Back to Journals » Breast Cancer: Targets and Therapy » Volume 18
Clonal Dynamics and Molecular Heterogeneity of Metaplastic Breast Cancer: Focus on TP53 and PIK3CA Truncal Mutations
Authors Ye R ![]() , Dai X, Yang Z, Zhang D, Hu Z
, Dai X, Yang Z, Zhang D, Hu Z ![]() , Liao Y, Yang Z
, Liao Y, Yang Z
Received 3 August 2025
Accepted for publication 3 February 2026
Published 17 February 2026 Volume 2026:18 558033
DOI https://doi.org/10.2147/BCTT.S558033
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Pranela Rameshwar
Rupei Ye,1,2,* Xinzhi Dai,3,* Zihan Yang,1,2 Dandan Zhang,1,2 Zhimin Hu,4 Yehui Liao,5 Zhihui Yang1,2
1Department of Pathology, The Affiliated Hospital, Southwest Medical University, Luzhou, Sichuan, 646000, People’s Republic of China; 2Precision Pathology Diagnosis for Serious Diseases Key Laboratory of LuZhou, Luzhou, Sichuan, 646000, People’s Republic of China; 3Department of Pathology, Ruijin Hospital Shanghai Jiao Tong University School of Medicine, Shanghai, 200025, People’s Republic of China; 4School of Basic Medical Sciences, Southwest Medical University, Luzhou, Sichuan, 646000, People’s Republic of China; 5Department of Orthopedics, The Affiliated Hospital, Southwest Medical University, Luzhou, Sichuan, 646000, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Yehui Liao, Email [email protected] Zhihui Yang, Email [email protected]
Background: Metaplastic breast carcinoma (MBC) is a rare and aggressive subtype of triple-negative breast cancer with distinct molecular features that remain incompletely characterized, hindering the development of effective therapies.
Methods: We integrated clinicopathological data with next-generation sequencing (NGS) of 437 cancer-related genes performed on 25 tumor samples (16 primary, 8 lymph node metastases, 1 distant metastasis) from 17 MBC patients. Functional enrichment analysis was conducted to identify key signaling pathways.
Results: Recurrent alterations were identified in TP53 (14/16, 87.5%), PIK3CA (9/16, 56.2%), and MCL1 (10/16, 62.5% amplified). TP53 mutations (primarily frameshift and missense) showed consistent variant types between primary and metastatic sites. PIK3CA hotspot mutations (eg, H1047R, E545K) persisted across metastases. In contrast, MCL1 amplification exhibited dynamic evolution, being lost in some primary tumors but acquired de novo in lymph node metastases. Functional enrichment analysis revealed the PI3K-Akt signaling pathway as the most significantly altered pathway in MBC.
Conclusion: This study delineates the distinct mutational landscape and clonal evolution patterns of MBC, underpinned by truncal mutations in TP53 and PIK3CA alongside dynamic MCL1 amplification. The persistent activation of the PI3K-Akt pathway presents a key therapeutic vulnerability. Our findings emphasize the potential of PI3K-Akt inhibition and metastasis-specific targeting strategies for this aggressive disease.
Keywords: metaplastic breast carcinoma, next-generation sequencing, clinicopathological characteristics, molecular characteristics, functional enrichment analysis, TP53, PIK3CA
Introduction
Metaplastic breast carcinoma (MBC) is a rare (accounting for 0.2–1% of breast cancers) yet aggressive subtype of triple-negative breast cancer, characterized by heterologous differentiation into squamous, spindle, or mesenchymal elements.1 The 2019 WHO classification recognizes six histological variants, ranging from indolent (eg, fibromatosis-like) to highly aggressive (eg, spindle cell or mesenchymal-differentiated) forms. Clinically, MBCs demonstrate distinct biological behavior-typically presenting as larger tumors with higher metastatic potential and poorer outcomes than conventional triple-negative breast cancers (TNBCs), while exhibiting marked resistance to standard chemotherapy.2 This therapeutic challenge is exacerbated in MBC, underscoring a critical unmet need beyond the limitations of current TNBC regimens. Despite recent advances in molecular profiling, MBC remains genetically enigmatic due to its rarity and histological diversity. While foundational NGS studies by Tray N3 and Zhai J4 have confirmed recurrent alterations in TP53 and the PI3K pathway in primary MBCs, comprehensive genomic characterization—particularly comparing paired primary and metastatic lesions to elucidate evolutionary patterns—is still lacking. This knowledge gap has hindered the development of subtype-specific therapies, leaving radical surgery as the primary treatment modality for most patients.5
The emergence of next-generation sequencing (NGS) now enables systematic investigation of MBC’s molecular landscape. We hypothesized that MBC is driven by a core set of truncal driver mutations (eg, in TP53 and PIK3CA) that are conserved during progression, while metastatic sites acquire distinct alterations contributing to their aggressive phenotype. To test this, we performed deep genomic profiling of 17 MBC cases using a 437-gene panel, with three primary objectives: (1) to define the recurrent genomic alterations driving MBC pathogenesis, (2) to characterize the evolutionary patterns during metastatic progression, and (3) to identify actionable therapeutic targets for this treatment-resistant disease. Our findings provide the most comprehensive molecular portrait of MBC to date, establishing a framework for precision medicine approaches in this challenging malignancy.
Materials and Methods
Patients and Samples
Seventeen consecutive MBC cases diagnosed between January 2010 and September 2022 were selected from surgical archives at our institution. The cohort consisted of East Asian patients. Inclusion criteria required: (1) histologically confirmed MBC per WHO 2019 classification, (2) complete tumor resection without neoadjuvant therapy, and (3) available clinicopathological data. Two expert breast pathologists independently reviewed all cases to verify diagnosis and histological subtype. The final cohort comprised 26 FFPE samples (17 primary tumors, 8 matched lymph node metastases, and 1 distant metastasis) from 17 patients, with complete clinicopathological annotation including tumor size, nodal status, and AJCC 8th edition staging. One primary tumor sample (from a case of fibromatosis-like metaplastic carcinoma) had insufficient material for DNA extraction. Therefore, high-quality DNA was obtained and next-generation sequencing (NGS) was successfully performed on 25 of the 26 samples (16 primary tumors, 8 lymph node metastases, and 1 distant metastasis). All procedures involving human participants and patient samples were conducted in compliance with the ethical standards of the Ethics Committee of the Affiliated Hospital of Southwest Medical University. This study was conducted and reported in accordance with the REMARK guidelines for tumor marker prognostic studies.
Immunohistochemistry (IHC)
Tumor receptor status was determined using standardized IHC protocols: ER/PR positivity (≥1% stained nuclei) and HER2 (IHC with FISH confirmation for equivocal cases) per ASCO/CAP guidelines Ki-67 proliferation index was quantified as the percentage of positively stained tumor nuclei.6,7
Next-Generation Sequencing and Bioinformatics Analysis
Tumor Content Assessment and Enrichment. Prior to DNA extraction, tumor content was assessed on hematoxylin and eosin (H&E)-stained sections by a board-certified breast pathologist. Macrodissection was routinely performed, particularly on lymph node and distant metastasis samples, to enrich for tumor cells and ensure a minimum estimated tumor purity of 20% for all samples subjected to sequencing. The post-enrichment estimated tumor purity for each of the 25 successfully sequenced samples is detailed in Supplementary Table S1.
Genomic DNA was extracted from FFPE tissues (≥20% tumor content) using the QIAamp Kit (Qiagen). Libraries were prepared using a customized 437-gene panel (Geneseeq Technology Inc), an upgraded version of GENESEEQPRIME® (425-gene panel) approved by both the US Food and Drug Administration (FDA) and China’s National Medical Products Administration (NMPA). 437-gene panel is designed for comprehensive genomic profiling, providing full exonic coverage for 355 key cancer-related genes, complemented by targeted sequencing of established mutational hotspot regions in the remaining 82 genes. All libraries were sequenced on an Illumina HiSeq 4000 platform to achieve a mean coverage of ≥1000×. Somatic variants were called using VarScan2 (v2.4.4) with stringent thresholds: variant allele frequency (VAF) ≥2%, a level chosen to balance the high sensitivity required for detecting low-frequency variants due to tumor heterogeneity or low sample purity, against the specificity needed to minimize false positives arising from the sequencing platform’s background error profile. To distinguish somatic mutations from germline polymorphisms, all called variants were filtered against public databases (gnomAD, 1000 Genomes, ExAC), and those with a population allele frequency >1% were excluded. Copy number variations (CNVs) were detected using an in-house pipeline, the ratio cut-off for copy number gain was defined as 2.0 for tissue samples. A ratio cut-off of 0.60 was used for copy number loss detection in all sample types. The thresholds were determined from previous assay validation using the absolute CNVs detected by droplet digital PCR (ddPCR). Gene fusions were identified by FACTERA.8,9 Tumor mutational burden (TMB) was calculated as somatic coding mutations per megabase (including synonymous alterationsand excluding known driver mutations) to focus on the background mutation burden that may be more reflective of immunogenicity, with TMB-high defined as >10 muts/Mb [doi: 10.1186/s13073-017-0424-2].

Microsatellite instability (MSI) status was assessed via 52 microsatellite loci (MSI-H threshold: >40% unstable sites).10
Variant Annotation and Functional Analysis
Somatic variants were annotated using VEP (v105, GRCh37/hg19) and classified by clinical significance (OncoKB v3.0, COSMIC v92). Protein-altering variants were further analyzed for: Functional impact: Missense variants assessed via PolyPhen-2/SIFT; truncating mutations evaluated for domain disruption (UniProtKB). Hotspot visualization: Mutation distributions mapped to protein domains using ggbio (R) and illustrated in Adobe Illustrator.
Pathway Enrichment
GO and KEGG analyses were performed using ClusterProfiler (R) to identify enriched biological processes (eg, epithelial proliferation), molecular functions (eg, kinase activity), and pathways (eg, PI3K-Akt). Results were considered significant after adjustment for multiple testing using the Benjamini-Hochberg false discovery rate (FDR) method, with an adjusted *p*-value (FDR) < 0.05.
Results
Clinicopathological Characteristics of the MBC Cohort
Our cohort comprised 17 female MBC patients with a median age of 48 years (range: 27–60). Tumors ranged from 1.5 to 15 cm in diameter (median: 4.6 cm), with the majority (88.2%, 15/17) classified as high-grade (Nottingham grade III). Histological subtyping revealed squamous cell carcinoma (SCC) as the most frequent variant (35.3%, 6/17), followed by metaplastic carcinoma with heterologous mesenchymal differentiation (MCHMD; 23.5%, 4/17), spindle cell carcinoma (SpCC; 17.6%, 3/17), and other rare subtypes (Figure 1). All cases except one (94.1%) were triple-negative, exhibiting high proliferative activity (mean Ki-67: 50.3%).
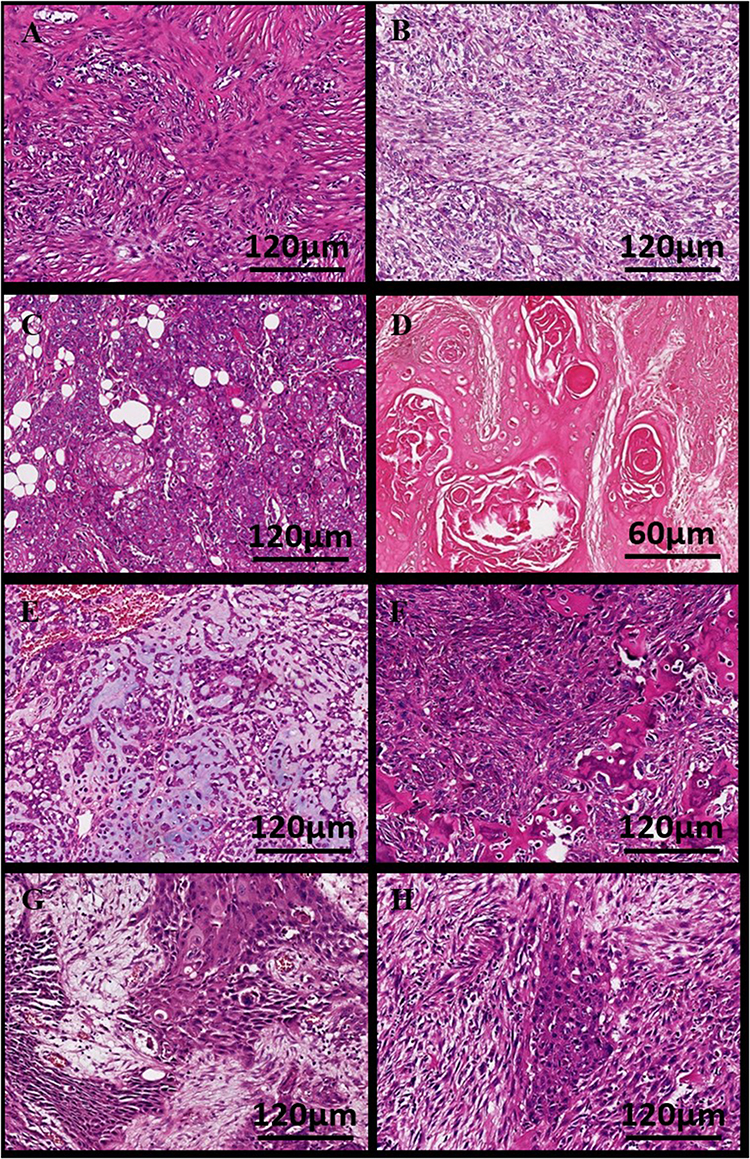 |
Figure 1 Representative histopathological features of metaplastic breast carcinoma subtypes (n=17 patients). All sections were stained with hematoxylin and eosin (H&E). Scale bars are provided for each image. All histopathological assessments were independently verified by two expert breast pathologists. (A) Fibromatosis-like metaplastic carcinoma: bland, spindled morphology with collagenized stroma. Scale bar: 120 μm. (B) Spindle cell carcinoma: highly atypical spindle cells with frequent mitotic figures (arrows). Scale bar: 120 μm. (C and D) Squamous cell carcinoma showing characteristic keratinization (arrowheads) and intercellular bridges. Scale bars: C: 120 μm, D: 60 μm. (E and F) Metaplastic carcinoma with heterologous mesenchymal differentiation: carcinoma cells transition to chondroid matrix (E) or osteosarcoma differentiation with bone matrix (F). Scale bars: E: 200 μm, F: 120 μm. (G and H) Metaplastic carcinoma with mixed squamous and spindle cell differentiation. Scale bars: G: 120 μm, H: 120 μm. |
At diagnosis, 53.3% (8/15) of evaluated patients had lymph node metastases, with SCC cases representing half of these metastatic events. One SCC patient developed lumbar metastases during follow-up. Initial AJCC 8th edition staging distributed cases across stage I (29.4%), stage II (41.1%), and stage III (17.5%). Treatment primarily involved mastectomy (70.6%) with adjuvant chemotherapy (64.7%).
With a median follow-up of 49 months (range: 16–77), one cancer-related death occurred at 21 months post-surgery among 13 evaluable patients (76.4% follow-up rate). The remaining patients showed no evidence of disease progression during surveillance (Table 1). The flow of patient and sample selection throughout the study is summarized in Figure 2.
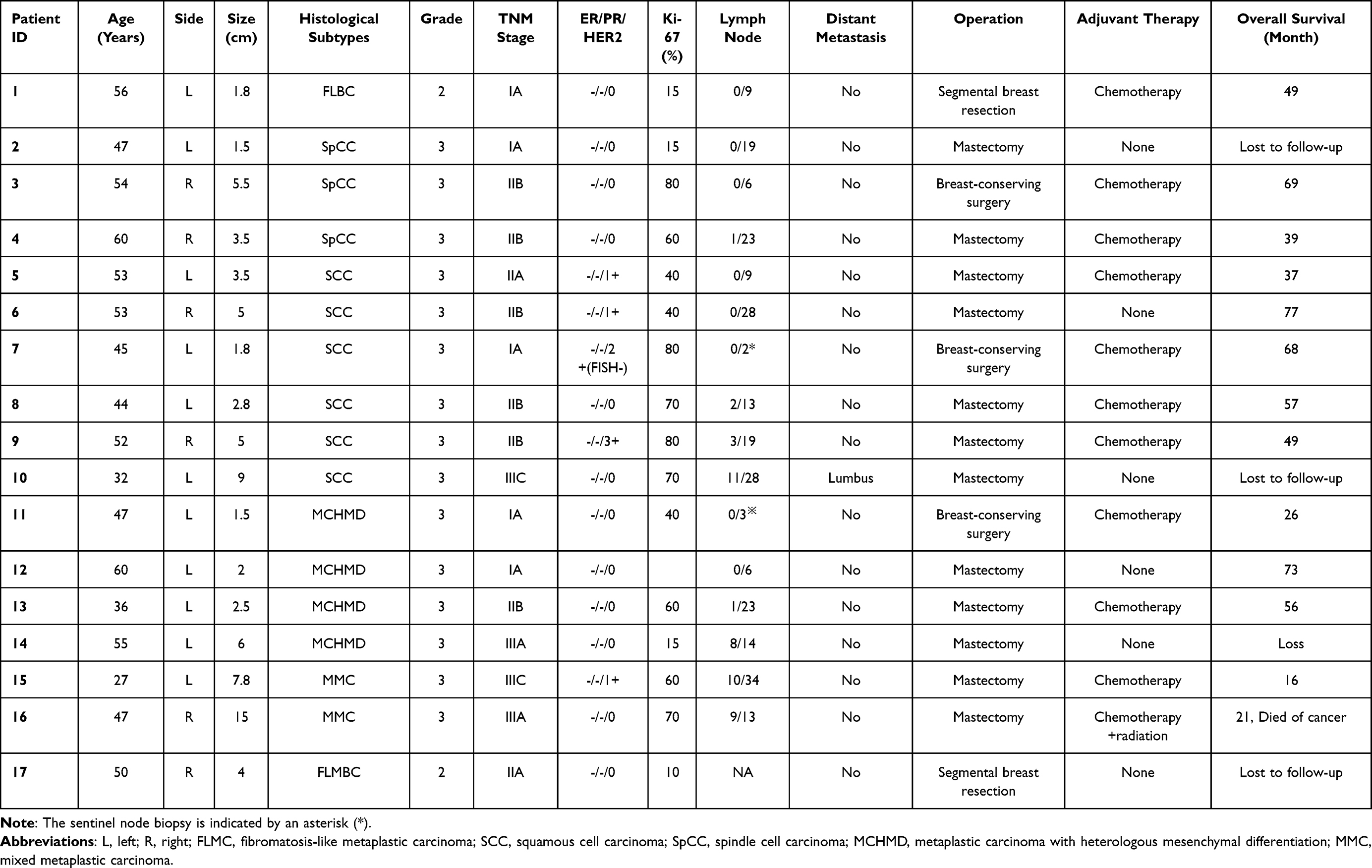 |
Table 1 Clinicopathological Characteristics of the Metaplastic Breast Carcinoma (MBC) Cohort (n = 17) |
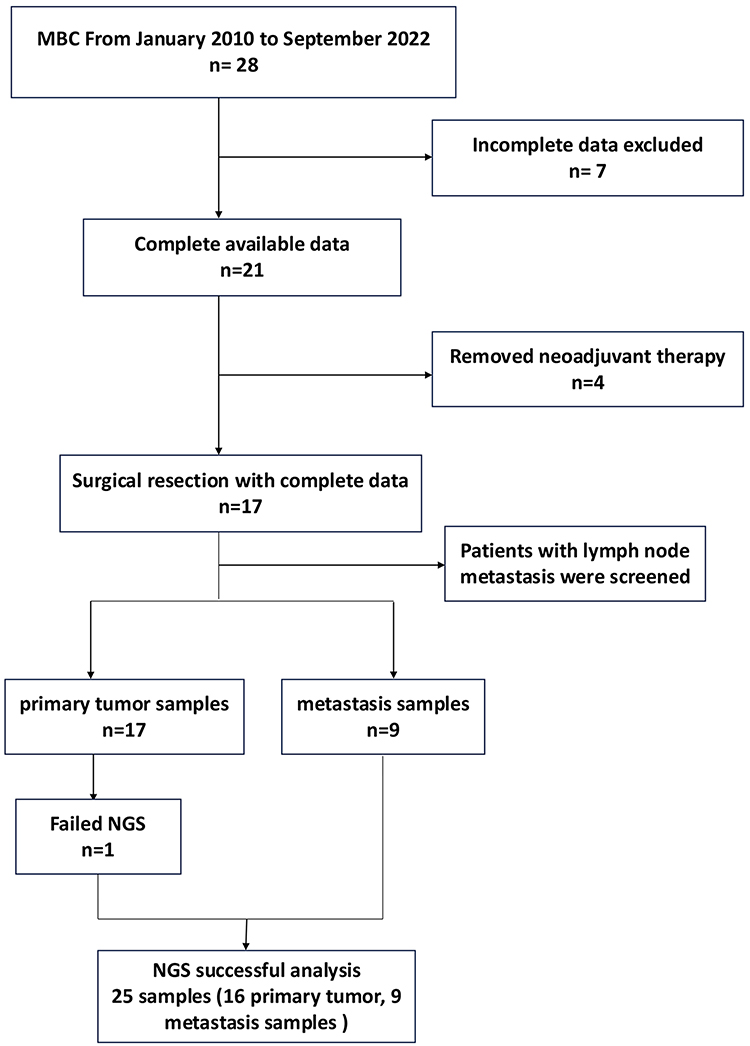 |
Figure 2 Flowchart of cohort selection. |
Genomic Landscape of Primary MBC Tumors
The genomic landscape of primary MBC tumors, based on NGS data from 16 primary samples (see Figure 2 for cohort flow), is summarized in Figure 3. The top 30 gene alterations identified in 16 primary tumor samples from patients with MBC. The top five genes were TP53 (14/16, 87.5%), MCL1 (10/16, 62.5%), PIK3CA (9/16, 56.2%), MYC (6/16, 37.5%) and PTEN (5/16, 31.2%). The most common mutation types were single nucleotide variants (SNVs), followed by copy number variations (CNVs) and structural variants (SVs).
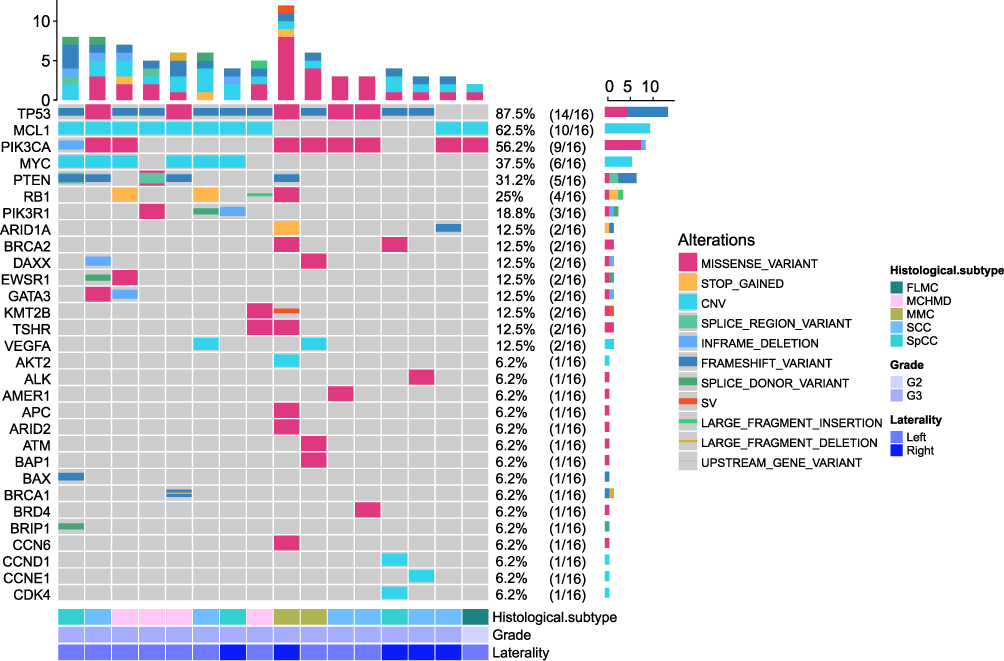 |
Figure 3 Genomic landscape of primary MBC (n=16 primary tumors). Heatmap displays the top 30 significantly altered genes in 16 primary MBC cases, with mutation frequencies indicated as percentages. Alteration types are color-coded. Samples are annotated by histological subtype (FLMC, MCHMD, MMC, SCC, SpCC), grade (G2/G3), and laterality (Left/Right). TP53 (87.5%), PIK3CA (62.5%), and MCL1 (62.5%) were the most frequently mutated genes. Abbreviations: CNV, copy number variation; SV, structural variant. |
TP53 exhibited the highest mutation frequency in this cohort at 87.5% (14/16), with the majority of mutations being frameshift variants and missense variants. For PIK3CA, the mutation frequency was 56.2% (9/16), including 8 missense variants and 1 inframe deletion. PTEN mutations were observed in 31.2% (5/16) of cases, including two heterozygous mutations, so there were seven mutations in total, including four frameshift variants, two splice region variants and one missense variant.
Beyond these key driver genes, several other notable mutations were identified. RB1 was mutated in 25% (4/16) of samples, mainly through stop gained and missense mutations. PIK3R1 mutations were detected in 18.8% (3/16) of cases, involving missense variants, splice donor variants, and inframe deletions. ARID1A alterations occurred in 12.5% (2/16) of samples, characterized by stop-gained and frameshift variants, while BRCA2 mutations (12.5%, 2/16) exclusively comprised missense variants.
Although the sample in this study was small, some associations between genomic alterations and histological subtypes can still be seen in Figure 3. FLMC had the lowest number of genetic changes and a lack of P53 mutations, compared with all other metaplastic cancers (100% for SpCC, 83% for SCC, 100% for MCHMD, and 100% for MMC), possibly reflecting underrepresentation of the sample. Further sample exploration is needed. PIK3CA mutation was detected in all metaplastic cancer subtypes, with the highest mutation rates in MMC (100%, 2/2) and SCC (66.6%, 4/6), and the lowest mutation rate in MCHMD (25%, 1/4).
Figure 4 illustrates the location of the pathogenic mutations on the protein structure of the frequently mutated genes TP53, PIK3CA, and PTEN. 14 TP53 mutations were observed in 16 samples. Nine frameshift mutations were TP53 inactivation mutations, which would lead to premature translation termination. Five missense mutations were reported, and no duplication mutations occurred (Figure 4A). All mutations in the PIK3CA gene were known pathogenic mutations (Figure 4B), with p.H1047R being the most frequent, followed by p.E545K, p.P104_G106delinsR, p.C420R, p.K111N, p.H1047L, and p.E726K. 11 PIK3CA variants were observed in 16 samples; all 10 mutations were reported as missense mutations, and 1 was a non-frameshift deletion (not involved in drawing pictures). Notably, two cases exhibited double mutations - one with the oncogenic hotspots p.E545K and p.H1047Y, and another with p.H1047R coupled with p.K111N. Inframe deletion were not included in Figure 4B. In addition, seven mutations werThese results provide a definitive molecular framework detected in the PTEN gene and two splice region variants are not plotted in Figure 4C. The PTEN p.N184Efs*6, p.V9Sfs*27 and p.G143Efs*2 mutations have not been reported in the ClinVar database.
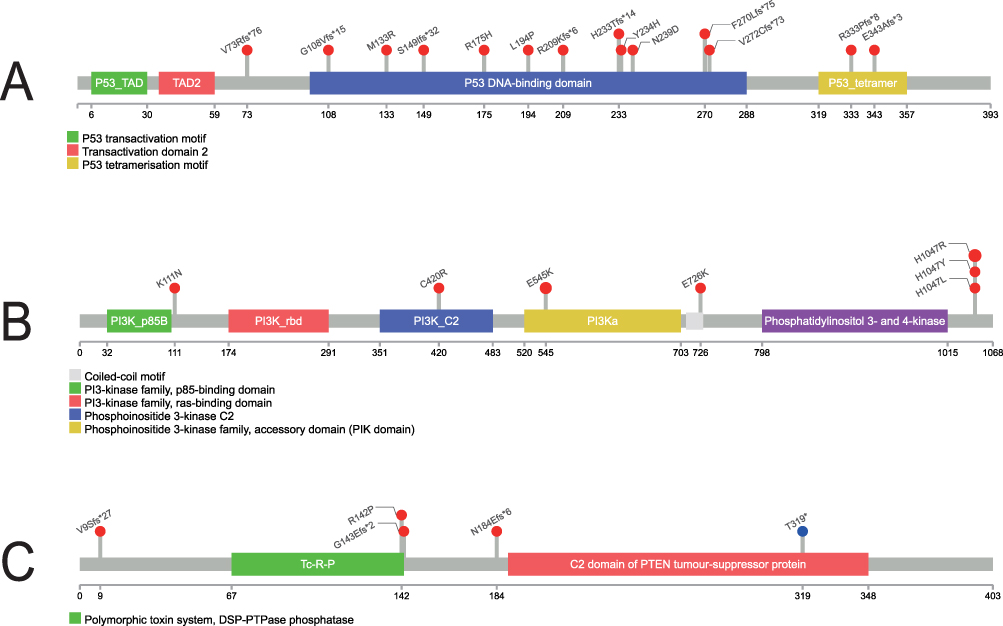 |
Figure 4 Protein domain structures and mutation hotspots of p53, PI3K, and PTEN. (A) p53 functional domains with selected mutation sites shown. (B) PI3K domains with common activating mutations indicated. (C) PTEN domains with frequent mutation sites marked. Numbered residues correspond to amino acid positions. Two additional splice region variants identified in the cohort are not depicted in this domain schematic. |
CNVs and SV Analyses
MBCs exhibited a relatively high burden of chromosomal copy number variations (CNVs). The most frequently amplified gene was MCL1 (62.5%, 10/16), observed in 2 of 3 SpCC, 3 of 6 SCC, 4 of 4 MCHMD, and the single FLMC. MYC ranked as the second most amplified gene (37.5%, 6/16), followed by recurrent amplifications of VEGFA (2/16), and single-case amplifications of AKT2, CCND1, CCNE1, and CDK4. Notably, a structural variant (SV) involving *ZNF845-KMT2B* was identified in one patient with mixed metaplastic carcinoma.
Clonal Dynamics in Paired Primary and Metastatic Lesions
NGS was performed on paired 8 axillary lymph node metastasis samples and 1 distant metastasis sample, and a total of 20 genetic alterations were detected (Figure 5). Notably, TP53 and PIK3CA alterations present in the primary tumors were maintained in the majority of matched metastatic lesions (77.8% and 44.4%, respectively). This conservation across sites suggests they may represent early clonal driver events, although formal clonality analysis (eg, using paired variant allele frequencies and phylogenetic reconstruction) was not performed in this study. TP53 mutations remained the most frequently altered gene, observed in 77.8% (7/9) of metastatic samples, predominantly consisting of missense variants and frameshift variants. PIK3CA mutations were detected in 44.4% (4/9) of cases, primarily as missense variants, with recurrent hotspot mutations such as H1047R. Other notable alterations included MCL1 amplification (44.4%, 4/9), MYC amplification (22.2%, 2/9), and PTEN mutations (22.2%, 2/9). The median tumor mutational burden (TMB) was 4.0 mutations per megabase (Mut/Mb) (range: 0–9.3 Mut/Mb), and all samples were microsatellite-stable (MSS), indicating no evidence of microsatellite instability (MSI).
 |
Figure 5 Mutational profile of metastatic MBC (n=9 metastatic samples). The heatmap illustrates the top 20 altered genes in 9 metastatic MBC cases, with mutation frequencies shown as percentages. Color-coded bars represent different variant types including CNVs, frameshift variants, and missense mutations. |
Comparison of Genetic Variations Between Primary Tumors and Metastases
The genomic landscape comparison between primary tumors and paired metastases revealed both conserved and divergent alterations (Table 2). TP53 mutations were generally maintained at the vast majority of metastatic sites (Cases 4, 9, 10, 13, 14, 15), demonstrating its role as a major tumor driver. Although based on a limited number of pairs (n=4), the MCL1 amplification patterns appeared dynamic: while present in primary tumors of Cases 4, 8, 13 and 14, only Cases 8 and 13 maintained these amplifications in lymph node metastases. Interestingly, Cases 9 and 10 acquired MCL1 amplifications in lymph node metastases despite their absence in matched primary tumors. A systematic comparison of key driver genes across all paired samples is provided in Table 2. Distant metastases (Case 10) lack MCL1 amplification but show additional co-amplifications (MYC amplification, SOX2 amplification), indicating progressive genomic evolution at the metastatic site. PIK3CA alterations in the primary lesions (cases 8, 10, 15, and 16) remained largely unchanged in the matched lymph node metastases, except in case 16. Case 16 demonstrated remarkable genomic divergence between primary and metastatic lesions. The primary tumor harbored complex alterations including variants of canonical cancer genes (TP53, PIK3CA, PTEN, RB1) along with 20 additional oncogenes/tumor suppressors, two gene fusions (ITPKC-DOT1L and ZNF845-KMT2B), and an elevated tumor mutational burden (TMB=28.8 muts/Mb). In striking contrast, the paired lymph node metastasis exhibited extreme genomic simplification, retaining only PALLD amplification while losing all other primary tumor alterations, accompanied by complete TMB reduction (0 muts/Mb). It is noteworthy that the sequencing quality control metrics for this metastatic sample, including DNA fragment size and median coverage depth, were within acceptable limits.
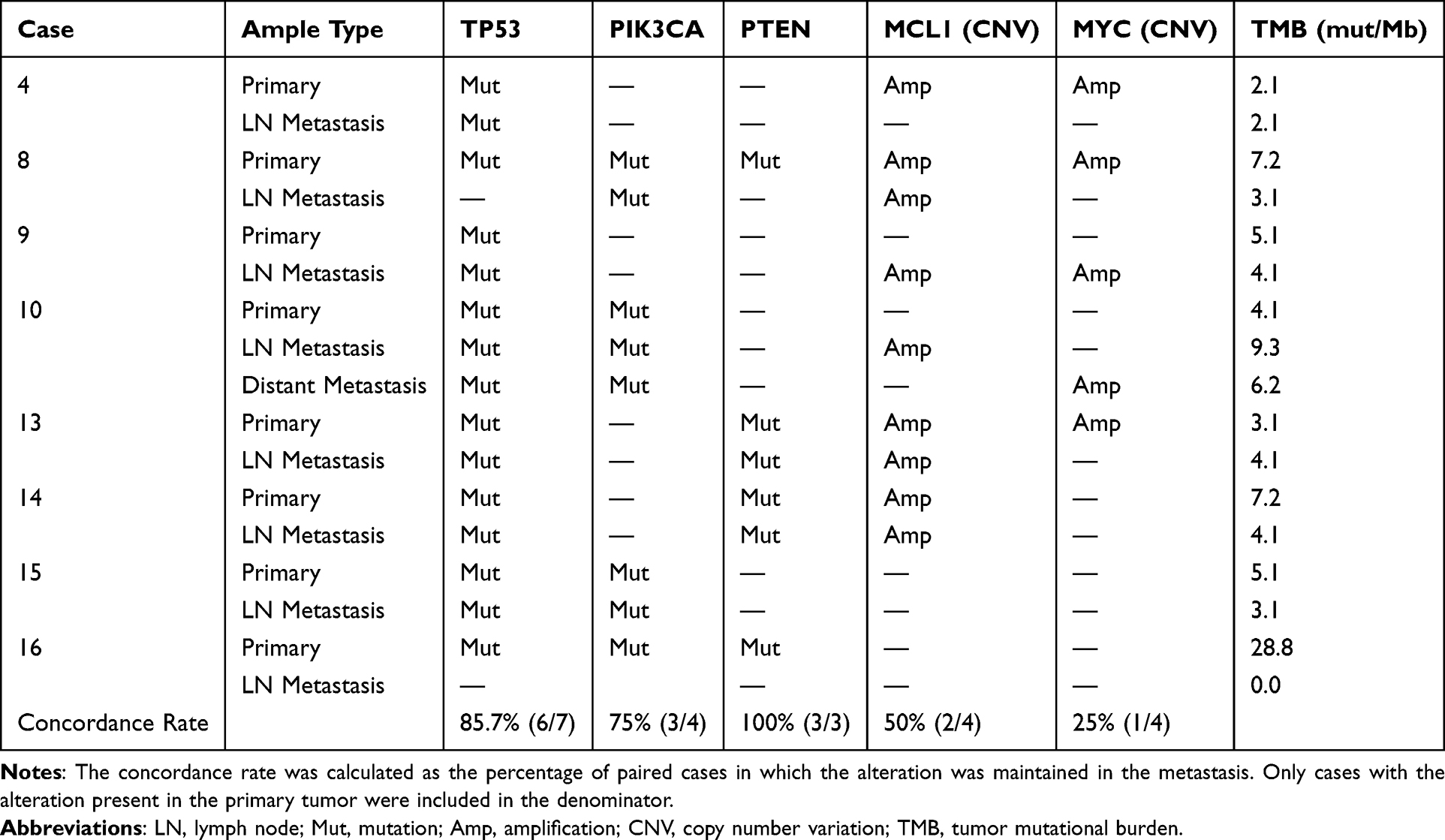 |
Table 2 Comparison of Key Genomic Alterations Between Primary Tumors and Paired Metastases |
Functional Enrichment Analysis of Mutated Genes
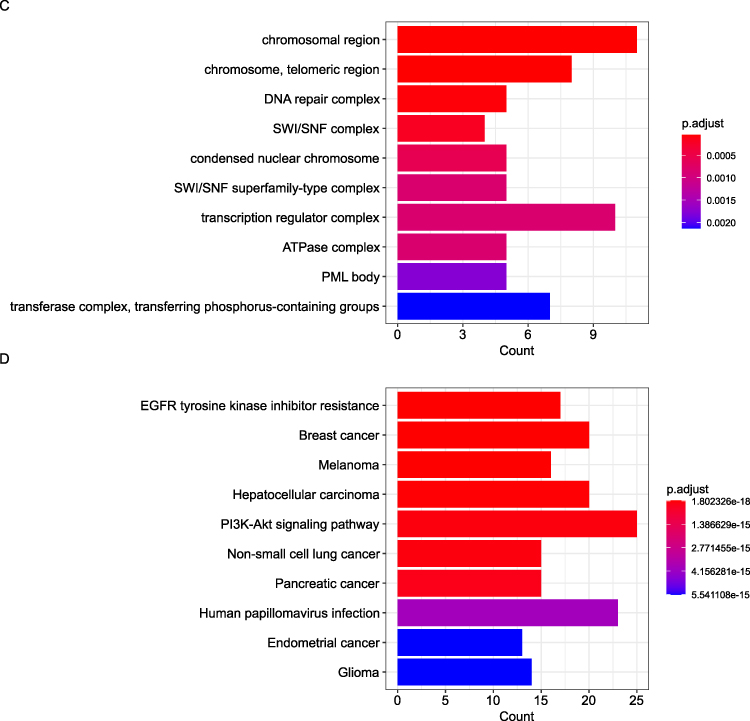
To elucidate the biological mechanisms driving metaplastic breast cancer (MBC), Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway and Gene Ontology (GO) functional enrichment analyses were performed on all candidate genes. Gene Ontology (GO) analysis revealed enriched terms across three hierarchical categories (Figure 6A–C). Biological Processes: Epithelial cell proliferation regulation, peptidyl-tyrosine phosphorylation, and radiation response (eg, EGFR, PIK3CA). Molecular Functions: Protein kinase activities (serine/threonine/tyrosine) and p53 binding (eg, TP53, ATM). Cellular Components: Chromatin remodeling (SWI/SNF complex) and DNA repair machinery (eg, BRCA1, PTEN). Figure 6D illustrates the pathway results of top10. GO/KEGG enrichment highlighted biologically coherent pathways, including PI3K-Akt signaling and EGFR resistance, aligned with the mutational spectrum. These results underscore the roles of proliferative signaling, kinase dysregulation, and genomic instability in MBC pathogenesis.
Figure 6 continued. Figure 6 Functional enrichment analysis of mutated genes in 16 metaplastic breast carcinomas. (A) Top enriched biological processes (BP) related to cell proliferation and radiation response. (B) Molecular functions (MF) highlighting kinase and transcription factor activities. (C) Cellular components (CC) including chromatin remodeling complexes. (D) KEGG pathways showing cancer-related signaling (PI3K-Akt) and therapy resistance mechanisms. Dot size represents gene count; color intensity indicates statistical significance (-log10[p.adjust]).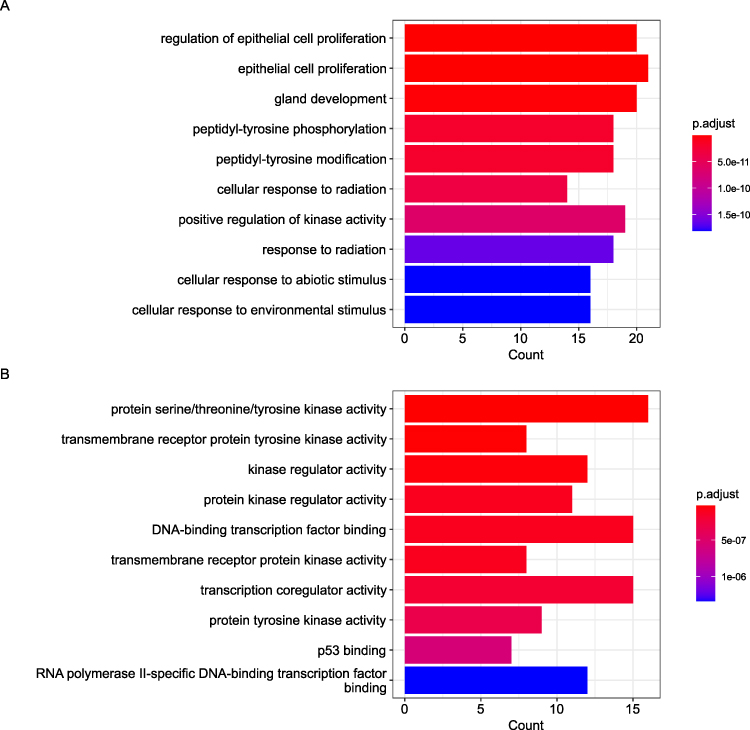
Analysis of Exploratory Biomarkers: TMB and MSI
The median TMB was 7.27 mut/Mb (range: 2.1–28.8), with 18.8% (3/16) cases exceeding 10 mut/Mb. These TMB-high cases included one spindle cell carcinoma (SpCC), one squamous cell carcinoma (SCC), and one mixed metaplastic carcinoma (MMC). Notably, the only TMB-high case with lymph node metastases showed complete TMB loss (0 mut/Mb) in metastatic lesions. All samples were microsatellite-stable (MSS), including the TMB-high cases.
Discussion
The genomic and clinicopathological heterogeneity of metaplastic breast carcinoma (MBC) revealed in this study provides critical insights into its aggressive behavior and therapeutic resistance. Below, we delve deeper into the implications of our findings, contextualizing them within the broader landscape of breast cancer biology and precision oncology.
Core Driver Events: TP53 and PIK3CA Mutations
The near-universal presence of TP53 mutations (87.5% in primary tumors, 77.8% in metastases) establishes it as the cornerstone genetic alteration in MBC. Notably, the mutation types (frameshift and missense variants) remained consistent between primary and metastatic lesions, suggesting TP53 inactivation occurs early in tumorigenesis and is stably maintained throughout progression. This finding differs from some reports in conventional triple-negative breast cancer where TP53 mutation subtypes may shift during metastasis,11 highlighting a unique aspect of MBC biology. The predominance of frameshift mutations (9/14 cases) indicates complete loss of p53 function is particularly important in MBC development, potentially explaining the characteristic genomic instability and aggressive behavior of these tumors.
The PIK3CA gene, encoding the catalytic subunit of phosphatidylinositol 3-kinase (PI3Kα), is a pivotal oncogenic driver in metaplastic breast carcinoma (MBC).3,4 In our cohort, PIK3CA mutations occurred in 56.2% (9/16) of cases, predominantly as hotspot mutations (H1047R and E545K), which constitutively activate the PI3K-Akt-mTOR pathway. These mutations destabilize the autoinhibitory interaction between the p110α catalytic subunit and the p85 regulatory subunit, leading to hyperactivation of downstream survival and metabolic pathways.12 Strikingly, PIK3CA mutations persisted in 44.4% (4/9) of metastatic lesions.13
The functional significance of these findings is underscored by our pathway analysis, which identified PI3K-Akt signaling as the most significantly enriched pathway. These mutations promote glucose metabolism, cell survival, and epithelial-mesenchymal transition (EMT)—hallmarks of MBC’s spindle and squamous differentiation. Intriguingly, PI3K-Akt activation coexisted with TP53 inactivation in 80% of cases, a combination known to accelerate genomic instability and metastasis in murine models.14 Clinically, this synergy may underlie MBC’s resistance to PI3K inhibitors alone, as seen in TNBC trials. While direct preclinical or clinical data in MBC are currently lacking, evidence from other cancer types suggests that dual targeting of PI3K and mutant p53 (eg, using alpelisib + APR-246) could disrupt this cooperative oncogenic axis,15 offering a compelling rationale for future combinatorial trials in MBC. The concurrent presence of PTEN mutations in 31.2% of cases further amplifies pathway activation, creating a potent oncogenic stimulus that likely contributes to MBC’s characteristic resistance to conventional therapies.
The observed frequencies of TP53 and PIK3CA mutations in our cohort are on the higher end of the spectrum reported in prior MBC studies,16 potentially attributable to our cohort’s enrichment for high-grade, advanced-stage tumors and the deep coverage of our targeted sequencing panel. The persistent presence of identical mutations from primary to metastatic sites strongly suggests their role as potential early, truncal drivers. However, we acknowledge that our study has not performed formal clonality analysis (eg, phylogenetic reconstruction integrating paired variant allele frequencies, tumor purity, and copy-number states with tools such as PyClone). Therefore, while our observational data strongly support the hypothesis, the definitive designation of these alterations as “truncal” remains to be validated. This represents an important direction for future research in larger, paired-sample cohorts.
Dynamic Adaptations in Disease Progression
Although our observation is based on a small subset of paired samples (n=4), the high frequency of MCL1 amplification (62.5% in primaries) and its variable patterns in metastases suggest potential important insights into MBC progression. MCL1, an anti-apoptotic BCL-2 family protein, is often amplified in therapy-resistant cancers to counteract pro-apoptotic signals from chemotherapy.17 In MBC, MCL1 overexpression may explain the limited efficacy of anthracyclines, which rely on apoptosis induction via BAX/BAK activation.18 Although not yet tested in MBC, preclinical studies in TNBC models demonstrate that MCL1 inhibitors synergize with DNA-damaging agents to overcome resistance,19 suggesting a viable strategy for biomarker-selected MBC patients. While Cases 8 and 13 maintained MCL1 amplification in lymph node metastases, Cases 9 and 10 acquired it de novo, suggesting context-dependent selection for this anti-apoptotic mechanism. Most strikingly, the single distant metastasis (Case 10) lost MCL1 amplification but gained SOX2 amplification, indicating a possible switch to stemness-related survival mechanisms in distant sites. These findings directly inform clinical trial design. MCL1 amplification could stratify patients for trials of MCL1 inhibitors in primary/lymph node disease. Conversely, SOX2 amplification presents a candidate target for trials in advanced, treatment-resistant metastases.
An Immunosuppressive Microenvironment and Biomarker Challenges
Our study reveals MBC had a low TMB with only 18.8% (3/16) of cases exhibiting high tumor mutational burden (TMB-H, >10 muts/Mb), significantly lower than the prevalence in immunotherapy-responsive cancers like melanoma (40–50%).20 This frequency is consistent with previous reports in MBC (eg, Tray et al), further confirming its immunogenically quiet nature. All cases were microsatellite-stable (MSS), consistent with their triple-negative status,3 and this “immune desert” phenotype likely contributes to MBC’s poor response to checkpoint inhibitors, as evidenced by recent single-cell studies MBCs showed a significant reduction in the proportion of TILS compared to TNBC.21,22
The utility of TMB as a biomarker in MBC, however, may be limited by its instability, as dramatically illustrated by Case 16. In this case, a TMB-high primary tumor (28.8 mut/Mb) gave rise to a lymph node metastasis with a TMB of 0 mut/Mb. Although stringent quality control metrics ruled out general technical failures, the most plausible explanation for this observation is a confluence of factors. We speculate that the metastatic lesion was dominated by a vastly simplified subclone that underwent a severe population bottleneck. The complete absence of other variants could be further exacerbated by a tumor purity that, while meeting the nominal inclusion threshold (≥20%), was sufficiently low to render the majority of subclonal mutations undetectable This case highlights that TMB can be a dynamic and context-dependent metric in MBC, and its interpretation requires careful consideration of tumor purity and clonal dynamics.
Therapeutic Implications and Future Perspectives
Our findings point to several promising therapeutic strategies: Combined PI3K and mutant p53 targeting to address the two most common genetic alterations; MCL1 inhibitors for primary tumors and lymph node metastases with amplifications; Exploration of SOX2 inhibition for distant metastatic disease. The small cohort size also precluded more robust statistical analyses, such as the calculation of confidence intervals for mutation prevalences. Additionally, the uniformly microsatellite-stable status and low TMB in most cases indicate that immunotherapy may have limited utility unless combined with strategies to overcome the immune-desert phenotype.
Thus, immunotherapy trials should prioritize combinations—eg, with PI3K inhibitors or epigenetic modulators—to inflame the microenvironment. TMB heterogeneity (Case 16) cautions that biomarker assessment should prioritize metastatic lesions.
While our study provides comprehensive genomic characterization, several limitations warrant mention. The rarity of MBC limited our cohort size, particularly for certain histological subtypes. Additionally, the retrospective nature precluded uniform treatment and follow-up data. As noted, the lack of formal clonality analysis means the evolutionary trajectories we propose require validation. Future studies should: Validate our findings in larger, prospective cohorts; Develop patient-derived xenograft models to test therapeutic combinations; Explore epigenetic contributors to the observed phenotypic plasticity.
Conclusion
While this study provides key genomic insights, its findings must be interpreted in the context of its limitations, including the retrospective, single-center design and limited cohort size inherent to this rare disease. Despite these constraints, metaplastic breast carcinoma (MBC) emerges as a genomically distinct triple-negative subtype defined by recurrent TP53 and PIK3CA driver mutations with dynamic MCL1 amplification patterns. Our findings reveal two fundamental biological characteristics: (1) remarkable stability of core oncogenic drivers (TP53/PIK3CA) throughout disease progression, contrasting with the plasticity of secondary adaptations (MCL1/SOX2), and (2) a consistently immunosuppressive microenvironment unresponsive to conventional checkpoint inhibition. The persistent PI3K-Akt pathway activation, present in 56.2% of cases, coupled with TP53 inactivation creates a unique therapeutic vulnerability that may be targeted through combinatorial approaches (eg, PI3K inhibitors with mutant p53 reactivators). These results provide a foundational molecular framework for developing precision strategies against this aggressive disease, emphasizing the need for simultaneous targeting of truncal drivers, epigenetic modifiers, and niche-specific adaptations.
Data Sharing Statement
The datasets generated and analyzed during the current study are not publicly available due to patient privacy concerns but are available from the corresponding authors, Rupei Ye or Zhihui Yang, upon reasonable request.
Ethics Approval and Consent to Participate
This case report was approved by the ethics committee of Southwest Medical University, which approved the study and procedures of data collection. All procedures in studies involving human participants were in accordance with the ethical standards of the institutional and/or national research committee and with the 1964 Declaration of Helsinki and its amendments, or comparable ethical standards. Informed written consent was obtained from all prospectively enrolled participants after full explanation of the study purpose and procedures. For retrospective cases where consent could not be obtained, the IRB granted a waiver, as this research involved minimal risk and used de-identified data.
Acknowledgments
We thank Charlesworth Author Services for linguistic assistance during manuscript preparation. We also acknowledge the technical support from Geneseeq Technology Inc. for NGS sequencing.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
This work was financially supported by Sichuan Science and Technology Program (No.2022YFS0636), Luzhou Science and Technology Program (No.2022-SYF-39); Science and Technology Strategic Cooperation Project between the People’s Government of Luzhou City and Southwest Medical University (No.2023LZXNYDJ038); Southwest Medical University applied basic research (2023QN070, 2025JC154).
Disclosure
The authors declare that they have no competing interests.
References
1. Schroeder MC, Rastogi P, Geyer CE, et al. Early and locally advanced metaplastic breast cancer: presentation and survival by receptor status in surveillance, epidemiology, and end results (SEER) 2010-2014. Oncologist. 2018;23(4):481–15. doi:10.1634/theoncologist.2017-0398
2. El Zein D, Hughes M, Kumar S, et al. Metaplastic carcinoma of the breast is more aggressive than triple-negative breast cancer: a study from a single institution and review of literature. Clin Breast Cancer. 2017;17(5):382–391. doi:10.1016/j.clbc.2017.04.009
3. Tray N, Taff J, Singh B, et al. Metaplastic breast cancers: genomic profiling, mutational burden and tumor-infiltrating lymphocytes. Breast. 2019;44:29–32.
4. Zhai J, Giannini G, Ewalt MD, et al. Molecular characterization of metaplastic breast carcinoma via next-generation sequencing. Human Pathol. 2019;86:85–92. doi:10.1016/j.humpath.2018.11.023
5. Corso G, Frassoni S, Girardi A, et al. Metaplastic breast cancer: prognostic and therapeutic considerations. J Surg Oncol. 2021;123(1):61–70. doi:10.1002/jso.26248
6. Hammond ME, Hayes DF, Wolff AC, et al. American society of clinical oncology/college of American Pathologists guideline recommendations for immunohistochemical testing of estrogen and progesterone receptors in breast cancer. J Oncol Pract. 2010;6(4):195–197. doi:10.1200/JOP.777003
7. Wolff AC, Hammond MEH, Allison KH, et al. Human epidermal growth factor receptor 2 testing in breast cancer: American society of clinical oncology/college of American pathologists clinical practice guideline focused update. Archiv Pathol Lab Med. 2018;142(11):1364–1382. doi:10.5858/arpa.2018-0902-SA
8. Koboldt DC, Zhang Q, Larson DE, et al. VarScan 2: somatic mutation and copy number alteration discovery in cancer by exome sequencing. Genome Res. 2012;22(3):568–576. doi:10.1101/gr.129684.111
9. Newman AM, Bratman SV, Stehr H, et al. FACTERA: a practical method for the discovery of genomic rearrangements at breakpoint resolution. Bioinformatics. 2014;30(23):3390–3393. doi:10.1093/bioinformatics/btu549
10. Chalmers ZR, Connelly CF, Fabrizio D, et al. Analysis of 100,000 human cancer genomes reveals the landscape of tumor mutational burden. Genome Med. 2017;9(1):34. doi:10.1186/s13073-017-0424-2
11. Bertucci F, Ng CKY, Patsouris A, et al. Genomic characterization of metastatic breast cancers. Nature. 2019;569(7757):560–564. doi:10.1038/s41586-019-1056-z
12. Soong TR, Dillon DA, Rice-Stitt TL, et al. Invasive lobular carcinoma with extracellular mucin (ILCEM): clinicopathologic and molecular characterization of a rare entity. Modern Pathol. 2022;35(10):1370–1382. doi:10.1038/s41379-022-01084-w
13. Garrido-Castro AC, Spurr LF, Hughes ME, et al. Genomic characterization of de novo metastatic breast cancer. Clin Cancer Res. 2021;27(4):1105–1118. doi:10.1158/1078-0432.CCR-20-1720
14. Martinez JD, Mo Q, Xu Y, et al. Common genomic aberrations in mouse and human breast cancers with concurrent P53 deficiency and activated PTEN-PI3K-AKT pathway. Int J Bio Sci. 2022;18(1):229–241. doi:10.7150/ijbs.65763
15. Ganci F, Pulito C, Valsoni S, et al. PI3K inhibitors curtail MYC-dependent mutant p53 gain-of-function in head and neck squamous cell carcinoma. Clin Cancer Res. 2020;26(12):2956–2971. doi:10.1158/1078-0432.CCR-19-2485
16. Dieci MV, Smutná V, Scott V, et al. Whole exome sequencing of rare aggressive breast cancer histologies. Breast Cancer Res Treat. 2016;156(1):21–32. doi:10.1007/s10549-016-3718-y
17. Karpel-Massler G, Ishida CT, Bianchetti E, et al. Inhibition of mitochondrial matrix chaperones and antiapoptotic Bcl-2 family proteins empower antitumor therapeutic responses. Cancer Res. 2017;77(13):3513–3526. doi:10.1158/0008-5472.CAN-16-3424
18. Lu X, Liu YC, Orvig C, et al. Discovery of a copper-based Mcl-1 inhibitor as an effective antitumor agent. J Med Chemistry. 2020;63(17):9154–9167. doi:10.1021/acs.jmedchem.9b02047
19. Torres-Adorno AM, Lee J, Kogawa T, et al. Histone deacetylase inhibitor enhances the efficacy of MEK inhibitor through NOXA-mediated MCL1 degradation in triple-negative and inflammatory breast cancer. Clin Cancer Res. 2017;23(16):4780–4792. doi:10.1158/1078-0432.CCR-16-2622
20. Goodman AM, Kato S, Bazhenova L, et al. Tumor mutational burden as an independent predictor of response to immunotherapy in diverse cancers. Mol Cancer Ther. 2017;16(11):2598–2608. doi:10.1158/1535-7163.MCT-17-0386
21. Kalaw E, Lim M, Kutasovic JR, et al. Metaplastic breast cancers frequently express immune checkpoint markers FOXP3 and PD-L1. British Journal of Cancer. 2020;123(11):1665–1672. doi:10.1038/s41416-020-01065-3
22. González-Martínez S, Pérez-Mies B, Pizarro D, et al. Epithelial mesenchymal transition and immune response in metaplastic breast carcinoma. Int J Mol Sci. 2021;22(14):7398. doi:10.3390/ijms22147398
 © 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.