.v.); group 3, gel (ACE-MKT) (t.d.); and group 4, MTX-MKT (s.c.). Combination therapy groups were as follows: group5, gel-(ACE-NLCs) (t.d.) + MTX-LPHNP (i.v.); group 6, gel-(ACE-MKT) (t.d.) + MTX-MKT (s.c.); group 7, untreated control; and group 8, untouched control. Drug formulations were topically applied to paw and knee joints of experimental RA animals at 1.5% concentration of ACE once per day. ACE was injected through subcutaneous and intravenous routes, and corresponded to 10 mg/kg MTX once per week.
The mean arthritis score and changes in paw thickness were recorded for each animal at different time intervals, and the inflammation in each paw was measured. Arthritis index (AI) was calculated as follows:

Histopathological Analyses
Tissue sections of the paw and knee joints were prepared for the histological assessment of the ankle and interphalangeal joints. Extracted organs were fixed with 10% formalin, decalcified with formic acid, and embedded in the paraffin, and sections of paraffin-embedded tissues were prepared and stained with Hematoxylin and Eosin (H and E). Severity of arthritis was assessed and scored under blinded conditions with three parameters: extent of infiltration of mononuclear and polymorpho-nuclear cells, extent of hyperplasia of synovium, and pannus formation.46 Histopathological changes in the skin of CIA animals receiving combination drug regime treatment and sections of joints, bones of paw and ankles were presented in accordance with our published findings.17 Arthritis mean score was quantified by determining histological parameters: hyperkeratosis (thickening of stratum corneum), parakeratosis, spongiosis and exocytosis.
Human Ethics Permission
The present study was approved by the Institutional Ethics Committees of the CSIR Institute of Microbial Technology (IEC approval no: IEC (Dec 2021) #2) and the Postgraduate Institute of Medical Education and Research (PGIMER) (IEC-12/2021-2271), Chandigarh. Written consent was obtained from patients with RA prior to sampling and the patients provided informed consent, in accordance with the Declaration of Helsinki.
Inclusion and Exclusion Criteria
Twelve healthy individuals and 29 RA patients were recruited for this study. All patients included in the study fulfilled the 2010 European League Against Rheumatism (EULAR) classification criteria for RA. Patients included in the study were aged between 18 and 70 years and were on DMARD therapy. Patients with any severe, progressive, or uncontrolled cardiac, hepatic, renal, HIV, or any infections such as tuberculosis, malaria, hepatitis B or C, or cancer were excluded from the study.
PBMC Selection and Determination of Immune Markers by the qRT-PCR
About 10–12 mL whole blood was collected in EDTA tubes from patients and healthy individuals under aseptic conditions. PBMCs were isolated by density gradient centrifugation using Ficoll Histopaque-1077 (Sigma, USA) according to the manufacturer’s recommendations. Isolated PBMCs were used to extract total RNA, synthesize cDNA, and quantify mRNA expression, following the methods described above.
Serum Estimation of Immune Markers in the Experimental Animals and Patients with RA
Blood samples of 3–4 mL were collected from drug-treated Wistar rats with experimental RA, patients with RA, and healthy controls, and centrifuged at 2500 RPM for 10 min to separate the serum, and stored at −80°C. Serum samples were used for sandwich ELISA according to the manufacturer’s recommendations. Briefly, the standard solution and samples were added to antibody-coated wells and incubated for 2 h at room temperature, followed by washing three times with washing buffer (PBS and Tween-80). Biotin-conjugated secondary antibodies were added and incubated for 2 h at room temperature. After washing, streptavidin-horseradish peroxidase was added and incubated for 1 h at room temperature. Following this, TMB substrate was added and incubated for 10 min in the dark, and the reaction was stopped by the stop solution. The absorbance was read at 450 nm using an ELISA plate reader (BioTek, EPOCH/2 microplate reader). The concentrations of the immune markers were calculated using their respective standards.
Statistical Analysis
Data are shown as mean ± SD, and statistical analysis was carried out by one-way analysis of variance (ANOVA) with Tukey–Kramer multiple comparison post-test using the GraphPadInStat™ software (GraphPad Software Inc., San Diego, California). Statistical differences are represented as *p< 0.05, **p< 0.01, ***p< 0.001, and ns= non-significant (p> 0.05). The unpaired t-test (non-parametric test, Mann–Whitney test) was used to analyze the data presented for the inflammatory cytokines in THP-I macrophages, patient samples, and paw, synovial fluid, and serum of RA-induced rats.
Results
Characterization of Prepared Drug Nanoparticles
Based on the drug solubility in lipids (Cetyl alcohol-CA, Gelucire), surfactant (Tween), co-surfactant (Ethanol, IPA), and lipid oils (Transcutol P) (SI Tables 1 and 2), the NLCs were prepared for the efficient use of ACE through the subcutaneous administration. Tween-80 and PLS-100 were used as the surfactant and co-surfactant, respectively, during the preparation of NLCs (SI Table 1). Furthermore, the ratios of lipids, surfactants, and co-surfactants were optimized based on the microemulsion (ME) region. The ME region was defined by the pseudoternary-phase diagram drawn by titration assays wherein branched series with serial changes in the compositional analysis were studied (SI Figure 1A).17 The ME region with the maximum solid lipid mixture was chosen to formulate NLCs for achieving greater drug loading and optimized characterization parameters (particle size, poly-dispersity index; PDI; data not shown). In addition, NLC formulations with respect to particle size, drug entrapment, and loading efficiency were optimized using the quality by design (QbD) approach.21 The optimized NLC formulations prepared by the solid-lipid (cetylalcohol+Gelucire), surfactant (Tween 80+PL S 100+Ethanol), and liquid-lipid (Transcutol P) at 12%, 9%, and 3% (w/w) were conjugated with the chemical enhancers (CEs) incorporated in the hydrogel for an efficient transdermal delivery of ACE for its maximum therapeutic effects.
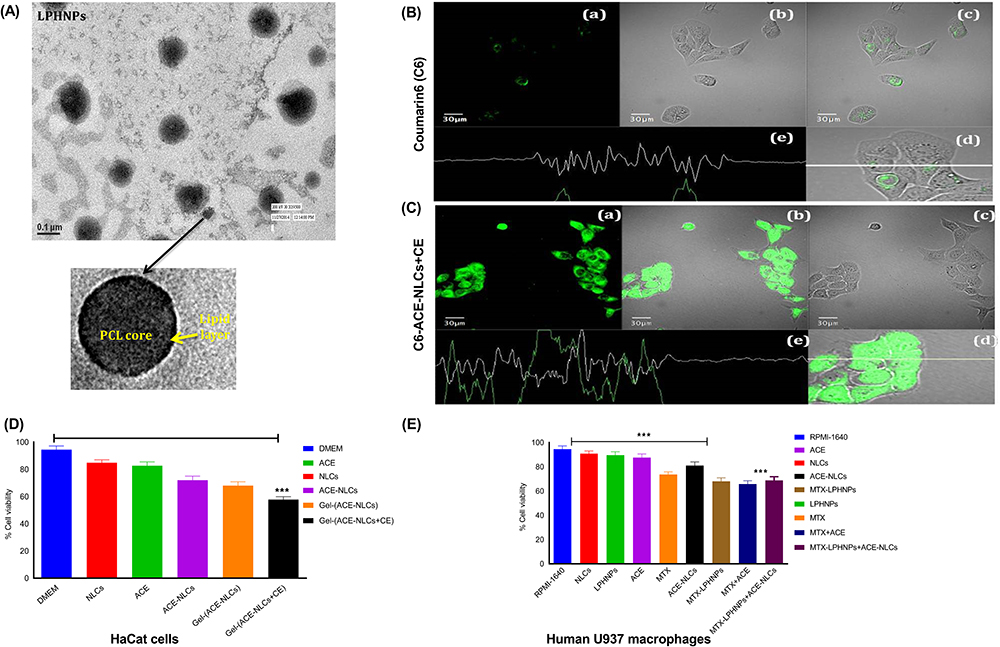 |
Figure 1 Characterization of methotrexate-loaded lipid-polymer hybrid nanoparticles (MTX-LPHNPs). Aceclofenac loaded nanostructured lipid carriers (ACE-NLCs) and MTX-LPHNPs treated Ha-Cat and U937 cells to confirm the cell uptake and viability. (A) High resolution-transmission electron microscopy (HR-TEM) of MTX-LPHNPs to characterize the spherical shape, qualitative cell uptake of (B) free Coumarin-6 (C-6) and (C) chemical enhancers (CE) conjugated NLCs (ACE-NLCs/CE) loaded C6 (1 µg/mL) was assessed with the HaCaT cells upon incubation for 3 h. The confocal fluorescence microscopy based analysis where each panel of this figure represents (a) images taken with green fluorescence channel; (b) Superimposition of figures. Figure (c) corresponding differential interface contrast (DIC) images of HaCaT cells; (d) and (e) show horizontal line series analysis of fluorescence along the white line (D) Cell viability of ACE-NLCs formulation assessed by the MTT assay in HaCat cells (E) Cell toxicity evaluated by the MTT based cell viability of MTX-LPHNPs, ACE-NLCs and combination of drugs (ACE & MTX) s well as their nanoformulations treatment in the human U937 cells. The statistical data is expressed as mean ± SE (n = 3). (***p<0.001 (highly significant). |
LPHNPs were prepared by the modified single-step method for PCL-mediated MTX encapsulation and coated with the lipid layer.27 The average size and PDI of MTX-LPHNPs and ACE-NLCs formulations were calculated as 168.5±8.5 and 189.87±7.2 and 0.11 and 0.19, respectively (SI Table 3). Furthermore, the increased size of the prepared MTX-LPHNPs and ACE-NLC formulations compared with the blank formulations suggested the higher drug loading (SI Table 3). The high-resolution transmission electron microscopy (HR-TEM) confirmed the intact spherical shape and surface morphology with the smooth edges in the nanometric size range (≤200 nm), with a narrow size distribution of the prepared MTX-LPHNPs (Figure 1) and ACE-NLCs formulations (Figure 2A). The negative zeta potential (−4.0 mV to −18.0 mV) induced repulsion prevents the formation of LPHNP aggregates. The particle size, PDI, and drug loading of the nano-formulations remained unchanged when stored at the different temperatures and relative humidity for over three months. This confirmed the stability of the prepared drug nanoparticles (SI Table 4). In addition, X-ray diffractometry (X-RD) analysis was carried-out to characterize the nanoformulations (SI information) to determine the impact of the encapsulation process on the state of prepared nanoparticles, ie crystalline/amorphous. X-RD profile of MTX showed characteristic crystalline peaks at 2θ positions of 8°, 9.2°, 11.8°, 13.0°, 14.8°, 19.8°, 22.5°, and 27° (SI Figure 1Ba). The blank and MTX-loaded LPHNPs showed similar characteristic lipid peaks at 18.4°and 23.53°, and 19°and 23.2°. Therefore, the absence of any characteristic crystalline peaks of MTX confirmed the efficient encapsulation of MTX into LPHNPs. Furthermore, the crystalline nature of aceclofenac was confirmed by the sharp peaks observed at 2θ = 8.8°, 14.5°, 18.5°, 19.5°, 22.5°, 24.5 and 26° (SI Figure 1Bb). The XRD profile of blank NLCs showed characteristic crystalline peaks at 2θ positions (6.15°,19.57°, 21.8°, 22.1°,23.29°, 23.6°, and 24.51°), similar to those with the ACE-NLCs. The similarity observed in the peaks could be attributed to the lipid material used to prepare the ACE-NLCs. X-RD profiling of ACE-NLCs showed the characteristic lipid peaks, whereas no crystalline peak was observed with the ACE-NLC formulation, and hence suggested the complete encapsulation of ACE.
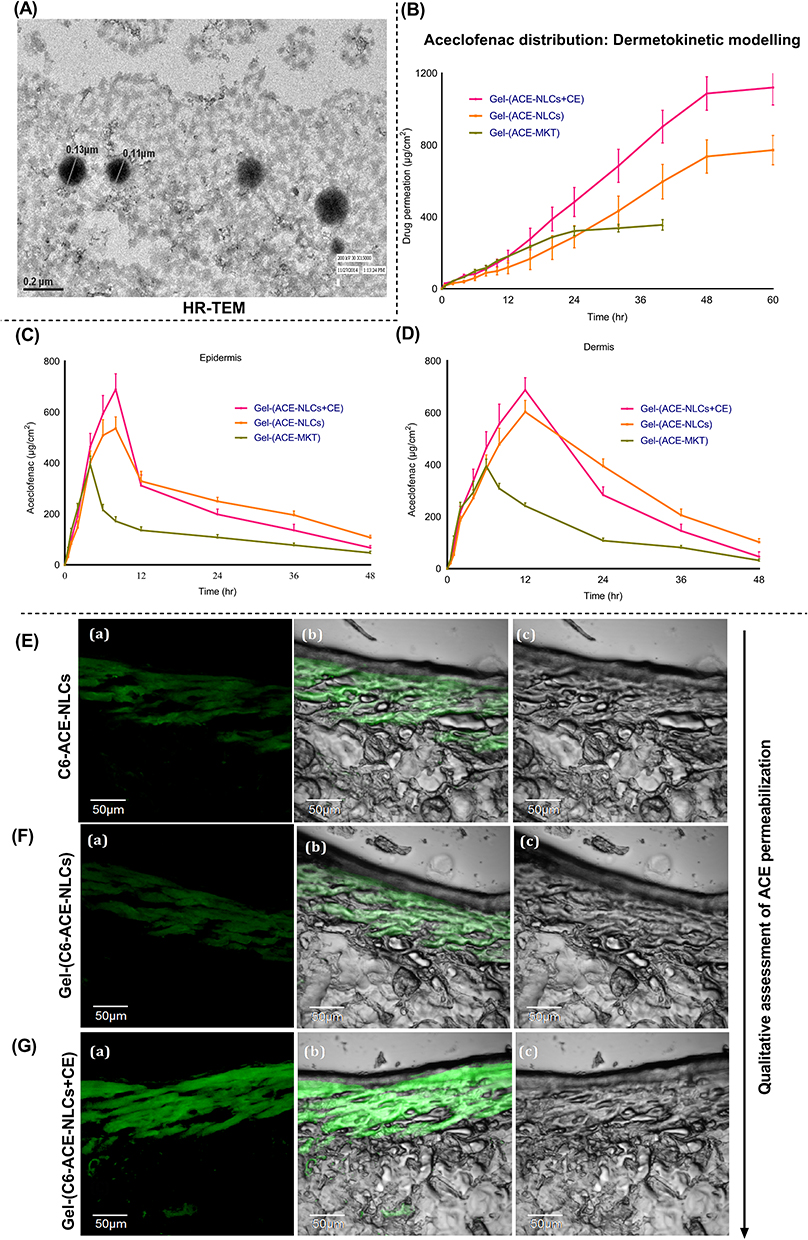 |
Figure 2 Confocal laser scanning microscopy (CLSM) based ex-vivo skin permeation of the ACE-NLCs formulation by dermatokinetic modeling. (A) HR-TEM analysis confirms the intact spherical shape and morphology of ACE-NLCs formulations (B) Ex-vivo quantitation of total aceclofenac permeated across skin of the ear pinnae of pig. ACE permeated across, (C) epidermis, and (D) dermis for 48 h of treatment with ACE-NLCs/CE formulations was quantified, CLSM images of skin permeation of (E) coumarin-6 (C6) labelled and ACE encapsulated NLC, coumarin-6 (C6) labelled and ACE encapsulated NLC-based hydrogel (F) without CE, and (G) with CE conjugation (Gel-(C6-ACE-NLCs/CE)). Individual figure shows, (a) Image under the green fluorescence channel (b) Superimposition of figures. (c) corresponds differential interface contrast (DIC) images. The statistical data is expressed as mean ± SE (n = 6). |
In vitro Drug Release
The cumulative in vitro release pattern of ACE and MTX by the NLC and LPHNP formulations exhibited rapid release (>60%) during the first 6 h, followed by a slow, biphasic and sustained release for up-to 48 h (SI Figure 2A) and 168 h (SI Figure 2B) when compared to the market formulations. The initial burst release of ACE and MTX could be explained by the absorbance of the drugs on the surface of the nano-carriers, leading to higher drug entrapment and significantly higher release of drugs. Furthermore, nearly 60% of MTX was released by the LPHNPs during the first 6 h, followed by sustained release for up-to 168 h (SI Figure 2B). The extended drug release demean our controlled by the drug diffusion was attested by the lipid and polymeric matrix of LPHNPs.27,47
Rheological and Texture Profile of ACE-Loaded NLC Gel
ACE-loaded NLC formulations were incorporated into Carbopol 940 using a chemical enhancer (PEG 200 dissolved in ethanol). Carbopols readily absorb water, swell, and exhibit different rheological properties, depending on their grade. The topical application of 1% carbopolsis essentially non-toxic and non-irritant materials hardly cause hypersensitivity in humans.21,48
Our earlier findings17 on the textural properties of gel-(ACE-NLCs+CE), including spreadability, gel strength, and extrusion force of the developed formulation, were satisfactory. As in other studies,49 we fitted the developed gel formulation (ACE-NLC) in the Herschel–Bulky model (SI Figure 3) and established a correlation between the decreased viscosity and increased shear stress in terms of the shear rate (0–100/sec with NLC-incorporated gel). The shear stress was directly proportional to the shear rate, and a sharp initial increase followed a slow but steady increase in the shear stress. Thus, NLC gel formulation exhibited pseudoplastic flow resulting from a colloidal network structure that deformed and adjusted in the direction of flow. Pseudoplastic flow and thixotropy were observed in the NLC gel because of their importance in transdermal applications. The results obtained from the amplitude LVR test showed a higher storage modulus (G’) than the loss modulus (G’). This conferred higher elasticity to the gel that dissipated less energy, and the loss modulus was higher for the viscous samples. The outcomes of the frequency sweep study helped determine the internal alterations in the gel structure, with no cross-over observed at the ambient temperature in the logarithmic graph. Further, the changes observed in the shear stress might alter their viscosity, stability, and structure (SI Figure 3).49,50
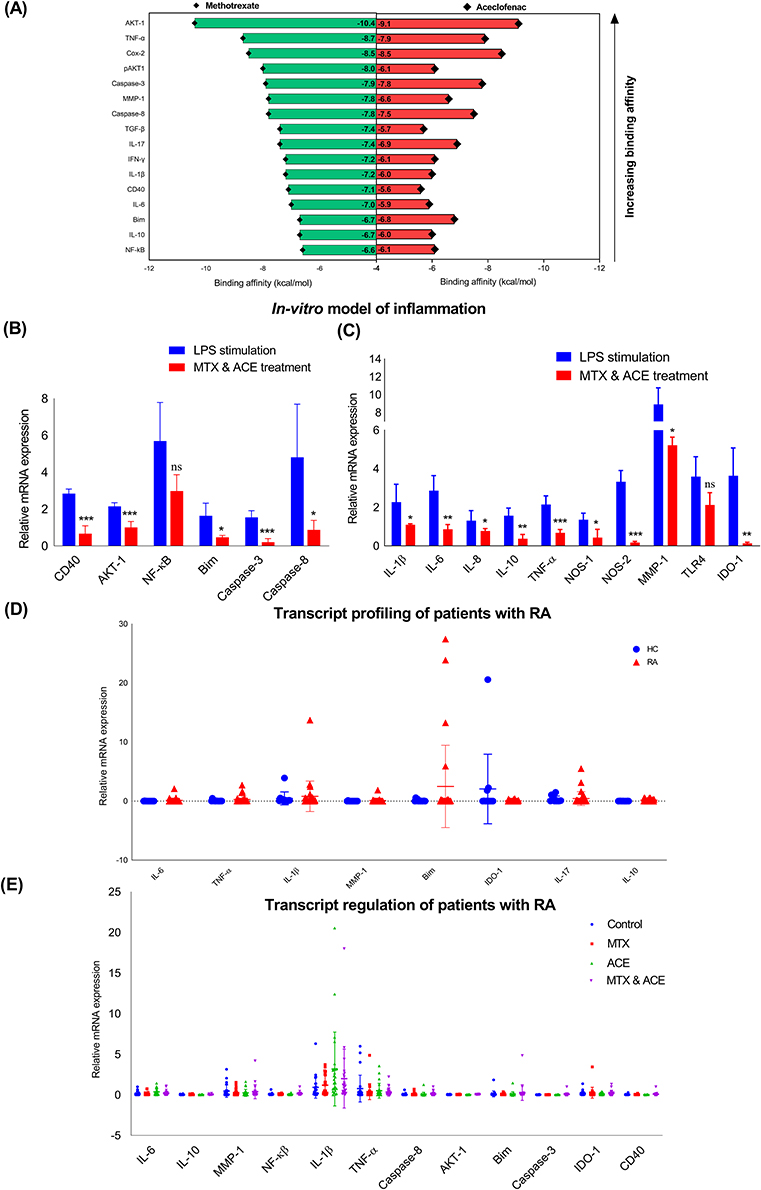 |
Figure 3 The assessment of the immune and signalling markers, (A) In silico molecular docking analyses based on the binding affinity of proteins (AKT-1, TNF-α, Cox-2, pAKT-1, Caspase-3, MMP-1, Caspase-8, TGF-β, IL-17, IFN-γ, IL-1 β, CD40, IL-6, Bim, IL-10, NF-kBp50) confirmed their interaction with ACE and MTX. The quantitation of the fold changes of the relative mRNA expression of (B) PI3K-Akt signalling (CD40, AKT-1, NF-kB, Bim, Caspase-3, 8) and (C) inflammatory and immunoregulatory markers (IL-1β, IL-6, IL-8, IL-10, TNF-α, NOS, MMP-1, TLR-4, IDO-1) in the stimulated macrophages receiving treatment with the ACE and MTX, (D) The transcript profiling of the inflammatory and immunoregulatory genes was carried out by the qRT-PCR on the PBMCs isolated from the patients with RA. PBMCs isolated from the healthy individuals served as healthy control (HC), (E) Gene transcription regulation (FR) of the inflammatory, immunoregulatory, signalling and apoptotic gene expression in the ACE (100 µg/mL), MTX (100 µg/mL) and combination of ACE (50 µg/mL) and MTX (50 µg/mL) treated PBMCs (1×106 PBMCs/mL) was determined (n=3, all experiments were performed in triplicates). (*p< 0.05 (significant), **p< 0.01 (moderately significant, ***p< 0.001 (highly significant). |
Cell Uptake and Biocompatibility of ACE-NLCs in the Human-Keratinocyte Epidermal (HaCaT) Cells
The in vitro internalization efficiency of NLCs and chemical enhancer (CE)-based gel formulations was assessed using HaCaT cells. The fluorescence emission of the hydrophobic fluorescent dye from the C-6-labelled ACE-NLC formulations was qualitatively determined (Figure 1B and C (a-e)).
The CE-conjugated ACE-NLC formulation suggested higher internalization to retain C-6as confirmed by extended (for 3h) emission of the fluorescence by the C-6 loaded nano-formulation taken up by the HaCaT cells when treated with C6-ACE-NLCs+CE. NLCs gained access to the cell surface with the minimal steric hindrance, and hence facilitated the entry of ACE-NLCs into HaCaT cells. The unmodified surfaces of NLCs exhibited greater endocytic internalization. The CE conjugation increased the ACE permeation in the C-6(ACE-(NLC+CE) formulation after incubation for 2–3 h in HaCaT cells (Figure 1B and C (a-e). We determined the cytotoxicity of ACE-NLCs and MTX-LPHNPs by the MTT cell viability assay in the HaCaT (Figure 1D) and human U937 cells (Figure 1E). ACE-NLCs+CE gel exhibited lower cell viability than the free and ACE-NLC formulations. Further, the completeness of the hydrogel formulation was determined with and without CE conjugation, and incorporation of NLCs into the hydrogel showed slightly higher cytotoxicity in HaCaT (Figure 1D) and U937 (Figure 1E) cells. The higher cytotoxicity seen in HaCaT cells was attributed to the irritant property of the CE-conjugated nanoparticles (Figure 1D).5 The vehicle control and aceclofenac-loaded NLCs showed no toxicity (p≤0.001), whereas ACE delivery via CE-conjugated NLCs exhibited significant cytotoxicity (Figure 1D). Also, the cytotoxicity of the nanoformulations in U937 cells was confirmed (Figure 1E). The combination of MTX-LPHNPs and ACE-NLCs saw a reduced cell viability, and higher cell viability was seen with the plain NLCs, LPHNPs, and ACE vehicle controls (RPMI-1640) (Figure 1E).
Ex-Vivo Skin Permeation of Aceclofenac Nanoparticles
The permeation parameters of our gel-loaded and CE-conjugated ACE-NLCs were investigated and compared with ACE market gel-(ACE-MKT) formulations (Table 1). Our data showed a significant (p<0.001) permeation of aceclofenac with gel-(ACE-NLC+CE) when compared to the gel-(ACE-NLC) without CE conjugation as well as the market gel-(ACE-MKT) formulations at 24, 48, and 60 h (Figure 2B, Table 1). Higher cumulative skin permeation of ACE using gel-(ACE-NLC) (772 µg) as compared to the lower (391 µg) permeation estimated with the market gel formulation (gel-(ACE-MKT)) 60 h following the treatment (Figure 2B, Table 1). The gel-(ACE-NLC+CE) conjugated with the CE exhibited 0.7 and three-fold permeation and was higher than that with the ACE-NLC-incorporated gel and market gel-(ACE-MKT) formulations (Table 1). A higher steady-state permeation flux (Jss) (µg/cm2/h) of ACE was estimated using the gel-(ACE-NLC+CE) (25.23). The latter was compared with the gel-(ACE-NLC) (17.09) and gel-(ACE-MKT) (12.82) formulations using the slope of the regression lines to fit the linear segment of the permeability profiles (Figure 2B, Table 1).51
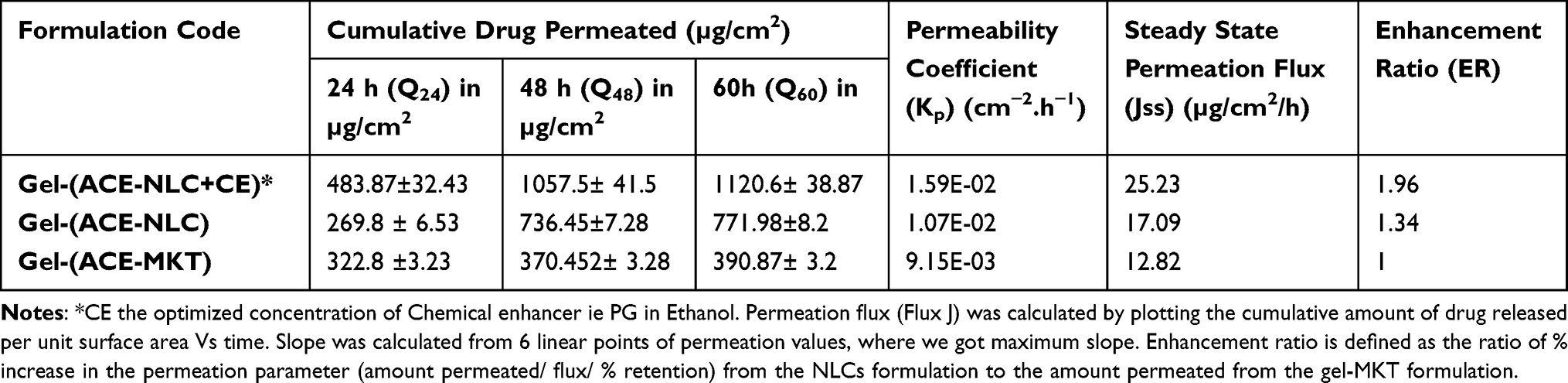 |
Table 1 Characteristic Skin Transport Parameters of ACE-Nanoparticles and the Market Formulation |
Dermatokinetic Modeling to Assess the Permeability of ACE-NLCs Formulation
Aceclofenac permeation across the skin layers was assessed by the ex-vivo assay performed with the pig’s ear pinnae (Figure 2B–D). The distribution of ACE in the epidermis (Figure 2C) and dermis (Figure 2D) was assessed by the one-compartment open body model (1-CBM) at the different time points using dermatokinetic modeling (Table 2). The arbitrary values for the dermatokinetic parameters (AUC0-48hrs, Ke, Kp) for gel-(ACE-NLs) and gel-(ACE-NLCs+CE) were higher (p<0.001) than the commercially available gel formulation. About 550–700 µg in the epidermis (Figure 2C) and 600–750 µg of aceclofenac in the dermis (Figure 2D) were seen deposited 6–8 h following treatment with the gel-(ACE-NLCs+CE) formulation (Figure 2C and D). The equilibrium of aceclofenac permeation was observed 9–10 h following the sustained delivery of ACE mediated by the NLCs (Figure 2C and D). The significant ACE deposition confirmed the penetration and permeation of ACE-NLCs across the skin layers in order to reach the dermal vasculature.
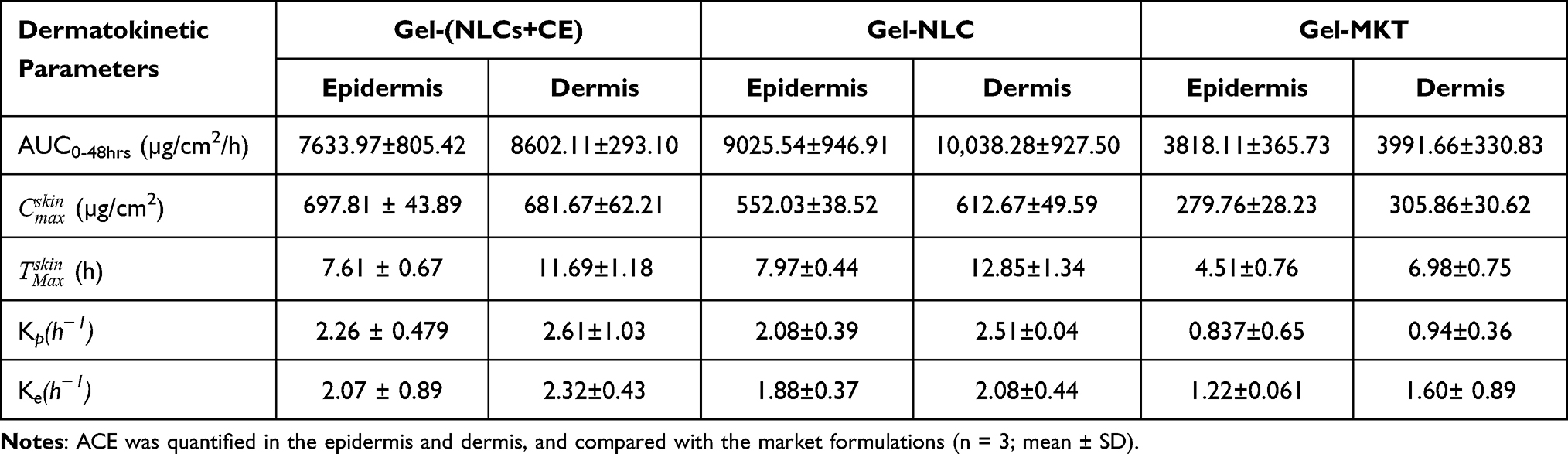 |
Table 2 Quantification of Aceclofenac Distribution Across Skin Layers Following the Topical Application of ACE-NLCs/CE Formulations |
Higher 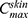 (µg/cm2) was observed with ACE-NLCs+CE and ACE-NLCs when compared with the market formulation (Table 2). The higher AUC0-48hrs and
(µg/cm2) was observed with ACE-NLCs+CE and ACE-NLCs when compared with the market formulation (Table 2). The higher AUC0-48hrs and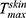 with the NLC gel formulation than that with the CE conjugated gel-(ACE-NLCs) formulation is due to the greater permeation of ACE-NLCs upon conjugation with the CEs. Therefore, the maximum ACE may have permeated the ACE-NLCs+CE gel formulation, leading to a higher AUC0-48hrs and
with the NLC gel formulation than that with the CE conjugated gel-(ACE-NLCs) formulation is due to the greater permeation of ACE-NLCs upon conjugation with the CEs. Therefore, the maximum ACE may have permeated the ACE-NLCs+CE gel formulation, leading to a higher AUC0-48hrs and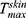 with the ACE-NLC gel formulation than the gel-(ACE-NLCs+CE) and market gel formulations. Our study design and composition of NLCs allowed higher transportation of ACE across the epidermis and dermis with NLCs than does with the existing commercially available gel formulation (Table 2). We believe that favorable interaction between NLCs, lipid/phospholipid-conjugated CE, and skin lipids may have synergized the interaction with ACE. It could lead to the transport of ACE mediated by the NLCs across skin layers and saw higher drug deposition at the diseased site.
with the ACE-NLC gel formulation than the gel-(ACE-NLCs+CE) and market gel formulations. Our study design and composition of NLCs allowed higher transportation of ACE across the epidermis and dermis with NLCs than does with the existing commercially available gel formulation (Table 2). We believe that favorable interaction between NLCs, lipid/phospholipid-conjugated CE, and skin lipids may have synergized the interaction with ACE. It could lead to the transport of ACE mediated by the NLCs across skin layers and saw higher drug deposition at the diseased site.
Next, we assessed the penetration/permeability of the prepared NLC formulations ex-vivo across the skin layers using CLSM (Figure 2E–G). Therefore, C-6 co-encapsulated NLCs were prepared and qualitatively determined to test the skin penetration ability of lipid nano-carriers. The CE-conjugated NLC-ACE showed higher skin uptake and greater permeation across the skin layers (Figure 2E–G). The CE conjugated and C-6 loaded gel-(ACE-NLCs) formulations showed better skin uptake (Figure 2G) compared to that seen with the C-6 loaded gel-(ACE-NLCs) formulation with no CE conjugation (Figure 2F). The bright fluorescence signals lasted longer with C-6 co-encapsulated gel-(C6-ACE-NLCs-CE) (Figure 2G) than that with gel-(C6-ACE-NLCs) (Figure 2F) and C6-ACE-NLCs (Figure 2E).
Signaling and Immune Mediators in the Macrophage Model of Inflammation
We next decided to determine the interactions of pro-inflammatory, apoptotic, bone degradation, and signaling protein markers to that with their ligands/drugs (ACE and MTX) by the molecular docking analysis (Figure 3A). Our docking analysis suggested an ascending order of binding affinity for the proteins that interact with ACE and MTX (Figure 3A). We saw the robust interaction between both ligands and Akt1, and the weakest interaction with IL-6 (Figure 3A). The detailed molecular docking analyses of drugs with immune and signaling markers have been illustrated in SI Figures 4–7.
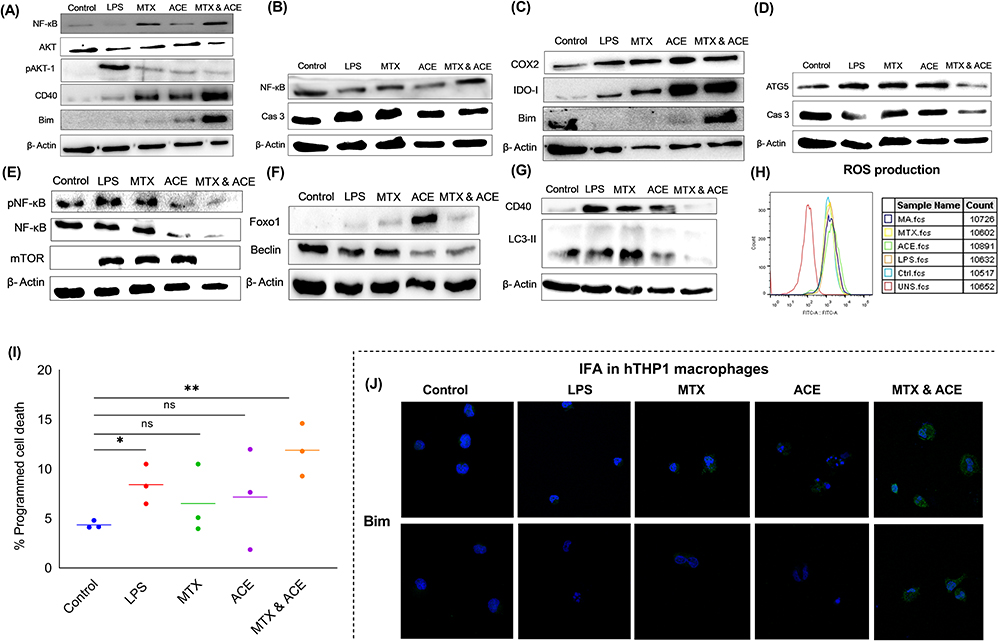 |
Figure 4 Protein expression of signalling/cell death (Bim, NF-kBp65, Akt-1, pAkt-1, CD40, Caspase-3), inflammatory (Cox-2), autophagy (Atg5, mTOR, Beclin, LC3II) and immunoregulatory (IDO-1) markers in the individual (ACE, MTX) and combination (ACE & MTX) drug treated macrophages stimulated with LPS (1 µg/mL) (A–G). The LPS (1 µg/mL for 24h) stimulated macrophages best mimicked the chronic inflammation induced by the RA. The drug treated cells were lysed and proteins were detected by the immunoblotting. β-actin served as a loading control. This data represents three independent experiments (n = 3), (H) the stimulated macrophages were treated with drugs to quantify the reactive oxygen species (ROS) production (I) The death of the stimulated macrophages was detected by the fluorescently labelled Annexin V staining kit. 7-aminoactinomycin (7-AAD) or propidium iodide (PI) stained cells were seen to identify the apoptotic stages by C6 accuri flow cytometer (n=3). (*p< 0.05 (significant), **p< 0.01 (moderately significant), (J) The confocal microscopy-based immunofluorescence assay (IFA) for the qualitative expression of Bim in the LPS stimulated macrophages (n=3, experiments were performed in triplicates). |
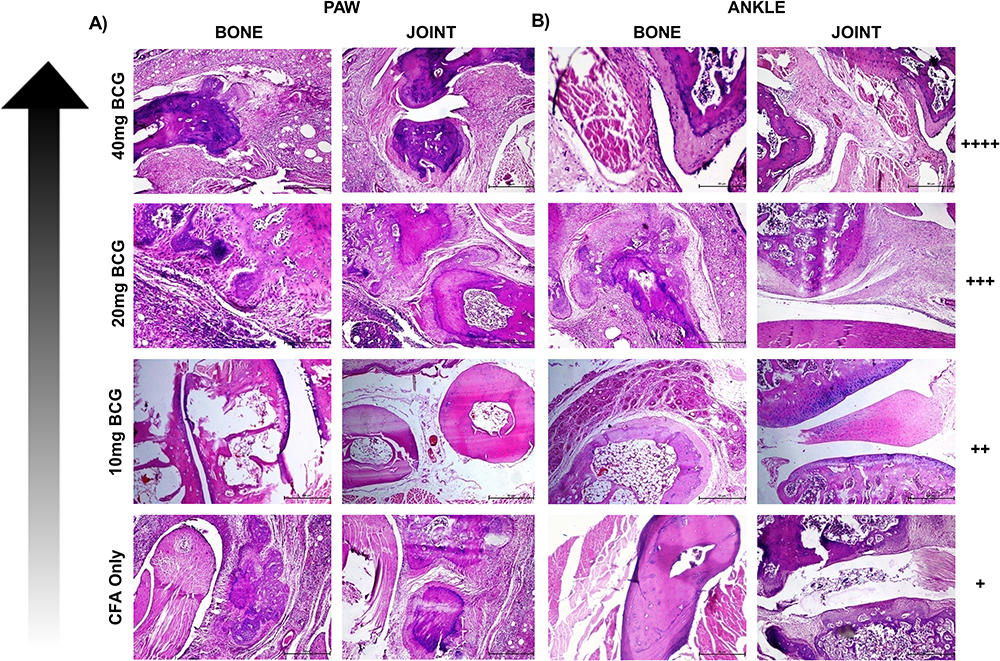 |
Figure 5 The induction of the experimental rheumatoid arthritis in, (A) paw and (B) ankle by the Complete Freund’s adjuvant (CFA) and Mycobacterium tuberculosis (Mtb; BCG) in the female rats. The color tonality (bottom to top) of arrow is in the order of the increasing severity of rheumatoid arthritis induced by the CFA and BCG 10mg (I), 20mg (II), 40mg (III). Top panels of (A and B) showing higher immune-cells infiltration is directly proportional to the severity of the induced RA in the paw and ankle. Each panel shows H &E stained images of bone and joints of paw and ankle. The degree of severity is presented as + equivocal; ++ mild; +++ moderate; and ++++ severe. (Scale bar: 50 μm). |
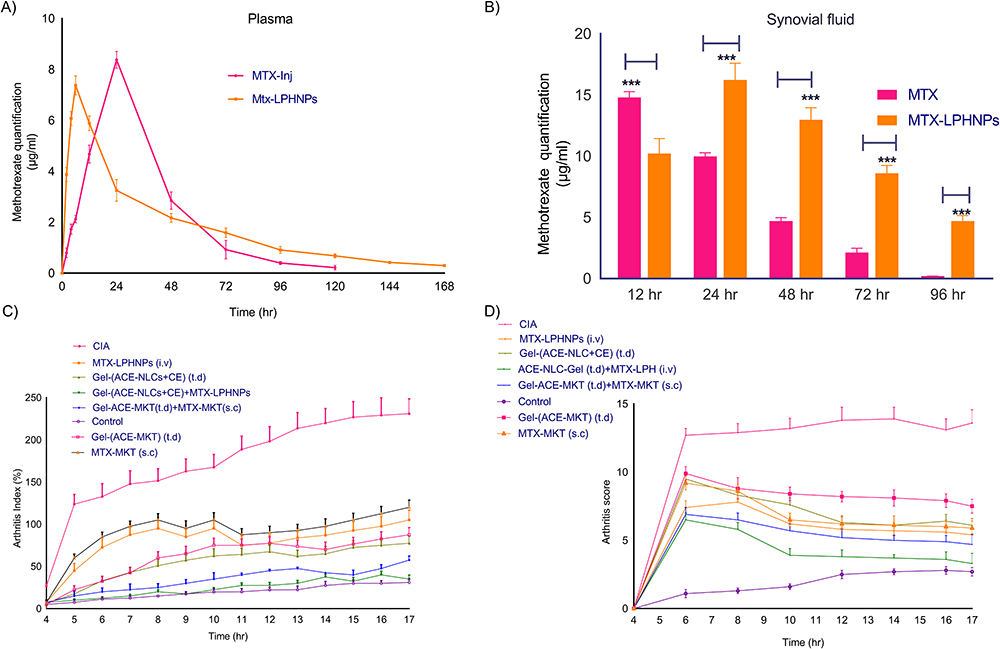 |
Figure 6 The bio-distribution of MTX (free and LPHNPs mediated delivery) was estimated in the systemic (plasma) and local (synovial fluid) tissues of CFA and BCG induced rheumatoid arthritis in rats. Assessment of severity of the experimental RA induced inflammation following the treatment with the mono- and combination drug therapy based on ACE and MTX. (A) plasma levels of MTX upon administered as “free” or mediated by LPHNPs, (B) synovial fluid levels of the MTX when administered as “free” or mediated by LPHNPs. Determination of the (C) arthritis index and, (D) arthritis score upon following the drug nanoparticles treatment and compared with the market formulations. The CFA induced arthritis (CIA) and untreated experimental animals served as positive and experimental/healthy controls. ACE was applied transdermally once a day and MTX administered weekly via the subcutaneous/intravenous route to mimic the existing market drug regimen. The healthy control animals treated with CFA and served as “untouched” control. The statistical data is expressed as mean ± SE (n = 6 animals/group). (***p<0.001 (highly significant). |
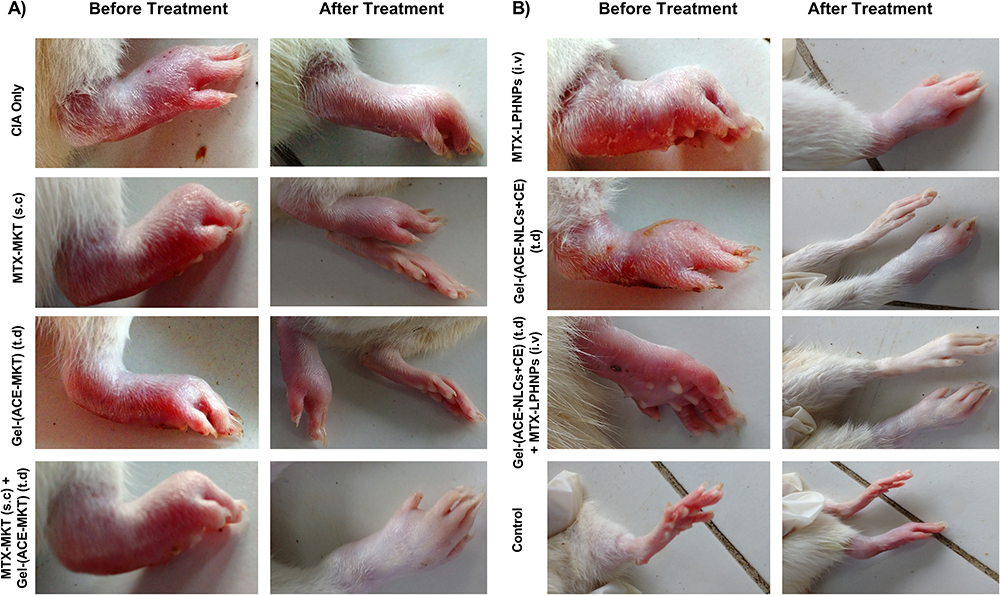 |
Figure 7 The therapeutic efficacy of “Mono” and “Combination” drug regimen in the rats with the experimental RA. The CIA served as a positive control and untouched controls were taken as the healthy/experimental control. (A) MTX-MKT (s.c.), Gel-(ACE-MKT) (t.d.) and combination of these formulations were administered to the experimental animals, (B) mono (MTX-LPHNPs (i.v.), Gel-(ACE-NLCs+CE) t.d.) and combination [MTX-LPHNPs (i.v.) and Gel-(ACE-NLCs+CE) t.d.)] drug therapy via the intravenous and transdermal route saw a significantly higher reduction of the inflammation induced by the RA. The experimental animals regained the paw structure similar to those with the untouched healthy control. |
We then determined the anti-inflammatory and signalling activities of ACE and MTX as well as combination of ACE and MTX treatment in LPS (1 µg/mL) stimulated macrophages (MMI) (Figure 3B and 3C, SI Figure 8A–8I). However, we sought to determine the gene transcription of pro-and anti-inflammatory, and signalling markers in the LPS stimulated macrophages with no drug treatment (SI Figure 9A). The LPS stimulation drove the higher expression of inflammatory immune markers compared to that seen with the unstimulated experimental control (SI Figure 9A). This data led us calculate the fold regulation (FR) of the relative mRNA expression of immune and signalling (PI3K-Akt) markers in the stimulated macrophages receiving treatment with ACE and MTX (SI Figure 8) as well as the combination of ACE and MTX (Figure 3B and C). Our data showed the inhibited mRNA expression of signaling (CD40, Akt1, NF-kB, Bim, Caspase-3, 8), pro-inflammatory (IL-1β, IL-6, TNF-α, TLR-4), apoptosis regulatory pathway (iNOS), and bone degradation (MMP-1) markers in the stimulated macrophages upon treated with the combination of drugs (Figure 3B and C). Our data showing the reduced expression of the metabolic enzyme (IDO-1) may imply the reduced production of IDO-competent inducible regulatory T cells (Tregs) RA (Figure 3C).
Ex-Vivo Gene Expression Profile of the Immune Markers from the Samples Collected on the Patients with RA
We analyzed the gene expression pattern of pro- and anti-inflammatory, bone degradation, apoptotic, and immunoregulatory markers (IL-6, IL-1β, TNF-α, IL-17, IL-10, MMP-1, Bim, and IDO-1) in the un-stimulated PBMCs selected from the blood collected on the patients with RA and the healthy controls (Figure 3D). Information regarding the primer sequences for all genes is provided in SI Table 5. We saw a higher expression of IL-1β, many-fold expression of Bim, and slightly higher expression of IL-17 in the PBMCs isolated from RA patients and healthy controls. There was no difference seen in the expression of the immunoregulatory metabolic enzyme (IDO-1) in the PBMCs isolated from patients with RA compared to that with the healthy controls (Figure 3D). Following this observation, we treated the PBMCs with ACE (100 µg/mL), MTX (100 µg/mL), and their combination (ACE and MTX, 50 µg/mL each) to quantitate the FR in the expression of relative mRNA of the immune/signaling markers (Figure 3E). Since the samples collected from patients already on drug therapy, we hardly saw the major changes in the FR of the individual and combination drug-treated PBMCs as compared to the untreated controls (Figure 3E). We quantify the slight difference in the transcript expression of IL-1β, MMP-1, TNF-α, Bim, and IDO-1, this difference did however not reach the statistical significance (Figure 3E).
Combination Drug Treatment Modulates the Inflammation to Establish the Immune Eubiosis
We sought to determine the protein expression of signaling and inflammatory markers in the in vitro model of inflammation (MMI) upon treated with the individual and combination of drugs (Figure 4A–G). The expression of Akt1, Bim, CD40, iNOS, TLR-4, NF-kβ, IDO1, and COX-2 was determined in the LPS stimulated macrophages without any drug treatment compared to the unstimulated control (SI Figure 9B). We then treated the inflamed macrophages with the individual treatment of ACE and MTX as well as the combination of ACE and MTX to determine the expression of immune-mediators and PI3K-Akt signaling markers (Figure 4A, B, C, E and F). The combination of ACE and MTX led to the Akt1 mediated death of stimulated macrophages driven by the induced CD40 expression (Figure 4A). Our findings suggest the death of antigen stimulated macrophages was controlled by the NF-β (Figure 4E) and dependent upon the non-phosphorylated/active FOXO1 (Figure 4F), which, in turn, promoted the expression of pro-apoptotic protein (Bim) in the nucleus (Figure 4A and C).52,53 Also, the caspase-3 independent death of antigen stimulated macrophages receiving treatment with the combination of drugs was seen (Figure 4B and D). The IDO-1 competent inducible regulatory T cells (iTregs) are crucial to maintain the auto-tolerance to be able to check RA progression.54–56 Our findings indicating the higher expression of immunoregulatory (IDO1) metabolic enzyme seen with the treatment of the combination of drugs suggested to establish the immune homeostasis (Figure 4C). The autophagy-like survival mechanisms may disrupt the auto-tolerance that could be detrimental to the death of the diseased cells. Hence, we investigated whether antigenic stimulation leads to the activation of the autophagy in the macrophages to withstand the drug pressure. The immunoblot analysis (Figure 4E–G) did not seen the expression of mTOR signaling (Figure 4E) in the macrophages that is associated with the synovitis and inflammatory arthritis. Further, the reduced expression of apoptosis inhibitors (survivin family, Beclin) (Figure 4F) and autophagy markers (LC3II) (Figure 4G) was seen in the antigenic stimulated and drug-treated macrophages. Collectively, our data suggest that the induced cell death and suppression of autophagy could have circumvented the therapeutic effects of ACE and MTX.
Estimation of the Reactive Oxygen Species (ROS) and Nitric Oxide (NO)
ROS production was determined in stimulated macrophages upon receiving treatment with the combination of ACE and MTX (Figure 4H). The right shift in the peak of stained, stimulated, and drug-treated macrophages compared to the unstimulated control suggests the increased ROS production. We opine that combination drug treatment augmented the ROS production that leads to the death of the stimulated cells (Figure 4H).
Further, the higher levels of NO (five-fold) suggest the greater expression of cartilage-destroying osteoclasts, leading to the recruitment of immune effecters. The immune effecters augment the RA-induced inflammation.57,58 Hence, the NO production was quantified in LPS (1 and 5 µg) stimulated THP-1 macrophages (SI Figure 9C) and consistent with the others,59–61 findings, we confirmed the higher production of NO.
Cell Death Programmed by the Combination Drug Treatment
ROS and NO production data led us confirm the role of the combination of ACE and MTX treatment in to induce the death of antigenic-stimulated macrophages (Figure 4I). The antigen stimulated macrophages are shown to enhance the death seen by the Annexin V staining following the drugs (mono- and combination) treatment. The increased death of stimulated macrophages was significantly higher (p<0.001) with the combined (ACE and MTX) treatment of drugs when compared with the individual drug (ACE, MTX) treatment (Figure 4I). Also, we confirmed the cell death by the immunofluorescence assay (IFA). The inactive transcription factor pFOXO1 (phosphorylated FOXO1) and pro-apoptotic protein (Bim) act in the tight regulation of CD40 mediated PI3K-Akt pathway.52,53 Therefore, we assessed the qualitative (Figure 4J) and quantitative (SI Figure 10) expression of Bim in the stimulated macrophages by the confocal microscopy-based IFA. The Bim expression was seen higher in the stimulated macrophages compared to those receiving treatment with the combination and individual drug treatment (Figure 4J, SI Figure 10).
Bio-Distribution of Methotrexate in Serum and Synovial Fluid
We developed the experimental RA in the Wistar rats by the treatment with CFA and BCG in order to validate the in vitro and ex-vivo findings. The severity of Complete Freund’s Adjuvant (CFA) induced rheumatoid arthritis was augmented by the Mtb (BCG) treatment. We observed that the severity of the disease was inhibited by the treatment with combination drug regimen. The chronic inflammation was seen mounted in the bones and joints of both paws and ankles of female rats following the administration with the 40 mg BCG along with CFA (Figure 5A and B). The 40 mg BCG was shown to best mimic the natural RA better than that seen with the 10 and 20 mg Mtb/BCG dosages (Figure 5A and B). Hence, 40 mg BCG was used to induce acute rheumatoid arthritis to carry out the animal assays.
The bio-distribution of MTX was determined in the plasma of experimental animals upon injection with the existing market formulation of MTX and prepared MTX-LPHNPs formulations via subcutaneous and intravenous routes of administration, respectively (Figure 6A). Maximum plasma concentration (Cmax) of the intravenously injected MTX mediated by LPHNPs was calculated 7.3 µg/mL 6 h post-injection. The maximum quantity of MTX was released by the LPHNPs in the plasma for 12 h and followed a slow release 168th h. In contrast, subcutaneously administered free MTX exhibited maximum concentration at 24 h, followed by a sustained release for up-to 120 h (Figure 6A). Additionally, we determined the availability of MTX (free or MTX-LPHNPs) in local tissues (synovial fluid) using HPLC and quantitated the maximum MTX release 12 h following the injection lasted for up-to 72 h (Figure 6B). A consistent decrease in the concentration of MKT-MTX was seen commencing from the 24 h post-injection lasting for up-to 96 h (Figure 6B). The LPHNPs mediated delivery of MTX showed the maximum deposition of MTX at the diseased site. The maximum MTX deposition was (16.22 µg/mL) after 24 h and then a sustained release for up-to 96 h (4.78 µg/mL MTX) (Figure 6B).
Therapeutic Efficacy of “Mono” and “Combination” Drug Regimen in the Rats with Experimental RA
The therapeutic efficacy of gel-(ACE-NLCs+CE) and MTX-LPHNP formulations as “mono” and “combination” drug regimens was tested in the female Wistar rats with the experimental RA. Mono- and combination therapy achieved their maximum therapeutic effect by transdermal application of ACE-NLCs and intravenous injection of the MTX-LPHNP formulation. Our prepared drug formulations were compared with the conventional aceclofenac gel applied transdermally, and commercially available formulations of MTX (MTX-MKT) administered through the subcutaneous route of administration (Figure 6C and D). The anti-arthritis activity of drugs was expressed as “arthritis index (AI)” (Figure 6C) and “arthritis score (AS)” (Figure 6D). Moreover, the reduced AI and AS were quantified with the combination drug therapy and exhibited a significantly reduced severity of RA (Figure 6C and D). The severely inflamed joints of experimental rats upon receiving treatment with the market MTX (s.c.), ACE-MKT gel (t.d.), and their combination (Figure 7A) showed a significant reduction in inflammation. However, this reduction in the inflammation could not restore the structure of paw and joints similar to the untouched control. Whereas, animals receiving treatment with our drug nanoformulations (gel-[ACE-NLCs+CE, MTX-LPHNPs]) to deliver ACE and MTX via transdermal and intravenous routes, respectively, as monotherapy and combination therapy not only showed a steep reduction in RA-induced chronic inflammation but also these drug nanoparticles restored the shape of the paw and joints, similar to the healthy/untouched control (Figure 7B).
Clinical Assessment of Disease Severity by Histopathology of Inflamed Tissues and Skin
The sections of bone and joint tissues of the paws (SI Figure 11A and B) and ankles (SI Figure 12A and B) of the Wistar rats with experimental RA were stained with hematoxylin and eosin (H and E). These animals were treated with the mono-drug therapy of market MTX (s.c.) and ACE-MKT gel (t.d.), and the combination therapy (ACE and MTX) exhibited the reduced disease severity, measured and graded from severe (++++) to normal (0) and compared with the healthy tissues. Inflammation was observed as severe osteomyelitis, bone destruction, and inflammation extending to the joint synovium (SI Figure 11 and 12). The animals receiving transdermal application of the market gel formulation of aceclofenac gel-(ACE-MKT) and subcutaneous injection of the MTX-MKT formulation exhibited the infiltration of the inflammatory cells, synovial hyperplasia, and partial destruction of bone and joints of paw when compared to the untreated control and the animals with the arthritis induced by the complete Freund’s (CIA) animals. Our data showed a reduced cell infiltration when the experimental animals were treated with the combination drug therapy (MTX-MKT [s.c.] and gel-[ACE-MKT] [t.d.]). However, this drug combination could not regain the structure or shape of the joints of the paw and ankles of the animals similar to the untreated control (Figure 7, SI Figure 11A and 12A). The experimental CIA and BCG-augmented CIA animals treated with the prepared nano-formulations via the transdermal application of gel-(ACE-NLCs+CE) and intravenous injection of MTX-LPHNPs saw a higher reduction in the severity of RA as well as pathological changes in the animals with experimental RA than the healthy controls (SI Figure 11B and 12B). Based on these investigations, we confirm the “therapeutic efficacy” of our drug nano-formulations as mono- and combination drug therapies to reduce the RA-induced inflammation and manage the disease (SI Fig 11A and 12A). The inhibition in the cell infiltration with the combination drug therapy treatment and resulting significant reduction in the arthritis index (Figure 6C) and arthritis score (Figure 6D) compared to CIA animals surface the importance of our drug formulation in RA therapeutics. The experimental rats with chronic RA receiving treatment with the combination of ACE and MTX nano-formulations showed faster recovery from RA. This recovery was comparable to that seen with the commercially available drug formulations for treating RA. Therefore, reduced arthritis parameters and paw thickness of the rats with experimental RA (SI Figure 11 and 12), regaining the normal shape and structure of paw and joints similar to healthy control (Figure 7B) suggested the “therapeutic potential” of the prepared nanoformulations (Figures 6 and 7).
Examination of Skin Histopathology
The transdermal application of gel-(ACE-MKT) and subcutaneous injection of market MTX-MKT formulations as mono- and combination drug regimens exhibited sub-optimal damage repair due to RA pathology by the restoration of the structural integrity. This investigation was validated by the increased number of mast cells than the severely inflamed, damaged, and dense inflammatory cell infiltration observed in the untreated control (SI Figure 13). The therapeutic effects of the mono-drug regimen with the transdermal application of gel-(ACE-NLCs+CE) and intravenous injection of MTX-LPHNPs showed better recovery (SI Figure 13) from RA pathology. In contrast, the combination drug regimen with our nanoformulations (gel-[ACE-NLCs+CE] [t.d.] and MTX-LPHNPs [i.v.] resulted in the complete recovery from arthritis and associated complications (SI Figure 13).
Serum Levels of Immune Biomarkers in Experimental Rats
The inflammatory mediators (IL-6, TNF-α, IL-1β, MMP-1, COX-2) and joint-destroying enzymes (iNOs) play a crucial role in the pathogenesis of RA.62 We used the prepared nano-formulations to confirm their therapeutic potential with respect to the anti-inflammatory and immunomodulatory activities. These activities were determined in the local and systemic fluids of the rats with the experimental RA (Figure 8 and 9). We observed a greater reduction in the RA-induced inflammation (local and systemic) than that seen with the existing market formulations (Figure 8A, B and 9A). We measured the pro-inflammatory cytokines (TNF-α, IL-1β, IL-6, and MMP-1), bone degradation enzyme (iNOS), and metabolic enzyme (cyclo-oxygenase-2; COX-2) in paw tissues (Figure 8A), synovial fluid (Figure 8B), and serum (Figure 9A) collected from the experimental animals receiving treatment with the drug nanoparticles. The mono-and combination drug regimens with the subcutaneously applied gel-(ACE-NLCs+CE) formulations and intravenously injected MTX-LPHNPs showed a significant (P≤0.05) reduction in immune mediators as well as bone degrading enzymes in paw tissues (Figure 8A) than the individual market formulations [gel-(ACE-MKT) (t.d.), MTX-MKT (s.c.)], and combination of the gel-(ACE-MKT) (t.d.) and MTX-MKT (s.c.) formulations. The combination of our nanoformulation exhibited significantly (P≤0.05) reduced inflammation in the paw tissues and synovial fluid in the experimental RA and CIA animals when compared to the commercial ACE and MKT-MTX formulations (Figure 8A and B). Also, systemic inflammation induced by RA in the experimental animals saw a significant reduction when our drug nano-formulations were used as mono- and combination drug regimen. In the end, reduced severity of RA was characterized by a marked reduction in TNF-α, IL-6, MMP-1, and IL-1β secretions in the sera of experimental animals upon treated with our drug nano-formulations (Figure 9A).
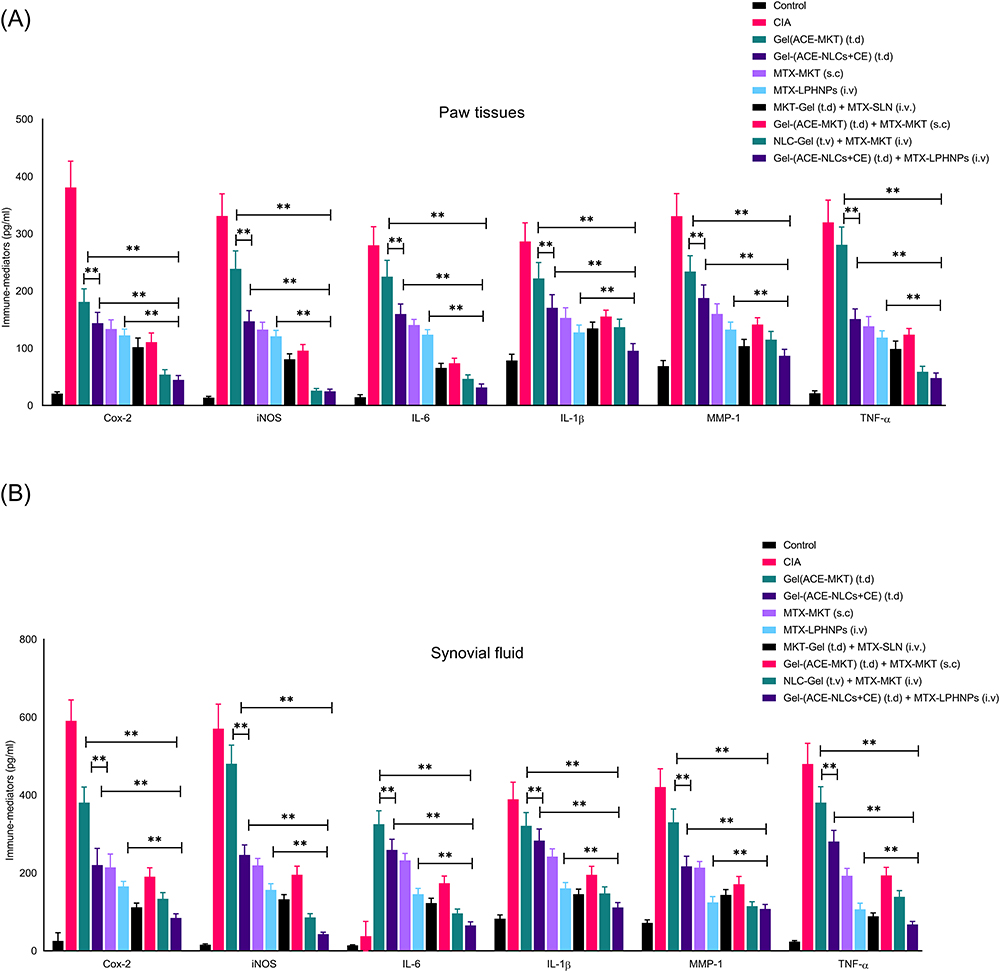 |
Figure 8 Determination of the “immune-mediators” and joint damage markers (TNF-α, IL-1β, IL-6, MMP-1, iNOS andCOX-2) in the, (A) paw tissues (B) synovial fluid of animals with experimental RA upon receiving treatment with the mono-[MTX-LPHNPs, gel-(ACE-NLCs+CE)] and combination (MTX-LPHNPs & gel-(ACE-NLCs+CE) drug therapy. Combination drug therapy with the transdermal application of gel-(ACE-NLCs+CE) and intravenous administration of MTX-LPHNPs saw the steep reduction in the local inflammation characterized by the reduced expression of the immune mediators as compared to the monotherapy and combination therapy of the market formulations (ACE gel-[ACE-MKT]) administered transdermally along with the subcutaneous injection of MTX-MKT formulation. Statistical data is expressed as mean ± SE (n = 6 animals per group). (**p< 0.001; highly significant). |
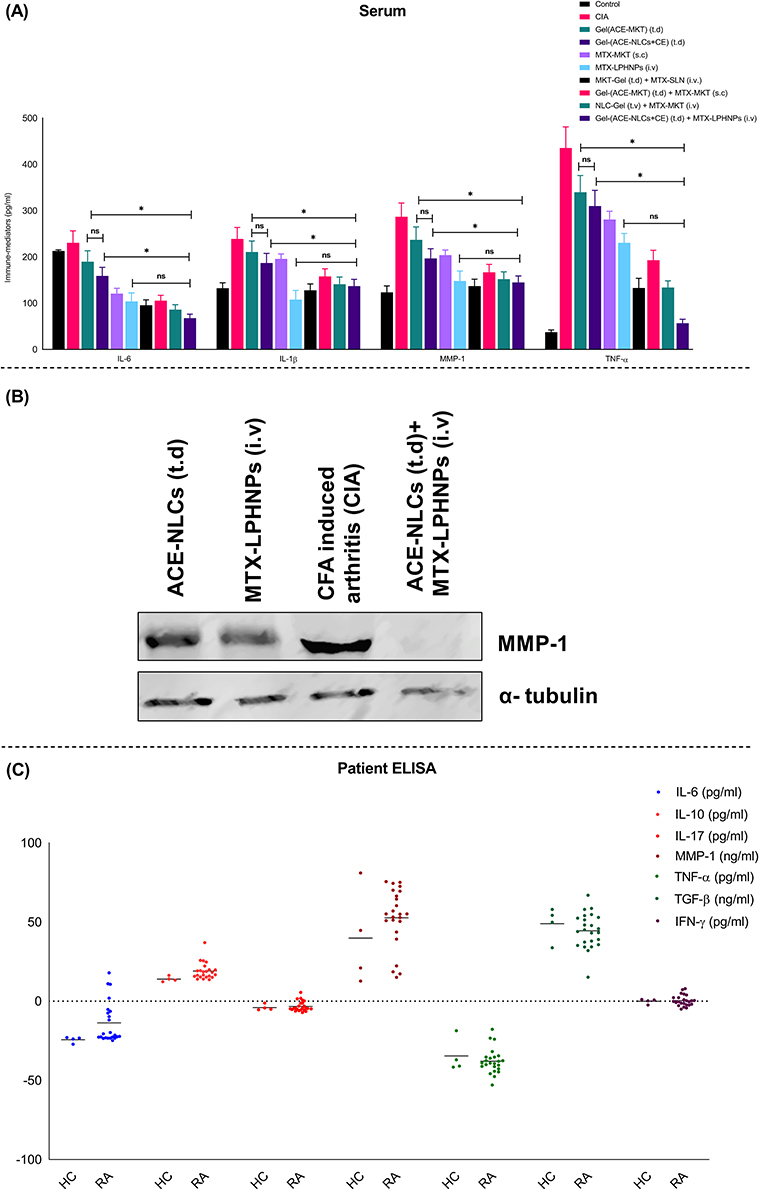 |
Figure 9 Determination of the “immune-mediators” and joint damage markers (TNF-α, IL-1β, IL-6, MMP-1, iNOS andCOX-2) in theA) sera of the animals with experimental RA upon receiving treatment with the mono-[MTX-LPHNPs, gel-(ACE-NLCs+CE)] and combination (MTX-LPHNPs & gel-(ACE-NLCs+CE) drug therapy. Combination drug therapy with the transdermal application of gel-(ACE-NLCs+CE) and intravenous administration of MTX-LPHNPs saw the steep reduction in the systemic inflammation characterized by the reduced expression of the immune mediators as compared to the monotherapy and combination therapy of the market formulations (A) CE gel-[ACE-MKT]) administered transdermally along with the subcutaneous injection of MTX-MKT formulation, (B) The co-delivery of ACE-NLCs+CE and MTX-LPHNPs following the transdermal and intravenous routes exhibited no expression of MMP-1 in the synovial fluid compared to the CIA animals. Cells from synovial fluid were washed with RPMI followed by lysing to detect the MMP-1 expression by the Western blots. α-Tubulin, loading control (n=3). Statistical data is expressed as mean ± SE (n = 6 animals per group). (*p< 0.01; significant)) (ns; not significant) (C) The estimation of inflammatory (IL-6, TNF-α, IFN-γ, IL-17), immunoregulatory (TGF-β, IL-10) and joint damage (MMP-1) markers in the serum samples collected from the patients with RA (n=24 patients). The statistical analysis was performed using one-way analysis of test. |
Quantification of MMP-1 in Synovial Fluid
MMP-1 interstitial and fibroblast collagenase (an enzyme encoding for the MMP1 gene) is known to degrade all components of the extracellular matrix during RA pathology.63 The in vitro (Figure 3B, SI Figure 8H and 9A) and ex-vivo studies carried out with the PBMCs isolated from the patients with RA (Figure 3D and E), and molecular docking of signaling and inflammatory proteins showing interaction with ACE and MTX (Figure 3A) confirmed the reduced mRNA expression of MMP-1 following the drug treatment. The gene transcription profiling in vitro, in silico and ex-vivo assays led us confirm the expression of MMP-1 in the synovial fluid of rats receiving treatment with the mono- and combination drug regimen (Figure 9B). No expression of MMP-1 was seen in the synovial fluid of animals treated with the combination of our drug nanoformulations (transdermal application of gel-(ACE-NLCs+CE) and intravenous injection of MTX-LPHNPs, and a lower expression was observed in the animals receiving treatment with the mono-drug regimen and CIA. In the end, absence of MMP-1 expression in the animals receiving treatment with the combination drug formulations confirmed the therapeutic potential of the prepared drug nanoparticles during RA pathology (Figure 9B).
Serum Profiling of Immune Mediators in the Patients with RA
We next confirmed the expression of pro-inflammatory (IL-6, IL-17, TNF-α, IFN-γ), bone degradation (MMP-1), and anti-inflammatory/immunoregulatory (IL-10, TGF-β) markers in sera samples collected from patients with RA (n=24) (Figure 9C). Since the patients were on drug therapy, a little reduction in the expression of anti-inflammatory cytokines (IL-6, IL-17), many-fold downregulation in the secretion of TNF-α, and almost no difference in the expression of IFN-γ was compared the healthy individuals (Figure 9C) in the end, higher expression of IL-10, TGF-β and MMP-1 was seen in the RA patients in comparison to the healthy controls (Figure 9C).
Discussion
MTX inhibits proliferation of the immune cells crucial in to elicit the inflammation and joint damage during RA pathogenesis. ACE suppresses RA-induced inflammation by inhibiting the COX-2. The CE-conjugated NLCs mediate the efficient delivery of ACE to the local tissues and greatly reduce the disease induced inflammation. The intravenous injection of MTX delivered via LPHNPs repairs the immune cells to check the progression of RA. Therefore, we took advantage of the ACE-NLC-based gel to increase its permeability with CE conjugation to achieve better deposition of the drugs at the diseased site.20,21,64 Further, a comparative study of the texture profile of ACE-NLCs in carpool gel formulations and rheological properties was performed using the MKT-gel formulation. As reported earlier,65,66 our data showed better affinity and biocompatibility of ACE nanoformulations tested in the skin model (ex-vivo), with respect to higher permeation, better cell uptake of C-6 loaded NLC formulations as well as better cytoplasmic distribution. The nano-formulations prepared from solid lipids exhibited significant cell viability in placebo controls (NLCs and LPHNPs) as compared to the other formulations tested.
The higher permeability of the gel-(ACE-NLC+CE) formulation compared to the commercial gel confirmed the higher deposition in order to achieve the maximum therapeutic effect of ACE (Table 1). The higher lipophilicity of the dermal layer circumscribes the partitioning of hydrophobic drugs. Furthermore, the enhanced permeability and retention (EPR) effect is attributed to the formation of a micro-reservoir of drug molecules within the skin (dermis) interior by virtue of the integration of phospholipids with skin lipids for the drug retention.67 The skin retention and subsequent permeation of ACE was seen greater with CE-conjugated NLC formulations than the market formulation gel-(ACE-MKT).68 We think that NLC formulations could effectively make the ACE accessible within the skin layers (stratum corneum, epidermis and dermis) due to the higher skin permeability for ACE. As previously published,20,69 the NLC formulation exhibited higher penetration and subsequent skin permeation allowed the higher drug deposition at the diseased site. The enhanced permeation (p < 0.001) of drug across the skin layers could be due to the increased contact time between the CE and stratum corneum.
MTX delivered through LPHNPs showed maximum systemic (plasma) and local (synovial fluid) deposition lasting longer than that seen with the market formulation at the diseased site (Figure 6A and B).70 In vitro macrophage model of inflammation (MMI) best mimicked the RA-induced inflammation upon stimulation with higher concentration of LPS for longer duration. The stimulated macrophages upon receiving mono- and combination drug treatment controlled the transcriptional regulation and protein expression of immune events and signaling pathways (Figures 3 and 4). As earlier,52,53 our findings suggest that Akt1 could mediate the death of antigen-stimulated macrophages (Figure 4). Further, treatment with the drugs lead to the suppression of the inflammation as confirmed by molecular profiling of immune biomarkers in PBMCs collected from patients with RA as well as MMI. The elevated serum levels of the inflammatory and regulatory markers from the patients with RA are in agreement with the published findings.71,72 The in vitro and ex-vivo findings were well supported by the in vivo findings performed in Wistar rats with experimental RA. Hence, our findings advocate the use of drug nanoparticles for the efficient delivery of ACE and MTX for achieving better drug therapeutics in the treatment/management of RA.
The iNOS pathway is known to regulate the apoptosis dependent upon the higher production of NO.61 The antigen-stimulated macrophage upon receiving treatment with mono- (SI Figure 8F) and combination of drugs (Figure 3B) showed a reduced expression of iNOS-mediated NO production. This suggested that our drug formulations could be crucial in inducing death of the diseased cells.
The selective death of the antigen-stimulated cells by the combination drug treatment attested our proposition whereby the compromised death of RA cells may have led to disease progression.73,74
Further, therapeutic efficacy of the combination drug treatment in the experimental rats showed erythema/inflammation or cell infiltration, and controlled arthritis index and arthritis score. Interestingly, co-delivery of the gel-(ACE-NLCs+CE) and MTX-LPHNP formulations via transdermal and intravenous routes, respectively, was shown to alleviate pain and suffering of patients with RA (Figures 5–7) (Tables 1, 2, and Figures 8 and 9). The prepared drug nano-formulations proved better than existing market formulations as far as the reduction in the inflammation and/or treatment/management of RA is concerned. Absence of MMP-1 expression in the synovial fluid of experimental animals once again confirmed the value of drug nanoparticles in overcoming the bone damage inflicted by the RA pathology.75–77
Histopathological analyses of the paw, ankle, and skin of experimental animals treated with these nano-formulations suggested the modulation of RA-induced inflammation. Moreover, short plasma life of intravenously injected MTX advocated for the need of LPHNP-mediated controlled MTX delivery for a better therapeutic effect,78 to manage/treat the RA with the minimal damage to the healthy tissues. Therefore, we are at the opinion that the drug nanoparticles could be crucial to address the serious health complications observed with the extended drug therapy.16 Also, we determined the therapeutic efficacy of the developed formulations: transdermal application of gel-(ACE-NLCs+CE) at the diseased site every-day and weekly intravenous injection of MTX-LPHNPs. The prepared drug formulations were compared with the market formulation gel-(ACE-MKT) applied through the transdermal route (everyday) and MTX-MKT injected subcutaneously (weekly) as the mono- and combination drug regimens. Our stability and safety data suggest that these nanoformulations are safe and could be licensed for the pilot patient trials. Finally, 14-3-3 zeta (an autoantigen) has been shown to suppress inflammatory arthritis,79 suggesting its potential as a potential vaccine candidate. Its antigenic potential should be however studied in the animal model(s). Therefore, collagen-induced arthritis (CIA) in humanized immune system (HIS) mice may be a better option to validate its antigenic potential for a development of vaccine for RA. Overall, the greater therapeutic efficacy of the combination regimen of these drug nanoformulations may be effectively used as a therapeutic approach to treat/manage RA (Figure 10).
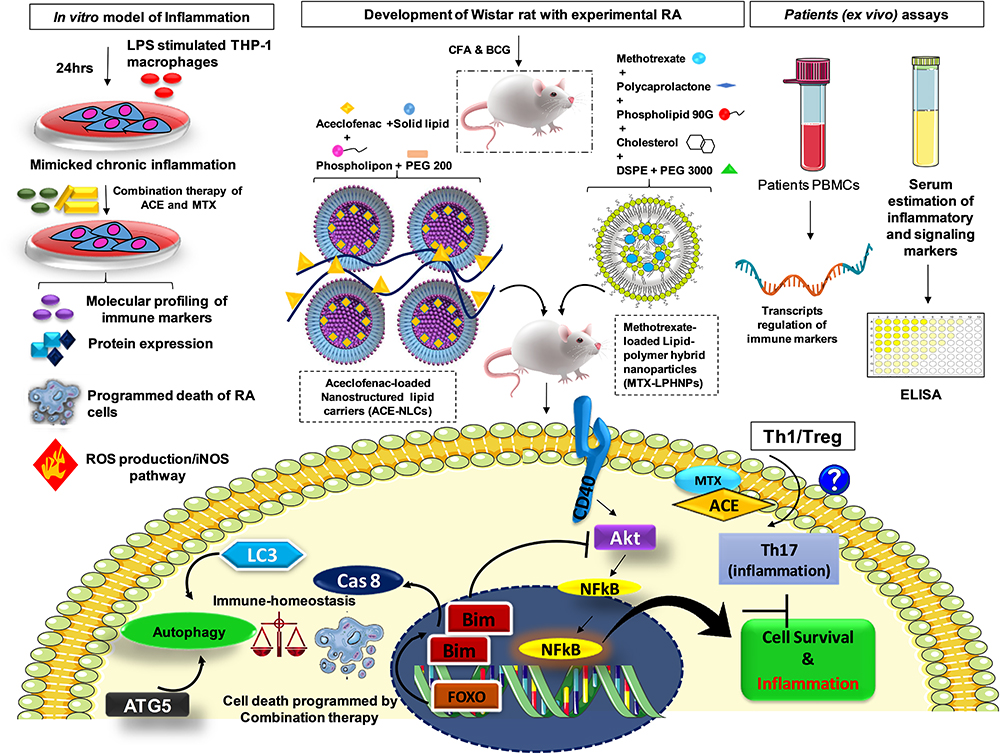 |
Figure 10 The in vitro model of macrophage inflammation (macrophage model of inflammation; MMI) (Antigen/LPS stimulated THP-1 macrophages), CFA and BCG induced acute rheumatoid arthritis in vivo model. The ex-vivo investigations were performed with the PBMCs isolated from blood samples drawn from the patients with RA. This confirms the importance of drug nanoparticles for the increased deposition of drugs at the diseased site. The assays were carried out with MMI to confirm the gene transcription and protein expression of the inflammatory and signaling pathways to establish the eubiosis. The drug nanoparticles influence the death and autophagy axis of the stimulated cells to favor the death of inflamed cells induced by the drug combination regimen. The in vitro and ex-vivo characterization of ACE-NLCs and MTX-LPHNPs formulations and the development of the Wistar rat model for RA confirmed the importance of the combination regimen of drug nanoparticles. The molecular profiling (PBMCs) and protein level (from serum) confirmation of the therapeutic potential of mono- and combination drug regimen from the samples collected on the patient with RA. In the end, the model indicating a mechanism whereby cell–cell interaction could induce death of the stimulated cells. CD40 mediated activation of Akt1 controlling the activation of NF-Kb is associated with its inhibitor IkB to retain it in the cytoplasm. The FOXO1 inside the nucleus regulates the expression of pro-apoptotic protein (Bim) that program the death of the diseased cells. |
Conclusion
Aceclofenac and methotrexate nanoformulations when used as a combination regimen showed greater “therapeutic potential” to treat rheumatoid arthritis. Present work is of immense clinical relevance since no effective treatment based on drugs or vaccines is available for RA. We are also studying the underlying mechanism for the modulation of RA-induced inflammation in collagen-induced arthritis-humanized immune system (CIA-HIS) mice. We propose to dissect the mechanism by which the combination drug regimen could influence the Th1/Th17 axis to inhibit inflammation-dependent conversion of Th1 to Th17 phenotype.80
Abbreviations
ACE, Aceclofenac; ACE-NLCs, Aceclofenac loaded-nanostructured lipid nanocarriers; CA, Cetyl alcohol; CE, Chemical enhancer; CFA, Complete Freund’s adjuvant; CIA, CFA-induced arthritis; CLSM, Confocal laser scanning microscopy; COX, Cyclooxygenase; CP, Cetyl Palmitate; DMARDs, Disease modifying anti-rheumatic drugs; GMS, Glyceryl monostearate; IFA, Immunofluorescence assay; iNOS, inducible nitric oxide synthase; LPHNPs, Lipid polymer hybrid Nanoparticles; MMPs, Matrix-metalloproteinases; Mtb, Mycobacterium tuberculosis; MTX, Methotrexate; MTX-LPHNPs, Methotrexate loaded-lipid polymer hybrid Nanoparticles; NLCs, Nanostructured lipid nanocarriers; NO, Nitric oxide; NSAIDs, Non-steroidal anti-inflammatory drugs; PCL, Polycaprolactone; PDI: Polydispersity index; PFA, Paraformaldehyde; RA, Rheumatoid arthritis; SA, Stearic acid; TEM, Transmission electron microscopy; TPA, Texture profile analysis; XRD, X-ray differactometry.
Acknowledgments
All authors would like to express their sincere gratitude to the instrumentation facility of CSIR-IMTECH, Panjab University, Chandigarh, and Nirma University, Ahmedabad. Rajeev Tyagi would like to offer sincere thanks to DBT, New Delhi and the Government of India for financially supporting this study in the form of the Ramalingaswami Re-entry Fellowship-2019 (D.O. NO. BT/HRD/35/02/2006) Sanction order (BT/RLF/Re-entry/27/2018). The Indian Council of Medical Research (ICMR) and the New Delhi extramural grant (35/1/2020-Nano/BMS) generously supported this study. Nikunj Tandel thanks the Indian Council of Medical Research (ICMR), New Delhi, Government of India, for providing a fellowship for his research (ICMR SRF No.2020-7623/CMB-BMS). Rajeev Tyagi expresses his gratitude to Dr. Pradip Sen and Dr. Manish Mishra to help us perform an experiment showing the expression of molecular markers. The funders had no role in the study design, collection, analyses, interpretation of data, writing of the manuscript, or decision to publish the results.
Disclosure
Current affiliation: Neeraj K. Garg: FR & D Non Orals, Sun Pharmaceuticals Industries Ltd. Tandalja, Vadodara, Gujarat, India; Sheetal Saini: Infectious Diseases Labs, A*STAR, Singapore; Praveen Sharma: Department of Molecular Medicine, Long School of Medicine, University of Texas Health Science Center at San Antonio, TX 78229, USA. The authors declare that they have no conflicts of interest for this work.
References
1. Mangalea MR, Paez-Espino D, Kieft K., et al. Individuals at risk for rheumatoid arthritis harbor differential intestinal bacteriophage communities with distinct metabolic potential. Cell Host Microbe. 2021;29(5):726–739 e725. doi:10.1016/j.chom.2021.03.020
2. Smolen JS, Landewe RBM, Bergstra SA, et al. EULAR recommendations for the management of rheumatoid arthritis with synthetic and biological disease-modifying antirheumatic drugs: 2022 update. Ann Rheum Dis. 2023;82(1):3–18. doi:10.1136/ard-2022-223356
3. Brennan FM, McInnes IB. Evidence that cytokines play a role in rheumatoid arthritis. J Clin Invest. 2008;118(11):3537–3545. doi:10.1172/JCI36389
4. Kondo N, Kuroda T, Kobayashi D. Cytokine networks in the pathogenesis of rheumatoid arthritis. Int J Mol Sci. 2021;22:20 doi:10.3390/ijms222010922.
5. Jeong JC, Chung YH, Park T, et al. Safety and effectiveness of 4-week therapy with aceclofenac controlled release once a day. Sci Rep. 2022;12(1):16519. doi:10.1038/s41598-022-20633-6
6. Cutolo M, Sulli A, Pizzorni C, Seriolo B, Straub RH. Anti-inflammatory mechanisms of methotrexate in rheumatoid arthritis. Ann Rheum Dis. 2001;60(8):729–735. doi:10.1136/ard.60.8.729
7. Hamed KM, Dighriri IM, Baomar AF, et al. Overview of Methotrexate Toxicity: a Comprehensive Literature Review. Cureus. 2022;14(9):e29518. doi:10.7759/cureus.29518
8. Xu M, Wu S, Wang Y, et al. Association between high-dose methotrexate-induced toxicity and polymorphisms within methotrexate pathway genes in acute lymphoblastic leukemia. Front Pharmacol. 2022;13:1003812. doi:10.3389/fphar.2022.1003812
9. Kremer JM, Genovese MC, Cannon GW, et al. Concomitant leflunomide therapy in patients with active rheumatoid arthritis despite stable doses of methotrexate: a randomized, double-blind, placebo-controlled trial. Ann Internal Med. 2002;137(9):726–733. doi:10.7326/0003-4819-137-9-200211050-00007
10. Capell HA, Madhok R, Porter DR, et al. Combination therapy with sulfasalazine and methotrexate is more effective than either drug alone in patients with rheumatoid arthritis with a suboptimal response to sulfasalazine: results from the double-blind placebo-controlled MASCOT study. Ann Rheumatic Dis. 2007;66(2):235–241. doi:10.1136/ard.2006.057133
11. Verschueren P, De Cock D, Corluy L, et al. Methotrexate in combination with other DMARDs is not superior to methotrexate alone for remission induction with moderate-to-high-dose glucocorticoid bridging in early rheumatoid arthritis after 16 weeks of treatment: the CareRA trial. Ann Rheumatic Dis. 2015;74(1):27–34. doi:10.1136/annrheumdis-2014-205489
12. Zanfirescu A, Nitulescu G, Stancov G, et al. Evaluation of topical anti-inflammatory effects of a gel formulation with plantago lanceolata, Achillea millefolium, Aesculus hippocastanum and Taxodium distichum. Sci Pharm. 2020;88(2):26. doi:10.3390/scipharm88020026
13. Smolen JS, Avila JC, Aletaha D. Tocilizumab inhibits progression of joint damage in rheumatoid arthritis irrespective of its anti-inflammatory effects: disassociation of the link between inflammation and destruction. Ann Rheum Dis. 2012;71(5):687–693. doi:10.1136/annrheumdis-2011-200395
14. Allison MC, Howatson AG, Torrance CJ, Lee FD, Russell RI. Gastrointestinal damage associated with the use of nonsteroidal antiinflammatory drugs. N Engl J Med. 1992;327(11):749–754. doi:10.1056/NEJM199209103271101
15. Mazaud C, Fardet L. Relative risk of and determinants for adverse events of methotrexate prescribed at a low dose: a systematic review and meta‐analysis of randomized placebo‐controlled trials. Br J Dermatol. 2017;177(4):978–986. doi:10.1111/bjd.15377
16. Onda K, Honma T, Masuyama K. Methotrexate-related adverse events and impact of concomitant treatment with folic acid and tumor necrosis factor-alpha inhibitors: an assessment using the FDA adverse event reporting system. Front Pharmacol. 2023;14:1030832. doi:10.3389/fphar.2023.1030832
17. Garg NK, Tyagi RK, Singh B, et al. Nanostructured lipid carrier mediates effective delivery of methotrexate to induce apoptosis of rheumatoid arthritis via NF-kappaB and FOXO1. Int J Pharm. 2016;499(1–2):301–320. doi:10.1016/j.ijpharm.2015.12.061
18. Garg NK, Singh B, Kushwah V, et al. The ligand (s) anchored lipobrid nanoconstruct mediated delivery of methotrexate: an effective approach in breast cancer therapeutics. Nanomed Nanotechnol Biol Med. 2016;12(7):2043–2060. doi:10.1016/j.nano.2016.05.008
19. Garg NK, Tyagi RK, Sharma G, et al. Functionalized lipid-polymer hybrid nanoparticles mediated codelivery of methotrexate and aceclofenac: a synergistic effect in breast cancer with improved pharmacokinetics attributes. Mol Pharmaceut. 2017;14(6):1883–1897. doi:10.1021/acs.molpharmaceut.6b01148
20. Garg NK, Tandel N, Bhadada SK, Tyagi RK. Nanostructured lipid carrier-mediated transdermal delivery of aceclofenac hydrogel present an effective therapeutic approach for inflammatory diseases. Front Pharmacol. 2021;12:713616. doi:10.3389/fphar.2021.713616
21. Garg NK, Sharma G, Singh B, et al. Quality by Design (QbD)-enabled development of aceclofenac loaded-nano structured lipid carriers (NLCs): an improved dermatokinetic profile for inflammatory disorder(s). Int J Pharm. 2017;517(1–2):413–431. doi:10.1016/j.ijpharm.2016.12.010
22. Zewail M, Nafee N, Helmy MW, Boraie N. Coated nanostructured lipid carriers targeting the joints–An effective and safe approach for the oral management of rheumatoid arthritis. Int J Pharm. 2019;567:118447. doi:10.1016/j.ijpharm.2019.118447
23. Gu Y, Tang X, Yang M, Yang D, Liu J. Transdermal drug delivery of triptolide-loaded nanostructured lipid carriers: preparation, pharmacokinetic, and evaluation for rheumatoid arthritis. Int J Pharm. 2019;554:235–244. doi:10.1016/j.ijpharm.2018.11.024
24. Tchetverikov I, Lohmander LS, Verzijl N, et al. MMP protein and activity levels in synovial fluid from patients with joint injury, inflammatory arthritis, and osteoarthritis. Ann Rheum Dis. 2005;64(5):694–698. doi:10.1136/ard.2004.022434
25. Gossage DL, Cieslarova B, Ap S, et al. Phase 1b study of the safety, pharmacokinetics, and disease-related outcomes of the matrix metalloproteinase-9 inhibitor andecaliximab in patients with rheumatoid arthritis. Clin. Ther. 2018;40(1):156–165. e155. doi:10.1016/j.clinthera.2017.11.011
26. Raeeszadeh-Sarmazdeh M, Do LD, Hritz BG. Metalloproteinases and their inhibitors: potential for the development of new therapeutics. Cells. 2020;9(5):1313. doi:10.3390/cells9051313
27. Garg NK, Singh B, Sharma G, et al. Development and characterization of single step self-assembled lipid polymer hybrid nanoparticles for effective delivery of methotrexate. RSC Adv. 2015;5(77):62989–62999. doi:10.1039/C5RA12459J
28. Jain S, Chauhan DS, Jain AK, et al., Inventors. A universal step-wise freeze drying process for lyophilization of pharmaceutical products; 2011.
29. Garg NK, Sharma G, Singh B, Nirbhavane P, Katare OP. Quality by Design (QbD)-based development and optimization of a simple, robust RP HPLC method for the estimation of Methotrexate. J Liquid Chromatogr Related Technol. 2015;38(17):1629–1637. doi:10.1080/10826076.2015.1087409
30. Garg NK, Dwivedi P, Campbell C, Tyagi RK. Site specific/targeted delivery of gemcitabine through anisamide anchored chitosan/poly ethylene glycol nanoparticles: an improved understanding of lung cancer therapeutic intervention. Eur. J. Pharm. Sci. 2012;47(5):1006–1014. doi:10.1016/j.ejps.2012.09.012
31. Jain AK, Jain A, Garga NK, et al. Adapalene loaded solid lipid nanoparticles gel: an effective approach for acne treatment. Colloids Surf B. 2014;121:222–229. doi:10.1016/j.colsurfb.2014.05.041
32. Chen B, Li H, Ding Y, Suo H. Formation and microstructural characterization of whey protein isolate/beet pectin coacervations by laccase catalyzed cross-linking. LWT – Food Sci Technol. 2012;47(1):31–38. doi:10.1016/j.lwt.2012.01.006
33. de Ávila MDR, Cambero MI, Ordóñez JA, de la Hoz L, Herrero AM. Rheological behaviour of commercial cooked meat products evaluated by tensile test and texture profile analysis (TPA). Meat Sci. 2014;98(2):310–315. doi:10.1016/j.meatsci.2014.05.003
34. Tyagi RK, Garg NK, Jadon R, et al. Elastic liposome-mediated transdermal immunization enhanced the immunogenicity of P. falciparum surface antigen, MSP-1. Vaccine. 2015;33:4630–4638. doi:10.1016/j.vaccine.2015.06.054
35. Kilfoyle BE, Sheihet L, Zhang Z, Laohoo M, Kohn J, Michniak-Kohn BB. Development of paclitaxel-TyroSpheres for topical skin treatment. J Control Release. 2012;163(1):18–24. doi:10.1016/j.jconrel.2012.06.021
36. Raza K, Singh B, Singla S, et al. Nanocolloidal carriers of isotretinoin: antimicrobial activity against Propionibacterium acnes and dermatokinetic modeling. Mol Pharmaceut. 2013;10(5):1958–1963. doi:10.1021/mp300722f
37. Ramachandran S, Kota P, Ding F, Dokholyan NV. Automated minimization of steric clashes in protein structures. Proteins. 2011;79(1):261–270. doi:10.1002/prot.22879
38. Trott O, Olson AJ. AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J Comput Chem. 2010;31(2):455–461. doi:10.1002/jcc.21334
39. Liu Y, Grimm M, Dai WT, Hou MC, Xiao ZX, Cao Y. CB-Dock: a web server for cavity detection-guided protein-ligand blind docking. Acta Pharmacol. Sin. 2020;41(1):138–144. doi:10.1038/s41401-019-0228-6
40. DeLano WL. Pymol: an open-source molecular graphics tool. CCP4 Newsl. Protein Crystal. 2002;40(1):82–92.
41. Biovia D Discovery Studio Modeling Environment, Dassault Syst. Release, San Diego; 2015:4.
42. Sharma P, Singh S. Combinatorial effect of DCA and Let-7a on Triple-Negative MDA-MB-231 cells: a metabolic approach of treatment. Integr Cancer Ther. 2020;19:1534735420911437. doi:10.1177/1534735420911437
43. Salter M, Duffy C, Garthwaite J, Strijbos PJ. Ex vivo measurement of brain tissue nitrite and nitrate accurately reflects nitric oxide synthase activity in vivo. J Neurochem. 1996;66(4):1683–1690. doi:10.1046/j.1471-4159.1996.66041683.x
44. Chillingworth NL, Donaldson LF. Characterisation of a Freund’s complete adjuvant-induced model of chronic arthritis in mice. J Neurosci Methods. 2003;128(1–2):45–52. doi:10.1016/s0165-0270(03)00147-x
45. Gomes RP, Bressan E, Silva TM, Gevaerd MS, Tonussi CR, Domenech SC. Standardization of an experimental model suitable for studies on the effect of exercise on arthritis. Einstein. 2013;11(1):76–82. doi:10.1590/s1679-45082013000100014
46. Geboes L, Klerck BD, Balen MV, et al. Freund’s complete adjuvant induces arthritis in mice lacking a functional interferon‐γ receptor by triggering tumor necrosis factor α–driven osteoclastogenesis. Arthritis Rheum. 2007;56(8):2595–2607. doi:10.1002/art.22791
47. Zhang L, Chan JM, Gu FX, et al. Self-assembled lipid− polymer hybrid nanoparticles: a robust drug delivery platform. ACS nano. 2008;2(8):1696–1702. doi:10.1021/nn800275r
48. Chawla V, Saraf SA. Rheological studies on solid lipid nanoparticle based carbopol gels of aceclofenac. Colloids Surf B Biointerfaces. 2012;92:293–298. doi:10.1016/j.colsurfb.2011.12.006
49. Ghica MV, Hîrjău M, Lupuleasa D, Dinu-Pîrvu C-E. Flow and thixotropic parameters for rheological characterization of hydrogels. Molecules. 2016;21(6):786. doi:10.3390/molecules21060786
50. Chen MH, Wang LL, Chung JJ, Kim Y-H, Atluri P, Burdick JA. Methods to assess shear-thinning hydrogels for application as injectable biomaterials. ACS Biomater Sci Eng. 2017;3(12):3146–3160. doi:10.1021/acsbiomaterials.7b00734
51. Xu Y, Shrestha N, Preat V, Beloqui A. An overview of in vitro, ex vivo and in vivo models for studying the transport of drugs across intestinal barriers. Adv Drug Deliv Rev. 2021;175:113795. doi:10.1016/j.addr.2021.05.005
52. Tyagi RK, Miles B, Parmar R, et al. Human IDO-competent, long-lived immunoregulatory dendritic cells induced by intracellular pathogen, and their fate in humanized mice. Sci Rep. 2017;7:41083. doi:10.1038/srep41083
53. Riol-Blanco L, Delgado-Martin C, Sanchez-Sanchez N, et al. Immunological synapse formation inhibits, via NF-kappaB and FOXO1, the apoptosis of dendritic cells. Nat Immunol. 2009;10(7):753–760. doi:10.1038/ni.1750
54. Jiang Q, Yang G, Liu Q, Wang S, Cui D. Function and role of regulatory T cells in rheumatoid arthritis. Front Immunol. 2021;12:626193. doi:10.3389/fimmu.2021.626193
55. Kanjana K, Chevaisrakul P, Matangkasombut P, Paisooksantivatana K, Lumjiaktase P. Inhibitory activity of FOXP3+ regulatory T cells reveals high specificity for displaying immune tolerance in remission state rheumatoid arthritis. Sci Rep. 2020;10(1):19789. doi:10.1038/s41598-020-76168-1
56. Yan S, Kotschenreuther K, Deng S, Kofler DM. Regulatory T cells in rheumatoid arthritis: functions, development, regulation, and therapeutic potential. Cell Mol Life Sci. 2022;79(10):533. doi:10.1007/s00018-022-04563-0
57. Van’t Hof R, Hocking L, Wright P, Ralston S. Nitric oxide is a mediator of apoptosis in the rheumatoid joint. Rheumatology. 2000;39(9):1004–1008. doi:10.1093/rheumatology/39.9.1004
58. Ralston SH. Nitric oxide and bone. Immunology. 2001;103(3):255–261. doi:10.1046/j.1365-2567.2001.01261.x
59. Nagy G, Koncz A, Telarico T, et al. Central role of nitric oxide in the pathogenesis of rheumatoid arthritis and systemic lupus erythematosus. Arthritis Res Therapy. 2010;12(3):1–6. doi:10.1186/ar3045
60. Weitoft T, Lind A, Larsson A, Ronnelid J, Hogman M. Exhaled nitric oxide in early rheumatoid arthritis and effects of methotrexate treatment. Sci Rep. 2022;12(1):6489. doi:10.1038/s41598-022-10334-5
61. Lee YM, Lee S, Kim WJ. Nitric oxide scavengers based on o-phenylenediamine for the treatment of rheumatoid arthritis. Biomater Sci. 2023;11(7):2395–2404. doi:10.1039/d2bm01994a
62. Kokkonen H, Soderstrom I, Rocklov J, Hallmans G, Lejon K, Rantapaa Dahlqvist S. Up-regulation of cytokines and chemokines predates the onset of rheumatoid arthritis. Arthritis Rheum. 2010;62(2):383–391. doi:10.1002/art.27186
63. Peake N, Khawaja K, Myers A, et al. Levels of matrix metalloproteinase (MMP)-1 in paired sera and synovial fluids of juvenile idiopathic arthritis patients: relationship to inflammatory activity, MMP-3 and tissue inhibitor of metalloproteinases-1 in a longitudinal study. Rheumatology. 2005;44(11):1383–1389. doi:10.1093/rheumatology/kei025
64. Czajkowska-Kosnik A, Szymanska E, Winnicka K. Nanostructured Lipid Carriers (NLC)-based gel formulations as etodolac delivery: from gel preparation to permeation study. Molecules. 2022;28:1 doi:10.3390/molecules28010235.
65. Silva E, Barreiros L, Segundo MA, Lima SAC, Reis S. Cellular interactions of a lipid-based nanocarrier model with human keratinocytes: unravelling transport mechanisms. Acta Biomater. 2017;53:439–449. doi:10.1016/j.actbio.2017.01.057
66. Cordenonsi LM, Faccendini A, Catanzaro M, et al. The role of chitosan as coating material for nanostructured lipid carriers for skin delivery of fucoxanthin. Int J Pharm. 2019;567:118487. doi:10.1016/j.ijpharm.2019.118487
67. Wu J. The enhanced permeability and retention (epr) effect: the significance of the concept and methods to enhance its application. J Pers Med. 2021;11:8 doi:10.3390/jpm11080771.
68. Rosenblum D, Joshi N, Tao W, Karp JM, Peer D. Progress and challenges towards targeted delivery of cancer therapeutics. Nat Commun. 2018;9(1):1410. doi:10.1038/s41467-018-03705-y
69. Gao S, Tian B, Han J, et al. Enhanced transdermal delivery of lornoxicam by nanostructured lipid carrier gels modified with polyarginine peptide for treatment of carrageenan-induced rat paw edema. Int j Nanomed. 2019;14:6135. doi:10.2147/IJN.S205295
70. Garg NK, Singh B, Tyagi RK, Sharma G, Katare OP. Effective transdermal delivery of methotrexate through nanostructured lipid carriers in an experimentally induced arthritis model. Colloids Surf B. 2016;147:17–24. doi:10.1016/j.colsurfb.2016.07.046
71. Dissanayake K, Jayasinghe C, Wanigasekara P, Sominanda A. Potential applicability of cytokines as biomarkers of disease activity in rheumatoid arthritis: enzyme-linked immunosorbent spot assay-based evaluation of TNF-alpha, IL-1beta, IL-10 and IL-17A. PLoS One. 2021;16(1):e0246111. doi:10.1371/journal.pone.0246111
72. Yamada S, Nagafuchi Y, Wang M, et al. Immunomics analysis of rheumatoid arthritis identified precursor dendritic cells as a key cell subset of treatment resistance. Ann Rheum Dis. 2023. doi:10.1136/ard-2022-223645
73. Zhu C, Zhou J, Li T, Mu J, Jin L, Li S. Urocortin participates in LPS-induced apoptosis of THP-1 macrophages via S1P-cPLA2 signaling pathway. Eur. J. Pharmacol. 2020;887:173559. doi:10.1016/j.ejphar.2020.173559
74. Suzuki T, Kobayashi M, Isatsu K, et al. Mechanisms involved in apoptosis of human macrophages induced by lipopolysaccharide from actinobacillus actinomycetemcomitans in the presence of cycloheximide. Infect Immun. 2004;72(4):1856. doi:10.1128/IAI.72.4.1856-1865.2004
75. Green MJ, Gough AK, Devlin J, et al. Serum MMP-3 and MMP-1 and progression of joint damage in early rheumatoid arthritis. Rheumatology. 2003;42(1):83–88. doi:10.1093/rheumatology/keg037
76. Tchetverikov I, Ronday HK, van El B, et al. MMP profile in paired serum and synovial fluid samples of patients with rheumatoid arthritis. Ann Rheumatic Dis. 2004;63(7):881. doi:10.1136/ard.2003.013243
77. Hussein R, Aboukhamis I. Serum matrix metalloproteinase-3 levels monitor the therapeutic efficacy in Syrian patients with rheumatoid arthritis. Heliyon. 2023;9(3):e14008. doi:10.1016/j.heliyon.2023.e1400
78. Rubbert-Roth A. Methotrexate in rheumatoid arthritis—another brick in the wall. Lancet Rheumatol. 2023;5(4):e173–e175. doi:10.1016/S2665-9913(23)00069-3
79. Kim J, Chun K, McGowan J, et al. 14-3-3zeta: a suppressor of inflammatory arthritis. Proc Natl Acad Sci U S A. 2021;118:34 doi:10.1073/pnas.2025257118.
80. Negi S, Tandel N, Sharma P, Kumar R, Tyagi RK. Aceclofenac and methotrexate combination therapy could influence Th1/Th17 axis to modulate rheumatoid-arthritis-induced inflammation. Drug Discov Today. 2023;28(8):103671. doi:10.1016/j.drudis.2023.103671
 © 2024 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2024 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.