Back to Journals » International Journal of Nanomedicine » Volume 18
Co-Delivery Nanomicelles for Potentiating TNBC Immunotherapy by Synergetically Reshaping CAFs-Mediated Tumor Stroma and Reprogramming Immunosuppressive Microenvironment
Authors Zhang Y, Han X, Wang K, Liu D, Ding X, Hu Z, Wang J
Received 21 April 2023
Accepted for publication 17 July 2023
Published 31 July 2023 Volume 2023:18 Pages 4329—4346
DOI https://doi.org/10.2147/IJN.S418100
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Yan Shen
Yue Zhang,1,* Xue Han,1,* Ke Wang,1 Da Liu,1 Xiaoyun Ding,2 Zhiqiang Hu,2 Jing Wang1,3
1School of Pharmacy, Ningxia Medical University, Yinchuan, 750004, People’s Republic of China; 2Oncology Hospital, General Hospital of Ningxia Medical University, Yinchuan, 750004, People’s Republic of China; 3Key Laboratory of Ningxia Minority Medicine Modernization, Ministry of Education, Yinchuan, 750004, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Zhiqiang Hu, Oncology Hospital, General Hospital of Ningxia Medical University, 804 Shengli Street, Yinchuan, 750004, People’s Republic of China, Tel/Fax +86 951 674 3771, Email [email protected] Jing Wang, School of Pharmacy, Ningxia Medical University, 1160 Shengli Street, Yinchuan, 750004, People’s Republic of China, Tel/Fax +86 951 408 1046, Email [email protected]
Purpose: Immune checkpoint inhibitors (ICI) have received the most attention for triple negative breast cancer (TNBC), while the response rate to ICI remains limited due to insufficient T cell infiltration. It is therefore essential that alternative strategies are developed to improve the therapeutic outcomes of ICI in non-responsive TNBC cases. The efficacy of pH-responsive nanomicelles (P/A/B@NM) co-loaded with paclitaxel (PTX), CXCR4 antagonist AMD3100, and PD-1/PD-L1 inhibitor BMS-1 activating the T cell-mediated antitumor immune response were evaluated using a 4T1 antiPD-1-resistance breast tumor model.
Methods: In vitro, pH-responsive antitumor effect of P/A/B@NM was investigated by assessing cell viability, migration and invasion. In vivo, the distribution of P/A/B@NM was visualized in 4T1 orthotopic TNBC model using an IVIS spectrum imaging instrument. The efficacy of the co-delivery nanocarriers was evaluated by monitoring mouse survival, tumor growth and metastasis, cancer-associated fibroblasts (CAFs)-mediated tumor stroma and immunosuppressive microenvironment components, and the recruitment and infiltration of CD8+ T cells.
Results: The prepared P/A/B@NM in acid microenvironment demonstrates remarkable cytotoxicity against MDA-MB-231 cells, with an IC50 of 105 μg/mL. Additionally, it exhibits substantial inhibition of tumor cell migration and invasion. The P/A/B@NM based on co-delivery nanocarriers efficiently accumulate at the tumor site and release the drugs in a pH-responsive controlled manner. The nanomedicine-PTX, AMD3100, and BMS-1 formulation significantly inhibits tumor growth and lung/liver metastasis by inducing antitumor immune responses via CXCL12/CXCR4 axis blockade, and immunogenic cell death to reprogramme both tumor stroma and immunosuppressive microenvironment. As a result, CD8+ T cell infiltration is triggered into the tumor site, boosting the efficacy of ICI therapy synergistically.
Conclusion: These results demonstrate that combination therapy using P/A/B@NM reshapes CAFs-mediated tumor stroma and immunosuppressive microenvironment, which can enhance the infiltration of CD8+ T cells, thereby reactivating anti-tumor immunity for non-responsive TNBC cases.
Keywords: TNBC, immunotherapy, co-delivery nanomicelles, CAFs, immunosuppressive microenvironment
Introduction
Triple negative breast cancer (TNBC), the most aggressive subtype of breast cancer, is characterized by high intratumoral heterogeneity, a lack of established molecular targets, low overall survival rates, and high recurrence rates.1 While some metastatic TNBC patients have experienced clinical benefits from programmed cell death-1 (PD-1) or programmed cell death-ligand 1 (PD-L1) inhibitors through the innovation of T cell-mediated immunotherapies, the overall response rate to immune checkpoint inhibitors (ICI) remains limited due to lack of known biomarkers, insufficient T cell infiltration, the immunosuppressive tumor microenvironment, and innate and acquired drug resistance.2,3 These limitations motivate the development of new or alternative strategies to improve the therapeutic outcomes of ICI in non-responsive TNBC cases.
Substantial evidence has established that the tumor microenvironment (TME) is a self-regulating ecosystem, where tumor cells, stromal cells, tumor vasculature, and immune cells are interwoven with noncellular components. This TME not only drives tumor progression but also profoundly alter the therapeutic response of tumors to ICI.4,5 Currently, it is widely believed that the stromal matrix and suppressive immunocellular composition are the two primary factors causing immunosuppression in solid tumors.5,6 In invasive human breast cancer, CXCL12 secreted by cancer-associated fibroblasts (CAFs) and its specific receptor CXCR4, regulate tumor biological behaviors such as tumor angiogenesis, invasion and metastasis, and promote desmoplasia and solid stress in the tumor tissue, which serves as a physical barrier for the penetration of therapeutics and T cells.7–9 Furthermore, the CXCL12/CXCR4 axis leads to intra-tumoral recruitment of immunosuppressive lymphocytes, such as myeloid-derived suppressor cells (MDSCs), M2-phenotype macrophages, and regulatory T cells (Tregs).10,11 Preclinical trial results show that CXCR4 antagonists alone or in combination with ICI alter tumor-stroma interactions and reduce tumor growth and metastasis burden.12 However, this approach also leads to immune system risks and side effects of hematopoietic dysfunction in patients.
The utilization of nanotechnology to construct drug delivery systems is an effective approach in cancer treatment.13,14 Combining cancer immunotherapies using polymer nanocarriers has emerged as a promising strategy to enhance treatment efficacy while mitigating immune-related toxicity caused by off-target effects.15–17 Polymer carriers offer various drug loading options, including covalent coupling to form polymer-drug conjugates, encapsulation of drugs within polymer micelles, incorporation of drugs into polymer vesicles, dispersion of drugs within polymer gels, and more. Among these approaches, polymer nanomicelles are particularly noteworthy, which are formed through the self-assembly of amphiphilic block copolymers, exhibiting a unique core-shell structure that provides excellent drug loading capabilities, as well as targeting ability, biodegradability, and stealth properties during circulation in the bloodstream. Furthermore, multifunctional nanomicelles can be designed by incorporating targeting ligands and sensitive chemical bonds/groups, enhancing their accumulation at tumor sites and internalization into specific cells. These nanomicelles can respond to endogenous/exogenous stimuli (such as pH, enzymes, redox, hypoxia, light, heat, and ultrasound) at the tumor site, allowing for controlled drug release.18
pH-responsive block polymer nanomicelles can co-deliver multiple drug combinations into tumor sites to achieve multi-targeted spatiotemporal and sequential drug release, thereby optimizing therapeutic effect.19,20 In this study, we propose a potential immunotherapeutic strategy based on three-drug co-delivery nanomicelles that respond to the acid tumor microenvironment. Paclitaxel (PTX), a chemotherapy drug, exhibits dual effects in the treatment of cancer. It not only effectively hinders the proliferation of cancer cells by stabilizing microtubules and inhibiting cell mitosis but also induces immunogenic cell death (ICD). AMD 3100, a CXCR4 antagonist, blocks CXCL12/CXCR4 axis to interfere with the cross-talk between CAFs, tumor cells, and immune cells, thereby exerting immunomodulatory effects that synergistically amplify the antitumor immunity of PD-1/PD-L1 inhibitor BMS-1, as illustrated in Scheme 1. The co-delivery nanomicelles (named P/A/B@NM) can reduce tumor angiogenesis, remodel extracellular matrix (ECM), decrease the chemotaxis and trafficking of immunosuppressive cells, and enhance the response of infiltrating CD8+ T cells, thus improving the therapeutic effect of TNBC.
 |
Scheme 1 Schematic illustration of P/A/B@NM structure and strategy for enhancing antitumor immunity. |
Materials and Methods
Chemicals and Materials
PTX was procured from Shanghai Yuanye Bio-Technology Co., Ltd (Shanghai, China), while AMD3100 and BMS-1 (with a purity of ≥ 98%) were obtained from MCE (NJ, USA). Amphiphilic block copolymer methyl-polyethylene glycol-hyd-poly (lactic-co-glycolic acid) (mPEG-hyd-PLGA) (PEG, Mw: 5000 Da; PLGA, Mw: 5000 Da) and Cy5-PEG5000-hyd-PLGA5000 were purchased from Xi’an Ruixi Biological Technology Co., Ltd (Xian, China). Polyvinyl alcohol (PVA, Mw: 9000–10000Da, 80% hydrolyzed) was sourced from Sigma-Aldrich (MO, USA). Matrigel was obtained from Corning (NY, USA). Collagenase type I, collagenase type IV, DNAase I, and trypsin were purchased from Invitrogen (CA, USA). Purified water was produced using a Milli-Q system obtained from Millipore (MA, USA).
Preparation and Characterization of P/A/B@NM
Self-assembly-solvent evaporation method was used with minor modifications to prepare P/A/B@NM. Initially, 5 mg of block copolymer carrier mPEG-hdy-PLGA or Cy5-PEG-hdy-PLGA was mixed with PTX (1 mg), AMD3100 (1 mg), and BMS-1 (1 mg) in 1 mL of dichloromethane to form the oil phase. Simultaneously, 100 mL of 1% PVA solution was heated at 95 °C to form the water phase. Afterwards, the oil phase was slowly added to the water phase by ultrasonic emulsification under ice bath condition to form a uniform milky emulsion, of which the organic solvent was removed with magnetic stirrers for 4 h. Thereafter, the unincorporated PTX, AMD3100 and BMS-1 were removed by centrifugation at 3000 rpm for 30 min at 4 °C using ultrafiltration tubes (Millipore, USA) to obtain the nanomicelles (P/A/B@NM). The morphology, particle diameter, zeta potential and dispersion index (PDI) of prepared P/A/B@NM were measured by transmission electron microscope (TEM) and dynamic light scattering analyzer. The encapsulation efficiency (EE) and drug loading efficiency (DLE) of PTX, AMD3100, and BMS-1 were determined by high-performance liquid chromatography (HPLC) with a UV detector at a wavelength of 280 nm, 230 nm, and 280 nm, respectively.21,22
pH-Sensitive Drug Release of P/A/B@NM
The pH-sensitive drug release profile of P/A/B@NM under different conditions (pH 7.4, pH 6.5, and pH 5.0) was evaluated using the dialysis method in vitro. Briefly, three batches of fresh P/A/B@NM suspension were placed in cellulose dialysis bags (8000Da, Solarbio, Beijing, China). After tightening both ends of the dialysis bags, the test samples were placed in a corked conical flask containing 50 mL of dissolution medium and stirred at 37 °C, 120 rpm. At predetermined time points (0.5, 1, 2, 4, 6, 8, 12, 24, and 48 h), 1 mL of release solution was collected, and an equal volume of fresh dissolution medium was added. Then drug release of P/A/B@NM was analyzed by HPLC, and the cumulative release curves of PTX, AMD3100, and BMS-1 were calculated.
Cell Lines and Experimental Animals Model
The human TNBC cell line (MDA-MB-231) and the murine TNBC cell line (4T1) were both procured from the Chinese Academy of Sciences Committee on Type Culture Collection Cell Bank (Shanghai, China). MDA-MB-231 and 4T1 cells were cultured in DMEM and RPMI 1640 medium, respectively, and supplemented with 10% fetal bovine serum (FBS) and 1% penicillin-streptomycin. The cells were maintained in a 37 °C incubator with a 5% CO2 atmosphere.
Female BALB/C mice with a weight range of 16 ± 2 g and aged 6 to 8 weeks were obtained from Charles River (Beijing, China) and were housed and cared for at the Laboratory Animal Centre in accordance with the Animal Facility Guidelines of Ningxia Medical University. To establish the murine orthotopic TNBC model, 4T1 cells (5×105/mouse) were injected into the right fourth pair of mammary fat pads of BALB/C mice under isoflurane anesthesia. All animal procedures were performed in compliance with the Institutional Animal Care and Use Committee at Ningxia Medical University (Permit Number: IACUC-NYLAC-2021-156).
In vitro Evaluation of Cell Viability, Migration and Invasion
For the cell viability assay, MDA-MB-231 cells were seeded and cultured overnight in 96-well plates. Subsequently, the cells were exposed to escalating concentrations of free PTX, free AMD3100, free BMS-1, and P/A/B@NM cultured solution under varying conditions (pH 7.4 and pH 6.5) for a duration of 48 h. The final concentrations of PTX, AMD3100, and BMS-1 in each group were set at 0.1, 0.25, 0.5, 1, 2.5, 5, and 10 µg/mL. Following this treatment, the cell viabilities were accessed according to the Cell Counting Kit-8 (CCK-8) standard protocol.
For the migration assay, firstly, MDA-MB-231 cells were cultured and treated with 20 μg/mL of P/A/B@NM (containing 0.65 μg/mL of PTX, 0.67 μg/mL of AMD3100, and 0.69 μg/mL of BMS-1) cultured solution under pH 7.4 and pH 6.5 for 24 h. After that, 200 μL of cell suspension was added to the upper chamber of Transwell, while the lower chamber contained a medium containing 15% FBS and was cultured in an incubator for an additional 24–48 h. Finally, the culture solution in the upper chamber was poured out, the cells in the upper chamber were gently wiped with a cotton swab, and the chamber was fixed in 4% paraformaldehyde and stained with crystal violet. The migrating cells at the bottom of the chamber were photographed and the number of migrating cells was counted using Image J software. For the invasion experiment, 100 μL of Matrigel matrix was coated on the upper part of Transwell chamber in advance and placed in an incubator for 12 h. The remaining steps were similar to the aforementioned migration assay.
In vivo Biodistribution Study
The 4T1 orthotopic TNBC model was utilized to investigate the distribution of P/A/B@NM through in vivo imaging. Tumor-bearing mice were administered intravenous injections of free Cy5 dye and Cy5-labeled nanocarriers (Cy5-P/A/B@NM) (100 μL, 2 mg/mL). The mice were anesthetized and imaged at 2, 8, 12, 24, and 48 h using an IVIS spectrum imaging instrument (Caliper PerkinElmer, USA) with excitation/emission wavelengths of 630/680 nm. Subsequently, the mice’s major organs and tumor tissues were excised for ex vivo imaging, and the imaging system analysis software was used to analyze all of the results obtained.
In vivo Antitumor Efficacy Study
On the ninth day following the construction of the murine orthotopic TNBC model, the mice were randomized into five groups (n=6 per group): (i) Saline group; (ii) B@NM group (BMS-1 delivery nanomicelles); (iii) P/B@NM group (PTX and BMS-1 co-delivery nanomicelles); (iv) A/B@NM group (AMD3100 and BMS-1 co-delivery nanomicelles); and (v) P/A/B@NM group (PTX, AMD3100 and BMS-1 co-delivery nanomicelles). The mice were intravenously injected with 200 μL of Saline or nanomicelles (containing 5 mg/kg of PTX, 5 mg/kg of AMD3100, and 5 mg/kg of BMS-1) on days 9, 12, 15, 18, 21 and 26, respectively. In the survival experiment, the time points for intravenous administration were extended to 33, 40, and 47 days. During treatment, tumor volumes were recorded every three days, and calculated using the formula 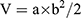 , where a and b represent the largest and the smallest diameter of the tumors. After day 30, the mice were sacrificed and the plasma was collected for further liver and kidney function analysis. The main organs and tumor tissues were excised, and tumor weights and lung metastatic tumor nodule counts were recorded. Some of these tissues were fixed in 4% paraformaldehyde, while others were stored at −80 °C for follow-up histological experiments.
, where a and b represent the largest and the smallest diameter of the tumors. After day 30, the mice were sacrificed and the plasma was collected for further liver and kidney function analysis. The main organs and tumor tissues were excised, and tumor weights and lung metastatic tumor nodule counts were recorded. Some of these tissues were fixed in 4% paraformaldehyde, while others were stored at −80 °C for follow-up histological experiments.
Immunofluorescence (IF), Immunohistochemistry (IHC) and Multi-Color IHC (m-IHC) Assay
For IF analysis, 4T1 and MDA-MB-231 cells were cultured and treated with 40 μg/mL of P/A/B@NM cultured solution under pH 7.4 and pH 6.5 for 24 h, while 1.3 μg/mL of free PTX was applied as positive control. Afterwards, the treated cells were incubated with the primary antibody against calreticulin (CRT) (1:200, Proteintech) and FITC-conjugated secondary antibody. Finally, the cells were stained with 4′, 6-diamidino-2-phenylindole (DAPI) and imaged using a fluorescence microscope.
For IHC and m-IHC assay, the paraffin sections of tumor tissues were first dewaxed and the antigens were repaired with sodium citrate (pH 6.0) using a microwave. The samples were then incubated with primary antibodies, including Ki-67 (1:100, Affinity Biosciences), CXCL12 (1:200, Proteintech), CXCR4 (1:200, Proteintech), VEGF-A (1:200, Proteintech), CD31 (1:1500, Proteintech), CRT (1:250, Proteintech), α-SMA (1:1000, Proteintech), Collagen Type Ι (1:800, Proteintech), CD8a (1:2000, Abcam), and FoxP3 (1:500, Abcam). The sections were then incubated with the horseradish peroxidase-conjugated secondary antibodies. For IHC assay, diaminobenzidine was used as the chromogen and the sections were counterstained with hematoxylin. For m-IHC analysis, the sections were incubated with TSA® substrate conjugated with fluorophores (Absin, Shanghai, China), and then the sections were sealed with DAPI-containing sealing solution before imaging with a confocal laser scanning microscope.
Enzyme-Linked Immunosorbent Assay (ELISA)
After fully homogenizing the isolated tumor tissues with a homogenizer, the samples were centrifuged for 20 min at 2000 rpm to collect the supernatant, which was passed through a 400-mesh cell sieve to perform content detection of cytokines and proteins, including CXCL12 (RK00168, ABclonal), CCL2 (EK287, MULTISCIENCES), VEGF-A (EK283, MULTISCIENCES), fibronectin (FN1) (EK03516, BOSTER), osteopontin (OPN) (EK2135, MULTISCIENCES), MMP9 (EK0466, BOSTER), HMGB1 (SEKM-0145, Solarbio), INF-γ (EK280, MULTISCIENCES), IL-2 (EK0932, BOSTER), IL-4 (EK0405, BOSTER), IL-10 (EK210, MULTISCIENCES), and TGF-β1 (EK981, MULTISCIENCES) using ELISA, according to the manufacturer instructions.
Flow Cytometry Analysis
For flow cytometry analysis, tumor tissues and spleen tissues from the mice were harvested, cut up and digested in hank’s balanced salt solution with collagenase type I (200 U/mL), collagenase type IV (200 U/mL), and DNAase I (100 μg/mL) to generate a single cell suspension. The resulting cells were stained with fluorescence-conjugated antibodies after the lysis of red blood cells and incubated with FcR Block (553141, BD Pharmingen) following the manufacturer’s protocol. For cell surface staining, the single cell suspension was incubated with fluorescence-conjugated antibodies including CD3-PerCP-Cy5.5 (551163, BD Pharmingen), CD4-PE (557308, BD Pharmingen), CD8a-FITC (553030, BD Pharmingen), CD25-BV421 (562606, BD Pharmingen), CD11b-FITC (557396, BD Pharmingen), Gr-1-PE (553128, BD Pharmingen), F4/80-PE (123110, Biolegend), CD86-PE-Cy7 (105014, Biolegend), and CD206-Alexa Fluor 647 (141712, Biolegend). For intracellular FoxP3 staining, cells were fixed and permeabilized in Cytofix/Cytoperm solution (562574, BD Pharmingen) for 15 min at 4 °C and then stained with FoxP3-Alexa Fluor 647 (560401, BD Pharmingen) in the dark for 30 min in staining buffer. Afterwards, the distribution and infiltration of immune cells including lymphocytes, MDSCs, macrophage subsets and Tregs cells in tumor tissues and spleen tissues were analyzed on flow cytometry. And all the data were analyzed using FlowJo software.
Statistical Analysis
All experiments were conducted in triplicate, and representative results are presented as mean ± SEM. Data analysis was performed using Origin 8.0 or SPSS 24.0 software. A P-value less than 0.05 (*), 0.01 (**), or 0.001 (***) was considered statistically significant.
Results
Characterization and Anti-Tumor Evaluation of P/A/B@NM in vitro
The preparation of P/A/B@NM was carried out by a one-step self-assembly-solvent evaporation method with optimized process conditions. The pH-responsive polymer micelles successfully encapsulated three hydrophobic drugs (PTX, AMD3100, and BMS-1) in their core. The size and morphology of the obtained P/A/B@NM were determined by TEM with a uniform spheroid shape and a size of 145 ± 4.5 nm (Figure 1A), while the size of unloaded nanomicelles presented 46 ± 5.4 nm (Figure 1B). Moreover, the hydrodynamic diameter of P/A/B@NM determined by dynamic light scattering was 202 ± 5.6 nm, with zeta potential of −1.14 ± 0.20 mV and PDI of 0.197 ± 0.007 (Figure 1C and D). P/A/B@NM exhibited good colloidal stability, maintaining their diameter within 24 h in RPMI1640 medium containing 10% FBS. The EE of PTX, AMD3100, and BMS-1 was calculated by HPLC analysis and reached 65.90 ± 1.47%, 66.03 ± 0.42%, and 67.00 ± 1.14%, respectively, for P/A/B@NM. The DLE of PTX, AMD3100, and BMS-1 was 3.92 ± 0.13%, 4.03 ± 0.05%, and 4.17 ± 0.03% for P/A/B@NM, respectively (Figure 1E).
 |
Figure 1 Characterization and anti-tumor evaluation in vitro of P/A/B@NM. (A and B) TME observation of P/A/B@NM and unloaded nanomicelles. (C and D) Size distribution, zeta potential and PDI determination by dynamic light scattering (n=3). (E) Encapsulation efficiency and drug loading efficiency measure by HPLC (n=3). (F–H) pH-responsive release of PTX, AMD3100 and BMS-1. (I) Cell viability assay of free PTX, free AMD3100, free BMS-1, and P/A/B@NM in normal physiological environment and acidic microenvironment (n=5). (J) Acidic microenvironment-responsive cell migration and invasion assay of P/A/B@NM (n=3). Error bars represent means ± SEM. ***P < 0.001. |
The acid tumor microenvironment can be used as endogenous stimulation to make drugs controllably release. Accordingly, we investigated the pH-sensitive drug release of P/A/B@NM in vitro at pH values of 7.4, 6.5, and 5.0. As depicted in Figure 1F–H, the release rates of PTX, AMD3100 and BMS-1 from P/A/B@NM were significantly affected by the pH values. Compared to 23.1% at pH 7.4, the cumulative release of PTX from P/A/B@NM was 49.9% at pH 6.5 and 58.7% at pH 5.0 after 48 h release. Similarly, the cumulative release of AMD3100 and BMS-1 from P/A/B@NM at pH 6.5 was 82.4% and 83.4%, respectively, with a lower release rate at pH 7.4, and no burst release was observed, indicating comparative stability under neutral conditions. The release of PTX, AMD3100, and BMS-1 was accelerated upon reducing the pH values to 6.5 and 5.0, suggesting that hydrazone bond breakage occurred in acidic conditions. Moreover, the pH-responsive antitumor effect of P/A/B@NM on MDA-MB-231 cells was assessed. As depicted in Figure 1I, compared to the cell viability of approximately 64.1 ± 1.5% observed with free PTX treatment, P/A/B@NM at pH 7.4 and pH 6.5 reduced cell viabilities to 49.8 ± 1.9% and 40.1 ± 0.8%, respectively, following 48 h of incubation at an equivalent PTX concentration of 10 μg/mL. These findings suggest that the nano-delivery system effectively increased the intracellular concentration of PTX, while the acidic microenvironment promoted the cytotoxicity mediated by AMD3100. In contrast, when compared to the cell viability of approximately 39.2 ± 1.3% observed with P/A/B@NM (pH 6.5) treatment, the diminished cytotoxic effects of free AMD3100 and P/A/B@NM (pH 7.4) were attributed to the concentration saturation effect of AMD3100 at a dosage of 10 μg/mL. Additionally, BMS-1 did not exhibit significant cytotoxic effects on MDA-MB-231 cells due to the absence of immune cells. Based on the aforementioned results, it can be concluded that P/A/B@NM, in an acidic microenvironment, demonstrates the most potent cytotoxicity against MDA-MB-231 cells, with an IC50 value of 105 μg/mL. Furthermore, P/A/B@NM at pH 6.5 significantly inhibited the migration and invasion ratio of MDA-MB-231 cells with 41.3 ± 3.8% and 35.9 ± 4.1%, respectively, compared to 75.4 ± 1.3% and 76.8 ± 2.6% of P/A/B@NM at pH 7.4 (Figure 1J). These results demonstrate the prepared P/A/B@NM has pH-sensitive drug release properties, providing a foundation for its application in vivo to respond to the acidic tumor microenvironment.
Co-Delivery Nanomicelles Based P/A/B@NM Inhibited Tumor Growth and Metastasis with Therapeutic Safety
Efficient delivery of nanomedicine to tumor sites is a crucial factor in evaluating their therapeutic efficacy. Thus, we first examined the distribution and accumulation of Cy5-P/A/B@NM at tumor sites over time. As depicted in Figure 2A and B, the fluorescence intensity or relative radiant efficiency of Cy5-P/A/B@NM increased significantly with time compared to free dye Cy5, indicating that Cy5-P/A/B@NM selectively accumulated in solid tumors through enhanced permeability and retention effect, while Cy5 alone had difficulty accumulating at the tumor site due to its free molecular diffusion. Furthermore, the images and relative radiant efficiency of Cy5-P/A/B@NM in isolated organs and tumor tissues highly demonstrated major accumulation in the tumor site, albeit with some nonspecific distribution in the liver due to undergoing opsonation (Figure 2C and D).
 |
Figure 2 Distribution and in vivo imaging of Cy5-P/A/B@NM in 4T1 orthotopic TNBC mice. (A and B) Distribution and relative radiant efficiency of free dye Cy5 and Cy5-P/A/B@NM at various times in 4T1 orthotopic TNBC mice (n=3). (C and D) Ex vivo imaging and relative radiant efficiency of Cy5 and Cy5-P/A/B@NM in isolated major organs and tumor (n=3). Error bars represent means ± SEM. *P < 0.05, **P < 0.01, ***P < 0.001. |
Furthermore, we evaluated the therapeutic efficacy of P/A/B@NM based combination therapy using a murine 4T1 orthotopic tumor model (Figure 3A). The percent survival results revealed that P/B@NM, A/B@NM and P/A/B@NM based combination therapy significantly prolonged the survival time of tumor-bearing mice, with percent survival of 20%, 40%, and 70% at 51 d, respectively. In contrast, the percent survival of B@NM based monotherapy was 20% at 45 d and 0% at 51 d (Figure 3B). In comparison to the Saline group and the B@NM-treated group, treatment with P/A/B@NM exhibited a substantial inhibition of primary tumor growth. Notably, the tumor volume and tumor weight of mice in the P/B@NM-treatment group and the A/B@NM-treatment group were 1.6-fold, 1.4-fold, 1.5-fold, and 1.2-fold higher, respectively, than those in the P/A/B@NM group (Figure 3C–F). Moreover, the results of tumor histopathological analysis showed that tumor cells in the Saline and B@NM-treated groups exhibited pleomorphism with an imbalance of nucleoplasmic ratio and hyperchromatic nuclei, while tumor cells in the P/A/B@NM-treated group showed typical apoptosis characteristics, such as cytoplasmic shrinkage and nuclear chromatin condensation, with the lowest cell proliferation index by Ki-67 analysis (Figure 3G and H). In comparison to the Saline group and the B@NM-treated group, treatment with P/A/B@NM exhibited a significant suppression of metastasis formation in the lung tissues of tumor-bearing mice (Figure 4A and B). Remarkably, the number of pulmonary metastatic nodules in the P/B@NM-treatment group and the A/B@NM-treatment group was 2.5-fold and 2-fold higher, respectively, than that in the P/A/B@NM-treated group. Additionally, histopathological analysis demonstrated a significant reduction in the size of metastatic lesions in the lungs, as well as the number and size of metastatic lesions in the liver upon treatment with P/A/B@NM combination therapy (Figure 4C). Notably, pathological analysis of the heart, spleen, and kidney in P/A/B@NM-treated groups showed no remarkable pathological lesions compared to those in the Normal group (Figure S1A). Moreover, a serum biochemical analysis indicated that after treatment with P/A/B@NM, all of the major indicators of liver and kidney function, including glutamic aminotransferase (ALT), aspartate aminotransferase (AST), serum creatinine (SCR), and blood urea nitrogen (BUN), remained within the normal range (Figure S1B). These results demonstrate that P/A/B@NM combination therapy achieves optimal therapeutic efficacy with high therapeutic safety.
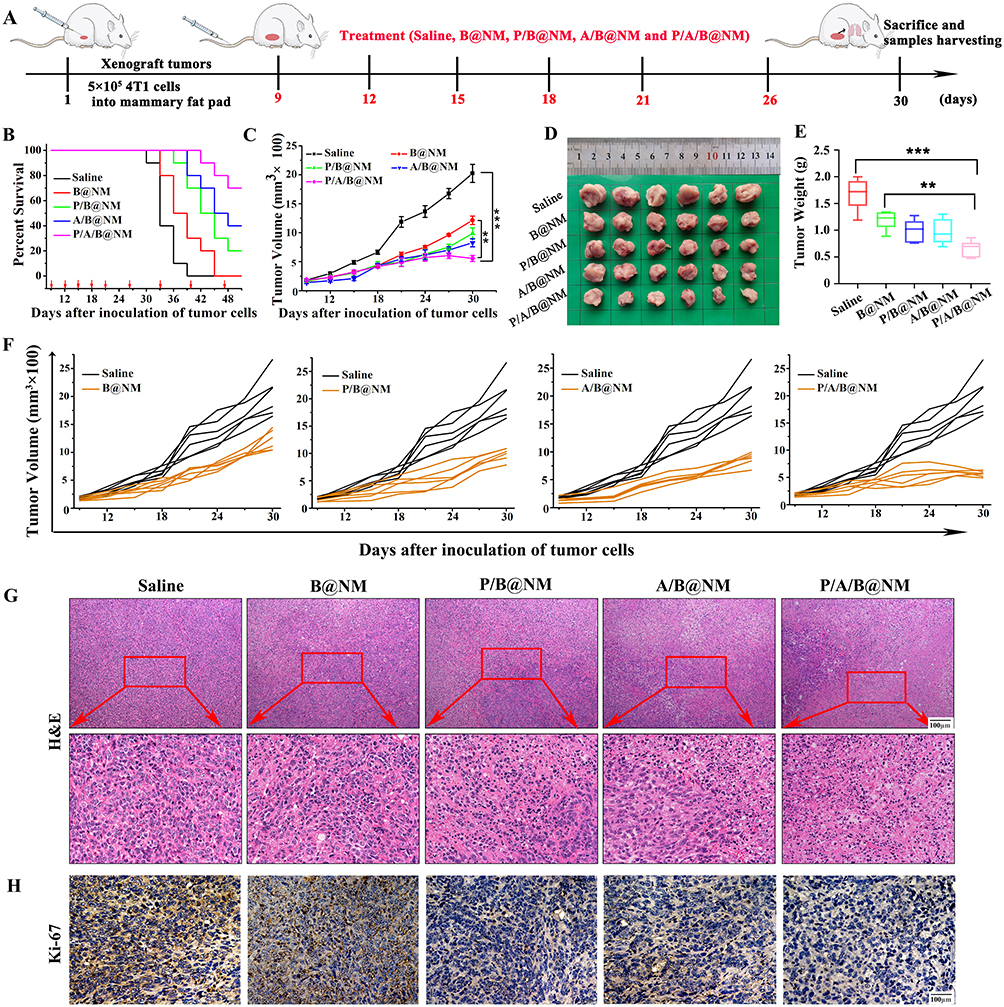 |
Figure 3 In vivo antitumor efficacy and survival study of co-delivery nanomicelles based combination therapy. (A) Establishment of murine orthotopic TNBC model and drug treatment. (B) Survival time monitoring of tumor-bearing mice after treatment. (C–F) Tumor volume and weight measure in different treatment groups (n=6). (G) Histological change of tumor tissues in different treatment groups. (H) Proliferation of tumor cells by Ki-67 of tumor tissues in different treatment groups. Error bars represent means ± SEM. **P < 0.01, ***P < 0.001. |
 |
Figure 4 Co-delivery nanomicelles based combination therapy inhibit the lung and liver metastasis of tumor cells in 4T1 orthotopic TNBC mice. (A) The representative photographs of tumor metastasis in lungs of mice. (B) Effect of co-delivery nanomicelles on lung metastasis nodules in mice (n=6). (C) Pathological analysis of metastasis in lung tissues and liver tissues after co-delivery nanomicelles treatment in orthotopic TNBC mice. Error bars represent means ± SEM. **P < 0.01, ***P < 0.001. |
Co-Delivery Nanomicelles Based P/A/B@NM Potentiated TNBC Immunotherapy by Reshaping CAFs-Mediated Tumor Stromal Microenvironment
Abundant CAFs in TME play a dual role in promoting tumor metastasis and inhibiting immune cell function by secreting various cytokines. As depicted in Figure 5A, P/A/B@NM-based combination therapy significantly downregulated the expression of CXCL12, CXCR4, and VEGF-A, which is extensively secreted by CAFs to trigger and magnify tumor angiogenesis. Consequently, the nanomedicine-PTX, AMD3100, and BMS-1 formulation markedly reduced tumor microvascular density, as evidenced by CD31 analysis (Figure S2). Moreover, the ELISA results revealed that the production of CXCL12, CCL2, and VEGF-A originating from CAFs was significantly decreased in the P/A/B@NM-treated group compared to the Saline group and B@NM-treated group (Figure 5B).
 |
Figure 5 Co-delivery nanomicelles based combination therapy disturbed the function of CAFs by reducing the cytokines and reshaping ECM. (A) Representative images of CXCL12, CXCR4, and VEGF-A by IHC in different treatment groups. (B). ELISA result showing intratumoral content of CXCL12, IL-6, and VEGF-A in different treatment groups (n=3). (C) Representative images of collagen fibers by Masson and Sirius red staining in different treatment groups. (D) ELISA result showing intratumoral content of FN1, OPN, and MMP9 in different treatment groups (n=3). Error bars represent means ± SEM. **P < 0.01, ***P < 0.001. |
Furthermore, we investigated whether targeting the CXCL12/CXCR4 axis could improve ICI responsiveness by reshaping the dense collagen matrix produced by CAFs. The results of Masson staining and Sirius Red staining in Figure 5C showed that the volume and density of collagen fibers were significantly decreased after administration of P/B@NM, A/B@NM, and P/A/B@NM, compared to Saline group and B@NM-treatment group. Additionally, the content of FN1, OPN, and MMP9 in primary tumor stroma of P/A/B@NM was downregulated, with 4.8-fold, 3.3-fold, and 4.3-fold lower levels than those in the B@NM-treated group, respectively (Figure 5D). To further investigate the role of CAFs on tumor-infiltrating CD8+ T cells, we assessed whether P/A/B@NM based combination therapy could revitalize CD8+ T cells by reducing α-SMA+ CAFs. As presented in Figure 6, the TME in the Saline group presented abundant α-SMA+ CAFs and strong deposition of Collagen I, which resulted in a CD8+ T cell exclusion phenomenon. Even after B@NM treatment, CD8+ T cell infiltration did not improve. Although treatment with P/B@NM and A/B@NM lowered the expression levels of ECM components (Collagen I) and increased the expression of CD8a, P/A/B@NM based combination therapy significantly enhanced the infiltrating level of CD8+ T cells by decreasing the activation of α-SMA+ CAFs.
 |
Figure 6 Co-delivery nanomicelles based combination therapy disturbed the function of α-SMA+ CAFs to increase intratumoral infiltration of CD8+ T cells. |
The data above illustrates that combination therapy using P/A/B@NM has the ability to reshape stromal microenvironment by disturbing the secretion and functions of CAFs. This is evidenced by a reduction in tumor stroma formation, and decompression of blood vessels. As a result, there is a decrease in immunosuppression and an increase in the infiltration of CD8+ T cells into the TME, thereby reactivating anti-tumor immunity.
Co-Delivery Nanomicelles Based P/A/B@NM Potentiated TNBC Immunotherapy by Reprogramming the Immunosuppressive Microenvironment
In TNBC, tumor progression and therapeutic outcome are influenced by complex immunosuppressive microenvironment. To investigate whether the nanomedicine-PTX, AMD3100 and BMS-1 formulation could promote immune infiltration of CD8+ T cells into the tumor, we first observed distribution of regulatory T cells and effector T cells. As illustrated in Figure 7A, the m-IHC analysis revealed a significant increase in the recruitment and infiltration of CD8+ T cells accompanied by a visible reduction infiltration of FoxP3+ Tregs cells in P/A/B@NM-treated group. Additionally, compared to B@NM-treated group, the percentage change of CD8+ T cells and FoxP3+ Tregs cells followed the same trend after P/A/B@NM-based combination therapy, as observed in the flow cytometry analysis (Figure 7B). Moreover, PTX, often used in combination with PD-1/PD-L1 inhibitors in the treatment of metastatic TNBC, has been reported to induce ICD of tumor cells, which may enhance the sensitivity of tumors to immunotherapy. Our results indicate that P/A/B@NM not only promotes the release of adenosine triphosphate (ATP) and high mobility group protein B1 (HGMB1) (Figure S3A and S3B), but also promotes the exposure of calreticulin (CRT) to the surface of 4T1 and MDA-MB-231 cells (Figure S3C). Similarly, P/A/B@NM significantly increased the exposure of CRT to the surface of tumor cells in primary tumor tissue, compared to the Saline group and B@NM group (Figure S3D). These biomarkers suggest that ICD induced by P/A/B@NM may promote the recruitment and activation of CD8+ T cells.
 |
Figure 7 Co-delivery nanomicelles based combination therapy decreased the number of FoxP3+ Tregs cells to increase intratumoral infiltration of CD8+ T cells. (A) Representative images of FoxP3 and CD8a using m-IHC analysis in different treatment groups. (B) Representative cytometric dot plots of FoxP3+ Tregs cells and CD3+CD8+ T cells in tumors (left) and percentage of intratumoral Tregs cells and CD8+ T cells in different treatment groups (right) (n=3). Error bars represent means ± SEM. **P < 0.01, ***P < 0.001. |
Furthermore, we investigated the infiltration of immune cells and the secretion of key immunomodulatory cytokines. As illustrated in Figure 8A, there was a notable reduction in the proportion of M2-TAMs and MDSCs among total live cells in the group treated with P/A/B@NM, compared to the Saline and B@NM-treated groups. In contrast, the populations of CD4+ T and M1 macrophages (F4/80+CD86hi) in tumors treated with B@NM were significantly lower than in the P/A/B@NM-treated group. These changes in the immune cell infiltration also led to decreased secretion levels of downstream cytokines, such as TGF-β1, IL-10, and IL-4, which can inhibit antigen presentation and T cell activation. Additionally, the secretion levels of IFN-γ and IL-12, which promote antitumor immune responses, were increased in tumor tissues after P/A/B@NM treatment (Figure 8B). The significant increase in IL-12 expression was consistent with the abundance of CD8+ T cells (Figure 7). Similarly, we observed a comparable proportion change of immunosuppressive cells (Tregs and MDSCs) and effector cells (CD8+ T and CD4+ T) in spleens after P/A/B@NM administration (Figure S4A and S4B).
 |
Figure 8 Co-delivery nanomicelles based combination therapy reprograms the tumor immunosuppressive microenvironment to enhance intratumoral T-cell infiltration and effector T-cell function. (A) Representative cytometric dot plots of CD3+CD4+ T cells, F4/80+CD86+M1-TAMs cells, F4/80+CD206+M2-TAMs cells, and CD11b+Gr-1+ MDSCs in tumors (left) and the proportions of CD4+ T cells, M1-TAMs cells, M2-TAMs cells, and MDSCs in tumors in different treatment groups (right) (n=3). (B) ELISA results showing TGF-β1, IL-10, IL-4, INF-γ, and IL-12 content in the tumors (n=3). Error bars represent means ± SEM. *P < 0.05, **P < 0.01, ***P < 0.001. |
These findings demonstrate that P/A/B@NM-based combination therapy efficiently reverses the immunosuppressive tumor microenvironment and activates the T cell-mediated antitumor immune response.
Discussion
Combinatorial cancer immunotherapies using tumor microenvironment responsive nanocarriers have emerged as a promising strategy to achieve potentiated treatment efficiency. Our prepared co-delivery nanomicelles (P/A/B@NM) has pH-sensitive drug release properties, which is evidenced by the accelerated release of PTX, AMD3100, and BMS-1 upon reducing the pH values to 6.5 and 5.0, and the enhanced capabilities of inducing cytotoxicity and inhibiting cell migration and invasion under pH 6.5 culture condition. pH-responsive polymer micelles containing hydrazone bonds remain stable in blood circulation, but their nanocarrier structure breaks down in the acidic microenvironment of tumors (pH 5.8~7.2), leading to effective drug release at tumor sites and improved therapeutic efficacy.23,24
Furthermore, we evaluated the antitumor efficacy of P/A/B@NM using a murine 4T1 orthotopic tumor model, which is reported to be resistant to anti-PD-1/PD-L1-immunotherapy.25 Our findings revealed that B@NM-based monotherapy has a disadvantage in inhibiting primary tumor growth and metastasis, while P/A/B@NM-based combination therapy significantly improves the survival time and inhibits tumor growth and lung/liver metastasis. Clinical data shows that TNBC is more sensitive to ICI therapy than other breast cancer subtypes. However, ICI monotherapy is not recommended due to its low therapeutic response rate.26 Therefore, combination therapy with ICI and chemotherapy or targeted therapy is preferred to enhance the efficacy of TNBC immunotherapy.27 Our co-delivery nanomicelles were designed to achieve optimal therapeutic efficacy with high therapeutic safety by combining PTX, AMD3100, and BMS-1 therapies to produce much stronger antitumor effects through multitarget synergism.
In TME, stromal cells interact with immune cells and ECM to promote the formation and stability of immunosuppressive microenvironment. As an important part of TME, CAFs participates in ECM remodeling and stimulates the differentiation and function of immune cells by secreting cytokines, ultimately affecting the immune response process.28 Previous studies have suggested that the exclusion of T lymphocytes by CAFs may be driven in part by CXCL12/CXCR4 signaling axis.9,29 Our results confirmed that targeting the CXCL12/CXCR4 axis using P/A/B@NM co-delivery nanomicelles increased the infiltration of CD8+ T cells into the TME through disturbing the secretion function of CAFs. It is found that CAFs secrete CXCL12, TGF-β and VEGF-A to promote Tregs recruitment, migration, and FoxP3+ Tregs differentiation, while FoxP3+ Tregs promote CD8+ T cell dysfunction by secreting IL-35 and IL-10.30 Additionally, CAFs produces CXCL12 and CCL2 to promote monocyte recruitment and induce TAMs to differentiate into M2 phenotype, which damages effector T cell function.31 Moreover, the complete infiltration of T cells into tumors depends not only on the recruitment of appropriate chemokines but also on the regulation of tumor blood vessels. Excessive production of VEGF-A by tumors and CAFs stimulates endothelial cell proliferation, leading to new angiogenesis, often accompanied by impaired tissue perfusion and increased vascular permeability, ultimately preventing T cell migration to TME.32,33
Furthermore, the dense collagen matrix produced by CAFs may act as a physical permeability barrier to the infiltration of T lymphocytes, reducing the efficacy of tumor immunotherapy.9,34 CAFs secretes TGF-β, collagen, MMPs and hyaluronic acid to reshape ECM, thereby increasing rigidity.35 It has been reported that excessive pressure of ECM components on blood vessels may hinder T-cell infiltration and interaction with tumor cells in space.36 Moreover, activated fibroblasts that produce dense matrix are indicative of a poor response to ICI in cancer patients.37 Our results verified that treatment with P/A/B@NM lowered the expression levels of ECM components and increased the infiltrating level of CD8+ T cells by decreasing the activation of α-SMA+ CAFs. These results confirmed that targeting the CXCL12/CXCR4 axis using P/A/B@NM can enhance ICI responsiveness by a reduction in tumor stroma formation, decompression of blood vessels and a decrease in immunosuppression.
The complex tumor immunosuppressive microenvironment is coordinated by multiple immunosuppressive signals in the regulatory network. The presence of immunoregulatory cytokines in the TME can recruit immunosuppressive cells. Tregs, TAMs, and MDSCs are the primary sources of regulatory cytokines. Studies have shown that Tregs inhibit DC antigen presentation and CD4+ T helper cell function, while TAMs restrict CD8+ T cell motility from infiltrating into tumor islets through long-lasting contact.38,39 MDSCs inhibit the trafficking of effector T lymphocytes into the tumors and release reactive oxygen species that remove the key nutrients necessary for T cell proliferation, thereby suppressing T cell function.40 Among various immunosuppressive cytokines, TGF-β, IL-10 and IL-4 play a significant role in the TME, primarily responsible for suppressing the immune response. In our study, combinatorial cancer immunotherapies using P/A/B@NM facilitates M1 macrophages polarization, eliminates immunosuppressive cells (such as Tregs and MDSCs), downregulates the expression of immunosuppressive cytokines (TGF-β1, IL-10, and IL-4), and upregulates the expression of immunostimulatory cytokines (IFN-γ and IL-12). Altogether, the prepared nanomedicine-PTX, AMD3100 and BMS-1 formulation, in which PTX enhances cytotoxicity and induces ICD, AMD 3100 blocks CXCL12/CXCR4 axis to alter the TME and revert the tolerogenic polarization of the TME by interfering with the cross-talk between CAFs, tumor cells and immune cells, and BMS-1 blocks PD-1/PD-L1 signaling to reduce T cell depletion, synergistically activates and amplifies the T cell-mediated antitumor immunity.
Conclusion
To overcome the poor therapeutic outcomes of ICI in non-responsive TNBCs, we designed and prepared pH-responsive nanomicelles (P/A/B@NM) co-loaded with PTX, AMD3100, and BMS-1. The P/A/B@NM demonstrates a pH-responsive antitumor effect in vitro by effectively inhibiting the proliferation of tumor cells, with an IC50 value of 105 μg/mL. Additionally, it significantly suppresses the migration and invasion of tumor cells, achieving inhibition ratios of 57% and 65%, respectively. The co-delivery nanomicelles efficiently accumulate at the tumor site and release the agents in a controlled manner. Our in vivo findings unequivocally illustrate that P/A/B@NM notably prolongs the survival time, effectively inhibits tumor growth with a remarkable tumor inhibition rate of approximately 57%, and suppresses lung metastasis by an impressive inhibition rate of 83%. These outcomes are attributed to the induction of robust antitumor immune responses in a murine model of 4T1 breast tumors. Importantly, our study shows that the blockade of CXCL12/CXCR4 axis and induction of ICD via the nanomedicine-PTX, AMD3100, and BMS-1 formulation leads to the reprogramming of both stromal and immune microenvironments, subsequently triggering CD8+ T cell infiltration into the tumor site. This synergistically enhances the efficacy of PD-1/PD-L1 therapy.
Abbreviations
ATP, adenosine triphosphate; AST, aspartate aminotransferase; BUN, blood urea nitrogen; CRT, calreticulin; CAFs, cancer-associated fibroblasts; DLE, drug loading efficiency; DAPI, 4′, 6-diamidino-2-phenylindole; ECM, extracellular matrix; EE, encapsulation efficiency; ELISA, Enzyme-Linked Immunosorbent Assay; FBS, fetal bovine serum; FN1, fibronectin 1; ALT, glutamic aminotransferase; HPLC, high-performance liquid chromatography; HGMB1, high mobility group protein B1; ICI, Immune checkpoint inhibitors; ICD, immunogenic cell death; IF, Immunofluorescence; IHC, Immunohistochemistry; m-IHC, Multi-color IHC; MDSCs, myeloid-derived suppressor cells; PD-1, programmed cell death-1; PD-L1, programmed cell death-ligand 1; PTX, paclitaxel; PVA, Polyvinyl alcohol; PDI, potential and dispersion index; SCR, serum creatinine; Tregs, regulatory T cells; TNBC, triple negative breast cancer; TME, tumor microenvironment; TEM, transmission electron microscope.
Acknowledgments
This work was supported by the National Natural Science Foundation of China (NSFC Grant No. 81960559) and the Natural Science Foundation of Ningxia (Grant No. 2022AAC05026).
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Disclosure
The authors report no conflicts of interest.
References
1. Bianchini G, Balko JM, Mayer IA, et al. Triple-negative breast cancer: challenges and opportunities of a heterogeneous disease. Nat Rev Clin Oncol. 2016;13(11):674–690. doi:10.1038/nrclinonc.2016.66
2. Keenan TE, Tolaney SM. Role of immunotherapy in triple-negative breast cancer. J Natl Compr Canc Netw. 2020;18(4):479–489. doi:10.6004/jnccn.2020.7554
3. Henriques B, Mendes F, Martins D. Immunotherapy in breast cancer: when, how, and what challenges? Biomedicines. 2021;9(11):1687. doi:10.3390/biomedicines9111687
4. Park M, Kim D, Ko S, et al. Breast cancer metastasis: mechanisms and therapeutic implications. Int J Mol Sci. 2022;23(12):6806. doi:10.3390/ijms23126806
5. Zhang Y, Zhang Z. The history and advances in cancer immunotherapy: understanding the characteristics of tumor-infiltrating immune cells and their therapeutic implications. Cell Mol Immunol. 2020;17(8):807–821. doi:10.1038/s41423-020-0488-6
6. Rømer AMA, Thorseth ML, Madsen DH. Immune modulatory properties of collagen in cancer. Front Immunol. 2021;12:791453. doi:10.3389/fimmu.2021.791453
7. Teicher BA, Fricker SP. CXCL12 (SDF-1)/CXCR4 pathway in cancer. Clin Cancer Res. 2010;16(11):2927–2931. doi:10.1158/1078-0432.CCR-09-2329
8. Mhaidly R, Mechta-Grigoriou F. Role of cancer-associated fibroblast subpopulations in immune infiltration, as a new means of treatment in cancer. Immunol Rev. 2021;302(1):259–272. doi:10.1111/imr.12978
9. Chen IX, Chauhan VP, Posada J, et al. Blocking CXCR4 alleviates desmoplasia, increases T-lymphocyte infiltration, and improves immunotherapy in metastatic breast cancer. Proc Natl Acad Sci U S A. 2019;116(10):4558–4566. doi:10.1073/pnas.1815515116
10. Mezzapelle R, Leo M, Caprioglio F, et al. CXCR4/CXCL12 activities in the tumor microenvironment and implications for tumor immunotherapy. Cancers. 2022;14(9):2314. doi:10.3390/cancers14092314
11. Zhou W, Guo S, Liu M, et al. Targeting CXCL12/CXCR4 axis in tumor immunotherapy. Curr Med Chem. 2019;26(17):3026–3041. doi:10.2174/0929867324666170830111531
12. Zeng Y, Li B, Liang Y, et al. Dual blockade of CXCL12-CXCR4 and PD-1-PD-L1 pathways prolongs survival of ovarian tumor-bearing mice by prevention of immunosuppression in the tumor microenvironment. FASEB J. 2019;33(5):6596–6608. doi:10.1096/fj.201802067RR
13. De Vita A, Liverani C, Molinaro R, et al. Lysyl oxidase engineered lipid nanovesicles for the treatment of triple negative breast cancer. Sci Rep. 2021;11(1):5107. doi:10.1038/s41598-021-84492-3
14. Gabellone S, Piccinino D, Filippi S, et al. Lignin nanoparticles deliver novel thymine biomimetic photo-adducts with antimelanoma activity. Int J Mol Sci. 2022;23(2):915. doi:10.3390/ijms23020915
15. Xue J, Li R, Gao D, et al. CXCL12/CXCR4 axis-targeted dual-functional nano-drug delivery system against ovarian cancer. Int J Nanomedicine. 2020;15:5701–5718. doi:10.2147/IJN.S257527
16. Kim D, Lee SS, Moon H, et al. PD-L1 targeting immune-microbubble complex enhances therapeutic index in murine colon cancer models. Pharmaceuticals. 2020;14(1):6. doi:10.3390/ph14010006
17. Zhao R, Liu J, Li Z, et al. Recent advances in CXCL12/CXCR4 antagonists and nano-based drug delivery systems for cancer therapy. Pharmaceutics. 2022;14(8):1541. doi:10.3390/pharmaceutics14081541
18. Uthaman S, Huh KM, Park IK. Tumor microenvironment-responsive nanoparticles for cancer theragnostic applications. Biomater Res. 2018;22(1):22. doi:10.1186/s40824-018-0132-z
19. Zhu SW, Ye M, Ma X, et al. pH-responsive nanoprodrugs combining a Src inhibitor and chemotherapy to potentiate antitumor immunity via pyroptosis in head and neck cancer. Acta Biomater. 2022;154:497–509. doi:10.1016/j.actbio.2022.10.051
20. Xie X, Chen Y, Chen Z, et al. Polymeric hybrid nanomicelles for cancer theranostics: an efficient and precise anticancer strategy for the codelivery of Doxorubicin/miR-34a and magnetic resonance imaging. ACS Appl Mater Interfaces. 2019;11(47):43865–43878. doi:10.1021/acsami.9b14908
21. Sokol M, Zenin V, Yabbarov N, et al. Validated HPLC method for paclitaxel determination in PLGA submicron particles conjugated with α-fetoprotein third domain: sample preparation case study. Ann Pharm Fr. 2021;79(5):500–510. doi:10.1016/j.pharma.2021.02.001
22. Bapatu HR, Maram RK, Murthy RS. Robust and rugged stability-indicating HPLC method for the determination of plerixafor and its related impurities in drug substances. J Chromatogr Sci. 2015;53(9):1432–1442. doi:10.1093/chromsci/bmv029
23. Liang Y, Su Z, Yao Y, et al. Preparation of pH sensitive pluronic-docetaxel conjugate micelles to balance the stability and controlled release issues. Materials. 2015;8(2):379–391. doi:10.3390/ma8020379
24. Su WT, Huang CC, Liu HW. Evaluation and preparation of a designed kartogenin Drug Delivery System (DDS) of hydrazone-linkage-based pH responsive mPEG-Hz-b-PCL nanomicelles for treatment of osteoarthritis. Front Bioeng Biotechnol. 2022;10:816664. doi:10.3389/fbioe.2022.816664
25. Mao C, Yeh S, Fu J, et al. Delivery of an ectonucleotidase inhibitor with ROS-responsive nanoparticles overcomes adenosine-mediated cancer immunosuppression. Sci Transl Med. 2022;14(648):eabh1261. doi:10.1126/scitranslmed.abh1261
26. Wang X, Wang F, Zhong M, et al. The biomarkers of hyperprogressive disease in PD-1/PD-L1 blockage therapy. Mol Cancer. 2020;19(1):81. doi:10.1186/s12943-020-01200-x
27. Hayashi H, Nakagawa K. Combination therapy with PD-1 or PD-L1 inhibitors for cancer. Int J Clin Oncol. 2020;25(5):818–830. doi:10.1007/s10147-019-01548-1
28. Kalluri R. The biology and function of fibroblasts in cancer. Nat Rev Cancer. 2016;16(9):582–598. doi:10.1038/nrc.2016.73
29. Feig C, Jones JO, Kraman M, et al. Targeting CXCL12 from FAP-expressing carcinoma-associated fibroblasts synergizes with anti-PD-L1 immunotherapy in pancreatic cancer. Proc Natl Acad Sci U S A. 2013;110(50):20212–20217. doi:10.1073/pnas.1320318110
30. Dituri F, Mancarella S, Serino G, et al. Direct and indirect effect of TGFβ on treg transendothelial recruitment in HCC tissue microenvironment. Int J Mol Sci. 2021;22(21):11765. doi:10.3390/ijms222111765
31. Timperi E, Gueguen P, Molgora M, et al. Lipid-associated macrophages are induced by cancer-associated fibroblasts and mediate immune suppression in breast cancer. Cancer Res. 2022;82(18):3291–3306. doi:10.1158/0008-5472.CAN-22-1427
32. Voron T, Marcheteau E, Pernot S, et al. Control of the immune response by pro-angiogenic factors. Front Oncol. 2014;4:70. doi:10.3389/fonc.2014.00070
33. Rahma OE, Hodi FS. The intersection between tumor angiogenesis and immune suppression. Clin Cancer Res. 2019;25(18):5449–5457. doi:10.1158/1078-0432.CCR-18-1543
34. Jenkins L, Jungwirth U, Avgustinova A, et al. Cancer-associated fibroblasts suppress CD8+ T-cell infiltration and confer resistance to immune-checkpoint blockade. Cancer Res. 2022;82(16):2904–2917. doi:10.1158/0008-5472.CAN-21-4141
35. Najafi M, Farhood B, Mortezaee K. Extracellular matrix (ECM) stiffness and degradation as cancer drivers. J Cell Biochem. 2019;120(3):2782–2790. doi:10.1002/jcb.27681
36. Freeman P, Mielgo A. Cancer-associated fibroblast mediated inhibition of CD8+ cytotoxic t cell accumulation in tumours: mechanisms and therapeutic opportunities. Cancers. 2020;12(9):2687. doi:10.3390/cancers12092687
37. Jensen C, Nissen NI, Von Arenstorff CS, et al. Serological assessment of collagen fragments and tumor fibrosis may guide immune checkpoint inhibitor therapy. J Exp Clin Cancer Res. 2021;40(1):326. doi:10.1186/s13046-021-02133-z
38. Vogel A, Martin K, Soukup K, et al. JAK1 signaling in dendritic cells promotes peripheral tolerance in autoimmunity through PD-L1-mediated regulatory T cell induction. Cell Rep. 2022;38(8):110420. doi:10.1016/j.celrep.2022.110420
39. Kersten K, Hu KH, Combes AJ, et al. Spatiotemporal co-dependency between macrophages and exhausted CD8+ T cells in cancer. Cancer Cell. 2022;40(6):624–638.e9. doi:10.1016/j.ccell.2022.05.004
40. Mo W, Liu S, Zhao X, et al. ROS scavenging nanozyme modulates immunosuppression for sensitized cancer immunotherapy. Adv Healthc Mater. 2023;8:e2300191. doi:10.1002/adhm.202300191
 © 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.