")
Back to Journals » Cancer Management and Research » Volume 15
Clinical Utility of Ivosidenib in the Treatment of IDH1-Mutant Cholangiocarcinoma: Evidence To Date
Authors Uson Junior PLS, Borad MJ
Received 27 April 2023
Accepted for publication 31 August 2023
Published 18 September 2023 Volume 2023:15 Pages 1025—1031
DOI https://doi.org/10.2147/CMAR.S326060
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Yong Teng
Pedro Luiz Serrano Uson Junior,1– 3 Mitesh J Borad1
1Mayo Clinic Cancer Center, Mayo Clinic, Phoenix, AZ, USA; 2HCOR, Hospital do Coração, Sao Paulo, Brazil; 3Center for Personalized Medicine, Hospital Israelita Albert Einstein, Sao Paulo, Brazil
Correspondence: Mitesh J Borad, Mayo Clinic Cancer Center, Mayo Clinic, Phoenix, AZ, USA, Tel +1 480 301 8000, Email [email protected]
Abstract: Ivosidenib is an isocitrate dehydrogenase 1 (IDH1) inhibitor that is FDA approved for patients with IDH1 mutation and acute myeloid leukemia and previously treated locally advanced or metastatic cholangiocarcinoma. In the Phase III trial ClarIDHy ivosidenib improved progression-free survival, 2.7 months versus 1.4 months (p < 0.0001) and overall survival (OS), median OS was 10.8 months for ivosidenib and 9.7 months for the placebo arm (p = 0.06) for patients with previously treated and IDH1 mutated cholangiocarcinoma. In this review article, we will address the mechanism of action of ivosidenib and data from early trials and safety from the randomized trial in cholangiocarcinoma. As a conclusion, future perspectives of IDH1 inhibition in IDH1 mutated tumors and possible strategies of sequencing and combinations will be reviewed and discussed.
Keywords: cholangiocarcinoma, biliary tract cancers, ivosidenib, IDH1
Introduction
Biliary tract cancers (BTC) are a set of malignant tumors that arise from the biliary tract ducts.1 A distinct group of malignant tumors are designed as BTC, including intrahepatic cholangiocarcinoma (iCCA), peri-hilar cholangiocarcinoma (pCCA), distal cholangiocarcinoma (dCCA) and gallbladder cancer (GC). Although it is a rare cancer, data has shown that the incidence of iCCA is on the rise globally.2 In 2020, it was estimated more than 900,000 new cases of primary liver cancers, with around 10–15% being iCCA.3
Systemic treatment for advanced disease has undergone significant improvements in recent years. Lately, the incorporation of an anti-programmed death-ligand 1 (anti-PD-L1) with chemotherapy has become the standard upfront option for metastatic BTCs.4 In the randomized phase III trial TOPAZ-1, the addition of the anti-PD-L1 durvalumab to chemotherapy cisplatin and gemcitabine improved progression-free survival (PFS), overall survival (OS) and objective response rate (ORR) compared to chemotherapy alone.4 However, the improvement in OS was just a few months (12.8 months versus 11.5 months).4 For second-line options chemotherapy regimens including fluorouracil associated with oxaliplatin5 or liposomal irinotecan6 are options, although of moderate benefit.5,6 Precision medicine incorporation in iCCA has brought new and effective opportunities for the patients that harbor targetable genomic alterations.7 It is estimated that more than 40% of iCCA will have some actionable targets; those include genomic alterations of FGFR 1–3, IDH 1 or 2, BRAF, HER2 and others.8
Isocitrate dehydrogenase 1 (IDH1) mutations are identified roughly in about 20% of patients with iCCA.7 Three isoenzymes; IDH1, IDH2 and 3 represent the IDH family. These enzymes in normal cells have the activity in the NADPH pathway to convert isocitrate to alpha-ketoglutarate (αKG) via oxidative decarboxylation, with concomitant production of NADPH.9,10 NADPH participates in important anabolic biosynthetic reactions and decreases reactive oxygen species (ROS)-related cellular damage.11 αKG generates glutamate and glutamine for the Krebs cycle.11 When IDH1 or IDH2 are mutated, the catalytic activity is altered, and converts αKG to an oncometabolite R (-)-2-hydroxyglutarate (2HG).9–11 2HG is oncogenic because it inhibits αKG-dependent enzymes such as prolyl hydroxylases (PHD), methylcytosine dioxygenase (TET2) and histone lysine demethylases (KDM), altering DNA methylation and preventing hypoxia-inducible factor 1 (HIF1) degradation.11,12 Those impaired detoxification mechanisms lead to genomic instability.13 Furthermore, in the presence of hypoxia and cell stress, the HIF1 increases fatty acid oxidation dependent on carnitine palmitoyl transferase 1C (CPT1C), providing alternative energy source to cancer cells promoting cancer proliferation.11,14 Reversing IDH mutations with IDH inhibitors is an effective method to control tumor growth in tumors harboring those mutations [Figure 1].
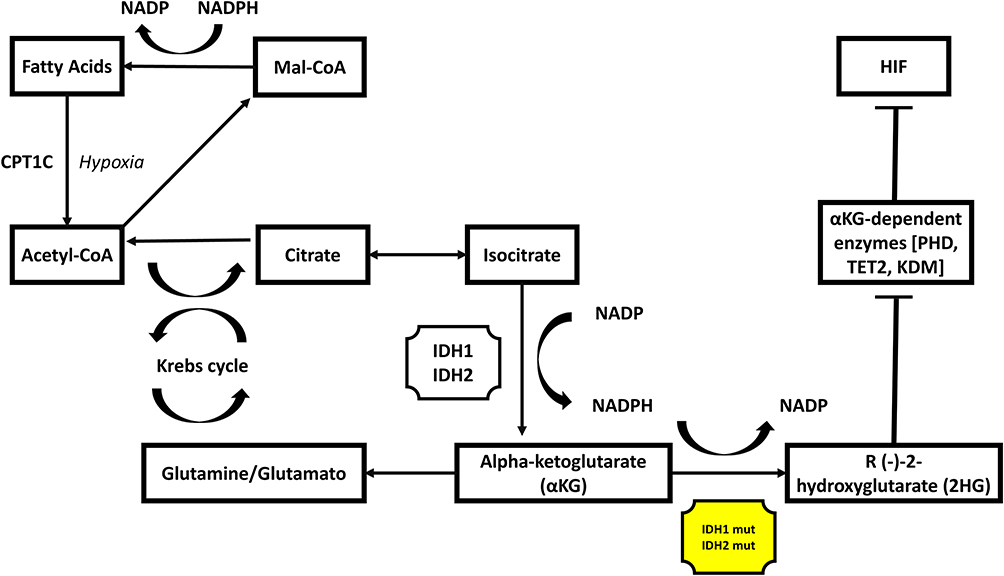 |
Figure 1 IDH 1 and 2 effects on cell metabolism. Notes: When IDH1 or IDH2 are mutated, the catalytic activity is altered, and converts αKG to an oncometabolite R (-)-2-hydroxyglutarate (2HG). 2HG is oncogenic because it inhibits αKG-dependent enzymes such as prolyl hydroxylases (PHD), methylcytosine dioxygenase (TET2) and histone lysine demethylases (KDM), altering DNA methylation and preventing hypoxia-inducible factor 1 (HIF1) degradation. HIF1 increases fatty acid oxidation dependence on carnitine palmitoyl transferase 1C (CPT1C), providing an alternative energy source to cancer cells promoting cancer proliferation. |
Mechanism of Action of Ivosidenib and Safety
Ivosidenib is an IDH1 inhibitor. It is an FDA-approved drug for patients with IDH1 mutation and: (1) newly diagnosed acute myeloid leukemia (AML) in monotherapy or in combination with azacytidine in adults 75 years or older; (2) refractory or relapsed AML; (3) previously treated locally advanced or metastatic CCA. In CCA, it was initially evaluated in a Phase I dose escalation and expansion trial including multiple solid tumors.15 In the trial, during the dose escalation phase, patients were treated with doses ranging between 200–1200 mg daily. A total of 73 patients with CCA with IDH1 mutations, who had received a median of two (range 1–5) previous systemic therapies, were included and received at least one dose of the study treatment.15 Around 90% of the patients had iCCA, and the rest were dCCA. The most common IDH1 mutation identified was R132C (77%), followed by R132L (11%), R132G (5%) and R132H and R132S both in 3%. The most common causes of adverse events (AE) included fatigue in 42% of the patients; two cases with grade ≥3, nausea in 34% of the patients; one case with a grade ≥3, diarrhea in 32%, abdominal pain in 27%; two cases with grade ≥3, anorexia in 27%; one case with grade ≥3 and vomiting in 23%. Worse AE were ascites in four cases and anaemia in three cases. Two reported on-treatment death (Clostridioides difficile infection and procedural hemorrhage) were assessed by the investigator and were not related to treatment. Just one serious AE was considered related to treatment (grade 2 supraventricular extrasystoles). Electrocardiogram QT prolongation was reported in eight patients (11%; grade 3 in one, grade 1 or 2 in ten). Overall ivosidenib was tolerable, with no dose-limiting toxicities and maximum tolerated dose reached. A dose of 500 mg daily was selected for the expansion phase. At 500 mg once daily, mean plasma 2HG concentrations decreased by 88% (71–92%), with no additional plasma reductions observed at doses greater than 500 mg once daily, further supporting this dose. An ORR was observed in four patients (5%, 95% CI 1.5–13.4). Around half of the patients treated had stable disease (56%).15 The median PFS of entire cohort was 3.8 months (95% CI 3.6–7.3), and the median OS was 13.8 months (95% CI 11.1–29.3). In most patients (n = 69), even those with disease progression, plasma 2HG decreased and remained at low concentrations, approximating the range seen in healthy volunteers. In this study, sixty-three patients had baseline genomic profiling information. A median of 2 (0–8) mutations were detected in addition to IDH1 mutation. The most observed commutation was PBRM1 (21%), other frequent mutations included ARID1A in 17%, PIK3CA in 13% and KRAS in 11%. No associations were observed between efficacy of ivosidenib with presence of specific co-mutations.15
Phase III Clinical Trial in Cholangiocarcinoma
The phase III trial ClarIDHy included patients from 49 hospitals in six countries (France, Italy, South Korea, Spain, UK, USA).16,17 From a total of 230 patients assessed for eligibility, 185 patients with advanced IDH1 mutant iCCA who had progressed on previous therapy and had up to two previous treatment regimens for advanced disease were randomized (2:1) to ivosidenib 500 mg once daily (n = 124) or placebo (n = 61). In cases of toxicities, dosing could be reduced to 250mg and re-escalated as investigator evaluation. The primary endpoint of the trial was PFS. Secondary endpoints included OS, ORR, duration, and time to response. It was assessed as well pharmacokinetics and pharma codynamics and quality of life.17 Regarding crossover, the rank-preserving structural failure time (RPSFT) method was used to reconstruct the survival curve (prespecified exploratory analysis) for patients receiving placebo as if crossover had never occurred.17 The RPSFT method is based on a common treatment assumption: the treatment effect of ivosidenib is the same for all individuals, regardless of when treatment is received.16,17 This method was proposed by Robins et al to correct non-compliance in randomized trials.18
Like the phase I study, R132C was the most prevalent IDH1 mutation (70%), more than 90% of the patients had metastatic disease, and about half had received two previous lines of therapy. Most patients had received a previous platinum-based therapy. By central independent radiology center (IRC), ivosidenib improved PFS, 2.7 months versus 1.4 months (HR 0.37; 95% CI 0.25–0.54; one-sided p < 0.0001) and improvements in OS were also observed, median OS was 10.8 months for ivosidenib and 9.7 months for the placebo arm (HR 0.69; 95% CI 0.44–1.1; one-sided p = 0.06). The RPSFT-adjusted median OS was 6.0 months for placebo (HR 0.46; 95% CI 0.28–0.75; p = 0.0008).17 The ORR of ivosidenib was low as 2%; however, half had stable disease (51%). Common treatment-related adverse effects with ivosidenib included nausea, diarrhea, fatigue, cough, abdominal pain, decreased appetite, ascites, vomiting, anemia, and constipation. Serious adverse events (AE) were reported for 30% of patients receiving ivosidenib and were deemed treatment related in just three (2%) patients (grade 3/4 hyperbilirubinemia, grade 2 electrocardiogram QT prolonged, and grade 3 pleural effusion). Quality of life assessment showed that the decline from baseline at cycle 2 on the EORTC QLQ-C30 physical functioning subscale was significantly less for patients in the ivosidenib group than for patients in the placebo group (p = 0·0059).17 Plasma 2HG decline was also observed after one cycle of ivosidenib in the patients treated.17
Final analysis of OS with final data cutoff of May 31, 2020, was published.16 One hundred and twenty-six patients were treated with ivosidenib 500 mg and 61 with placebo. As of the data cutoff date, 43 patients (70%) randomly assigned to placebo had crossed over to receive open label ivosidenib. Based on 150 OS events, the median OS was 10.3 months with ivosidenib and 7.5 months with placebo (HR, 0.79 [95% CI, 0.56–1.12]; 1-sided p = 0.09).16 The RPSFT-adjusted median OS was 5.1 months with placebo (HR, 0.49 [95% CI, 0.34–0.70]; 1-sided p < 0.001). The 1-year survival rate was 43% for ivosidenib group vs 36% for the placebo group. Data from the final results of the trial can be seen on Table 1, including updated safety data.16 One of the most important treatment-related AE, prolonged QT interval on electrocardiogram, was observed in only 8% of the patients exposed to ivosidenib, and most grade 1 or 2. Based on these results, on August 25, 2021, the FDA approved ivosidenib for the treatment of adult patients with unresectable locally advanced or metastatic IDH1 mutated CCA as detected by an FDA-approved test with disease progression after 1 to 2 prior lines of systemic therapy for advanced disease.
 |
Table 1 Ivosidenib Dosing, Trial Results and Adverse Events in Cholangiocarcinoma Patients |
Mechanisms of Resistance
New known or likely oncogenic mutations emerged at a variant allele frequency (VAF) of 5% or more during treatment in six patients in the phase I trial, spanning seven genes from multiple functional pathways, those alterations were detected by post-treatment tumor biopsies.15 One patient developed an IDH2-R172V (VAF = 10%) and one an IDH1-R132F (VAF = 37%) mutation at disease progression, and commutations in TP53, ARID1A, POLE, PIK3R1, and TBX3 emerged in four other patients.15 In another report, two patients with iCCA and IDH1 R132C mutations were treated with ivosidenib and evaluated for mechanisms of resistance.19 After disease progression, a new oncogenic IDH2 mutation was identified in one patient by tumor biopsy, and in the other patient an acquired IDH1 D279N mutation was detected.19 To evaluate those findings, colleagues utilized cells expressing the double-mutant variants. In vitro, IDH1 R132H/D279N produced 2HG with less efficiency than IDH1 R132H. However, the binding spot of ivosidenib was compromised, causing inefficiency of the drug as an inhibitor.19 Another IDH1 inhibitor LY3410738 was evaluated in those cells, and 2HG production and cell proliferation was blocked.19 Isoform switching as a mechanism of resistance to ivosidenib was already previously described in another manuscript, including patients with AML as well.20 The study reported two cases of patients with refractory IDH1 R132C-mutant AML who progressed on ivosidenib, had rising blood 2HG and emergence of IDH2 R140Q mutations. Another case of AML with IDH2 R140Q mutation achieved a durable remission with the IDH2 inhibitor enasidenib; at progression on therapy, it was detected emergence of a new IDH1 R132C mutation that was sensitive to AG-881, an IDH 1/2 inhibitor.20 All these studies strongly suggest that two important mechanisms of resistance related to refractory of ivosidenib are acquired mutations changing the binding spot of the drug and isoform switching, from mutant IDH1 to mutant IDH2 or vice versa. In order to achieve long-term benefit with IDH inhibition, alternating inhibitors with different molecular structures could overcome resistance. These new drugs are currently being evaluated in Phase II trials in AML and other advanced solid tumors.21
Discussion
The results of the ivosidenib randomized phase III trial are very important and practice changing for patients with metastatic iCCA due to multiple findings. First, ivosidenib reduced the risk of progression or death by 63% (HR, 0.37 [95% CI, 0.25–0.54]; 1-sided P < 0.001) compared to placebo.16 Second, ivosidenib improved numerically overall survival despite the high rate of crossover (70%), median OS of 10.3 with ivosidenib versus 7.5 months with placebo, this benefit was further confirmed after the crossover adjustment, with a median OS for the placebo arm of 5.1 months (p < 0.001).16 Furthermore, the median OS of ivosidenib compares favorably with other options of second-line treatments including FOLFOX and liposomal irinotecan with fluorouracil.5,6 Ivosidenib is nowadays included in the current ESMO guidelines as a recommended second-line option for patients with advanced biliary cancers that failed first-line chemoimmunotherapy treatment and harbors IDH1 mutations.22
IDH1 mutations are genomic alterations more common than FGFR2 fusions in iCCA.1 Furthermore, problems related to detection of FGFR2 genomic alterations are not observed in the case of IDH1 mutations.8,23 In the largest cell-free tumor DNA (ctDNA) data, 2068 samples from 1671 patients were analyzed with next-generation sequencing.8 Of patients with IDH1 mutations detected in tissue samples, 87% were also detected in ctDNA, showing a high concordance.8 Also, two cases had IDH1 mutations detected only in the ctDNA analysis. These findings confirm that ctDNA can be used to detect IDH1 mutations in advanced iCCA in cases where tissue is scarce or unavailable. Interestingly, the detection rates of IDH1 mutations differ for the time of ctDNA collection, with higher concordances when the collection is done before starting systemic treatment.8 In this study, 72.4% of IDH mutations were R132C; other mutations included: R132L (10.4%), R132G (6.7%), R132H (5.5%), R132S (3.1%), E85K (0.6%), I130V (0.6%) and G105D (0.6%).
In order to improve outcomes in patients with iCCA and IDH1 mutations, new strategies are being developed. Ivosidenib is safe when used in association with other regimens. Data for AML with ivosidenib and azacytidine improved outcomes in AML and is a new FDA approved option for those patients. Some trials with ivosidenib combinations are ongoing, as an example ivosidenib in combination with nivolumab in participants with IDH1 mutated advanced solid tumors [NCT04056910] and ivosidenib in combination with modified FOLFIRINOX in pancreatic cancers [NCT05209074]. The synergism of ivosidenib with other treatments is due to potential effects in the tumor microenvironment.24,25 In melanoma cell models, under low glucose conditions, ivosidenib impaired the basal oxygen rate and decreased the ATP production, suggesting a pharmacological action against wild-type IDH1.24 In pancreatic cancer models, similar effects were observed, with enhancement of chemotherapy efficacy and apoptosis, when combined with ivosidenib.25 A very interesting strategy is evaluating ivosidenib or pemigatinib with standard first-line chemotherapy regimen gemcitabine and cisplatin in advanced CCA. Currently, the phase I study is recruiting patients in US centers. Main objectives include safety, tolerability, maximum tolerated dose (MTD) and/or recommended Phase 2 dose. The study is active but not recruiting. No data has been published [NCT04088188].
Ivosidenib is an option for patients with advanced IDH1 mutated iCCA, it is a standard therapy for patients that failed or are intolerant to chemotherapy, and due safety, combinations with chemotherapy or immunotherapy are being investigated and may change the landscape of treatment of IDH1 mutated CCA soon.
Disclosure
Mitesh Borad has received a grant to the institution from Senhwa Pharmaceuticals, Adaptimmune, Agios Pharmaceuticals, Halozyme Pharmaceuticals, Celgene Pharmaceuticals, EMD Merck Serono, Toray, Dicerna, Taiho Pharmaceuticals, Sun Biopharma, Isis Pharmaceuticals, Redhill Pharmaceuticals, Boston Biomed, Basilea, Incyte Pharmaceuticals, Mirna Pharmaceuticals, Medimmune, Bioline, Sillajen, ARIAD Pharmaceuticals, PUMA Pharmaceuticals, Novartis Pharmaceuticals, QED Pharmaceuticals, Pieris Pharmaceuticals, consultancy from ADC Therapeutics, Exelixis Pharmaceuticals, Inspyr Therapeutics, G1 Therapeutics, Immunovative Therapies, OncBioMune Pharmaceuticals, Western Oncolytics, Lynx Group and travel support from Astra Zeneca. Pedro Luiz Serrano Uson Junior has no relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript. This includes employment, consultancies, honoraria, stock ownership or options, expert testimony, grants, patents received or pending, or royalties.
References
1. Uson Junior PLS, Bogenberger J, Borad MJ. Advances in the treatment of biliary tract cancers. Curr Opin Gastroenterol. 2020;36(2):85–89. doi:10.1097/MOG.0000000000000606
2. Saha SK, Zhu AX, Fuchs CS, et al. Forty-year trends in cholangiocarcinoma incidence in the US: intrahepatic disease on the rise. Oncologist. 2016;21(5):594–599. doi:10.1634/theoncologist.2015-0446
3. Sung H, Ferlay J, Siegel RL, et al. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2021;71(3):209–249. doi:10.3322/caac.21660
4. D-Y O, He AR, Qin S, et al. Durvalumab plus gemcitabine and cisplatin in advanced biliary tract cancer. NEJM Evidence. 2022;1:EVIDoa2200015.
5. Lamarca A, Palmer DH, Wasan HS, et al. Second-line FOLFOX chemotherapy versus active symptom control for advanced biliary tract cancer (ABC-06): a Phase 3, open-label, randomised, controlled trial. Lancet Oncol. 2021;22(5):690–701. doi:10.1016/S1470-2045(21)00027-9
6. Yoo C, Kim K-P, Jeong JH, et al. Liposomal irinotecan plus fluorouracil and leucovorin versus fluorouracil and leucovorin for metastatic biliary tract cancer after progression on gemcitabine plus cisplatin (NIFTY): a multicentre, open-label, randomised, phase 2b study. Lancet Oncol. 2021;22(11):1560–1572. doi:10.1016/S1470-2045(21)00486-1
7. Uson Junior PLS, Borad MJ. Precision approaches for cholangiocarcinoma: progress in clinical trials and beyond. Exp Op Investig Drugs. 2022;31(1):125–131. doi:10.1080/13543784.2022.2017882
8. Berchuck JE, Facchinetti F, DiToro DF, et al. The clinical landscape of cell-free DNA alterations in 1671 patients with advanced biliary tract cancer. Annals of Oncology. 2022;33(12):1269–1283. doi:10.1016/j.annonc.2022.09.150
9. Golub D, Iyengar N, Dogra S, et al. Mutant isocitrate dehydrogenase inhibitors as targeted cancer therapeutics. Front Oncol. 2019;9:417. doi:10.3389/fonc.2019.00417
10. Dang L, Yen K, Attar EC. IDH mutations in cancer and progress toward development of targeted therapeutics. Ann Oncol. 2016;27(4):599–608. doi:10.1093/annonc/mdw013
11. Cairns RA, Harris IS, Mak TW, et al. Cancer cell metabolism. In: Cold Spring Harbor Symposia on Quantitative Biology. Vol. 76. Cold Spring Harbor Laboratory Press; 2011:85–95.
12. Dang L, White DW, Gross S, et al. Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature. 2009;462(7274):739–744. doi:10.1038/nature08617
13. Bleeker FE, Atai NA, Lamba S, et al. The prognostic IDH1 R132 mutation is associated with reduced NADP+-dependent IDH activity in glioblastoma. Acta Neuropathol. 2010;119(4):487–494. doi:10.1007/s00401-010-0645-6
14. Chen T, Wu G, Hu H, et al. Enhanced fatty acid oxidation mediated by CPT1C promotes gastric cancer progression. J Gastrointest Oncol. 2020;11(4):695. doi:10.21037/jgo-20-157
15. Lowery MA, Burris HA 3rd, Janku F, et al. Safety and activity of ivosidenib in patients with IDH1-mutant advanced cholangiocarcinoma: a Phase 1 study. Lancet Gastroenterol Hepatol. 2019;4(9):711–720. doi:10.1016/S2468-1253(19)30189-X
16. Zhu AX, Macarulla T, Javle MM, et al. Final overall survival efficacy results of ivosidenib for patients with advanced cholangiocarcinoma with IDH1 mutation. JAMA Oncology. 2021;7(11):1669–1677. doi:10.1001/jamaoncol.2021.3836
17. Abou-Alfa GK, Macarulla T, Javle MM, et al. Ivosidenib in IDH1-mutant, chemotherapy-refractory cholangiocarcinoma (ClarIDHy): a multicentre, randomised, double-blind, placebo-controlled, phase 3 study. Lance Oncol t. 2020;21(6):796–807. doi:10.1016/S1470-2045(20)30157-1
18. Robins JM, Tsiatis AA. Correcting for non-compliance in randomized trials using rank preserving structural failure time models. Commun Stat. 1991;20(8):2609–2631. doi:10.1080/03610929108830654
19. Cleary JM, Rouaisnel B, Daina A, et al. Secondary IDH1 resistance mutations and oncogenic IDH2 mutations cause acquired resistance to ivosidenib in cholangiocarcinoma. NPJ Prec Oncol. 2022;6(1):1–8.
20. Harding JJ, Lowery MA, Shih AH, et al. Isoform switching as a mechanism of acquired resistance to mutant isocitrate dehydrogenase inhibition. Cancer Discov. 2018;8(12):1540–1547. doi:10.1158/2159-8290.CD-18-0877
21. Reinbold HIC, Rabe P, Rabe P, et al. Resistance to the isocitrate dehydrogenase 1 mutant inhibitor ivosidenib can be overcome by alternative dimer-interface binding inhibitors. Nat Comm. 2022;13(1):1–12. doi:10.1038/s41467-022-32436-4
22. Vogel A, Bridgewater J, Edeline J, et al. Biliary tract cancer: ESMO clinical practice guideline for diagnosis, treatment and follow-up☆. Annals of Oncology. 2023;34(2):127–140. doi:10.1016/j.annonc.2022.10.506
23. Junior U, Luiz Serrano P, Bogenberger JM, et al. ”FGFR2-IIIb expression by immunohistochemistry has high specificity in cholangiocarcinoma with FGFR2 genomic alterations. Dig Dis Sci. 2022;67(8):3797–3805. doi:10.1007/s10620-021-07303-9
24. Zarei M, Hajihassani O, Hue JJ, et al. Wild-type IDH1 inhibition enhances chemotherapy response in melanoma. J Exp Clin Cancer Res. 2022;41(1):1–18. doi:10.1186/s13046-022-02489-w
25. Zarei M, Hajihassani O, Hue JJ, et al. Targeting wild-type IDH1 enhances chemosensitivity in pancreatic cancer. bioRxiv. 2023;2023:3.
 © 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.